

Abstract
Neuropathic pain is a debilitating condition for which the development of effective treatments has been limited by an incomplete understanding of its molecular basis. The cationic current Ih mediated by hyperpolarization-activated cyclic nucleotide-gated (HCN) channels plays an important role in pain by facilitating ectopic firing and hyperexcitability in DRG neurons, however little is known regarding the role of Ih in supraspinal pain pathways. The medial prefrontal cortex (mPFC), which is reported to be involved in the affective aspects of pain, exhibits high HCN channel expression. Using the spared nerve injury (SNI) model of neuropathic pain in Long–Evans rats and patch-clamp recordings in layer II/III pyramidal neurons of the contralateral mPFC, we observed a hyperpolarizing shift in the voltage-dependent activation of Ih in SNI neurons, whereas maximal Ih remained unchanged. Accordingly, SNI mPFC pyramidal neurons exhibited increased input resistance and excitability, as well as facilitated glutamatergic mGluR5-mediated persistent firing, compared with sham neurons. Moreover, intracellular application of bromo-cAMP abolished the hyperpolarizing shift in the voltage-dependent activation of Ih observed in SNI neurons, whereas protein kinase A (PKA) inhibition further promoted this shift in both SNI and sham neurons. Behaviorally, acute HCN channel blockade by local injection of ZD7288 in the mPFC of SNI rats induced a decrease in cold allodynia. These findings suggest that changes in the cAMP/PKA axis in mPFC neurons underlie alterations to HCN channel function, which can influence descending inhibition of pain pathways in neuropathic conditions.
SIGNIFICANCE STATEMENT Recent studies investigating the role of the medial prefrontal cortex (mPFC) in neuropathic pain have led to an increased awareness of how affective and cognitive factors can influence pain perception. It is therefore imperative that we advance our understanding of the involvement of supraspinal pain pathways. Our electrophysiological and behavioral results support an important role for hyperpolarization-activated cyclic nucleotide-gated channels and the cAMP/protein kinase A signaling axis in promoting hyperexcitability and persistent firing in pyramidal neurons of the mPFC in neuropathic animals. These findings offer novel insights, with potential therapeutic implications, into pathophysiological mechanisms underlying the abnormal contribution of layer II/III prefrontal pyramidal neurons to chronic pain states.
Keywords: anterior cingulate cortex, hyperexcitability, allodynia, neuropathic pain, persistent firing
Introduction
Neuropathic pain is a debilitating condition resulting from disease or trauma to the nervous system. Patients suffering from this chronic pain state often experience hypersensitivity to peripheral stimuli and spontaneous pain that can persist for prolonged periods of time after the original injury (Zhuo, 2007; Sandkühler, 2009). These long-term nociceptive effects are believed to stem from maladaptive synaptic changes that occur in peripheral nerve endings, spinal cord, and cortical regions involved in the affective aspects of pain, including the prefrontal cortex, anterior cingulate cortex (ACC), and amygdala (Zhuo, 2004; Costigan et al., 2009).
The medial prefrontal cortex (mPFC) plays a vital role in cognitive and emotional functions including working memory and attention (Arnsten and Li, 2005; Zhang et al., 2013). Moreover, prefrontal subregions have been shown to be involved in pain processing. For example, the ACC subdivision of the mPFC mediates the affective components of pain responses and is involved in pain-related learning associated with prediction and avoidance of noxious stimuli (Malin et al., 2007). Neuroimaging studies in humans have demonstrated that the ACC is the most consistently activated structure during pain stimulation (Rainville et al., 1997; Apkarian et al., 2005). Accordingly, electrophysiological studies have reported that neurons in the ACC respond to noxious stimuli (Sikes and Vogt, 1992; Hutchison et al., 1999), exhibit enhanced spike frequency and intrinsic excitability after peripheral nerve injury (Metz et al., 2009; Gong et al., 2010; Blom et al., 2014), and can display synaptic long-term potentiation in rats following digit amputation (Wei and Zhuo, 2001). Therefore, strengthening of synaptic transmission and connections in supraspinal regions such as the ACC may underlie persistent pain (Zhuo, 2004).
Hyperpolarization-activated and cyclic nucleotide-gated (HCN) channels play an important role in the pathogenesis of neuropathic pain by facilitating spontaneous ectopic firing and hyperexcitability in DRG neurons (Chaplan et al., 2003; Luo et al., 2007). This ectopic activity is an important feature of neuropathic pain because it is believed to underlie the development of spontaneous pain and allodynia. Studies have revealed that blocking the cationic current mediated by HCN channels (Ih) with ZD7288 in different rat models of neuropathic pain provides significant analgesic effects and abolishes spontaneous discharges from injured nerve fibers (Chaplan et al., 2003; Jiang et al., 2008b). Conversely, it has also been reported that blockade of Ih in nodose sensory neurons with CsCl causes an increase in input resistance and neuronal firing (Doan et al., 2004). Hence, it remains unclear whether HCN channels contribute to pain hyperexcitability via enhanced cationic currents or increased input resistance. Furthermore, little is known regarding the role of Ih in supraspinal pain pathways, particularly in the mPFC, which exhibits high HCN channel expression. Therefore, the general aim of this study was to provide insights into the involvement of cortical HCN channels in pain processing. We investigated the cellular physiology of prefrontal pyramidal neurons during neuropathic pain conditions using the rat spared nerve injury (SNI) model and whole-cell patch-clamp recordings in layer II/III of the mPFC.
Materials and Methods
SNI model of neuropathic pain.
All experimental procedures were approved by the McGill University Animal Care Committee and were in compliance with the guidelines of the Canadian Council on Animal Care. The SNI surgery was performed on male Long–Evans (LE) rats 19–21 d of age (Charles River Canada). The animals were anesthetized with 1–3% isoflurane and 100% O2. To induce the SNI neuropathy, an incision was made through the lateral surface of the left thigh to expose the sciatic nerve at the level of the trifurcation into its three terminal branches: the common peroneal, tibial, and sural nerves. The common peroneal and tibial nerves were tightly ligated and severed, whereas the sural nerve was left intact. A second group of rats was used for the control sham surgery. In these rats, the left sciatic trifurcation was exposed but was not further manipulated (Decosterd and Woolf, 2000). This model of neuropathic pain allows for behavioral testing of noninjured skin territories by restricting the association of distal intact axons with degenerating axons. All experiments were performed on animals 3 weeks after surgery. However, to assess early and/or long-term changes associated with peripheral nerve injury, animals at 1 week and 5 weeks after surgery were also used.
Behavioral tests.
Pain-related behavior was assessed using the von Frey, acetone, and Hargreaves' tests on naive, sham-operated, and SNI LE rats 3 weeks after surgery. The behavioral tests were performed on the lateral plantar surface of the left (ipsilateral to nerve injury) and right (contralateral) hindpaw, the skin territory innervated by the intact sural nerve. Before any behavioral testing, animals were habituated to the testing environment for ∼45 min.
Mechanical allodynia was measured using a series of calibrated von Frey filaments (Stoelting) ranging from 0.6 to 26 g. Animals were placed in plastic cages situated over a wire-mesh floor. The von Frey filaments were applied in order of increasing stiffness to the lateral plantar surface of the hindpaw through the mesh floor. Each filament was applied to the foot until it bent. The time interval between consecutive filament applications was 5 s. The threshold was taken as the lowest force that evoked a brisk withdrawal response to one of three repetitive stimuli and not related to stepping (Tal and Bennett, 1994).
Cold allodynia was assessed using the acetone drop method (Choi et al., 1994). In the same test environment described above, a 40 μl droplet of acetone was applied to the lateral plantar region of the hindpaw using a transfer pipette. Responses within the first 20 s were scored as follows: 0 = no response, 1 = one rapid hindpaw flick/stamp, 2 = prolonged hindpaw withdrawal or repeated flicks/stamps, and 3 = periods of flicking/stamping with licking of the plantar hindpaw and/or vocalizations (Flatters and Bennett, 2004). Acetone application was repeated 3 times for each hindpaw, with a 5 min interval between each application. For each rat, the mean of the three scores for each hindpaw was calculated for data analysis.
Thermal hyperalgesia was assessed by measuring the pain threshold (withdrawal latency) in response to high temperature using the plantar test (Hargreaves Apparatus Type 7370; UgoBasile). Rats were placed into individual plastic cages over a glass floor 5 min before the experiment. A noxious thermal stimulus was focused through the glass onto the lateral plantar surface of the hindpaw until the animal briskly lifted the hindpaw away from the heat source. A cutoff latency of 20 s was used to avoid tissue damage. The heat stimulation was repeated 3 times for each hindpaw, at 5 min intervals, and the mean was calculated for data analysis.
Brain slice preparation.
