

Abstract
CNS neurons in adult mammals do not spontaneously regenerate axons after spinal cord injury. Preconditioning peripheral nerve injury allows the dorsal root ganglia (DRG) sensory axons to regenerate beyond the injury site by promoting expression of regeneration-associated genes. We have previously shown that peripheral nerve injury increases the number of macrophages in the DRGs and that the activated macrophages are critical to the enhancement of intrinsic regeneration capacity. The present study identifies a novel chemokine signal mediated by CCL2 that links regenerating neurons with proregenerative macrophage activation. Neutralization of CCL2 abolished the neurite outgrowth activity of conditioned medium obtained from neuron–macrophage cocultures treated with cAMP. The neuron–macrophage interactions that produced outgrowth-promoting conditioned medium required CCL2 in neurons and CCR2/CCR4 in macrophages. The conditioning effects were abolished in CCL2-deficient mice at 3 and 7 d after sciatic nerve injury, but CCL2 was dispensable for the initial growth response and upregulation of GAP-43 at the 1 d time point. Intraganglionic injection of CCL2 mimicked conditioning injury by mobilizing M2-like macrophages. Finally, overexpression of CCL2 in DRGs promoted sensory axon regeneration in a rat spinal cord injury model without harmful side effects. Our data suggest that CCL2-mediated neuron–macrophage interaction plays a critical role for amplification and maintenance of enhanced regenerative capacity by preconditioning peripheral nerve injury. Manipulation of chemokine signaling mediating neuron–macrophage interactions may represent a novel therapeutic approach to promote axon regeneration after CNS injury.
SIGNIFICANCE STATEMENT CNS axons do not regenerate spontaneously after injury. However, preconditioning peripheral nerve injury enables dorsal root ganglia sensory neurons to regenerate central axons beyond spinal lesion. The exact mechanism by which the conditioning injury enhances axon regeneration capacity remains elusive. We report here that neuronal CCL2 induced by conditioning injury mediates neuron–macrophage interactions, resulting in accumulation of perineuronal macrophages with a proregenerative phenotype. Genetic or immunological inhibition of CCL2 abolished conditioning effects in vitro and in vivo, indicating that CCL2-mediated activation of proregenerative macrophages is essential in the conditioning injury-induced enhanced regenerative capacity. Intraganglionic CCL2 gene delivery recapitulates conditioning effects after spinal cord injury, suggesting that a chemokine signal mediating neuron–macrophage interaction may be a novel target for axon regeneration therapeutics.
Keywords: axon regeneration, CCL2, chemokine, cAMP, macrophage, neuron–macrophage interaction
Introduction
CNS neurons in adult mammals do not spontaneously regenerate axons after disruptive injuries, such as stroke or spinal cord injury. This is in part because an inhospitable environment at the injury site actively inhibits axon growth by producing growth-inhibitory molecules (Filbin, 2003; Silver and Miller, 2004; Lang et al., 2015). Recent studies have emphasized that a lack or decline of intrinsic neuronal capacity in the mature CNS also contributes substantially to the failure of spontaneous axon regeneration (Moore et al., 2009; Park et al., 2010; O'Donovan et al., 2014). The intrinsic axon regeneration capacity may be constrained by the ability of neurons to express regeneration-associated genes (RAGs). The necessity of neuronal RAG expression for axon regeneration is exemplified in the conditioning injury model, in which a preceding injury to the peripheral nerve upregulates expression of RAGs in the dorsal root ganglia (DRG) sensory neurons and thereby enhances regeneration of their central branches in the spinal cord (Richardson and Issa, 1984; Smith and Skene, 1997; Neumann and Woolf, 1999). Recent studies have provided novel insights into how a peripheral nerve injury triggers RAG expression in the nuclei of the DRG sensory neurons. Axotomy generates various retrograde signals being transmitted to the cell bodies (Rishal and Fainzilber, 2014) and the retrograde injury signals elicit epigenetic modifications of RAGs, triggering initiation of their transcription (Cho et al., 2013; Finelli et al., 2013; Puttagunta et al., 2014). However, these mechanisms may not fully explain how over weeks or even months enhanced RAG expression can be maintained, thus allowing axons to achieve sustained elongation through and beyond the injury site (Neumann and Woolf, 1999; Ylera et al., 2009; Blesch et al., 2012).
There is growing evidence that innate immunity can boost endogenous mechanisms in axon regeneration (Gensel et al., 2012). Several studies have demonstrated that chemically activated macrophages near neuronal cell bodies can exert potent effects on axon regeneration (Yin et al., 2003; Hauk et al., 2010; Baldwin et al., 2015). However, it is not known whether endogenous signaling processes commensurate to those induced by chemical activation exist. Previous studies have shown that preconditioning peripheral nerve injury induces accumulation of macrophages around sensory neurons in the DRGs and that the activated macrophages may play a role in enhancing intrinsic regenerative capacity (Lu and Richardson, 1991, 1993; Niemi et al., 2013). Our previous study also suggested that a conditioning peripheral nerve injury may elicit neuron–macrophage interactions in the DRGs to drive macrophage activation toward a proregenerative phenotype (Kwon et al., 2013). The present study identifies CCL2 (also known as monocyte chemoattractant protein-1) as an endogenous signaling molecule that mediates neuron–macrophage interactions after conditioning injury. We provide evidence that CCL2-mediated neuron–macrophage interaction is required for amplification and maintenance, but not for initial induction of enhancement of regenerative capacity by conditioning injury. Furthermore, our finding that CCL2 signaling can mimic the conditioning effects suggests that chemokine-mediated neuron–macrophage interactions may be a novel target to promote axon regeneration after CNS injury.
Materials and Methods
Animals.
Adult female Sprague Dawley rats (250–300 g) and C57BL/6 wild-type (WT) mice were purchased from Orient Bio. CCL2-deficient and CCR2-deficient mice on C57BL/6 background were purchased from The Jackson Laboratory. The Institutional Animal Care and Use Committee of Ajou University School of Medicine approved all animal protocols.
Surgical procedures.
Animals were anesthetized with chloral hydrate (400 mg/kg, i.p.) during all the surgical procedures. After being dissected and exposed, the right sciatic nerve was ligated proximal to its trifurcation and cut below the ligation site with fine surgical scissors. To create a dorsal column lesion in the spinal cord, a dorsal laminectomy was performed at the T7 level and bilateral dorsal columns with adjacent lateral columns were cut out with iridectomy scissors. To visualize regenerating axons, cholera toxin subunit B (CTB; List Biological Laboratories) was injected using a protocol modified slightly from that in the previous report (Kwon et al., 2013). After the sciatic nerve between the thigh muscles was exposed, a small incision was made on the perineurium just proximal to the trifurcation site. Four microliters of unconjugated CTB solution (0.1% in PBS) were slowly injected using the Hamilton syringe through the perineural incision and animals were killed 5 d after the injection. For intraganglionic injection, the L5 DRG was exposed after removal of the lateral process of the L5 vertebral bone. A Hamilton syringe configured with a glass pipette was controlled using a micromanipulator, and the tip of the glass pipette was slowly advanced into the L5 DRG under a surgical microscope. Two microliters of recombinant chemokines (250 μg/ml; R&D Systems) or CCL2-neutralizing antibody (200 μg/ml; R&D Systems) were injected into the L5 DRG at a rate of 0.5 μl/min using a nanoinjector.
Preparation and injection of adeno-associated virus serotype 5 with a CCL2 expression cassette.
The recombinant adeno-associated virus (AAV) vector containing a CCL2 expression cassette was generated by replacing the GFP cassette of pAAV-CAG-GFP (Addgene) with the full-length cDNA for rat CCL2 (OriGene). AAV serotype 5 (AAV5) virus preparation was performed by the University of North Carolina at Chapel Hill Vector Core facility. Control AAV5-GFP was purchased from the same facility. The titer of both viruses was ∼1 × 1016 vector genome copies/ml. Two microliters of AAV5-GFP or AAV5-CCL2 were injected into the L5 DRG at a rate of 0.5 μl/min using the method described above for intraganglionic injection of recombinant chemokines or neutralizing antibodies.
Primary culture of dissociated adult DRG neurons and neurite outgrowth assay.
DRGs were freshly dissected and treated with 125 U/ml type XI collagenase (Sigma-Aldrich) dissolved in DMEM (Hyclone) for 90 min at 37°C with gentle rotation. After washing five times with DMEM, the cells were dissociated by trituration using a pipette tip and centrifuged at 1500 rpm for 3 min. Cell pellets were resuspended in Neurobasal-A (Invitrogen) supplemented with B-27 (Invitrogen) and plated onto eight-well culture slides (BD Biosciences) precoated with 0.01% poly-d-lysine (Sigma-Aldrich) and 3 μg/ml laminin (Invitrogen). Neurons from a single L5 DRG of one rat or from the L4, L5, and L6 DRGs of one mouse were cultured in a single well. The culture duration was strictly limited to 15 h, at which point there is minimal neurite outgrowth in the control condition (without conditioning injury). Neurite outgrowth was visualized by immunostaining with mouse anti-β III tubulin (1:1000; Promega) followed by an Alexa 594-conjugated anti-mouse secondary antibody (Invitrogen).
Primary macrophage culture and neuron–macrophage coculture.
To harvest peritoneal macrophages, adult rats or mice were anesthetized with an overdose of chloral hydrate. Immediately after sacrificing the animals by cervical dislocation, peritoneal macrophages were harvested by intraperitoneal lavage with 50 or 20 ml of ice-cold PBS for rats or mice, respectively. The lavage fluid was centrifuged at 1500 rpm for 10 min at 4°C to pellet the cells. The cell pellets were resuspended in 5 ml of red blood cell lysis buffer (Qiagen) for 3 min at room temperature and then washed three times using PBS. After the final wash, the cells were resuspended in RPMI-1640 (Hyclone) supplemented with 10% fetal bovine serum, plated onto a 100 mm culture dish or six-well plate (BD Biosciences) coated with 0.01% poly-d-lysine (Sigma-Aldrich), and maintained at 37°C in a humidified incubator with 5% CO2. To investigate phenotypic changes in macrophages in response to chemokine treatment, macrophage cultures were allowed to stabilize for 4 h after plating and then treated with recombinant chemokines at a concentration of 100 μm for 24 h. To establish neuron–macrophage cocultures, dissociated DRG neurons were incubated in a six-well plate with DMEM supplemented with B-27 for 4 h. Then, peritoneal macrophages were plated on top of neurons at a ratio of 1:5 (neurons to macrophages). Four hours after macrophage plating, the neuron–macrophage cocultures were treated with dibutyryl-cAMP (db-cAMP; 100 μm; Calbiochem) or PBS as a control. After 24 h, the culture medium was replaced with fresh DMEM supplemented with B-27. The cocultures were maintained for 72 h without changing the medium and then the conditioned medium (CM) was collected, centrifuged at 1500 rpm for 5 min, and passed through a 0.2 μm filter (BD Biosciences) to remove any remaining cellular debris. For neurite outgrowth assays with CM, primary dissociated DRG neuron cultures were established in eight-well culture slides as described above and maintained in culture for 2 h to allow for attachment to the culture dishes before replacing the medium with the collected CM. After 15 h in culture, the cells were fixed and immunostained for β III tubulin to visualize neurite outgrowth. To neutralize CCL2 in the coculture condition, CCL2-neutralizing antibodies or control nonimmune goat IgGs (20 ng/ml; R&D Systems) were added during both the 24 h treatment with db-cAMP and during the 72 h medium-conditioning period. To test whether CCR4 is involved in the CCL2-mediated neuron–macrophage interaction, 0.1 μm C 021 dihydrochloride (#3581, Tocris), CCR4 inhibitor, was added during the cocultures composed of WT neurons and CCR2-deficient macrophages.
