

Abstract
Expression of the neuronal K/Cl transporter KCC2 is tightly regulated throughout development and by both normal and pathological neuronal activity. Changes in KCC2 expression have often been associated with altered chloride homeostasis and GABA signaling. However, recent evidence supports a role of KCC2 in the development and function of glutamatergic synapses through mechanisms that remain poorly understood. Here we show that suppressing KCC2 expression in rat hippocampal neurons precludes long-term potentiation of glutamatergic synapses specifically by preventing activity-driven membrane delivery of AMPA receptors. This effect is independent of KCC2 transporter function and can be accounted for by increased Rac1/PAK- and LIMK-dependent cofilin phosphorylation and actin polymerization in dendritic spines. Our results demonstrate that KCC2 plays a critical role in the regulation of spine actin cytoskeleton and gates long-term plasticity at excitatory synapses in cortical neurons.
SIGNIFICANCE STATEMENT Changes in the expression of neuronal chloride transporters, such as KCC2, occur during postnatal development and are induced in a variety of neurological and psychiatric conditions. Such changes are expected to primarily impact GABA signaling because GABAA receptors are predominantly permeable to chloride ions. However, the KCC2 transporter forms clusters near glutamatergic synapses and interacts with several actin-related proteins. We show that KCC2 is strictly required for LTP expression at hippocampal excitatory synapses. This effect is due to KCC2 interaction with the Rac1/PAK signaling pathway that controls actin polymerization. Suppressing this interaction promotes actin polymerization thereby hindering AMPA receptor traffic upon KCC2 suppression. Alterations of KCC2 expression therefore impact not only GABAergic signaling but also glutamatergic synaptic function and long term plasticity.
Keywords: actin, AMPA receptor, KCC2, spine, STED microscopy, synaptic plasticity
Introduction
Fast synaptic inhibition in the brain relies on activation of chloride-permeant GABAA and glycine receptor channels. Hence, chloride homeostasis is critical to maintain the efficacy and polarity of signals mediated by these receptors. In mature neurons, transmembrane Cl gradients are primarily established by opposing actions of the NKCC1 and KCC2 cotransporters, resulting in a net efflux of chloride (Blaesse et al., 2009). Upregulation of KCC2 expression during development is associated with a progressive hyperpolarizing shift in the reversal potential of IPSCs (Rivera et al., 1999). KCC2 may also be rapidly downregulated by neuronal activity through post-translational modifications (Chamma et al., 2012) as well as modifications at the transcriptional level in a variety of neurological and psychiatric disorders (Coull et al., 2003; Cohen et al., 2002; Boulenguez et al., 2010; Tyzio et al., 2014). Because most of these conditions are associated with partial loss of inhibition, high expectations are put on diuretic drugs that may act to compensate for the loss of KCC2 function and thereby restore neuronal chloride homeostasis and synaptic inhibition (Kahle et al., 2008; Gagnon et al., 2013; Löscher et al., 2013).
KCC2, however, is not specifically enriched near GABAergic synapses but instead shows diffuse somatodendritic distribution with large clusters in dendritic spines near postsynaptic densities (Gulyás et al., 2001; Chamma et al., 2013), suggestive of interactions with glutamatergic signaling. Precocious expression of recombinant KCC2 in embryonic cortical neurons leads to exuberant spinogenesis and excitatory synapse density (Fiumelli et al., 2013). Conversely, genetic ablation of KCC2 precludes spine morphogenesis and excitatory synapse formation (Li et al., 2007), whereas chronic KCC2 suppression in mature neurons reduces the confinement of several transmembrane proteins, including AMPARs within dendritic spines (Gauvain et al., 2011). Interestingly, these effects are independent of ion transport and instead involve KCC2 interactions with intracellular partners. KCC2 has been shown to be engaged in variety of protein interactions that may impact its aggregation and function in dendritic spines (Medina et al., 2014). These include synaptic proteins, such as GluK2 (Mahadevan et al., 2014) and its interacting protein Neto2 (Ivakine et al., 2013), acting to enhance KCC2 membrane stability, as well as actin-related proteins, such as 4.1N (Li et al., 2007) and βPIX (Llano et al., 2015), which may influence KCC2 anchoring to spine cytoskeleton (Chamma et al., 2013) and actin polymerization (Saneyoshi et al., 2008), respectively. Such interactions are therefore predicted to influence spine actin cytoskeleton.