Acute coronal brain slices for electrophysiological recordings were obtained from sham-operated and SNI male LE rats 3 weeks after surgery according to a procedure described previously (Zhang and Séguéla, 2010). Briefly, animals were deeply anesthetized with a ketamine:xylazine mixture (60:5 mg/kg) and perfused transcardially with ice-cold choline chloride-based artificial CSF (cutting solution) consisting of the following (in mm): 110 choline-Cl, 1.25 NaH2PO4, 25 NaHCO3, 7 MgCl2, 0.5 CaCl2, 2.5 KCl, 7 glucose, 3 pyruvic acid, and 1.3 ascorbic acid bubbled with carbogen (O2 95%, CO2 5%). Brain slices (300 μm) containing the mPFC including the rostral dorsal ACC and prelimbic subdivisions (bregma 3.70 ∼ 1.70 mm in rats, Paxinos and Watson, 1998) were obtained using a vibratome (VT1000S; Leica) and allowed to recover for at least 1 h at room temperature in standard artificial CSF (extracellular solution, see below).
Electrophysiological recordings.
Individual brain slices were placed in a recording chamber mounted on the stage of an upright microscope Axioskop (Zeiss) equipped with a 63× water-immersion objective and differential contrast optics and constantly perfused with oxygenated (95% O2, 5% CO2) standard extracellular solution. The standard extracellular solution for voltage-clamp and current-clamp experiments contained the following (in mm): 125 NaCl, 2.5 KCl, 1.6 CaCl2, 2 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, 3 pyruvic acid, 1.3 ascorbic acid, and 10 glucose, pH 7.4. In voltage-clamp recordings investigating Ih, BaCl2 (200 μm) was routinely added to the extracellular solution to block K+ conductances. In current-clamp recordings, kynurenic acid (2 mm) and picrotoxin (100 μm) were routinely added to the extracellular solution to block ionotropic synaptic transmission mediated by glutamate and GABAA receptors, respectively, to assess persistent firing.
A near-infrared CCD camera (XC-75; Sony) was used to visualize the neurons. Slice perfusion was driven by gravity at a speed of 1 ml/min. The temperature of the perfusion solution was maintained at 32–33°C using a temperature controller (TC-324B; Warner Instruments). Brain slices were stabilized using a U-shaped stainless steel anchor with Lycra threads spaced 1.5 mm apart (Warner Instruments). Superficial layer II/III principal neurons with a typical pyramidal shape from the contralateral mPFC were chosen for electrophysiological recordings. Patch pipettes (5–9 MΩ) were pulled on a Brown Flaming puller (Model P-97; Sutter Instruments) using borosilicate glass electrodes and filled with the following (in mm): 120 K-gluconate, 20 KCl, 2 MgCl2, 0.2 EGTA, 10 HEPES, 7 di-tris phosphocreatine, 4 Na2-ATP, and 0.3 Na2-GTP pH adjusted to 7.3 with KOH. Tight seals (∼2–3 GΩ) were obtained by applying constant negative pressure on the patch pipette. Electrical signals were amplified using an Axopatch 200B amplifier (Molecular Devices), low-pass Bessel filtered at 10 kHz, sampled at 20 kHz via a Digidata 1322A interface (Molecular Devices), and stored on a Pentium computer using pClamp version 9.2.1.8 software (Molecular Devices) for offline analysis.
In the present study, all cells were quiescent at rest and had a resting membrane potential ranging from −60 to −80 mV. BaCl2 (200 μm) was added to the extracellular solution to inhibit inwardly rectifying K+ currents while recording the Ih mediated by HCN channels in voltage-clamp experiments. To determine the voltage dependence of Ih activation, whole-cell patch-clamp recordings were performed in voltage-clamp mode on superficial layer II/III pyramidal neurons. Membrane potential was varied by a series of hyperpolarizing voltage steps (duration of 2.5 s) decreasing in increments of −10 mV from a holding potential of −50 mV to a final voltage of −130 mV. For bromo-cAMP experiments, the membrane potential was held at −40 mV and hyperpolarizing voltage steps ranging from −40 to −130 mV were applied. Series resistance was compensated (40–90%). The current amplitudes were determined as the difference in current at the beginning of the voltage step, immediately after the capacitive transient, and at the end of the step. The amplitude and density of Ih were assessed from the −70 mV voltage step (physiologically relevant potential), whereas the maximal available current was determined from the −130 mV step. The tail currents were measured upon return to a fixed potential (−130 mV for 1.5 s) after the final voltage step to remove the effect of the driving force. Tail currents were normalized to the maximal tail current and plotted as a function of the corresponding voltage step to determine the fractional activation of Ih (i.e., activation curve) (Eq. 1). Data points were fitted to the Boltzmann function using Clampfit version 9.2 (Molecular Devices) to determine the half-activation potential (V1/2) (Eq. 2).
 |
Neuronal excitability was assessed using the input–output curve measured from the changes in membrane potential (presence of action potentials) evoked by current steps (0–140 pA, increasing in increments of 10 pA) in current-clamp mode. Input resistance was determined by injecting a negative current pulse (−300 pA), followed by subsequent 20 pA increments in current-clamp mode. The membrane input resistance value was measured from the current pulse that brought the membrane potential to ∼−70 mV. During the current-clamp recordings, the holding current was adjusted to obtain a holding membrane potential of −60 mV.
To investigate the influence of the changes in Ih on excitatory synaptic activity in the mPFC, miniature EPSCs (mEPSCs) were recorded in voltage-clamp mode in the presence of picrotoxin (100 μm) and tetrodotoxin (TTX, 1 μm) from sham and SNI pyramidal neurons before and after bath application of ZD7288 (15 μm). The membrane potential was held at −70 mV and series resistance was compensated (40–70%). For mEPSC analysis, 200 successive events were randomly selected and analyzed using the MiniAnalysis Program (Synaptosoft). The current threshold for event detection was set at 6 pA.
To determine whether SNI or sham-operated neurons display intrinsic persistent firing, a nonsynaptic cellular substrate for working memory and attention in the mPFC (Haj-Dahmane and Andrade, 1998; Sidiropoulou et al., 2009; Zhang and Séguéla, 2010; Zhang et al., 2013), current-clamp gap-free recordings were performed on mPFC pyramidal neurons in the presence of kynurenic acid (2 mm) and picrotoxin (100 μm). After obtaining whole-cell patch and adjusting the holding current to a membrane potential of −60 mV, a depolarizing current pulse (100 pA for 1 s) was applied before drug administration to induce repetitive spiking during the current pulse (control). The group I mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG) was then applied to determine whether long-lasting sustained firing (persistent firing), which outlasts the stimulation pulse, could be induced after the depolarizing pulse (Zhang and Seguela, 2010). The firing frequency is defined as the average spiking frequency within 20 s after the depolarizing current pulse. The plateau potential refers to the average membrane potential during long-lasting persistent firing or during short (<20 s) sustained firing [afterdepolarizing potentials (ADPs) with superimposed action potentials]. The amplitude of plateau potentials reflects the difference between the mean membrane potential (in millivolts) measured at baseline before the pulse and the mean membrane potential measured during the steady-state phase of persistent neuronal firing (excluding action potentials and ADPs).
Immunohistochemistry and imaging.
Three weeks after surgery, rats were deeply anesthetized with a ketamine:xylazine mixture (60:5 mg/kg, i.p.) injected with 0.5 ml of heparin (11.2 U/ml) and perfused transcardially with 100 ml of PBS followed by 500 ml of 4% PFA solution per rat. Cortical tissue was extracted from perfused sham (n = 4) and SNI (n = 4) rats and postfixed in 4% PFA overnight at 4°C. Brains were then cryoprotected in 30% sucrose in PBS at 4°C for 2 d. Coronal sections of 40 μm containing the mPFC were cut using a cryostat, collected as free-floating in PBS, and permeabilized with PBS plus 0.2% Triton X-100 (PBS-T).
For immunofluorescence labeling of HCN channels, sections were first incubated for 1 h at room temperature in 10% normal donkey serum (Jackson Laboratories) in PBS-T to block unspecific labeling and subsequently incubated overnight at 4°C with a rabbit HCN2 antibody (1:200; Alomone Labs). Sections were then incubated for 2 h at room temperature with secondary donkey anti-rabbit IgGs conjugated to Alexa Fluor 488 (1:800) in PBS-T (Invitrogen). Negative controls consisted of immunofluorescence labeling after preincubation of the primary antibodies in presence of the HCN2 peptide antigen (preadsorption) and in the absence of primary antibodies.