Real-time RT-PCR and ELISA.
Total RNAs were extracted from dissected DRGs or cultured cells using Trizol (Invitrogen) according to the manufacturer's protocol. To obtain the DRGs, animals were anesthetized with an overdose of chloral hydrate and briefly perfused with ice-cold saline to remove blood components from the tissue. The L4 and L5 DRGs were quickly dissected and the spinal nerves attached to the DRGs were carefully removed. Dissected DRGs were frozen rapidly and stored at −70. Samples were quantified using a Nanodrop 1000 instrument (Thermo Fisher Scientific). Two micrograms of RNA were reverse transcribed to cDNA using a standard reverse transcription protocol. One microliter of cDNA was added to a PCR-reaction premix (GenDepot) containing corresponding primer pairs (10 pm). The following primers were used for PCR: 18S ribosomal RNA (rRNA), 5′-CGGCTACCACATCCAAGGAA-3′ (forward), 5′-TGCTGGCACCAGACTTGCCCTC-3′ (reverse); CCL2, 5′-ATGCAGTTAATGCCCCACTC-3′ (forward), 5′-TTCCTTATTGGGGTCAGCAC-3′ (reverse); fractalkine, 5′-CGCTCTGAATAGCTCCAACC-3′ (forward), 5′-CTGCTCCTCAGGCCTACAAC-3′ (reverse); CD-16, 5′-TGTTTGCTTTTGCAGACAGG-3′ (forward), 5′-GCACCGGTATTCTCCACTGT-3′ (reverse); CD-32, 5′-TGTCGCTGGAATTGCTGTAG-3′ (forward), 5′-AGGTCCTGGCCTTACTGGTT-3′ (reverse); CD-86, 5′-CTCAGTGATCGCCAACTTCA-3′ (forward), 5′-ATCTGCATGTTGTCGCCATA-3′ (reverse); CD-163, 5′-GATGTGGCTCTGCACTTCAA-3′ (forward), 5′-ACCAGTCTGGGTTTCCTGTG-3′ (reverse); CD-206, 5′-CAAGGAAGGTTGGCATTTGT-3′ (forward), 5′-CAAAGGAACGTGTGCTCTGA-3′ (reverse); inducible nitric oxide synthase (iNOS), 5′-CACCTTGGAGTTCACCCAGT-3′ (forward), 5′-ACCACTCGTACTTGGGATGC-3′ (reverse); IL-2, 5′-AAACTCCCCATGATGCTCAC-3′ (forward), 5′-GAAATTTCCAGCGTCTTCCA-3′ (reverse); IL-10, 5′-CCTGCTCTTACTGGCTGGAG-3′ (forward), 5′-TGTCCAGCTGGTCCTTCTTT-3′ (reverse); arginase-1, 5′-TATCGGAGCGCCTTTCTCTA-3′ (forward), 5′-ACAGACCGTGGGTTCTTCAC-3′ (reverse). Quantitative real-time PCR was performed using an Applied Biosystems 7500 Real-Time PCR System. Cycling conditions were 94°C for 30 s, 55∼64°C for 31 s, and 72°C for 60 s for a total of 34 cycles. Melting curves were generated after the last extension step, and the Ct values were determined by the software. Target gene expression was normalized relative to expression of 18S rRNA as an internal control. The concentration of CCL2 in the culture media was measured using an ELISA kit for rat CCL2 (R&D Systems), according to the manufacturer's protocol. One hundred microliters of CM were used in each well. Three independent cultures were performed for each condition and each sample was measured in duplicate.
Chemokine PCR array.
The rat chemokines and receptors RT2 Profiler PCR Array kit (PARN-022Z, Qiagen) in a 96-well format was used to search for a chemokine recruiting proregenerative macrophages to DRGs after sciatic nerve injury (SNI). This PCR array kit profiles the expression of 84 genes that encode chemokines, chemokine receptors, and other genes related to chemotaxis. The L4 and L5 DRGs were quickly dissected 0, 1, 3, and 7 d after SNI. Dissected DRGs were frozen rapidly and stored at −70°C until use. Total RNA was purified using the RNeasy Tissue Mini kit (Qiagen) in accordance with the manufacturer's instructions. cDNA was obtained from purified RNA using the RT2 First Strand Kit (Qiagen) provided with the PCR array kit. Two micrograms of cDNA template mixed with PCR master mix were dispensed in each well and real-time PCR was performed using the Applied Biosystems 7500 system. Three independent arrays (three animals) were performed for each condition [0 (control), 1, 3, and 7 d after SNI] and the average array value compared with the control condition was obtained. The k-means clustering algorithm was used to cluster genes with a similar temporal expression pattern using the MultiExperiment Viewer software (Saeed et al., 2006).
Magnetic-activated cell sorting for isolation of macrophages from DRGs.
L4 and L5 DRGs were dissected and dissociated with collagenase in DMEM as described above for the neuron cultures. Cells were centrifuged at 1500 rpm for 3 min at 4°C and washed with magnetic-activated cell sorting (MACS) buffer (0.5% BSA, 2 mm EDTA in PBS without divalent ions, pH 7.2). Cell suspensions were incubated for 15 min on ice with biotin-conjugated primary antibodies against CD68 (1:100; Abcam), which is found in the cytoplasmic granules of the cells in the macrophage lineage, and then incubated with avidin-conjugated microbeads (Miltenyi Biotec) for 10 min on ice. Then, cell suspensions were allowed to flow through the minicolumn of the MACS Cell Separation kit (Miltenyi Biotec). The flow-through was collected as CD68-negative cells. CD68-positive cells adsorbed to the minicolumn were eluted using 500 μl of MACS buffer. Total RNA was extracted from each fraction using the methods described above.
Tissue processing and immunohistochemistry.
Rats were anesthetized with an overdose of chloral hydrate and perfused with heparinized PBS followed by 4% paraformaldehyde (PFA) in 0.2 m phosphate buffer. DRGs or spinal cord tissues containing the lesion site were dissected and postfixed in 4% PFA for 2 h, followed by cryoprotection in a graded series of sucrose solutions. DRGs were cryosectioned at a thickness of 20 μm. For spinal cord tissue, 20-μm-thick parasagittal cryosections were cut. Tissue sections were mounted onto SuperFrost Plus slides (Fisher Scientific) and stored at −20°C until use. For immunohistochemistry, tissue sections were treated with 10% normal serum and 0.3% Triton X-100 for 1 h, and then primary antibodies dissolved in the same blocking solution were applied at 4°C overnight. The primary antibodies were rabbit anti-Iba1 (1:500; Wako), rabbit anti-CCL2 (1:1000; Abcam), mouse anti-calcitonin gene-related peptide (anti-CGRP; 1:1000; Abcam), rabbit anti-GAP-43 (1:100; Millipore), rabbit anti-phospho c-Jun (1:100; Millipore), rabbit anti-phospho-STAT3 (1:100; Cell Signaling Technology), and goat anti-CTB (1:10,000; List Biological Laboratories). Tissue sections were washed thoroughly and then incubated with appropriate secondary antibodies tagged with Alexa Fluor 488 or 594 (1:500; Invitrogen) for 1 h at room temperature. For visualization of the CTB tracing signal in the spinal cord sections, biotinylated anti-goat IgGs (Vector Laboratories) followed by Alexa 594 streptavidin conjugates (Invitrogen) were applied after primary antibody application. The immunoreactive signals were amplified using a Tyramide Signal Amplification Kit (PerkinElmer) according to the manufacturer's protocol. The images were taken using an FV 300 confocal microscope (Olympus).
Quantitative image analysis.
For neurite outgrowth assays, neurite length was measured using the NeuronJ plugin for the image analysis software suite ImageJ. Each well in an eight-well culture slide was divided into four quadrants, and a 200× magnification image was obtained at the center of each quadrant (four images in each well). Neurites were traced for all neurons in each image and the number of DRG neurons per image was counted. There were ∼20 neurons per image and, therefore, ∼80 neurons were measured for each well. The average neurite length per neuron in each well was calculated by dividing the total neurite length by the number of neurons. The average group values for neurite length per neuron were obtained by averaging values from three to four independent cultures. To quantify the number of macrophages in DRGs, the number of macrophages in DRGs was quantified in images of Iba1-immunostained tissue sections taken at 200× magnification. Macrophages were counted in four images per animal and the total number was divided by the area of the imaged DRG to obtain the macrophage counts per unit area. To analyze the frequency of CCL2 expression in DRG neurons with different sizes, L5 DRG neurons were classified based on the greatest diameter as follows: neurons with the diameter <23 μm were counted as small, those with the diameter >32 μm as large, and those between 23 and 32 μm as medium-sized DRG neurons (Fang et al., 2006). L5 DRG sections were immunostained doubly with NeuN and CCL2, and the proportions of CCL2-positive neurons in different size categories were obtained.
To quantify the extent of dorsal column axon regeneration after injury, six consecutive parasagittal sections collected at a distance of 60 μm from the intersection with the caudal lesion border and containing CTB-traced axons were used for analysis. In each section, images were captured at 200× magnification covering from 5 mm caudal to 5 mm rostral to the lesion site. A montage image was generated using the image-montaging module installed in the Neurolucida system (MBF Biosciences). After the caudal lesion border was identified using GFAP staining, regenerating axons beyond the lesion border in each section were reconstructed using Photoshop software and overlaid onto a virtual section image comprising the spinal cord contour and lesion border reconstructed from the third or fourth parasagittal section. Then, the lengths of all reconstructed axons from the six sections in the composite image were summed to generate the total length of regenerating axons in each animal. In addition, the greatest distance of regenerating axon beyond the caudal lesion border was recorded for each animal and averaged for each group. The extent of regeneration was also evaluated by counting the number of axons at increasing distances from the lesion site. In each parasagittal section, lines perpendicular to the longitudinal axis were drawn at 200 μm intervals from the lower lesion border to delineate counting blocks, with blocks named according to their shortest distance to the lesion border (for example, the number of axons between +200 and +400 was recorded as the value in the +200 block). The number of CTB-positive axons between successive lines was counted and recorded as the number of axons in the block. The number of axons in each of the six sections was summed for each animal.
Measurement of pain-perception threshold.
The threshold for heat perception was quantified by measuring foot-withdrawal latency to heat stimulation (56°C) applied to the plantar surface of the hindpaw 14 and 28 d after intraganglionic injection of AAV5-GFP or AAV5-CCL2. Sham-operated animals were subjected to laminectomy, but did not receive contusion injury. Approximately 10 min before testing, rats were placed in a clear, open-topped acrylic cylinder (30 cm height × 15 cm diameter) on the unheated plate (JD-A-10A, Jeungdo Bio & Plant) to become familiarized with the environment. The maximum latency was set as 25 s to prevent potential injury. The withdrawal latency was defined as the elapsed time, in seconds, from stimulus onset to paw withdrawal. The latencies were averaged over three trials with each trial separated by a 5 min interval. The mechanical paw-withdrawal threshold was measured using a Von Frey test. Fourteen and 28 d after intraganglionic injection, animals were placed on an elevated wire grid and Von Frey hairs with forces in a log increment (0.45, 0.74, 1.26, 2, 3.3, 5, 8, and 15 g) were prepared. A Von Frey hair was applied six times for 10 s each to the region between the footpads in the plantar aspect of the hindpaw. The 2 g of stimulus hair was applied first and, if a negative response occurred in >50% of the applications, the Von Frey hair with the next higher force was applied. None of the animals in our study showed a response with the 2 g of stimulus. The flexor reflex withdrawal threshold was defined as the Von Frey hair force at which an animal showed a withdrawal response.