Dendritic spines exhibit various forms of activity-driven plasticity leading to coordinated modulation of their structure and synaptic function (Bosch and Hayashi, 2012). Long-term potentiation (LTP) of excitatory synapses involves synaptic translocation of proteins, including AMPARs (Malinow and Malenka, 2002) and a persistent increase in spine volume (Kopec et al., 2006; Harvey and Svoboda, 2007). The two processes are mechanistically correlated (Kopec et al., 2007) and rely on rapid remodeling of actin cytoskeleton within dendritic spines. This remodeling may be both permissive for activity-driven AMPA receptor membrane insertion (Gu et al., 2010) and lateral diffusion (Rust et al., 2010) as well as required for spine head enlargement (Okamoto et al., 2004). Here, we therefore tested whether the reciprocal interaction between KCC2 and spine actin cytoskeleton may impact LTP at excitatory synapses in hippocampal neurons. We show that KCC2 is strictly required for LTP expression and that suppressing KCC2 expression or protein interactions with its carboxy-terminal domain prevents activity-driven membrane insertion of AMPARs. This effect reflects remodeling of F-actin in dendritic spines due to Rac1/PAK- and LIMK-dependent inhibition of cofilin. Our results reveal that KCC2 plays a critical role at mature cortical excitatory synapses where it not only controls synaptic efficacy but also gates the expression of long-term plasticity.
Materials and Methods
DNA and lentiviral constructs.
Rat Slc12a5-specific and nontarget shRNA sequences (Gauvain et al., 2011) were inserted in a pGeneClip hMGFP vector (Promega) for neuronal transfection in vitro. For some experiments requiring massive transduction (see Figs. 1, 4E, 7A), the same sequences were introduced in pTRIP vector under U6 promoter and used to produce purified lentiviral particles (titer 7–9 × 109 IU/ml, UNC Vector Core facility). Knockdown efficiency was verified by both immunohistochemistry (see Fig. 1B) and Western blotting (−70%, p = 0.04). The pEGFP-N1 vector used in some experiments was from Clontech. The GluA1-superecliptic pHluorin (SEP) construct in pCI vector from the Malinow laboratory (Kopec et al., 2006) was obtained from Addgene. Rat KCC2-CTD (amino acids 637–1116) was cloned into the pEGFP-IRES vector as described previously (Gauvain et al., 2011). The LifeAct Venus construct was obtained by replacing GFP by Venus sequence from a pCMV LifeAct-GFP2 construct (Gauvain et al., 2011). mCherry-tCaMKII was obtained by replacing GFP by mCherry from GFP-tCaMKII plasmid from the Malinow laboratory. mCherry-tCaMKII was then cloned into a pCMV-Tet3G-TRE vector (Clontech) to allow for doxycycline-induced expression (TET-ON system). For rescue experiments, recombinant KCC2 4904–4924 nucleotide sequence AAC GAG GTC ATC GTG AAT AAA TCC was replaced with AAT GAA GTT ATT GTT AAC AAG TCT to preserve amino acid sequence. Rescue efficacy was verified both by KCC2 immunocytochemistry and in electrophysiological assays of chloride extrusion (data not shown).
Figure 1.

Chronic knockdown of KCC2 precludes LTP at perforant path inputs onto dentate gyrus granule cells. A, Maximal intensity projection of confocal micrographs showing EGFP (green) and DAPI (blue) staining in parasagittal hippocampal slices 2 weeks after stereotaxic injection of a lentivirus-expressing nontarget (shNT) shRNA sequence and EGFP. Arrowhead indicates mossy fibers from infected granule cells. Scale bar, 200 μm. B, Maximal intensity projection of confocal micrographs of dentate gyrus areas infected with lentiviruses expressing either nontarget (shNT) or KCC2-directed (shKCC2) shRNA, showing massive silencing of KCC2 expression in the latter (see in particular KCC2 immunostaining in st. granulosum [st.gr.]). Scale bar, 25 μm. C, Superimposed average of 60 consecutive fEPSPs recorded within st. moleculare (st. mol.) upon extracellular stimulation of perforant path inputs, before (gray) and 40 min after (black) high-frequency stimulation (HFS, 4 × 1 s at 100 Hz). D, Left, Time course of changes in fEPSP slope upon HFS protocol in slices from animals infected with lentiviruses expressing shNT (n = 8) or shKCC2 (n = 10). Blue bars represent the time windows used for averages. Right, Summary data of fEPSP slope 40 min after HFS, normalized to the mean of 10 min baseline. **p < 0.01.