Digital images were captured with a high-resolution CCD camera attached to a Zeiss Axioplan 2 imaging fluorescence microscope connected to a computer running Zeiss Axiovision version 4.8 software. Three coronal sections containing the mPFC were used to acquire images for each rat. Images were exported in TIFF for analysis using ImageJ version 1.47. Fluorescence density was measured as the mean gray value within the selection area (layer I and layer II/III of the mPFC), which reflects the sum of the gray values of all the pixels in the selection divided by the total number of pixels.
Stereotaxic surgery and acute cortical infusions.
Approximately 3–4 weeks after sham or SNI surgery (to provide sufficient recovery time), animals were deeply anesthetized (1–3% isoflurane in pure O2) and implanted with a guide cannula (26 gauge; Plastics One). Cannulas were implanted into the contralateral mPFC using the following stereotaxic coordinates: +2.9 mm from bregma (anteroposterior), −1 mm from midline (mediolateral), +4.1 mm from skull surface (dorsoventral), and the mediolateral angle set at 11°. Cortical infusions of saline (0.5 μl) or ZD7288 (0.2 μg/μl in 0.5 μl of saline) into the prefrontal cortex of sham and SNI rats were performed using a syringe pump (Harvard Apparatus) and a 50 μl Hamilton syringe on freely moving animals. The internal cannula (33 gauge; Plastics One) was attached to a connector assembly to hold it in place within the guide cannula during in vivo acute infusions.
The acetone test (cold sensitivity/allodynia) and von Frey test (mechanical sensitivity/allodynia) were used to determine the behavioral significance of inhibiting Ih in the mPFC in vivo. Before any behavioral testing, animals were habituated to the testing environment for ∼45 min. Pain-related behavior was assessed at baseline (before any drug infusion) and 10–15 min and 30–40 min after infusion of ZD7288 or saline in sham and SNI rats.
Drugs and solutions.
All reagents were purchased from Sigma-Aldrich except ZD7288, which was purchased from Tocris Bioscience; TTX, which was purchased from Alomone Labs; and H89, which was purchased from ApexBio Technology. DHPG and ZD7288 were dissolved in water and freshly diluted from stock to the desired concentrations before the experiments. The compound 8-bromoadenosine 3′,5′-cyclic monophosphate (bromo-cAMP) was dissolved in intracellular solution. The H89 compound was dissolved in 50% DMSO and then aliquoted into intracellular patch solution to obtain a final DMSO concentration of 0.1% and final H89 concentration of 5 μm.
Data analysis.
Electrophysiological data were analyzed using SigmaStat version 3.1, GraphPad Prism version 5.0, and Clampfit 9.2 (Molecular Devices) software. Values are expressed as means ± SEM. Two-sample unpaired t tests were used for comparison between two independent groups of neurons (sham-operated vs SNI), whereas two-sample paired t tests were used for comparison between two dependent groups. The Mann–Whitney rank-sum test was used when the normality test failed using SigmaStat. One-way ANOVA with Bonferroni correction was used for comparison of multiple independent groups. Kruskal–Wallis one-way ANOVA with Dunn's multiple-comparison test was used when the normality test failed. Two-way ANOVA with Bonferroni post-test was used for comparison of two independent variables each with multiple conditions. Differences were considered statistically significant when p < 0.05.
Results
Increased sensitivity of SNI LE rats to mechanical and cold stimulation
The SNI model of neuropathic pain has been well characterized and shown to consistently induce prolonged and robust sensory hypersensitivity in rats (Decosterd and Woolf, 2000). However, it has been reported that different strains of rats may develop and express different degrees of neuropathic pain behaviors (Yoon et al., 1999; Mills et al., 2001). For example, Yoon et al. (1999) demonstrated that LE rats display attenuated pain behaviors after peripheral nerve injury compared with other strains. For this reason, we assessed pain-related behavior in sham-operated and SNI LE rats 3 weeks after surgery to confirm the development of neuropathic pain. All SNI rats developed qualitative signs suggestive of sensory hypersensitivity of the hindpaw ipsilateral to the nerve injury, such as avoiding bearing weight on the afflicted paw and holding the paw in a flexed protective position. Quantitatively, SNI rats exhibited strong allodynia indicated by a significant increase in mechanical sensitivity (sham: 21.87 ± 2.01 g, n = 8; SNI: 4.85 ± 1.12 g, n = 8; p = 0.0008; Fig. 1A) and cold responsiveness (sham: 0.33 ± 0.12, n = 8; SNI: 1.41 ± 0.29, n = 8; p = 0.0044; Fig. 1B) in the ipsilateral (left) hindpaw compared with sham-operated and naive rats. However, in naive, sham-operated, and SNI animals, the withdrawal response latency to heat stimulation in the Hargreaves' test was not significantly modified (sham: 6.35 ± 0.56 s, n = 8; SNI: 5.06 ± 0.50 s, n = 8; p = 0.1108; Fig. 1C). Behavioral tests were also performed on the contralateral (right) hindpaw. No significant differences in sensitivity to mechanical and thermal stimulation were observed between the three groups of animals (Fig. 1A–C). These behavioral data are consistent with the findings initially reported by Decosterd and Woolf (2000).
Figure 1.

LE rats with peripheral nerve injury display nocifensive behaviors consistent with chronic neuropathic pain. A, Animals with SNI displayed a significantly decreased withdrawal threshold to mechanical stimulation using Von Frey filaments in the left (ipsilateral) hindpaw 3 weeks after surgery compared with sham-operated and naive rats. No significant difference was observed in mechanical sensitivity in the right (contralateral) hindpaw between the three groups. B, SNI rats also displayed increased cold sensitivity using the acetone test in the left hindpaw 3 weeks after surgery. No significant difference was observed in the right paw between the three groups. C, Withdrawal response latency to heat stimulation in the Hargreaves' test was not significantly different in SNI compared with sham and naive rats in both the left and right hindpaws. n = 8 in all groups. Values are mean ± SEM. ***p < 0.001; **p < 0.01.
Alteration in Ih in layer II/III pyramidal neurons of mPFC in SNI animals
Although the cationic current mediated by HCN channels, Ih, has been implicated in peripheral pain pathways (Chaplan et al., 2003; Yao et al., 2003; Doan et al., 2004; Emery et al., 2011), the exact role of Ih in supraspinal pain regions during neuropathic pain remains unclear. We investigated whether Ih is altered between sham and SNI conditions using whole-cell patch-clamp recordings in layer II/III pyramidal neurons of the contralateral mPFC (Fig. 2A,B). In the presence of Ba2+ to inhibit IKir, all pyramidal neurons tested in both groups (n = 21 sham; n = 31 SNI) expressed Ih currents in response to a series of hyperpolarizing voltage steps, with typical voltage- and time-dependent activation (Fig. 2C). Furthermore, bath application of the selective HCN channel antagonist ZD7288 (15 μm) for 15 min resulted in a 86.55 ± 5.84% and 88.23 ± 5.37% reduction in Ih from sham (n = 9) and SNI (n = 7) neurons, respectively (Fig. 2D,E). While Ih is robustly expressed in PFC cells from both sham and SNI animals, it is possible that the maximal available current may be altered after SNI injury. To test this, we assessed Ih from the −130 mV step and did not observe any significant difference in current amplitude (sham: 163.55 ± 14.77 pA, n = 21; SNI: 173.22 ± 12.21 pA, n = 30; p = 0.615; Fig. 2E, left) or density (sham: 11.7 ± 1.07 pA/pF, n = 21; SNI: 12.82 ± 1.13 pA/pF, n = 27; p = 0.489; Fig. 2E, right) between sham and SNI pyramidal neurons. These findings suggest that the HCN channel density at the surface of sham and SNI pyramidal neurons is comparable because Ih is maximal at membrane potentials negative to −110 mV (Pape, 1996). Consistently, using immunofluorescence for localization of HCN2 subunits in the mPFC, we did not observe any significant difference in expression levels between sham and SNI conditions (layer II/III density = sham: 33.40 ± 2.35, n = 4; SNI: 33.31 ± 2.10, n = 4; p = 0.9773; layer I density = sham: 18.30 ± 1.44, n = 4; SNI: 19.02 ± 1.65, n = 4; p = 0.7555). However, when Ih was measured at −70 mV, we observed a significant decrease in current amplitude (sham: 30.41 ± 4.14 pA, n = 21; SNI: 20.39 ± 1.88 pA, n = 31; p = 0.022) and current density (sham: 2.21 ± 0.32 pA/pF, n = 21; SNI: 1.52 ± 0.16 pA/pF, n = 28; p = 0.048) in pyramidal neurons from SNI animals compared with sham (Fig. 2F).
Figure 2.