Statistical analysis.
All numerical values and error bars in the quantification graphs are expressed as mean ± SEM. Statistical comparison of mean values was performed using unpaired Student's t tests or one-way ANOVA followed by Tukey's post hoc tests. Quantification graphs were generated using GraphPad Prism version 5.00 (GraphPad Software).
Results
Identification of CCL2 as a mediator for neuron–macrophage interactions after conditioning injury
We have previously shown that macrophage activation in DRGs is critically important in maintaining enhanced regenerative capacity after conditioning injury. We hypothesized that injury signals transmitted to DRG neurons lead to the production of molecules with chemokine activity that then recruit macrophages to DRGs. To identify potential chemokine molecules mediating these effects, we used a PCR array to examine the expression of 84 genes related to known chemokines and chemotactic activities or chemokine receptors in mRNAs obtained from DRGs at different time points after SNI. We performed the assay at 1, 3, and 7 d after SNI and more than two-thirds of the genes showed upregulation at any of the three time points. Using k-means clustering analysis, the 84 genes were classified into different clusters based on the similarity of their temporal patterns of expression. Because obvious increase in macrophage number was observed as early as 3 d after injury (Kwon et al., 2013), we reasoned that the relevant chemokine expression would occur earlier than the 3 d time point and focused on the cluster of genes upregulated 1 d after SNI (Fig. 1A). Of the genes belonging to this cluster, CCL2 showed the most robust expression. Real-time PCR confirmed the upregulation of the CCL2 gene at the 1 and 3 d time points (Fig. 1B). Immunohistochemistry revealed CCL2 protein expression predominantly localized in DRG neurons at 1 d after SNI (Fig. 1C,D). In contrast, CCL2 expression was not observed after injury to the central branches (dorsal column injury or rhizotomy; Fig. 1E,F). We found that upregulation of CCL2 occurred predominantly in medium or large DRG neurons. CCL2 expression was observed in 52.3 ± 0.5% and 38.5 ± 2.1% of medium and large DRG neurons, respectively. However, only 7.0 ± 0.7% of small DRG neurons were colocalized with CCL2 at 1 d after SNI.
Figure 1.
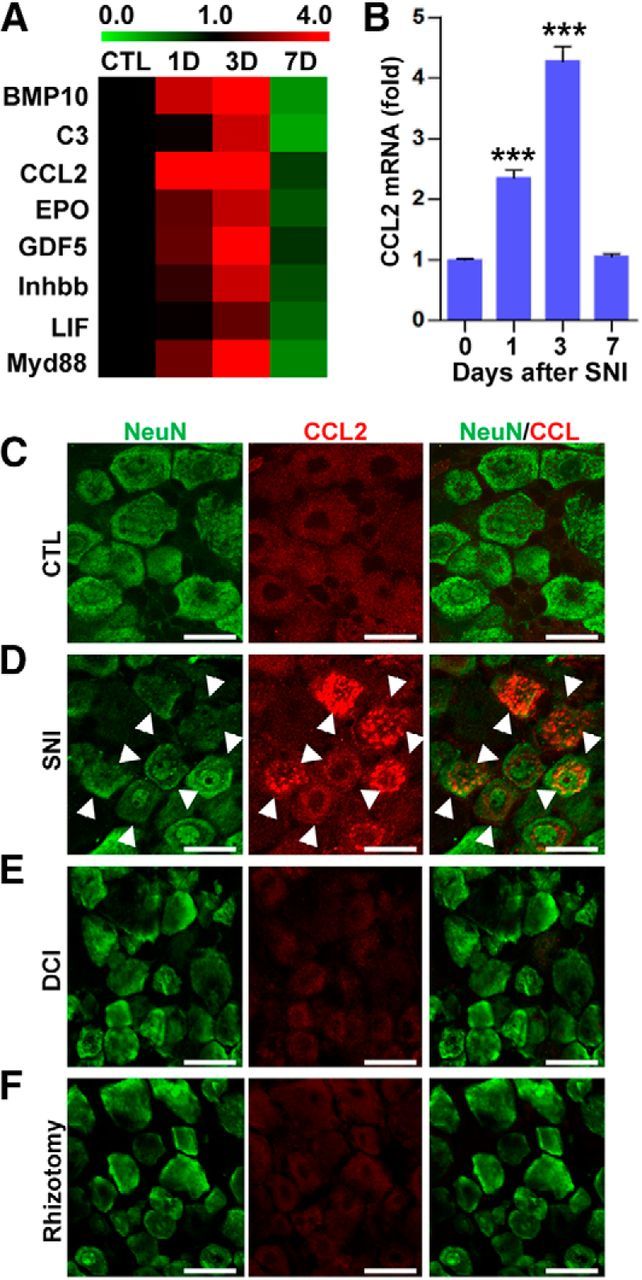
Identification of CCL2 as a potential chemokine in the DRGs after SNI. A, Color-coded heat maps of expression levels measured by chemokine PCR array. Fold changes in chemokine expression level relative to that in the control group were log2-transformed and color-coded based on the color scale shown at the top. The chemokines and chemotaxis-related molecules shown here belonged to the cluster of genes whose expression was already elevated 1 d after SNI. The signals from three independent arrays were averaged to obtain the expression level at each time point. B, A quantification graph of real-time RT-PCR results for CCL2 gene expression in DRG samples obtained before (0 d) or 1, 3, and 7 d after SNI. N = 3 animals for each time point. ***p < 0.001 compared with control (0 d) values by one-way ANOVA followed by Tukey's post hoc analysis. C–F, Representative immunofluorescence images of L5 DRG tissue sections from an uninjured control (CTL) animal (C) or obtained 1 d after SNI (D), dorsal column injury (DCI; E), or rhizotomy (F). Sections were stained with a CCL2 antibody (red) and a NeuN antibody (green). Arrows indicate cells positive for both markers. Scale bars, 50 μm.
In our previous study, intraganglionic injection of cAMP also resulted in an increase in macrophages (Kwon et al., 2013). Furthermore, CM from neuron–macrophage cocultures treated with cAMP robustly promoted neurite outgrowth. We found that cultured DRG neurons increased CCL2 mRNA in response to db-cAMP treatment (Fig. 2A) and the concentration of CCL2 protein in the media was increased accordingly (Fig. 2B). Treatment of db-cAMP in the neuron–macrophage cocultures increased the CCL2 protein concentration in the media to a comparable extent. However, cAMP treatment in cultured macrophages only slightly increased the CCL2 level, suggesting that, in the coculture condition, CCL2 is primarily induced in and secreted from neurons. Next, we examined whether CCL2 plays a functional role in mediating neuron–macrophage interaction in the coculture system. Most of the cultured DRG neurons treated with CM obtained from the cocultures treated with PBS did not grow neurites for 15 h of culture duration (Fig. 2C). However, DRG neurons treated with CM from db-cAMP-treated cocultures showed extensive neurite outgrowth. CCL2-neutralizing antibodies were added to the coculture system together with db-cAMP, and the CM was tested for neurite outgrowth activity. We found that treatment with CCL2-neutralizing antibodies profoundly attenuated the neurite outgrowth activity of the CM (Fig. 2C,D). Because the CCL2-neutralizing antibodies were not removed from the collected CM, the attenuation of neurite outgrowth activity could be due to a direct effect of CCL2 on neurite outgrowth of cultured DRG neurons. To rule out this possibility, CCL2-neutralizing antibodies were added to DRG neuron cultures grown in CM obtained from cocultures treated with db-cAMP alone. The addition of the CCL2-neutralizing antibodies did not affect the neurite outgrowth activity of the CM (Fig. 2C,E), indicating that CCL2 mediates neuron–macrophage interaction in the cocultures stimulated by cAMP rather than directly affecting neurite outgrowth.
Figure 2.
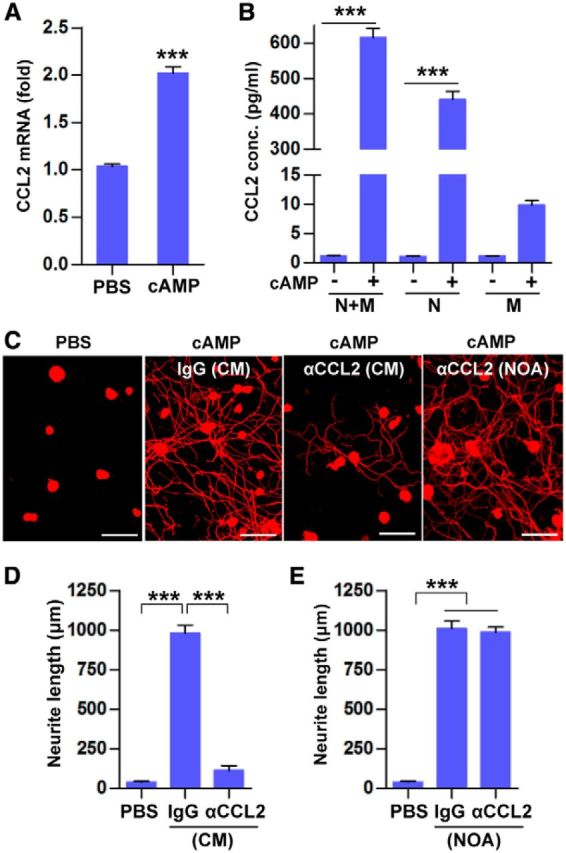
CCL2 plays an essential role in neuron–macrophage interactions in vitro. A, A quantification graph of real-time RT-PCR results for CCL2 expression in cultured DRG neurons treated with db-cAMP (cAMP) or PBS. N = 3 independent cultures for each condition. ***p < 0.001 compared with PBS by unpaired t tests. B, ELISA measurement of the CCL2 concentration in the cell culture media obtained from different culture conditions. N + M, Neuron–macrophage cocultures; N, neuron-only cultures; M, macrophage-only cultures. N = 3 independent cultures for each condition. ***p < 0.001 by unpaired t test. C, Representative images of β III tubulin-positive cultured DRG neurons grown for 15 h with CM obtained from neuron–macrophage cocultures. To obtain the CM, neuron–macrophage cocultures were treated first for 24 h with PBS or cAMP together with control IgG or CCL2-neutralizing antibodies (αCCL2) and then the IgG or CCL2-neutralizing antibodies were maintained in the cultures during the subsequent 72 h CM collection period (CM). In other outgrowth assays, CCL2-neutralizing antibodies were added directly to CM obtained from neuron–macrophage cocultures treated with cAMP so that the antibodies were present only during the neurite outgrowth assay (NOA). D, E, Quantification graphs of neurite length in the experiments in which the antibodies were present during the neuron–macrophage cocultures (CM collection period; D) or only during the NOA (E). N = 4 independent cultures using independent CMs for each condition. ***p < 0.001 compared with PBS values by one-way ANOVA followed by Tukey's post hoc analysis. Scale bars, 100 μm.