Figure 4.

Defect in activity-dependent GluA1 membrane delivery downstream αCaMKII activation and GluA1 S845 phosphorylation. A, Schematic representation of construct used for doxycycline-inducible, truncated (constitutively active) αCaMKII expression in hippocampal neurons. B, Confocal fluorescence micrographs of mCherry (red) and SEP-GluA1 (pseudocolors) in dendritic sections of neurons expressing mCherry, SEP-GluA1, and inducible truncated αCaMKII, in the absence (Control) or after 24 h exposure to 2 μg/ml doxycycline. No mCherry expression was detected in the absence of doxycycline. Scale bar, 2 μm. C, Overlay of confocal fluorescence micrographs of mCherry and SEP-GluA1 in neurons expressing mCherry, SEP-GluA1, and inducible truncated αCaMKII and either nontarget (shNT) or KCC2-directed (shKCC2) shRNA, in the absence or 24 h presence of doxycycline. Scale bar, 2 μm. D, Quantification of SEP fluorescence in neurons not exposed to doxycycline (Ctrl) or exposed to doxycycline for 24 h (+Dox), normalized to the mean SEP fluorescence in the absence of doxycycline (shNT: Ctrl, n = 105; +Dox, n = 121 shKCC2: Ctrl, n = 84; +Dox, n = 90). *p < 0.05. **p < 0.01. n.s., Nonsignificant. E, Representative immunoblots of total or Ser845-phosphorylated GluA1 and Tuj1 from hippocampal cultures infected with lentiviruses expressing either nontarget (shNT) or KCC2-directed (shKCC2) shRNA, before (Ctrl) or at different times of cLTP induction. F, Quantification from 3 or 4 independent experiments of pS845/total GluA1 ratios from immunoblots as in D, normalized to that in shNT-expressing neurons before cLTP induction. No significant difference in GluA1 S845 phosphorylation was observed between shNT- and shKCC2-expressing neurons, either in control or upon cLTP induction.
Figure 7.
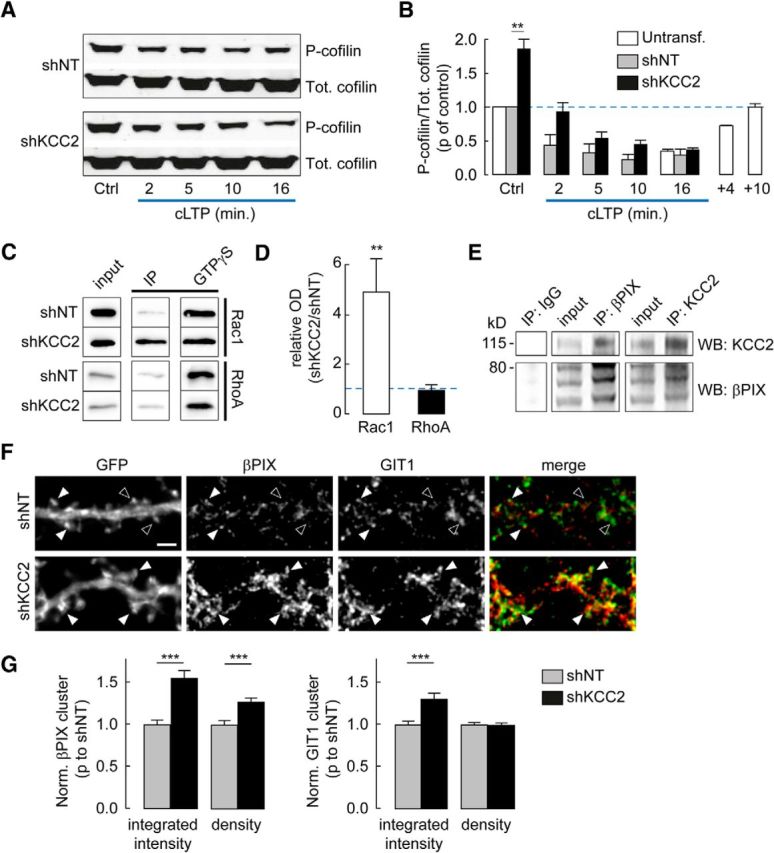
Phosphorylation of cofilin and recruitment of Rac1 signaling in neurons lacking KCC2. A, Representative immunoblots of total (Tot.) or Ser3-phosphorylated (P-) cofilin from hippocampal cultures transduced with lentiviruses expressing either nontarget (shNT) or KCC2-directed (shKCC2) shRNA, before (Ctrl) or at different times of cLTP induction. B, Quantification from three independent experiments of pSer3/total cofilin ratios from immunoblots as in A, normalized to that in shNT-expressing neurons before cLTP induction. Note the twofold increase in pSer3/total cofilin ratio in shKCC2-expressing neurons before cLTP induction. **p < 0.01. White bars represent data from uninfected neurons, showing a transient, ≈75% decrease in pSer3/total cofilin ratio during cLTP induction with complete recovery within 10 min. C, Western blots from cultures transduced with lentiviruses expressing shNT or shKCC2, probed with Rac1 (left) or RhoA (right) antisera after pull-down assays using GST-PAK (Rac1) or GST-Rhotekin (RhoA) in the absence (IP) or presence (GTPγS) of 200 μm GTPγS. D, Quantification from 4 (Rac1) and 3 (RhoA) experiments as in C, expressed as the ratio of activated Rho GTPase signal normalized to maximal activity in GTPγS, in