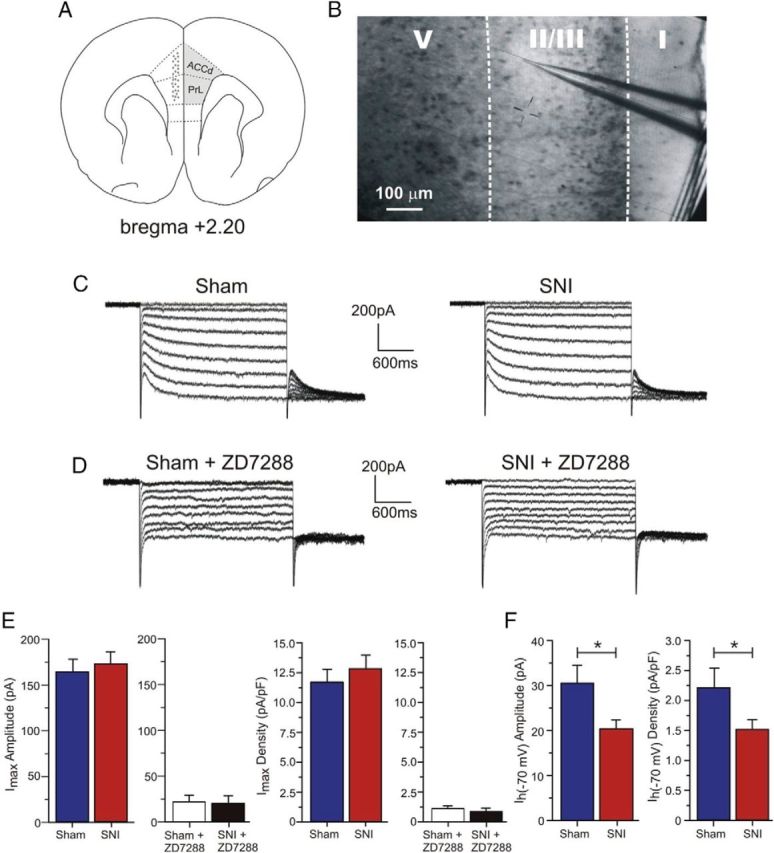
Maximal Ih is not significantly different in layer II/III mPFC pyramidal neurons between sham and SNI animals, but is reduced in SNI neurons at more physiological membrane potentials. A, Schematic diagram of a coronal section of rat brain showing the recorded area (gray dots) in the mPFC, including the rostral dorsal ACC (ACCd) and prelimbic cortex (PrL), contralateral to the site of nerve injury. Adapted from Paxinos and Watson (1998). B, Typical video image of an acute brain slice showing the patch electrode in layer II/III of the mPFC. C, Sample voltage-clamp traces illustrating Ih recordings in sham and SNI neurons. D, Addition of the HCN channel blocker ZD7288 (15 μm) to the bath solution for 15 min abolished Ih. E, Quantitative results illustrating the absence of significant difference in maximal Ih amplitude (n = 21 for sham; n = 30 for SNI) and density (n = 21 for sham; n = 27 for SNI) between sham-operated and SNI neurons measured at −130 mV. Application of ZD7288 abolished Ih measured in current amplitude (n = 9 for sham; n = 7 for SNI) and current density (n = 7 for sham; n = 6 for SNI). F, Significant decrease in current amplitude and density at −70 mV in SNI (n = 31 and 28) compared with sham neurons (n = 21). Values are mean ± SEM. *p < 0.05.
Hyperpolarizing shift in voltage dependence of HCN channel activation in SNI neurons
The decreased Ih measured at −70 mV in SNI prefrontal pyramidal neurons may be due to a reduction in open channel probability. To address this possibility, we investigated the voltage dependence of HCN channel activation in sham and SNI pyramidal neurons from the contralateral mPFC. The activation curve of Ih in both sham and SNI conditions was estimated by analyzing the tail currents evoked at a fixed potential of −130 mV (Fig. 2C). We observed a negative shift in the activation curve of Ih in SNI compared with sham neurons, exemplified by a significant −7.3 mV shift in the V1/2 in SNI prefrontal pyramidal cells (sham V1/2: −85.4 ± 0.75 mV, n = 21; SNI V1/2: −92.7 ± 0.71 mV, n = 31; p < 0.0001; Fig. 3A). This hyperpolarizing shift in the voltage dependence of Ih activation is indicative of a reduction in open channel probability of HCN channels in SNI conditions. Moreover, bath application of ZD7288 (15 μm) fully abolished this shift and overall channel availability in both sham (n = 9) and SNI conditions (n = 7), suggesting that the negative shift in open channel probability is mediated entirely by Ih (Fig. 3B). In addition, we performed similar experiments on SNI animals at 1 week and 5 weeks after nerve injury to investigate early and long-term changes in HCN channel properties. We observed no alterations in Ih voltage dependence at the 1 week post-SNI time point (sham V1/2: −89.36 ± 0.59 mV, n = 8; SNI V1/2: −90.24 ± 0.51 mV, n = 9; p = 0.2798). However, a negative shift in Ih voltage dependence (sham V1/2: −85.47 ± 0.90 mV, n = 8; SNI V1/2: −90.65 ± 0.71 mV, n = 7; p = 0.0007) and an increase in input resistance and excitability were observed at the 5 weeks post-SNI time point (data not shown), similar to the changes observed in SNI animals 3 weeks after peripheral nerve injury. These findings indicate that the plastic changes in HCN channel function that we observed at 3 weeks after nerve injury are maintained over long periods and contribute to the chronic neuropathic phenotype.
Figure 3.

SNI pyramidal neurons from the contralateral mPFC display a nerve injury-associated hyperpolarizing shift in the voltage dependence of HCN channels. A, Left, Analysis of tail currents from voltage-clamp recordings revealed a hyperpolarizing shift in the voltage dependence of HCN channel activation in SNI (n = 31) compared with sham neurons (n = 21), indicating a reduction in open channel probability in neuropathic conditions. Activation curves were fitted according to the Boltzmann equation to normalized and averaged tail currents. Right, V1/2 was significantly shifted −7.3 mV in SNI pyramidal neurons. B, Application of the HCN channel blocker ZD7288 (15 μm) abolished the shift between sham (n = 9) and SNI neurons (n = 7) and reduced the overall percentage of channel availability in both conditions. Values are mean ± SEM. ***p < 0.001; *p < 0.05.
Increased excitability in contralateral SNI pyramidal neurons
HCN channels are versatile regulators of neuronal excitability in the neocortex (Day et al., 2005; Wang et al., 2007; Zhang et al., 2013); thus, it is possible that the observed hyperpolarizing shift in Ih activation may affect the intrinsic excitability of prefrontal pyramidal neurons. We investigated this possibility by measuring the number of action potentials evoked by different current intensities (input–output curve) in sham and SNI neurons. We observed a significant increase in the number of action potentials elicited for current intensities ranging from 60 to 140 pA in SNI compared with sham layer II/III pyramidal neurons, indicating that neuronal excitability is enhanced in SNI conditions (sham: 6.62 ± 0.51 at 80 pA intensity, n = 29; SNI: 9.04 ± 0.66 at 80 pA intensity, n = 23; p = 0.005; Fig. 4A,B). Interestingly, we did not observe a significant change in the resting membrane potential of prefrontal pyramidal neurons after SNI (sham: −63.43 ± 1.74 mV, n = 14; SNI: −65.84 ± 1.17 mV, n = 19; p = 0.2417; Fig. 4C). Lastly, bath application of ZD7288 (15 μm) further elevated neuronal spiking activity in response to varying current intensities in both sham (n = 9) and SNI pyramidal neurons (n = 8), but a significant difference in neuronal responsiveness was no longer observed between groups (data not shown). This suggests that the increase in neuronal excitability observed in SNI conditions mainly stems from reduced HCN channel availability.
Figure 4.

Neuronal excitability is increased in SNI pyramidal neurons. A, Typical current-clamp traces (0 to 140 pA) illustrating increased action potential firing for any given current intensity in SNI compared with sham pyramidal neurons. Membrane potential was held at −60 mV. B, Quantitative data plotted as an input–output curve showing an increase in responsiveness in the SNI (n = 23) compared with the sham condition (n = 29). C, No significant difference was observed in the resting membrane potential of mPFC pyramidal neurons from sham (n = 14) and SNI (n = 19) animals. Values are mean ± SEM. **p < 0.01; *p < 0.05.