The in vitro neuron–macrophage interactions driving the proregenerative macrophage phenotype require CCL2 in neurons and CCR2 in macrophages
Various cells types can produce CCL2 in nervous tissue (Dewald et al., 2005; Zhang et al., 2005). We wanted to confirm that CCL2 derived from neurons mediates neuron–macrophage interactions leading to production of proregenerative factors. As was shown in cultures using rat cells, CM obtained from cocultures of neurons and macrophages from WT mice treated with db-cAMP induced robust neurite outgrowth activity (Fig. 3A,B). In contrast, CM obtained from cocultures of CCL2-deficient neurons and WT macrophages did not have neurite outgrowth activity (Fig. 3C,D). However, using CM from the reverse coculture condition (WT neuron plus CCL2-deficient macrophage) resulted in robust neurite outgrowth of cultured DRG neurons (Fig. 3E,F). This indicates that neuronal CCL2, but not CCL2 generated from macrophages, plays an essential role in mediating the neuron–macrophage interactions. We also set up cocultures of CCR2-deficient neurons and WT macrophages and found that their CM had robust neurite outgrowth activity (Fig. 3G,H). However, the CM from the reverse condition (WT neurons and CCR2-deficient macrophages) had substantially attenuated, but not totally abolished, neurite outgrowth activity (Fig. 3I,J). The remaining neurite outgrowth activity is probably meditated by CCR4, which can also bind CCL2 (Craig and Loberg, 2006). To test this possibility, cocultures composed of WT neurons and CCR2-deficient macrophages were treated with the CCR4 antagonist C 021 dihydrochloride. The CM obtained from these cultures almost completely abolished the remaining neurite outgrowth activity. Together, these data support the idea that neuron-derived CCL2 binds to CCR2/CCR4 receptors on macrophages, thus initiating signaling pathways that lead to the production of proregenerative factors.
Figure 3.

CCL2 in neurons and CCR2/CCR4 in macrophages are required for in vitro neuron–macrophage interactions to produce proregenerative activity. A, C, E, G, I, Representative images of neurite outgrowth in DRG neuron cultures treated with CM obtained from neuron–macrophage cocultures using WT, CCL2-deficient (CCL2−/−), or CCR2-deficient (CCR2−/−) neurons (N) or macrophages (M). Genotype conditions for the cocultures to obtain CM were indicated at the left of the representative images. All cocultures were treated with either PBS or db-cAMP (cAMP), and the CMs were collected for 72 h. I, C 021, CCR4 antagonist, was added at a concentration of 0.1 μm during the coculture period. WT neurons were used for the neurite outgrowth assays for all conditions. DRG neurons and their neurites were visualized by immunofluorescence staining for β III tubulin. Scale bars, 100 μm. B, D, F, H, J, Quantification graphs of neurite outgrowth in the presence of CM from cultures of the different neuron–macrophage genotype combinations treated with PBS or cAMP. N = 4 independent cultures using independent CMs for each condition. ***p < 0.001 and **p < 0.01 compared with PBS values by unpaired t test.
CCL2 is necessary for enhanced regenerative capacity of DRG neurons by conditioning injury
We tested whether conditioning effects are abolished in CCL2-deficient (CCL2−/−) mice. As expected, we could not observe increased macrophage accumulation in DRGs after SNI in CCL2−/− mice (Fig. 4A–C). L4 and L5 DRGs from WT or CCL2−/− mice were freshly dissected at different time points after SNI and cultured for 15 h. In cultures from animals without SNI, there was no neurite outgrowth during this short culture period (Fig. 4D). Neurite outgrowth was clearly observed in DRG neurons taken from WT animals 3 and 7 d after SNI (Fig. 4D,F). The conditioning effects were appreciable even 1 d after SNI; a small number of neurons isolated at this time point achieved substantial neurite outgrowth (Fig. 4D). DRG neurons obtained from CCL2−/− mice 3 and 7 d after SNI did not show any neurite outgrowth (Fig. 4E,F), indicating that conditioning effects were indeed abolished in CCL2−/− mice. Interestingly, a small subset of DRG neurons isolated from CCL2-deficient mice 1 d after SNI showed enhanced neurite outgrowth (Fig. 4E). The percentage of neurons that grew neurites longer than twice the cell diameter was comparable in neurons isolated from WT and CCL2−/− mice at 1 d after SNI (WT, 7.2 ± 0.4%; CCL2−/−, 6.6 ± 0.4%), indicating that CCL2 is dispensable for initial induction of conditioning effects. Conditioning effects were also substantially reduced in CCR2-deficient mice (data not shown). At 7 d after SNI, mean neurite length of dissociated DRG neurons from CCR2-deficient mice was 308.9 ± 50.0 μm, <50% of the length in WT neurons. However, the neurite outgrowth was more extensive than that of CCL2−/− neurons, which can also be explained by redundant binding of CCL2 to CCR4 (Craig and Loberg, 2006).
Figure 4.
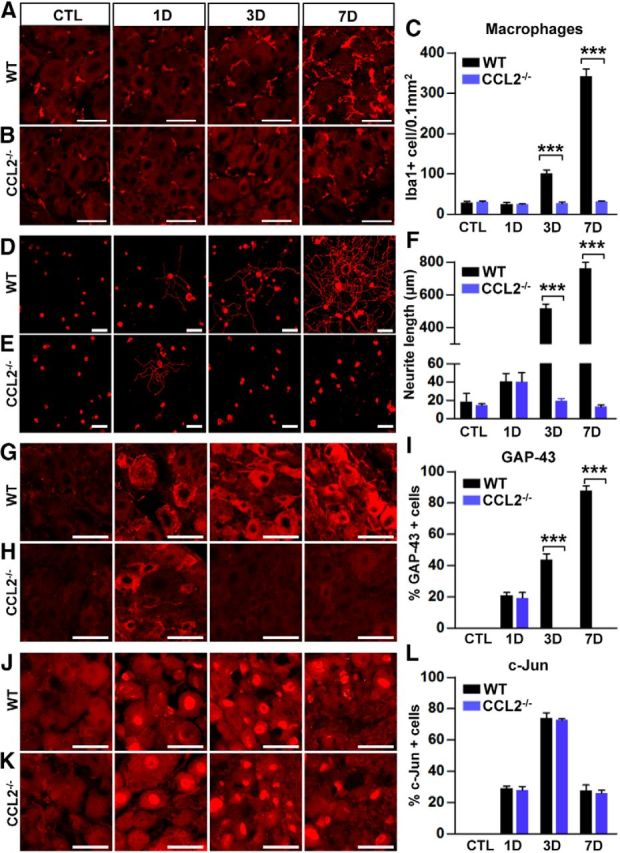
Changes in the number of macrophages in DRG, axon growth capacity, and expression of RAGs in CCL2-deficient mice. A, B, Representative images of Iba1-positive macrophages in DRGSs of WT (A) and CCL2−/− (B) mice at different time points after SNI. C, Quantification of the number of macrophages in WT and CCL2−/− mice 0 (CTL), 1, 3, and 7 d after SNI. N = 3 animals for each condition. Scale bars, 50 μm. D, E, Representative images of neurite outgrowth of DRG neurons taken from WT (D) and CCL2−/− (E) mice at different time points after SNI. Neurons from the L4, L5, and L6 DRGs were cultured for 15 h before being fixed for the immunofluorescent visualization of neurites with anti-β III tubulin. Scale bar, 100 μm. F, Comparison of the mean neurite length between cultures from WT and CCL2−/− mice 0 (CTL), 1, 3, and 7 d after SNI. N = 4 animals for each condition. ***p < 0.001 compared with WT values by unpaired t test. G, H, J, K, Representative immunofluorescence images of GAP-43 (G, H) or c-Jun (J, K) staining in L5 DRG sections obtained from WT (G, J) and CCL2−/− (H, K) mice at different time points after SNI. Scale bars, 50 μm. I, L, Quantification graphs of the percentage of GAP-43-positive (F) or c-Jun-positive (I) cells in WT and CCL2−/− mice 0 (CTL), 1, 3, and 7 d after SNI. N = 3 animals for each condition. ***p < 0.001 compared with WT values by one-way ANOVA followed by Tukey's post hoc analysis.
We examined the expression of RAGs after SNI in CCL2−/− mice. In WT mice, the percentage of DRG neurons expressing GAP-43 gradually increased after SNI (Fig. 4G), resulting in >80% of the neurons being positive for GAP-43 at the 7 d time point (Fig. 4I). In CCL2−/− mice, GAP-43 expression was not observed at the 3 and 7 d time points (Fig. 4H,I), the time points at which conditioning effects were completely abrogated (Fig. 4E). However, GAP-43 expression was clearly observed at the 1 d time point (Fig. 4H), the time point at which conditioning effects were observed to a limited degree. This result reinforces the idea that CCL2 is dispensable for initial regenerative responses triggered by preconditioning SNI and further suggests that CCL2 may be required to amplify and/or maintain the expression of a subset of RAGs associated with sustained promotion of axon growth potential. We also examined the expression of c-Jun, a RAG involved in early transcriptional events. In WT mice, c-Jun expression was induced 1 d after SNI, peaked at 3 d, then decreased by 7 d to a level similar to that at the 1 d time point (Fig. 4J,L). The percentage of neurons expressing c-Jun in CCL2-deficient mice was comparable to that in WT mice 1 d after SNI (Fig. 4K,L). Intriguingly, unlike GAP-43 expression, c-Jun expression in CCL2−/− mice was not reduced at the 3 and 7 d time points, when neurite outgrowth activity was almost completely absent. We also observed a similar pattern of expression for STAT3 (signal transducer and activator of transcription 3), another regeneration-associated transcription factor, in CCL2-deficient mice (data not shown). The preservation of c-Jun and STAT3 expressions at these time points suggest that expressions of a subset of RAGs different from that to which GAP-43 belongs are not influenced by CCL2-mediated proregenerative macrophage activation.
Constitutive deletion of CCL2 should affect inflammatory reactions not only in DRGs but also at the injury site in the sciatic nerve, which may potentially influence the regenerative capacity of DRG neurons (Barrette et al., 2008). Therefore, we also tested for an essential role of CCL2 in conditioning effects by locally injecting CCL2-neutralizing antibodies into L5 DRG immediately after SNI in rats. As expected, there was a robust increase of the macrophage number in the L5 DRG 7 d after SNI (Fig. 5A,B) and the L5 DRG neurons showed enhanced neurite outgrowth when isolated at this time point (Fig. 5C,D). In contrast, intraganglionic injection of CCL2-neutralizing antibodies almost completely abrogated the enhanced neurite outgrowth capacity (Fig. 5C,D) and the diminution of conditioning effects was accompanied by decreased numbers of Iba1-positive macrophages in the L5 DRG (Fig. 5A,B).
Figure 5.

Intraganglionic injection of CCL2 neutralizing antibodies abolishes conditioning effects. A, Representative images of Iba1 staining in L5 DRGs obtained 7 d after SNI of animals with intraganglionic injection of IgG or CCL2-neutralizing antibody (αCCL2). The injection was performed immediately after SNI. Scale bars, 50 μm. B, Comparison of the number of macrophages. N = 3 animals per group. ***p < 0.001. C, Representative images of neurons cultured from L5 DRGs with intraganglionic injection of IgG or αCCL2 of animals subjected 7 d previously to SNI. Scale bars, 100 μm. D, Quantification of neurite outgrowth. N = 3 animals per group. ***p < 0.001.
CCL2 enhances axon growth potential by driving M2 macrophage polarization
Next, we examined whether intraganglionic injection of CCL2 is sufficient to enhance growth capacity mimicking conditioning injury. Injection of recombinant CCL2 into rat L5 DRG substantially increased the number of macrophages in the DRG 7 d after injection (Fig. 6A,C). CCL2 injection was indeed sufficient to enhance neurite outgrowth of cultured L5 DRG neurons isolated 7 d after injection (Fig. 6B,D). The enhancement of regenerative capacity after SNI might be achieved by any chemokine molecule that can recruit macrophages to DRGs rather than by CCL2 specifically. This possibility was tested by injecting other chemokines with macrophage-recruiting activity into the L5 DRG. We chose CX3CL1 (fractalkine) and CCL3 (MIP1-α) because both of these chemokines can be produced by neurons and can recruit macrophages into the CNS (Olszewski et al., 2000; Tong et al., 2000; Kuijpers et al., 2010). In our chemokine PCR array experiment, CX3CL1 belonged to the chemokine cluster showing no increase or a slight decrease, and CCL3 belonged to the cluster showing increase in expression level 3 d after SNI (data not shown). The number of macrophages in L5 DRGs was increased by intraganglionic injection of recombinant CX3CL1 or CCL3 (Fig. 6A,C), to an extent comparable to that induced by SNI (Fig. 5B) or intraganglionic CCL2 injection (Fig. 6C). However, there was no neurite outgrowth in cultures of DRG neurons isolated 7 d after intraganglionic injection with either CX3CL1 or CCL3 (Fig. 6B,D). Therefore, the macrophages recruited by either CX3CL1 or CCL3 do not seem to have the ability to enhance the regenerative capacity of DRG sensory neurons.
Figure 6.
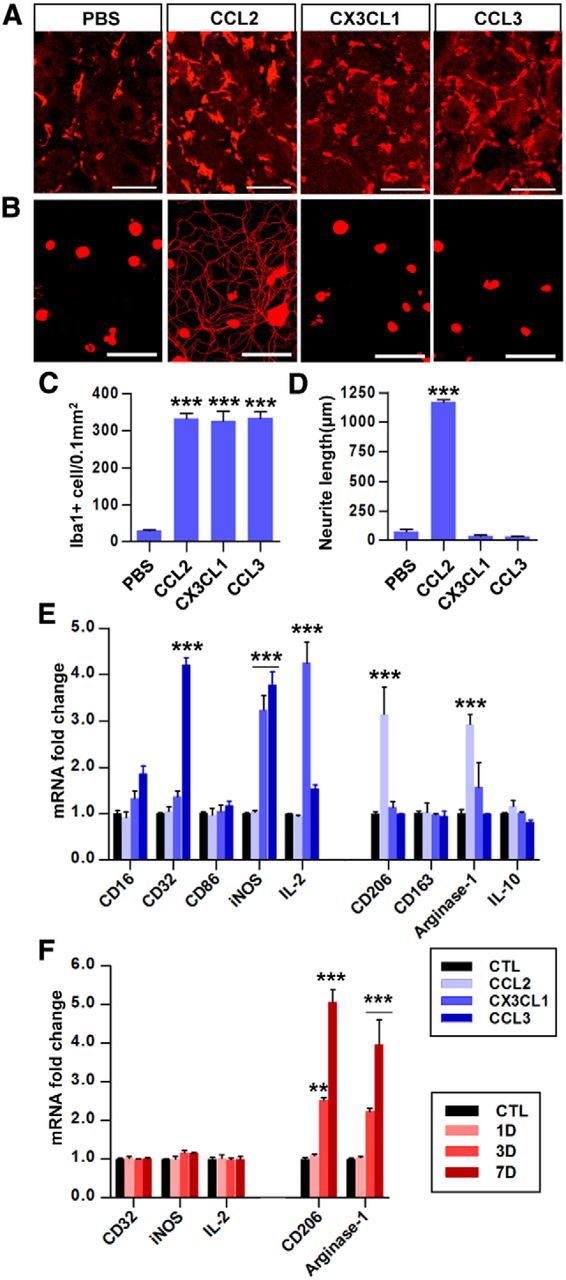
CCL2 is sufficient for enhancement of axon regenerative capacity by mobilizing M2-like macrophages. A, Representative images of Iba1 staining in L5 DRG sections obtained 7 d after intraganglionic injection of PBS or CCL2, CXCL1, and CCL3. Scale bars, 50 μm. B, Representative images of neurons cultured from L5 DRGs dissected from animals with intraganglionic injection of PBS, or CCL2, CXCL1, and CCL3. Scale bars, 100 μm. C, Quantitative comparison of the number of Iba1-positive macrophages in DRGs. N = 3 animals per group. ***p < 0.001. D, Quantitation of neurite outgrowth. The culture period was 15 h and DRG neurons and their neurites were visualized by immunofluorescence staining for β III tubulin. N = 3–4 animals per group. ***p < 0.001. E, Real-time RT-PCR results for M1 and M2 markers in cultured macrophages treated with CCL2, CX3CL1, or CCL3 for 24 h. N = 3 independent cultures per group. ***p < 0.001 compared with the control (untreated) condition. F, Real-time RT-PCR results for of M1 and M2 marker gene expression in MACS-separated (using CD68 antibody) macrophages from the L4 and L5 DRGs at the indicated time points after SNI (CD68-positive fraction). N = 4 animals per group. **p < 0.01 and ***p < 0.001 compared with control values, respectively, by one-way ANOVA followed by Tukey's post hoc analysis.
The above results suggest a possibility that the phenotype of macrophages induced by CCL2 is different from that induced by CX3CL1 or CCL3. To test this idea, cultured macrophages were treated with CCL2, CX3CL1, or CCL3 for 24 h and the expression of various markers of the two main macrophage polarization types, M1 and M2, was examined (Fig. 6E). CCL2-treated macrophages did not show an increase in the expression of any of the tested M1 markers (CD16, CD32, CD86, iNOS, and IL-2). In contrast, both CX3CL1-treated and CCL3-treated macrophages showed enhanced expression of some of these M1 markers: iNOS and IL-2 were elevated following CX3CL1 treatment and CD16, CD32, and iNOS increased after CCL3 treatment. When the expression of M2 markers was evaluated, only CCL2 increased the expression of any of these genes (CD206 and arginase-1). We also examined the expression of M1 and M2 markers in the MACS-separated macrophages in DRGs at different time points after SNI (Fig. 6F). The CD68-positive fraction was devoid of MAP-2 gene expression, while the CD68-negative fraction expressed MAP-2, indicating successful separation between macrophages and neurons (data not shown). SNI increased the expressions of the M2 markers CD206 and arginase-1 in CD68-positive macrophages, whereas the expression of M1 markers (CD32, iNOS, and IL-2) was not significantly affected. These data suggest that CCL2 produced by DRG neurons after SNI may not only recruit but also activate macrophages to take on the M2 phenotype.
Overexpression of CCL2 enhances axon regeneration in vivo
Finally, we sought to examine whether overexpression of CCL2 in DRGs could promote sensory axon regeneration in an in vivo spinal cord injury model. For overexpression of CCL2 in DRG sensory neurons, we chose to use AAV5 based on a previous study reporting that AAV5 is the most efficient serotype for transducing DRG neurons in vivo (Mason et al., 2010). The efficiency of AAV5 in transduction of DRG neurons was tested in our model. AAV5-GFP viral supernatant was injected into L5 DRGs and the expression of GFP was examined 7, 14, and 28 d after injection (Fig. 7A–C). At the 7 d time point, very few GFP+/NeuN+ cells were observed in the L5 DRG. However, the number of GFP+/NeuN+ cells was substantially increased 14 and 28 d after injection. We also confirmed that the majority of the L5 DRG neurons expressed CCL2 by 28 d after injection of AAV5-CCL2 (Fig. 7D–F). Intraganglionic injection of AAV5-CCL2 resulted in a significant increase in the number of macrophages at the 14 d time point compared with the macrophage number in animals with AAV-GFP injection (Fig. 8A,B). The increase was more robust 28 d after injection (Fig. 8A–C). Despite the robust increase in macrophages, neurofilament-positive DRG neurons seemed to be healthy without any sign of DRG tissue damage (Fig. 8D), and there was no immunoreactivity to caspase-3, the marker of cell death (data not shown). These data indicate that the macrophages activated by AAV5-CCL2 did not produce cytotoxic effects. Neurite outgrowth activity of DRG neurons cultured 28 d after intraganglionic AAV5-CCL2 injection was also dramatically enhanced (Fig. 8E,F). In contrast, there was no discernable neurite growth of DRG neurons injected with AAV5-GFP.
Figure 7.
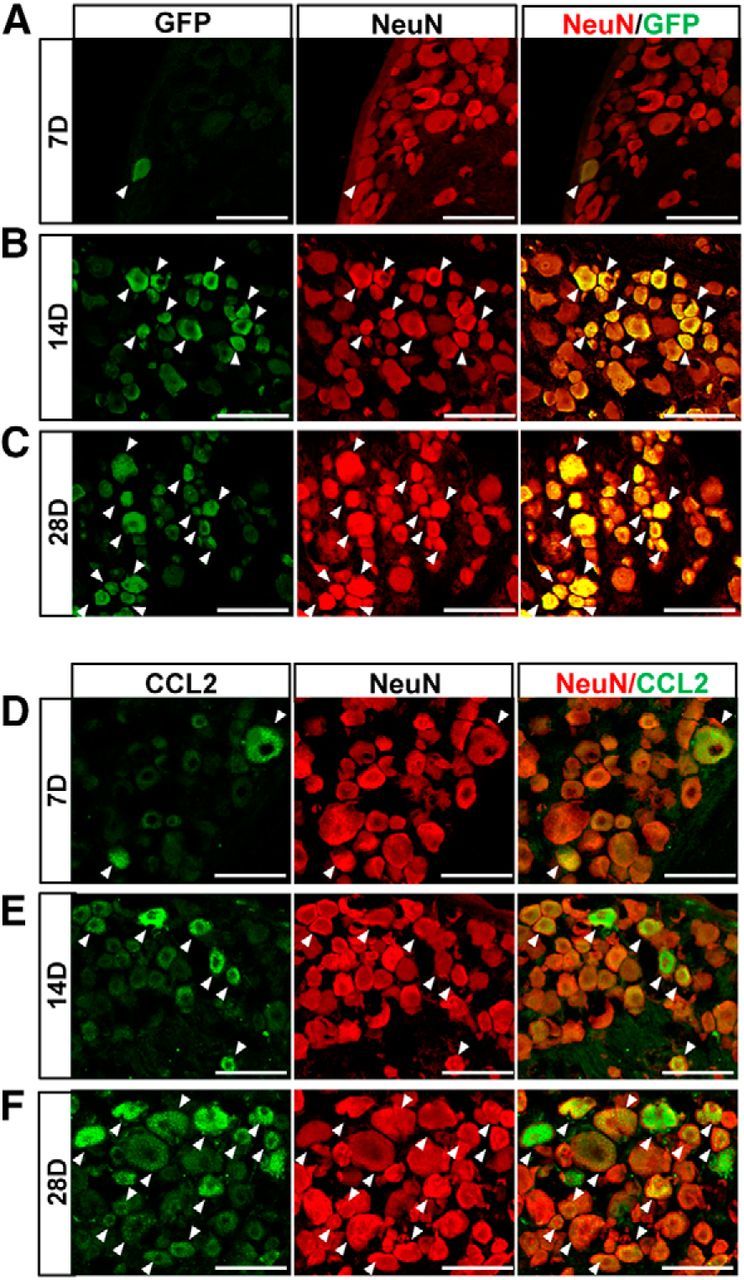
Intraganglionic gene delivery by AAV5. A–C, Confocal images of GFP-positive and NeuN-stained cells in the L5 DRG 7 d (A), 14 d (B), and 28 d (C) after intraganglionic injection of AAV5-GFP. Arrows indicate GFP+/NeuN+ cells. Scale bars, 100 μm. D–F, Confocal images of DRG sections double stained for CCL2 (green) and NeuN (red) 7 d (D), 14 d (E), and 28 d (F) after intraganglionic injection of AAV5-CCL2. Arrows indicate CCL2+/NeuN+ cells. Scale bars, 100 μm.