Increased membrane input resistance in contralateral SNI pyramidal neurons
It has been reported that blocking HCN channels enhances the ability of excitatory synaptic connections to generate action potentials by increasing membrane input resistance, which essentially makes the cell less leaky and less likely to shunt EPSPs (Doan et al., 2004; Wang et al., 2007). In the absence of Ih, any given positive input current would evoke a greater depolarizing change in the membrane potential (Jiang et al., 2008a; Wahl-Schott and Biel, 2009). To determine whether the observed increase in neuronal excitability in the SNI condition is accompanied by a change in membrane input resistance (Rin), we applied a series of hyperpolarizing current pulses. Rin was determined from the current pulse that brought the membrane potential to −70 mV. Interestingly, we observed a significant increase in the steady-state Rin in SNI neurons compared with sham neurons (sham: 166.6 ± 10.9 MΩ, n = 11; SNI: 226.1 ± 16.1 MΩ, n = 12; p = 0.0069; Fig. 5C). Moreover, the current pulse-evoked depolarizing membrane potential sag, which is a characteristic feature of HCN channel activation, was present in both sham and SNI conditions but appeared larger in the latter, which is consistent with an increase in Rin in SNI neurons (Fig. 5A,B). Bath application of ZD7288 (15 μm) completely eliminated the current pulse-evoked depolarizing sag (Fig. 5D,E) and increased the Rin in both sham and SNI pyramidal neurons to similar values (sham: 273.7 ± 21.7 MΩ, n = 6; SNI: 287.3 ± 22.7 MΩ, n = 6; p = 0.6743; Fig. 5F). The significant increase in membrane input resistance observed in SNI conditions appears to be due to decreased open HCN channel probability at physiologically relevant membrane potentials.
Figure 5.

SNI neurons display higher Rin than sham neurons. A, D, Sample current-clamp traces of sham and SNI neurons from the contralateral mPFC with and without the HCN channel blocker ZD7288. Membrane potential was held at −60 mV. B, Depolarizing sag, a typical feature of HCN channel activation, was observed in both sham and SNI neurons. C, Quantitative results illustrating a significant increase in Rin in SNI (n = 12) compared with sham neurons (n = 11) measured from the current step that brought the membrane potential to −70 mV. E, Depolarizing sag was abolished in both sham and SNI conditions after the application of ZD7288 (15 μm). F, Quantitative effect of ZD7288 on Rin in sham (n = 6) and SNI neurons (n = 6). Rin was increased to similar values in both conditions. Values are mean ± SEM. **p < 0.01.
Prefrontal synaptic hyperactivity in SNI conditions and contribution of HCN channels
It is likely that a reduction in Ih at physiologically relevant potentials, as seen in SNI conditions, not only enhances postsynaptic cellular excitability, but may also enhance synaptic transmission. To assess spontaneous glutamate release at synapses in the mPFC, we recorded mEPSCs in layer II/III pyramidal neurons in the presence of picrotoxin and TTX (Fig. 6). Neurons from SNI animals displayed increased excitatory synaptic activity reflected by a marked decrease in the mEPSC interevent interval compared with sham neurons, indicating an increase in glutamate release events (sham: 439.5 ± 12.63 ms, n = 7; SNI: 369.9 ± 11.59 ms, n = 10; p < 0.001; Fig. 6B,C). No significant change in mEPSC amplitude was detected (sham: 9.130 ± 0.09 pA, n = 7; SNI: 9.032 ± 0.08 pA, n = 10; p > 0.05; Fig. 6B,C). To determine specifically the influence of reduced Ih in SNI conditions on synaptic excitability, the HCN channel blocker ZD7288 (15 μm) was applied in the perfusion solution. Application of ZD7288 further decreased the mEPSC interevent interval (control: 369.9 ± 11.59 ms; ZD7288: 320.4 ± 11.03 ms, p < 0.01; n = 10; Fig. 6B,C), but had no effect on the mEPSC amplitude (control: 9.03 ± 0.08 pA; ZD7288: 9.01 ± 0.06 pA, p > 0.05; n = 10; Fig. 6B,C) in SNI neurons. In sham neurons, application of ZD7288 significantly enhanced the mEPSC interevent interval to a larger degree than in SNI neurons (control: 439.5 ± 12.63 ms; ZD7288: 351.7 ± 12.22 ms, p < 0.001; n = 7; Fig. 6B,C) and similarly had no significant effect on the mEPSC amplitude (control: 9.13 ± 0.09 pA; ZD288: 8.80 ± 0.08 pA, p > 0.05; n = 7; Fig. 6B,C). Thus, reduced Ih observed in SNI conditions directly affects synaptic activity and neuronal excitability via increased glutamate release.
Figure 6.
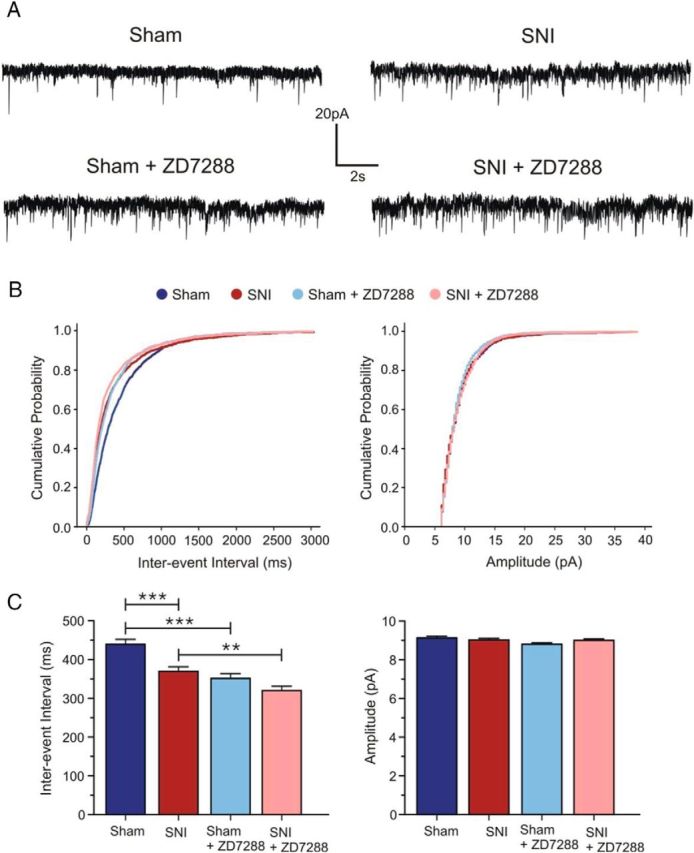
HCN channel blockade enhances glutamate release in layer II/III of the mPFC in both sham and SNI conditions. A, Top, Sample voltage-clamp traces (Vh = −70 mV) illustrating the increase in frequency, but not amplitude, of mEPSCs in SNI (n = 10) compared with sham pyramidal neurons (n = 7). Bottom, Application of ZD7288 (15 μm) further enhanced the mEPSC frequency in both conditions with no visible change in mEPSC amplitude (n = 7 for sham; n = 10 for SNI). B, Cumulative probability plots for interevent intervals and amplitudes of mEPSCs showing the ZD7288-induced increase in frequency with no change in amplitude. C, Quantitative effect of ZD7288 on interevent intervals and amplitudes of mEPSCs. Values are mean ± SEM. ***p < 0.001; **p < 0.01.
Facilitation of persistent firing in SNI pyramidal neurons
We have shown previously that application of low concentrations of the mGluR5 agonist DHPG (< 5 μm) are normally insufficient to evoke robust plateau potentials and persistent firing, a putative cellular mechanism for working memory, after a depolarizing current pulse in mPFC pyramidal neurons (Zhang et al., 2013). However, by inhibiting Ih and increasing input resistance via application either of ZD7288 or the α2 adrenoceptor agonist clonidine, long-lasting and sustained persistent firing can be induced by subthreshold doses of DHPG (Zhang et al., 2013). To determine whether the reduced open probability of HCN channels and related increase in input resistance and excitability could serve to prime SNI prefrontal pyramidal neurons for mGluR5-driven intrinsic persistent firing, we performed continuous current-clamp recordings at a membrane potential of −60 mV in the presence of 2.5 μm DHPG. The ionotropic synaptic blockers kynurenic acid (2 mm) and picrotoxin (100 μm) were applied to block glutamatergic and GABAergic transmission, respectively. After bath application of DHPG for 15 min, we observed ADPs or persistent firing in 8 of 14 SNI prefrontal neurons following a depolarizing current pulse (100 pA for 1 s). In contrast, bath application of DHPG failed to evoke persistent firing in response to the same current pulse in sham neurons (n = 8 of 8; Fig. 7A). Quantitatively, SNI neurons displayed significantly greater plateau potential amplitudes (sham: 1.55 ± 0.69 mV, n = 8; SNI: 7.11 ± 1.15 mV, n = 14; p = 0.0027; Fig. 7B) and firing frequency (sham: 0 Hz, n = 8; SNI: 0.87 ± 0.58 Hz, n = 14; p = 0.031; Fig. 7B) compared with sham neurons.
Figure 7.