Figure 8.
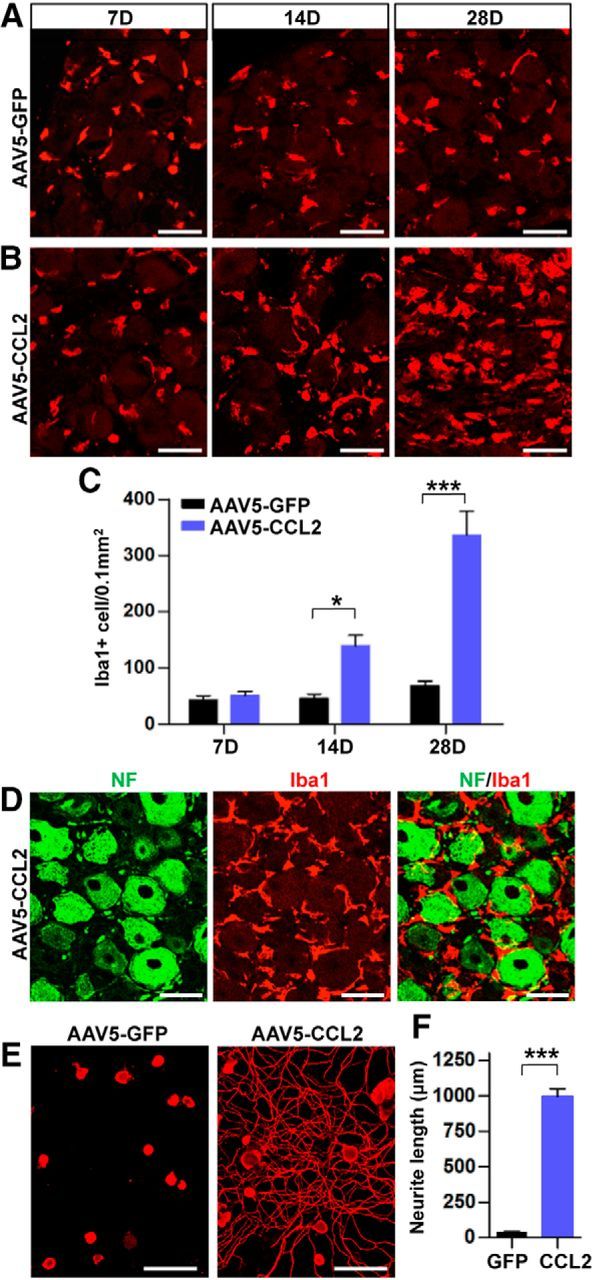
Intraganglionic AAV5-CCL2 injection increases the number of macrophages in the DRGs and enhances neurite outgrowth. A, B, Representative images of Iba1 staining in L5 DRG sections obtained 7, 14, and 28 d after intraganglionic injection of AAV5-GFP (A) or AAV5-CCL2 (B). Scale bars, 50 μm. C, A quantification graph comparing the mean number of Iba1-positive macrophages in DRGs injected with AAV5-GFP or AAV5-CCL2. N = 4 animals per group. *p < 0.05 and ***p < 0.001, respectively, by unpaired t test. D, Representative images of DRG sections stained with neurofilament (NF; green) and Iba1 (red) 28 d after intraganglionic injection of AAV5-CCL2. Scale bars, 50 μm. E, Representative images of neurons cultured from L5 DRGs freshly dissected from animals subjected 28 d previously to intraganglionic injection of AAV5-GFP or AAV5-CCL2. The culture period was 15 h and DRG neurons and their neurites were visualized by immunofluorescence staining for β III tubulin. Scale bars, 100 μm. F, A quantification graph comparing the mean neurite length. ***p < 0.001 by unpaired t test.
We also studied the effects of AAV5-CCL2 on sensory axon regeneration in an in vivo spinal cord injury model. Intraganglionic AAV5-GFP or AAV5-CCL2 injection was performed 7 d before spinal injury. In another group of animals, AAV5-CCL2 was injected 1 d after spinal lesion. Regenerating sensory axons traced by CTB were examined 1 month after the spinal injury. In animals with control AAV5-GFP injection, injured axons retracted a few hundred micrometers away from the caudal end of the injury site (as determined by GFAP staining; Fig. 9A) and they frequently had round and bulging tips resembling retraction bulbs (Fig. 9A, 2). Intraganglionic injection of AAV5-CCL2 7 d before the spinal cord hemisection prevented the axonal retraction and promoted axonal growth beyond the caudal injury border (Fig. 9B). Regenerating axonal fibers were sometimes observed several hundred micrometers rostral to the caudal injury border (Fig. 9B, arrows). Overexpression of CCL2 in DRG neurons did not significantly influence glial environment at the lesion site because intensity of GFAP (astrocytes) or Iba-1 (microglia) immunostaining signals were not changed by CCL2 overexpression (data not shown). Importantly, postinjury AAV5-CCL2 injection supported axonal regeneration to an extent similar to that achieved by preinjury injection (Fig. 9C). The extent of axon regeneration by CCL2 overexpression was comparable to that achieved by preconditioning SNI performed 7 d before the spinal lesion (Fig. 9D). For quantification, we reconstructed axons regenerating beyond the injury border from six parasagittal sections, overlaid them in a single image plane (Fig. 9A'–D'), and summed the length of all axons in the composite image (Fig. 9E). There was no significant difference in the total length of regenerating axons between the preinjury and postinjury injection groups. Similar results were obtained in the comparison of the length of the longest regenerating axons (Fig. 9F) or in the analysis of the number of axons at different distances from the lesion epicenter (Fig. 9G). These results demonstrate that overexpression of CCL2 can lead to robust sensory axon regeneration following spinal cord injury, mimicking conditioning effects.
Figure 9.
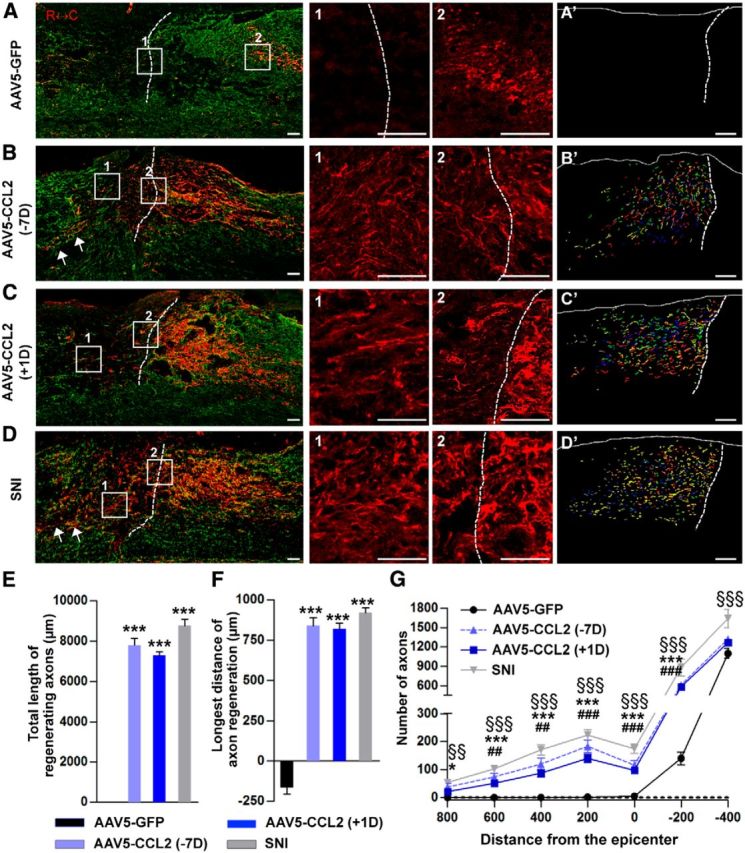
CCL2 overexpression promotes axon regeneration in a spinal cord injury model. A–D, Representative images of CTB-labeled axons (red) and GFAP-immunostained spinal cord sections (green) from animals injected with AAV5-GFP 7 d before injury (A), with AAV5-CCL2 7 d before injury (B, −7D), or with AAV5-CCL2 1 d after injury (C, +1D), and those subjected to preconditioning SNI before creating the spinal lesion (D). Dashed lines indicate caudal lesion borders as determined by GFAP immunostaining. The boxed regions in A–D are magnified in the center panels. Scale bars, 100 μm. A'–D', Axons regenerating beyond the caudal lesion border were reconstructed using Photoshop software from six consecutive parasagittal sections collected at a 60-μm intersection interval. The virtual section image uses different colors to distinguish the different sections. Scale bars, 100 μm. E, A quantification graph comparing the mean total axon length beyond the caudal lesion border measured in the composite images. ***p < 0.001 compared with the AAV5-GFP control value by one-way ANOVA followed by Tukey's post hoc analysis. F, A quantification graph of the mean longest distance of axon regeneration beyond the caudal lesion border. ***p < 0.001 compared with the AAV5-GFP control value by one-way ANOVA followed by Tukey's post hoc analysis. G, A quantification graph comparing the number of CTB-positive axons at different distances from the lesion epicenter. Negative values for distance indicate areas caudal from the epicenter. *p < 0.05 and ***p < 0.001 between animals injected with AAV5-GFP and those with AAV5-CCL2 7 d before the lesion (−7D); ##p < 0.01 and ###p < 0.001 between animals with AAV5-GFP and those with AAV5-CCL2 1 d after the lesion (+1D); §§p < 0.01 and §§§p < 0.001 between animals with AAV5-GFP and those subjected to SNI 7 d before the spinal lesion, by one-way ANOVA followed by Tukey's post hoc analysis. N = 8, 8, 9, and 6 animals for the AAV5-GFP, AAV5-CCL2 (−7D), AAV5-CCL2 (+1D), and SNI groups respectively.
Since CCL2 is known to be a mediator of microglial activation in the dorsal horn in neuropathic pain (Gao and Ji, 2010), we compared the extent of CGRP immunoreactivity in the dorsal horn of the lumbar spinal cord (Fig. 10A). There was no appreciable difference in the CGRP immunoreactivity in animals treated with AAV5-CCL2 either preinjury or postinjury and those treated with control AAV5-GFP. Moreover, intraganglionic AAV5-CCL2 injection, either preinjury or postinjury, did not induce thermal (Fig. 10B) or mechanical hypersensitivity (Fig. 10C).
Figure 10.

Overexpression of CCL2 did not induce neuropathic pain. A, Representative images of CGRP-immunostained lumbar spinal cord sections obtained from animals subjected to intraganglionic injection of AAV5-GFP or AAV5-CCL2 7 d before or 1 d after dorsal hemisection spinal cord injury. Scale bars, 200 μm. B, Graphs showing the withdrawal response latency (in seconds) to nociceptive heat stimulation 14 and 28 d after spinal cord injury in animals with intraganglionic injection of AAV5-GFP or AAV5-CCL2 7 d before or 1 d after the dorsal hemisection spinal injury. C, Graphs showing the flexor reflex withdrawal threshold (in grams) after stimulation of the plantar surface with a series of Von Frey hairs in the same animals. N = 8, 8, and 9 animals for the AAV5-GFP, AAV5-CCL2 (−7D), and AAV5-CCL2 (+1D) groups, respectively.
Discussion
Our previous study suggested that neuron–macrophage interactions may underlie activation of macrophages with a proregenerative phenotype following preconditioning peripheral nerve injury (Kwon et al., 2013). Strong evidence for this came from an in vitro neuron–macrophage coculture experiment in which treatment with cAMP, a substance well known to mimic conditioning effects (Neumann et al., 2002; Qiu et al., 2002), produced proregenerative CM only when the two cell types were cocultured. Using the same coculture system, the present study demonstrates that CCL2 is a key mediator of the neuron–macrophage interaction driving the proregenerative macrophage phenotype.
CCL2 was identified as a candidate mediator by screening for chemokines and related molecules whose expression was regulated before the peak of macrophage activation after preconditioning SNI. Confirming the activity of CCL2 as a neuron–macrophage interaction mediator in the coculture system, CCL2-neutralizing antibodies abolished the proregenerative activity of CM when applied during the coculture period, but not when used during the neurite outgrowth assay. Several lines of evidence indicated that the neurons were the source of CCL2 mediating the neuron–macrophage interactions. Treatment with cAMP resulted in a significant increase in the level of CCL2 protein in the neuron–macrophage coculture or in the neuron-only culture, but not in the culture with macrophages alone. More evidently, cAMP failed to produce proregenerative CM when CCL2-deficient neurons were cocultured with WT macrophages, while the CM obtained from the reverse coculture condition had vigorous neurite outgrowth activity. In vivo, CCL2 expression was confined to the DRG sensory neurons following SNI. These results are in accordance with previous studies demonstrating upregulation of CCL2 expression by neurons following injuries to the peripheral nerves in various chronic pain models (Tanaka et al., 2004; Zhang and De Koninck, 2006; Jeon et al., 2009). CCL2 signals through its cognate receptor, CCR2. Released CCL2 activates neuronal CCR2 receptors in neuropathic pain models (Jung and Miller, 2008; Jung et al., 2008). However, neuronal CCR2 was dispensable for the cAMP-triggered neuron–macrophage interaction in our system: cAMP-treated cocultures of CCR2-deficient neurons and WT macrophages still produced proregenerative CM. Instead, macrophage expression of CCR2 was required as proregenerative activity was significantly attenuated when cocultures of WT neurons and CCR2-deficient macrophages were treated with cAMP. Thus in our system, CCL2 released from neurons activates CCR2 in macrophages to drive them to a proregenerative phenotype.
In CCL2-deficient mice, conditioning effects were preserved 1 d after SNI but the enhanced regenerative capacity of neurons was almost completely abrogated at later time points, suggesting a role for CCL2 in amplification and/or maintenance rather than initiation of proregenerative mechanisms. At the very early time point, a very small number of neurons from CCL2-deficient mice grew significant neurites and the percentage of neurons bearing neurites was comparable to that of neurons from WT mice. This is consistent with our previous study in which infusion of a macrophage deactivator, minocycline, to the DRG was not effective in abrogating the regenerative response at 1 d after SNI (Kwon et al., 2013). These findings suggest that initial regenerative responses are triggered independently of macrophages and inflammatory reactions in the DRGs. In support of this notion, we found unambiguous activation of two RAGs, c-Jun and GAP-43, in DRG neurons at 1 d in both WT and CCL2-deficient mice. Interestingly, however, the expression patterns of the two RAGs were dissociated at the 3 d time point, when neurite outgrowth in cultures of neurons from the CCL2-deficient mice was almost absent. The preservation of c-Jun activation in CCL2-deficient mice suggests that c-Jun and related signaling processes function at a level upstream of CCL2. In contrast, the loss of GAP-43 expression and diminished capacity for axon growth at the later time points indicate that both require CCL2 signaling for amplification and/or maintenance after the 1 d time point.
Based on these observations, we propose a model in which a mechanism linking axotomy-induced retrograde injury signals and CCL2 upregulation plays a critical role in amplifying and/or maintaining regenerative capacity (Fig. 11). One or more RAGs (for example, c-Jun or STAT3) induce upregulation of CCL2 in response to the retrograde injury signals (Fig. 11A,B, RAGx). The initial regenerative responses or expression of a subset of RAGs (for example, GAP-43) are not sustained without CCL2 from injured DRG neurons. Macrophages activated by CCL2 render the microenvironment surrounding the DRG neurons supportive of amplification and/or maintenance of the expression of RAGs and the regenerative capacity of the neurons (Fig. 11C). This growth-supportive perineuronal microenvironment may persist for >1 month, since our previous study found an increase in macrophages up to ≥4 weeks after SNI (Kwon et al., 2013). Therefore, we postulate that axotomy-induced CCL2 upregulation and ensuing perineuronal macrophage activation plays a critical role in the long-term maintenance of the enhancement of regenerative capacity that occurs after conditioning injury (Ylera et al., 2009; Blesch et al., 2012).
Figure 11.
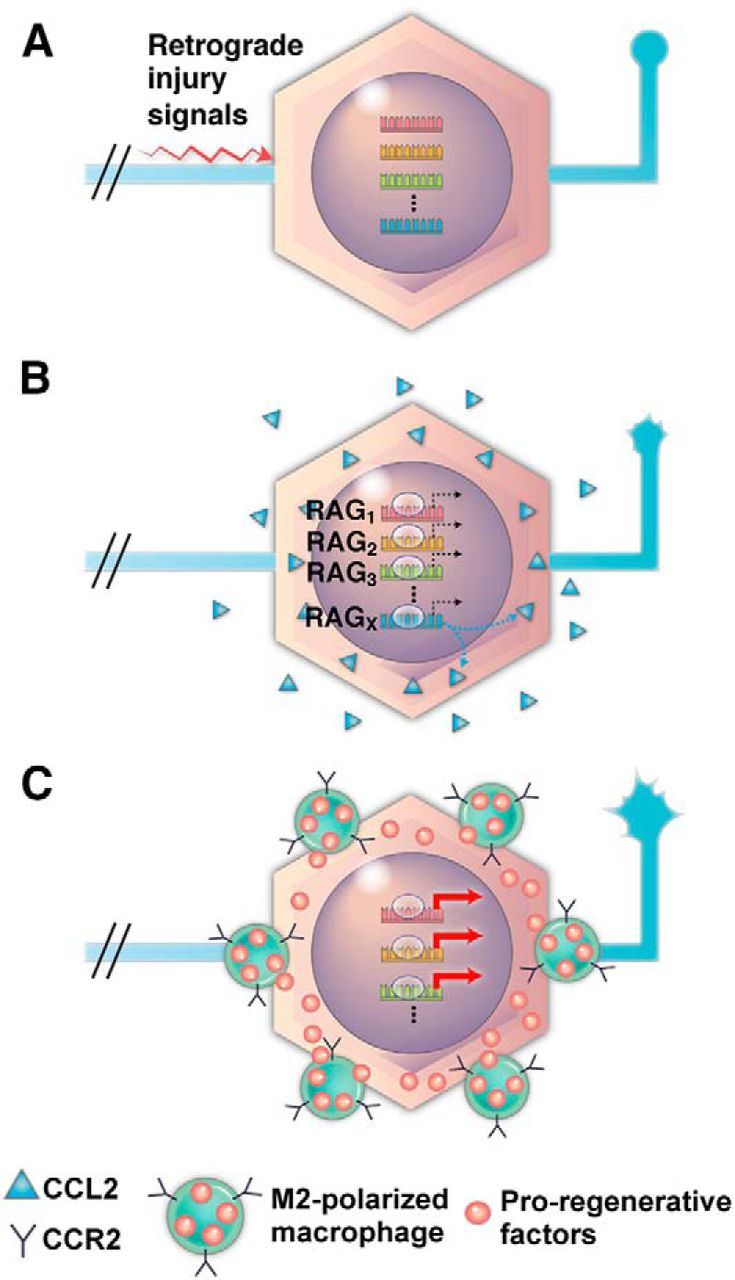
A diagram of the proposed model for the role of CCL2-mediated macrophage activation following preconditioning SNI. A, An axotomy at the sciatic nerve produces injury signals that are retrogradely transmitted to neurons in the DRGs. B, The injury signals induce initial activation (dotted black arrows) of multiple RAGs. Activation of a particular RAG (RAGX) may be specifically linked to the production of CCL2 by DRG neurons (dotted blue arrows). C, Activated macrophages (M2-polarized) expressing CCR2 provide proregenerative factors resulting in the amplification and maintenance (thick red arrows) of RAGs. Changes in the tip morphology of central axons reflect the enhanced regenerative capacity of DRG neurons.
Macrophages can be polarized into different functional phenotypes in response to different environmental cues (Murray et al., 2014; Wang et al., 2014). It has been proposed that M2-polarized macrophages make essential contributions to neural repair processes, such as neurogenesis, angiogenesis, remyelination, and axon regeneration (Kigerl et al., 2009; Hu et al., 2015). CCL2 can polarize macrophages to acquire M2 phenotypes in non-neural environments (Sierra-Filardi et al., 2014; Lee et al., 2015), as well as in the contused spinal cord (Gadani et al., 2015). A recent study also reported that CCL2, in combination with ectodomain of sialic acid-binding Ig-like lectin-9, promotes the M2-like differentiation of macrophages producing CM with robust neurite outgrowth activity (Matsubara et al., 2015). Here, we demonstrated that CCL2 treatment drove the M2 phenotype, whereas CCL3 or CXCL1, both of which failed to enhance axon growth capacity after intraganglionic injection, increased the expression of genes associated with the M1 phenotype. This observation indicates that CCL2 functions not only to recruit macrophages but also to stimulate an M2 phenotype and this M2 polarization may be essential in the regeneration-promoting effects of conditioning injury.
The present study demonstrated that chemokine-mediated macrophage activation has therapeutic potential to enhance axon regeneration following spinal cord injury. Most promising was the finding that postinjury CCL2 activation was as beneficial as preinjury intervention. This is in accordance with studies showing that conditioning injury enhances regenerative responses whether the conditioning injury procedure is performed before or after the central lesion (Kadoya et al., 2009; Ylera et al., 2009). Our finding that overexpression of CCL2 postinjury mimicked conditioning effects suggests that any therapeutic strategy aimed at boosting RAG expression would be effective when instituted after a central injury (as is the case in all clinical situations). Importantly, macrophage activation in the DRGs by chronic activation of CCL2 did not result in neuronal damages, whereas zymosan-induced macrophages produced cytotoxicity in a previous study (Gensel et al., 2009). This discrepancy implies that the CCL2-induced macrophage activation may be more physiologic than the chemically induced process in recapitulating the activation of macrophages and inflammatory reactions that occur following conditioning injury and that a therapeutic use of the physiologic mechanism would avoid inflammation-related cytotoxicity. In addition to an absence of cytotoxicity, CCL2 overexpression induced no changes in the threshold for pain perception. The absence of uncontrolled growth of pain fibers suggests that the enhanced regenerative capacity induced by CCL2-mediated macrophage activation may selectively influence regeneration of injured axons and not sprouting of uninjured axons terminating at the dorsal horns. Thus, the therapeutic effects of CCL2 modulation seem to be unaccompanied by undesirable side effects.