Facilitation of glutamatergic persistent firing in SNI but not in sham mPFC pyramidal neurons. A, Sample current-clamp traces demonstrating that subthreshold DHPG (2.5 μm) induces ADPs and persistent firing, a cellular substrate for working memory in the mPFC, in some SNI (n = 14) but not in any sham pyramidal neurons (n = 8) at a holding voltage of −60 mV. B, Quantitative results of plateau potential amplitude and frequency of the persistent firing response induced by DHPG in sham and SNI neurons as shown in A. Values are mean ± SEM. **p < 0.01; *p < 0.05.
Involvement of the cAMP/PKA signaling axis in HCN channel dysfunction in SNI neurons
Cyclic nucleotides (e.g., cAMP) are key modulators of HCN channel activity. Binding of cAMP to the cyclic nucleotide-binding domain on the C terminus of HCN channels favors channel opening and shifts the voltage dependence to more depolarized potentials (Robinson and Siegelbaum, 2003; Zhou and Siegelbaum, 2007; Wahl-Schott and Biel, 2009). It is possible that changes in cAMP levels after peripheral nerve injury, such as a decrease in cAMP (Walikonis and Poduslo, 1998), might underlie this reduction in open HCN channel probability observed in prefrontal pyramidal neurons from SNI animals. To address this, we applied bromo-cAMP (200 μm) intracellularly via the patch pipette to determine whether increasing cAMP levels could normalize the voltage activation curve. Tail current analysis in control experiments without bromo-cAMP confirmed the hyperpolarizing shift in voltage dependence of Ih activation in SNI compared with sham neurons (Fig. 8B). We observed a significant shift (−5.4 mV) in the V1/2 in SNI prefrontal pyramidal cells (sham V1/2: −86.5 ± 0.52 mV, n = 5; SNI V1/2: −91.9 ± 0.38 mV, n = 6; p < 0.0001; Fig. 8C). In the presence of bromo-cAMP, we observed a shift in the activation curve of both sham and SNI prefrontal pyramidal neurons to more depolarized potentials and the hyperpolarizing shift in the voltage dependence of Ih activation seen in SNI neurons was eliminated (Fig. 8A,B). The V1/2 of Ih after bromo-cAMP application was identical in sham and in SNI neurons (sham V1/2: −72.3 ± 0.26 mV, n = 6; SNI V1/2: −72.1 ± 0.43 mV, n = 7; p = 0.6913; Fig. 8C). Furthermore, studies have shown that PKA can enhance Ih and shift its voltage dependence to more depolarized potentials in neurons and sinoatrial myocytes (Mellor et al., 2002; Liao et al., 2010; Cheng and Zhou, 2013). To determine whether PKA may contribute to the decreased HCN channel availability observed in SNI conditions, we applied the PKA inhibitor H89 (5 μm) intracellularly via the patch pipette and investigated its effect on Ih voltage dependence. Inhibition of PKA caused a significant hyperpolarizing shift in the V1/2 of Ih in sham neurons (control V1/2: −82.84 ± 0.49 mV; H89 V1/2: −86.53 ± 0.79 mV, p = 0.0028; n = 6) and further shifted the voltage-dependent activation in SNI neurons to more negative potentials (control V1/2: −89.67 ± 0.4 mV; H89 V1/2: −92.56 ± 0.8 mV, p = 0.0136; n = 8; Fig. 8D,E). In addition, we observed an increase in input resistance after H89 application in sham (control: 195.3 ± 28.52 MΩ; H89: 270.2 ± 24.71 MΩ, p = 0.0194; n = 6) and a nonsignificant increasing trend in SNI neurons (control: 271.4 ± 18.64 MΩ; H89: 310.5 ± 31.09 MΩ, p = 0.1235; n = 8; Fig. 8F).
Figure 8.

The cAMP/PKA signaling axis contributes to the Ih voltage dependence shift observed in SNI mPFC pyramidal neurons. A, Representative voltage-clamp traces illustrating tail currents measured upon return to a fixed membrane potential (−130 mV) in sham and SNI neurons after bromo-cAMP application within the patch pipette. B, Analysis of tail currents from traces without bromo-cAMP revealed a consistent hyperpolarizing shift in the voltage dependence of HCN channel activation in SNI (n = 6) compared with sham neurons (n = 5). After bromo-cAMP application (200 μm), the activation curve of Ih in sham (n = 6) and SNI (n = 7) conditions was shifted to similar and more depolarized potentials. C, Quantitative results showing a significant positive shift in the V1/2 to similar values in both sham and SNI conditions after bromo-cAMP. D, Intracellular application of the PKA inhibitor H89 (5 μm) caused a hyperpolarizing shift in the Ih activation curve in both sham (n = 6) and SNI (n = 8) neurons. E, Quantitative results illustrating a significant hyperpolarizing shift in the V1/2 of Ih in sham and SNI neurons after H89 application. F, Input resistance was increased after H89 application in both sham (n = 6) and SNI (n = 8) neurons, but only reached statistical significance in sham conditions. Values are mean ± SEM. ***p < 0.001; **p < 0.01; *p < 0.05.
Analgesic effect of acute prefrontal HCN channel inhibition in SNI animals
To determine the behavioral significance of reduced Ih in the mPFC, in vivo evoked pain behavior was assessed after acute infusions of saline or the HCN channel blocker ZD7288 (0.2 μg/μl) into the mPFC of freely moving sham and SNI rats. We observed that local injection of ZD7288 in the mPFC significantly reduced cold allodynia on the hindpaw ipsilateral to the injury in SNI animals compared with the saline-infused group 30–40 min after infusion (saline cold score: 1.57 ± 0.20; ZD7288: 0.76 ± 0.23, n = 7; p < 0.05; Fig. 9A). In sham rats, ZD7288 microinfusions did not alter the cold sensitivity of the ipsilateral hindpaw compared with the saline-infused group 30–40 min after infusion (saline cold score: 0.33 ± 0.13; ZD7288: no detectable response, n = 4; p > 0.05; Fig. 9A). However, we did not observe any significant reduction in mechanical allodynia in SNI animals infused with ZD7288 compared with the saline-infused group 30–40 min after infusion (saline: 4.57 ± 1.04 g; ZD7288: 7.57 ± 1.60 g, n = 7; p > 0.05; Fig. 9B).
Figure 9.

Acute infusion of the HCN channel blocker ZD7288 into the mPFC produces analgesic effects in SNI rats. Acute infusions of ZD7288 (0.2 μg/μl in 0.5 μl) or saline (0.5 μl) were administered into the PFC using stereotaxic cannulation. A, Left, SNI animals (n = 7) displayed a significantly decreased withdrawal threshold to mechanical stimulation on the hindpaw ipsilateral to nerve injury compared with sham animals (n = 4) at baseline. No significant reduction in mechanical allodynia was observed in SNI animals infused with ZD7288 (n = 7) compared with the saline-infused SNI group (n = 7). In sham rats, ZD7288 (n = 4) did not alter mechanical sensitivity (n = 4). Right, No significant difference was observed in mechanical sensitivity in the contralateral hindpaw in sham and SNI animals before or after infusions. B, Left, SNI animals (n = 7) also displayed increased cold sensitivity in the ipsilateral hindpaw compared with sham animals (n = 4) at baseline. ZD7288 infusions into the PFC produced a significant decrease in cold allodynia in SNI animals (n = 7) compared with the control saline-infused SNI group (n = 7). In sham rats (n = 4), ZD7288 did not significantly alter cold sensitivity compared with the saline group (n = 4). Right, No significant difference was observed in cold sensitivity in the contralateral hindpaw in sham and SNI animals before and after infusions. Values are mean ± SEM. ***p < 0.001; **p < 0.01; *p < 0.05.
Discussion
Decreased open probability of ion channels is known to play a major role in intrinsic neural excitability. Therefore, our discovery of a hyperpolarizing shift in the voltage dependence of Ih activation in layer II/III pyramidal neurons of the contralateral mPFC after peripheral nerve injury provides insights into the contribution of HCN channels to prefrontal hyperexcitability during neuropathic pain processing. The involvement of Ih was further confirmed using the selective HCN channel blocker ZD7288, which abolished the shift between Ih activation curves from sham and SNI neurons.
Interestingly, no change in the maximal available current (measured at −130 mV) was observed between mPFC pyramidal neurons from sham and SNI animals. This suggests that the overall expression of HCN channels is unchanged between both groups because current density reflects functional protein levels. This is further supported by our semiquantitative analysis of HCN2 immunofluorescence in the mPFC, which revealed no significant difference in expression levels, either in density or pattern, between sham and SNI conditions. It has been postulated that changes in HCN channel function in DRG neurons after peripheral nerve injury might arise from posttranslational modifications including alterations in regulatory molecules, subunit assembly or accessory proteins (Chaplan et al., 2003; Jiang et al., 2008a). If the number of HCN channels is not significantly changed between the mPFC of sham and SNI rats, it is likely that the reduction in Ih at physiologically relevant potentials in SNI neurons stems from nerve injury-associated alterations in cAMP modulation of HCN channel activity (Walikonis and Poduslo, 1998; Wang et al., 2001; Biel et al., 2009).
All HCN channel subunits share a highly conserved cyclic nucleotide binding domain located on the cytosolic C terminus and binding of cAMP favors channel opening and shifts the voltage dependence to more depolarized potentials (Wang et al., 2001; Wahl-Schott and Biel, 2009). However, sensitivity to cAMP varies depending on the HCN channel subtype: cAMP strongly regulates HCN2 and HCN4 subtypes, but has little effect on HCN1 and HCN3 (Robinson and Siegelbaum, 2003; Wahl-Schott and Biel, 2009). HCN1 and HCN2 are the main subtypes detected in layers II and III of the neocortex (Monteggia et al., 2000; Notomi and Shigemoto, 2004). Furthermore, due to the considerable overlap of distribution between HCN1 and HCN2 isoforms, Ih can be carried by heteromeric HCN1 + 2 channels sensitive to cAMP (Ulens and Tytgat, 2001). We observed that bromo-cAMP application shifted the voltage dependence of Ih to more positive voltages in both sham and SNI neurons by +14 and +20 mV, respectively, and completely eliminated the hyperpolarizing shift in Ih activation detected in SNI conditions. In addition, the activation curve of Ih in SNI neurons exhibited a midpoint of activation similar to that of HCN2 (typically −95 mV; Wahl-Schott and Biel, 2009), suggesting a role for reduced cAMP modulation of postsynaptic HCN function in the neuropathic mPFC after peripheral nerve injury (Walikonis and Poduslo, 1998) and the potential involvement of HCN2 subunit-containing channels.
In addition, PKA could play a role in regulating HCN channel activity because it regulates cAMP levels (Vandamme et al., 2012) and inhibition of PKA significantly reduces the ability of Gs-coupled receptor agonists to shift the Ih voltage dependence to positive potentials (Liao et al., 2010). Inhibiting basal PKA activity using H89 caused a significant hyperpolarizing shift in the voltage-dependent activation of Ih in both sham and SNI neurons. This negative shift was greater in sham (−3.69 ± 0.93 mV) compared with SNI neurons (−2.89 ± 0.99 mV), likely due to higher functional availability of HCN channels in sham conditions. Moreover, the fact that we observed increases in input resistance in both sham and SNI neurons after H89 application further supports a role for PKA in regulating HCN channel activity. These results suggest that peripheral nerve injury-associated changes in the cAMP/PKA signaling axis in pyramidal neurons, in this case a decrease in cAMP levels and basal PKA activity, may affect HCN channel activity in SNI conditions and contribute to the observed changes in Ih voltage-dependent activation.
HCN channels are known to be strong regulators of neuronal excitability in the neocortex (Wang et al., 2007; Arnsten, 2009; Zhang et al., 2013). Therefore, the hyperpolarizing shift in the activation curve of Ih is expected to affect the electrophysiological properties of prefrontal neurons. Accordingly, we observed that SNI neurons displayed increased firing in response to a depolarizing current pulse. Previous reports have also demonstrated an increase in NMDA-driven synaptic currents, enhanced spiking and intrinsic excitability, and the development of long-term potentiation in synapses within the mPFC and ACC after peripheral nerve injury (Wei and Zhuo, 2001; Zhuo, 2008; Metz et al., 2009; Gong et al., 2010; Blom et al., 2014). Studies investigating peripheral pain pathways have shown that enhanced currents through HCN channels are implicated in enhanced nociceptive transmission (Chaplan et al., 2003; Luo et al., 2007). Moreover, a recent study from Koga et al. (2015) showed that HCN channels play a role in sustaining enhanced excitatory synaptic transmission after nerve injury because blocking HCN channels with ZD7288 can erase a presynaptic form of LTP at ACC synapses and produce both analgesic and anxiolytic effects. However, we report here that the increased excitability of prefrontal neurons may stem from a distinct mechanism involving a shunting effect whereby reduced open probability of postsynaptic HCN channels increases membrane input resistance. This phenomenon has also been observed in peripheral pain pathways (Doan et al., 2004) and is consistent with the role of Ih in regulating the efficiency of synaptic input and cortical network activity (Day et al., 2005; Wang et al., 2007; Arnsten, 2009; Poolos, 2012; Zhang et al., 2013). Interestingly, we did not detect any significant change in the resting membrane potential of SNI compared with sham neurons, suggesting that a larger membrane input resistance in SNI pyramidal neurons is sufficient to promote hyperexcitability by decreasing the threshold for spike initiation.
Activation of mGluR5 can induce slow ADPs or persistent firing in neurons from different brain regions including the entorhinal cortex, ACC, and mPFC (Yoshida et al., 2008; Zhang and Seguela, 2010; Zhang et al., 2013). Persistent firing is postulated to be a nonsynaptic cellular mechanism for working memory. Our results indicate that reduced open probability of HCN channels and the related increase in excitability in SNI neurons facilitate the expression of persistent firing induced by glutamatergic inputs. Therefore, SNI-facilitated persistent firing in the mPFC may contribute to the expression of certain behavioral or cognitive manifestations of the neuropathic pain condition, but may not necessarily promote more nociception. Enhanced excitatory transmission and presynaptic LTP in the ACC may reflect an anxious state and/or an attentional shift that may alter behavioral responses to chronic pain (Koga et al., 2015) because attention linked to ACC activation can greatly influence the affective and sensory aspects of pain perception (Bantick et al., 2002). For example, subjects engaging in meditation or attention-demanding cognitive tasks report significantly reduced pain unpleasantness and intensity (Bantick et al., 2002; Perlman et al., 2010).
In addition to neuronal hyperexcitability, we observed that peripheral nerve injury-associated alterations in HCN channel function lead to an increase in glutamate release since the mEPSC frequency was significantly increased in SNI prefrontal neurons. This mechanism may underlie the potentiation of mGluR5-dependent persistent firing observed in SNI neurons. Therefore, we propose that reduced Ih in SNI conditions contributes to increased neuronal excitability in the mPFC by presynaptically increasing glutamate release and by postsynaptically promoting intrinsic persistent firing (Zhang et al., 2013).
Lastly, we investigated the behavioral significance of reducing Ih in the mPFC by assessing in vivo evoked pain behavior after acute infusions of the HCN channel blocker ZD7288 into the mPFC of freely moving sham and SNI rats. Interestingly, ZD7288 cortical infusions caused an analgesic effect reflected by a significant reduction in cold allodynia. Therefore, in vivo acute inhibition of HCN channels in the mPFC provides some modality-specific pain relief from behavioral sensitization caused by peripheral nerve injury. This is in agreement with the results obtained by Koga et al. (2015), in which a ZD7288 microinfusion into the ACC produced an analgesic effect in nerve-injured animals.
In summary, we observed that peripheral nerve injury caused a hyperpolarizing shift in the voltage dependence of postsynaptic HCN channel activation resulting in a subsequent increase in membrane input resistance and excitability in layer II/III pyramidal neurons of the contralateral mPFC. This is consistent with previous reports indicating that persistent pain is associated with increased neuronal activity within the mPFC (Wei and Zhuo, 2001; Metz et al., 2009; Blom et al., 2014). Downregulation of cAMP synthesis and reduced basal PKA activity may underlie the decrease in open HCN channel probability observed in SNI conditions and may result from activation of Gi-coupled receptors (Benarroch, 2013; Zhang et al., 2013). Thus, our findings provide novel insights into supraspinal mechanisms underlying pathophysiological alterations of prefrontal functions in neuropathic pain states.
Footnotes
This work was supported by the Canadian Institutes of Health Research (Grant MOP-130239 to P.S.) and the Natural Sciences and Engineering Council of Canada (Grant DG-203061 to P.S.). S.C.M. holds a Fonds de recherche du Québec–Santé (FRQS) doctoral studentship. We thank our colleagues Julien Gibon and Stephen Glasgow (Montreal Neurological Institute) for their critical reading of the manuscript, as well as Magali Millecamps (Dentistry, McGill), Manon St-Louis (Pharmacology and Therapeutics, McGill), and Sophie Pawlowski (Pharmacology and Therapeutics, McGill) for expert advice and technical support.
The authors declare no competing financial interests.