Considering a previous report showing involvement of CCL2 in regulating macrophage responses to remove myelin debris from damaged axons (Perrin et al., 2005), it is conceivable that CCL2 overexpressed in DRGs by AAV in our study might enhance myelin breakdown at the injury site and thereby enhance regeneration of injured axons. However, we could not detect obvious CCL2 immunoreactivity in the lesion epicenter at the time point when the CCL2 immunoreactivity in DRGs was evidently observed (data not shown), suggesting that CCL2 overexpressed in the cell body may not be effectively transported to the axon terminals. Upregulation of CCL2 expression in the lesioned spinal cord or severed sciatic nerve is usually limited within several days after injury (Ma et al., 2002; Perrin et al., 2005). This suggests that acutely upregulated CCL2 mobilizes macrophages capable of degrading myelin debris at the lesion site. Therefore, though we cannot exclude a possibility that a small amount of CCL2 may be transported to the axon terminals, it is unlikely that the potential CCL2 upregulation at a later period would significantly function to enhance removal of myelin debris. In addition, we could not detect any significant changes in the intensity of GFAP or Iba-1 immunoreactivity at the lesion site in animals with CCL2 overexpression. These findings together indicate that the enhanced regeneration by CCL2 overexpression in DRGs cannot be explained by alterations in cellular and molecular environment at the lesion site.
In conclusion, we demonstrate that axotomy-induced CCL2 mediates neuron–macrophage interactions to drive activation of proregenerative macrophages after conditioning injury. CCL2-mediated neuron–macrophage interactions are not required for the induction of conditioning effects but play a critical role in amplifying and/or maintaining a subset of RAG expression and enhancing regenerative capacity. Finally, chronic overexpression of CCL2 in DRGs promotes sensory axon regeneration beyond the spinal lesion site without harmful side effects, highlighting the potential of chemokine-induced macrophage activation for axon-regeneration therapeutics.
Footnotes
This work was supported by National Research Foundation of Korea grants funded by the Korean government (MSIP; 2012R1A2A2A01013143, 2012R1A5A2051429, 2014M3A9B6034224). We thank the Vector Core Facility of the University of North Carolina at Chapel Hill for rAAV viral vector production.
The authors declare no competing financial interests.
References
- Baldwin KT, Carbajal KS, Segal BM, Giger RJ. Neuroinflammation triggered by β-glucan/dectin-1 signaling enables CNS axon regeneration. Proc Natl Acad Sci U S A. 2015;112:2581–2586. doi: 10.1073/pnas.1423221112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrette B, Hébert MA, Filali M, Lafortune K, Vallières N, Gowing G, Julien JP, Lacroix S. Requirement of myeloid cells for axon regeneration. J Neurosci. 2008;28:9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesch A, Lu P, Tsukada S, Alto LT, Roet K, Coppola G, Geschwind D, Tuszynski MH. Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to camp-mediated effects. Exp Neurol. 2012;235:162–173. doi: 10.1016/j.expneurol.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell. 2013;155:894–908. doi: 10.1016/j.cell.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig MJ, Loberg RD. CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer metastasis Rev. 2006;25:611–619. doi: 10.1007/s10555-006-9027-x. [DOI] [PubMed] [Google Scholar]
- Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- Fang X, Djouhri L, McMullan S, Berry C, Waxman SG, Okuse K, Lawson SN. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J Neurosci. 2006;26:7281–7292. doi: 10.1523/JNEUROSCI.1072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Finelli MJ, Wong JK, Zou H. Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci. 2013;33:19664–19676. doi: 10.1523/JNEUROSCI.0589-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron. 2015;85:703–709. doi: 10.1016/j.neuron.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Nakamura S, Guan Z, van Rooijen N, Ankeny DP, Popovich PG. Macrophages promote axon regeneration with concurrent neurotoxicity. J Neurosci. 2009;29:3956–3968. doi: 10.1523/JNEUROSCI.3992-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Kigerl KA, Mandrekar-Colucci SS, Gaudet AD, Popovich PG. Achieving CNS axon regeneration by manipulating convergent neuro-immune signaling. Cell Tissue Res. 2012;349:201–213. doi: 10.1007/s00441-012-1425-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk TG, Leibinger M, Müller A, Andreadaki A, Knippschild U, Fischer D. Stimulation of axon regeneration in the mature optic nerve by intravitreal application of the toll-like receptor 2 agonist Pam3Cys. Invest Ophthalmol Vis Sci. 2010;51:459–464. doi: 10.1167/iovs.09-4203. [DOI] [PubMed] [Google Scholar]
- Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. doi: 10.1038/nrneurol.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Lee KM, Cho HJ. Expression of monocyte chemoattractant protein-1 in rat dorsal root ganglia and spinal cord in experimental models of neuropathic pain. Brain Res. 2009;1251:103–111. doi: 10.1016/j.brainres.2008.11.046. [DOI] [PubMed] [Google Scholar]
- Jung H, Miller RJ. Activation of the nuclear factor of activated T-cells (NFAT) mediates upregulation of CCR2 chemokine receptors in dorsal root ganglion (DRG) neurons: a possible mechanism for activity-dependent transcription in DRG neurons in association with neuropathic pain. Mol Cell Neurosci. 2008;37:170–177. doi: 10.1016/j.mcn.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. 2008;104:254–263. doi: 10.1111/j.1471-4159.2007.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoya K, Tsukada S, Lu P, Coppola G, Geschwind D, Filbin MT, Blesch A, Tuszynski MH. Combined intrinsic and extrinsic neuronal mechanisms facilitate bridging axonal regeneration one year after spinal cord injury. Neuron. 2009;64:165–172. doi: 10.1016/j.neuron.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers M, van Gassen KL, de Graan PN, Gruol D. Chronic exposure to the chemokine CCL3 enhances neuronal network activity in rat hippocampal cultures. J Neuroimmunol. 2010;229:73–80. doi: 10.1016/j.jneuroim.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MJ, Kim J, Shin H, Jeong SR, Kang YM, Choi JY, Hwang DH, Kim BG. Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J Neurosci. 2013;33:15095–15108. doi: 10.1523/JNEUROSCI.0278-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li S, Karimi-Abdolrezaee S, Busch SA, Shen Y, Silver J. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature. 2015;518:404–408. doi: 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, Karpurapu M, Deng J, Qian F, Kelly EA, Jarjour NN, Ackerman SJ, Natarajan V, Christman JW, Park GY. Recruited alveolar macrophages, in response to airway epithelial-derived MCP-1/CCL2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol. 2015;52:772–784. doi: 10.1165/rcmb.2014-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Richardson PM. Inflammation near the nerve cell body enhances axonal regeneration. J Neurosci. 1991;11:972–978. doi: 10.1523/JNEUROSCI.11-04-00972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Richardson PM. Responses of macrophages in rat dorsal root ganglia following peripheral nerve injury. J Neurocytol. 1993;22:334–341. doi: 10.1007/BF01195557. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB. Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res. 2002;68:691–702. doi: 10.1002/jnr.10269. [DOI] [PubMed] [Google Scholar]
- Mason MR, Ehlert EM, Eggers R, Pool CW, Hermening S, Huseinovic A, Timmermans E, Blits B, Verhaagen J. Comparison of AAV serotypes for gene delivery to dorsal root ganglion neurons. Mol Ther. 2010;18:715–724. doi: 10.1038/mt.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara K, Matsushita Y, Sakai K, Kano F, Kondo M, Noda M, Hashimoto N, Imagama S, Ishiguro N, Suzumura A, Ueda M, Furukawa K, Yamamoto A. Secreted ectodomain of sialic acid-binding Ig-like lectin-9 and monocyte chemoattractant protein-1 promote recovery after rat spinal cord injury by altering macrophage polarity. J Neurosci. 2015;35:2452–2464. doi: 10.1523/JNEUROSCI.4088-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/S0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/S0896-6273(02)00702-X. [DOI] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernández L, Lindborg JA, Mandell D, Zigmond RE. A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J Neurosci. 2013;33:16236–16248. doi: 10.1523/JNEUROSCI.3319-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan KJ, Ma K, Guo H, Wang C, Sun F, Han SB, Kim H, Wong JK, Charron J, Zou H, Son YJ, He Z, Zhong J. B-RAF kinase drives developmental axon growth and promotes axon regeneration in the injured mature CNS. J Exp Med. 2014;211:801–814. doi: 10.1084/jem.20131780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski MA, Huffnagle GB, McDonald RA, Lindell DM, Moore BB, Cook DN, Toews GB. The role of macrophage inflammatory protein-1 alpha/CCL3 in regulation of T cell-mediated immunity to Cryptococcus neoformans infection. J Immunol. 2000;165:6429–6436. doi: 10.4049/jimmunol.165.11.6429. [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Kanter JL, He Z. PTEN/mTOR and axon regeneration. Exp Neurol. 2010;223:45–50. doi: 10.1016/j.expneurol.2009.12.032. [DOI] [PubMed] [Google Scholar]
- Perrin FE, Lacroix S, Avilés-Trigueros M, David S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain. 2005;128:854–866. doi: 10.1093/brain/awh407. [DOI] [PubMed] [Google Scholar]
- Puttagunta R, Tedeschi A, Sória MG, Hervera A, Lindner R, Rathore KI, Gaub P, Joshi Y, Nguyen T, Schmandke A, Laskowski CJ, Boutillier AL, Bradke F, Di Giovanni S. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat Commun. 2014;5:3527. doi: 10.1038/ncomms4527. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/S0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- Rishal I, Fainzilber M. Axon-soma communication in neuronal injury. Nat Rev Neurosci. 2014;15:32–42. doi: 10.1038/nrn3609. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Sierra-Filardi E, Nieto C, Domínguez-Soto A, Barroso R, Sánchez-Mateos P, Puig-Kroger A, López-Bravo M, Joven J, Ardavín C, Rodríguez-Fernández JL, Sánchez-Torres C, Mellado M, Corbí AL. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192:3858–3867. doi: 10.4049/jimmunol.1302821. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–469. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Tong N, Perry SW, Zhang Q, James HJ, Guo H, Brooks A, Bal H, Kinnear SA, Fine S, Epstein LG, Dairaghi D, Schall TJ, Gendelman HE, Dewhurst S, Sharer LR, Gelbard HA. Neuronal fractalkine expression in HIV-1 encephalitis: roles for macrophage recruitment and neuroprotection in the central nervous system. J Immunol. 2000;164:1333–1339. doi: 10.4049/jimmunol.164.3.1333. [DOI] [PubMed] [Google Scholar]
- Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1–m2 polarization balance. Front Immunol. 2014;5:614. doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylera B, Ertürk A, Hellal F, Nadrigny F, Hurtado A, Tahirovic S, Oudega M, Kirchhoff F, Bradke F. Chronically CNS-injured adult sensory neurons gain regenerative competence upon a lesion of their peripheral axon. Curr Biol. 2009;19:930–936. doi: 10.1016/j.cub.2009.04.017. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang N, Inan S, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci U S A. 2005;102:4536–4541. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]