References
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Li BM. Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol Psychiatry. 2005;57:1377–1384. doi: 10.1016/j.biopsych.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Bantick SJ, Wise RG, Ploghaus A, Clare S, Smith SM, Tracey I. Imaging how attention modulates pain in humans using functional MRI. Brain. 2002;125:310–319. doi: 10.1093/brain/awf022. [DOI] [PubMed] [Google Scholar]
- Benarroch EE. HCN channels: function and clinical implications. Neurology. 2013;80:304–310. doi: 10.1212/WNL.0b013e31827dec42. [DOI] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Blom SM, Pfister JP, Santello M, Senn W, Nevian T. Nerve injury-induced neuropathic pain causes disinhibition of the anterior cingulate cortex. J Neurosci. 2014;34:5754–5764. doi: 10.1523/JNEUROSCI.3667-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci. 2003;23:1169–1178. doi: 10.1523/JNEUROSCI.23-04-01169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Zhou Y. Novel role of KT5720 on regulating hyperpolarization-activated cyclic nucleotide-gated channel activity and dorsal root ganglion neuron excitability. DNA Cell Biol. 2013;32:320–328. doi: 10.1089/dna.2013.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Yoon YW, Na HS, Kim SH, Chung JM. Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain. 1994;59:369–376. doi: 10.1016/0304-3959(94)90023-X. [DOI] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ. Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and Kleak channels. J Neurosci. 2005;25:8776–8787. doi: 10.1523/JNEUROSCI.2650-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Doan TN, Stephans K, Ramirez AN, Glazebrook PA, Andresen MC, Kunze DL. Differential distribution and function of hyperpolarization-activated channels in sensory neurons and mechanosensitive fibers. J Neurosci. 2004;24:3335–3343. doi: 10.1523/JNEUROSCI.5156-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. 2011;333:1462–1466. doi: 10.1126/science.1206243. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain. 2004;109:150–161. doi: 10.1016/j.pain.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Gong KR, Cao FL, He Y, Gao CY, Wang DD, Li H, Zhang FK, An YY, Lin Q, Chen J. Enhanced excitatory and reduced inhibitory synaptic transmission contribute to persistent pain-induced neuronal hyper-responsiveness in anterior cingulate cortex. Neuroscience. 2010;171:1314–1325. doi: 10.1016/j.neuroscience.2010.10.028. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Ionic mechanism of the slow afterdepolarization induced by muscarinic receptor activation in rat prefrontal cortex. J Neurophysiol. 1998;80:1197–1210. doi: 10.1152/jn.1998.80.3.1197. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Davis KD, Lozano AM, Tasker RR, Dostrovsky JO. Pain-related neurons in the human cingulate cortex. Nat Neurosci. 1999;2:403–405. doi: 10.1038/8065. [DOI] [PubMed] [Google Scholar]
- Jiang YQ, Sun Q, Tu HY, Wan Y. Characteristics of HCN channels and their participation in neuropathic pain. Neurochem Res. 2008a;33:1979–1989. doi: 10.1007/s11064-008-9717-6. [DOI] [PubMed] [Google Scholar]
- Jiang YQ, Xing GG, Wang SL, Tu HY, Chi YN, Li J, Liu FY, Han JS, Wan Y. Axonal accumulation of hyperpolarization-activated cyclic nucleotide-gated cation channels contributes to mechanical allodynia after peripheral nerve injury in rat. Pain. 2008b;137:495–506. doi: 10.1016/j.pain.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Koga K, Descalzi G, Chen T, Ko HG, Lu J, Li S, Son J, Kim T, Kwak C, Huganir RL, Zhao MG, Kaang BK, Collingridge GL, Zhuo M. Coexistence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron. 2015;85:377–389. doi: 10.1016/j.neuron.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Lockhead D, Larson ED, Proenza C. Phosphorylation and modulation of hyperpolarization-activated HCN4 channels by protein kinase A in the mouse sinoatrial node. J Gen Physiol. 2010;136:247–258. doi: 10.1085/jgp.201010488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Chang L, Brown SM, Ao H, Lee DH, Higuera ES, Dubin AE, Chaplan SR. Role of peripheral hyperpolarization-activated cyclic nucleotide-modulated channel pacemaker channels in acute and chronic pain models in the rat. Neuroscience. 2007;144:1477–1485. doi: 10.1016/j.neuroscience.2006.10.048. [DOI] [PubMed] [Google Scholar]
- Malin EL, Ibrahim DY, Tu JW, McGaugh JL. Involvement of the rostral anterior cingulate cortex in consolidation of inhibitory avoidance memory: interaction with the basolateral amygdala. Neurobiol Learn Mem. 2007;87:295–302. doi: 10.1016/j.nlm.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA, Schmitz D. Mediation of hippocampal mossy fiber long-term potentiation by presynaptic Ih channels. Science. 2002;295:143–147. doi: 10.1126/science.1064285. [DOI] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV, Martina M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc Natl Acad Sci U S A. 2009;106:2423–2428. doi: 10.1073/pnas.0809897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD, Hains BC, Johnson KM, Hulsebosch CE. Strain and model differences in behavioral outcomes after spinal cord injury in rat. J Neurotrauma. 2001;18:743–756. doi: 10.1089/089771501316919111. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ. Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res. 2000;81:129–139. doi: 10.1016/S0169-328X(00)00155-8. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Perlman DM, Salomons TV, Davidson RJ, Lutz A. Differential effects on pain intensity and unpleasantness of two meditation practices. Emotion. 2010;10:65–71. doi: 10.1037/a0018440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP. Hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channelopathy in epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, DElgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies. Bethesda, MD: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- Rainville P, Duncan GH, Price DD, Carrier B, Bushnell MC. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968–971. doi: 10.1126/science.277.5328.968. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Sidiropoulou K, Lu FM, Fowler MA, Xiao R, Phillips C, Ozkan ED, Zhu MX, White FJ, Cooper DC. Dopamine modulates an mGluR5-mediated depolarization underlying prefrontal persistent activity. Nat Neurosci. 2009;12:190–199. doi: 10.1038/nn.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikes RW, Vogt BA. Nociceptive neurons in area 24 of rabbit cingulate cortex. J Neurophysiol. 1992;68:1720–1732. doi: 10.1152/jn.1992.68.5.1720. [DOI] [PubMed] [Google Scholar]
- Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–382. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Ulens C, Tytgat J. Functional heteromerization of HCN1 and HCN2 pacemaker channels. J Biol Chem. 2001;276:6069–6072. doi: 10.1074/jbc.C000738200. [DOI] [PubMed] [Google Scholar]
- Vandamme J, Castermans D, Thevelein JM. Molecular mechanisms of feedback inhibition of protein kinase A on intracellular cAMP accumulation. Cell Signal. 2012;24:1610–1618. doi: 10.1016/j.cellsig.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci. 2009;66:470–494. doi: 10.1007/s00018-008-8525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walikonis RS, Poduslo JF. Activity of cyclic AMP phosphodiesterases and adenylyl cyclase in peripheral nerve after crush and permanent transection injuries. J Biol Chem. 1998;273:9070–9077. doi: 10.1074/jbc.273.15.9070. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen S, Siegelbaum SA. Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. J Gen Physiol. 2001;118:237–250. doi: 10.1085/jgp.118.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Wei F, Zhuo M. Potentiation of sensory responses in the anterior cingulate cortex following digit amputation in the anaesthetised rat. J Physiol. 2001;532:823–833. doi: 10.1111/j.1469-7793.2001.0823e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Donnelly DF, Ma C, LaMotte RH. Upregulation of the hyperpolarization-activated cation current after chronic compression of the dorsal root ganglion. J Neurosci. 2003;23:2069–2074. doi: 10.1523/JNEUROSCI.23-06-02069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YW, Lee DH, Lee BH, Chung K, Chung JM. Different strains and substrains of rats show different levels of neuropathic pain behaviors. Exp Brain Res. 1999;129:167–171. doi: 10.1007/s002210050886. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Fransén E, Hasselmo ME. mGluR-dependent persistent firing in entorhinal cortex layer III neurons. Eur J Neurosci. 2008;28:1116–1126. doi: 10.1111/j.1460-9568.2008.06409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Séguéla P. Metabotropic induction of persistent activity in layers II/III of anterior cingulate cortex. Cereb Cortex. 2010;20:2948–2957. doi: 10.1093/cercor/bhq043. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Cordeiro Matos S, Jego S, Adamantidis A, Séguéla P. Norepinephrine drives persistent activity in prefrontal cortex via synergistic alpha1 and alpha2 adrenoceptors. PLoS One. 2013;8:e66122. doi: 10.1371/journal.pone.0066122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Siegelbaum SA. Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure. 2007;15:655–670. doi: 10.1016/j.str.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M. Neuronal mechanism for neuropathic pain. Mol Pain. 2007;3:14. doi: 10.1186/1744-8069-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M. Cortical excitation and chronic pain. Trends Neurosci. 2008;31:199–207. doi: 10.1016/j.tins.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zhuo M. Central plasticity in pathological pain. Novartis Found Symp. 2004;261:132–145. discussion 145–154. [PubMed] [Google Scholar]