protein extracts from neurons expressing shKCC2 over neurons expressing shNT. **p < 0.01. E, Coimmunoprecipitation (IP) experiment from uninfected neuron cultures at 21 DIV. Western blots (WB) show enrichment and detection of βPIX after KCC2 pull-down (middle) and reciprocally (left). F, Wide-field fluorescent micrographs of representative dendritic sections of neurons expressing EGFP and either nontarget (shNT) or KCC2-directed shRNA (shKCC2). Neurons were immunostained for GFP, βPIX (red), and GIT1 (green). Filled arrowheads indicate dendritic spines immunopositive for both βPIX and GIT1. Open arrowheads indicate spines with no colocalization. Scale bar, 2 μm. G, Quantification of βPIX (left) and GIT1 (right) cluster integrated intensity and density, normalized to that in shNT-expressing neurons. Data are from two independent experiments as in F, and 104 (shNT) and 110 (shKCC2) cells, respectively. ***p < 0.001.
Cell culture and transfection.
Hippocampal neurons were prepared as described previously (Gauvain et al., 2011) from embryonic day 19 Sprague Dawley rat pups. After dissociation, cells were plated on polyornithine-coated glass coverslips at a density of 3.4 × 104 cells/cm−2 and maintained in a CO2 incubator set at 37°C in a culture medium composed of Neurobasal supplemented with B27 (Invitrogen), 2 mm glutamine, and penicillin/streptomycin. After 2 weeks, neurons were transfected using transfectin (Bio-Rad) according to the manufacturer's instructions (with 1 μg DNA for 3 μl transfectin per well). Neurons were then used for biological assays within 7–10 d. KCC2 extinction by RNA interference was detected from 4 d after transfection (Gauvain et al., 2011). To maintain a similar timeframe, pharmacological blockade experiments were performed by applying the specific KCC2 antagonist VU0240551 (6 μm) for 3 days.
In some experiments shown in Figure 8, neurons were exposed to LIMK or GTPase inhibitors. Ser3 peptide was synthesized by Proteogenix and contained the 16 N-terminal amino-acid sequence of cofilin (MASGVAVSDGVIKVFN) fused after 3 glycine residues to the penetratin sequence (RQIKIWFQNRRMKWKK), as described previously (Gu et al., 2010). Ser3 peptide (20 μg/ml), LIMKi (10 μm in DMSO, Millipore), and Rhosin (30 μm, Millipore) were applied onto neuron cultures for 4–5 h before experiments. IPA-3 (5 μm, Millipore) was applied for 24 h. LIMKi, IPA-3, and Rhosin were also present during recordings.
Figure 8.

KCC2 controls actin depolymerization through a Rac1-PAK-LIMK pathway. A, Overlay of mCherry (red) and SEP (pseudocolors) spinning-disc fluorescence micrographs of dendritic sections of shNT or shKCC2-expressing neurons before (Control) and 35 min after cLTP induction. Neuron cultures were exposed either for 4 h to cofilin N-ter hexadeca-peptide fused to penetratin (Ser3 pept., 20 μg/ml) or the LIMK inhibitor LIMKi (10 μm), or for 24 h to the PAK inhibitor IPA-3 (5 μm). Filled arrowheads indicate cLTP-induced SEP fluorescence spots. Open arrowheads indicate spines with unchanged or reduced SEP fluorescence. Scale bar, 2 μm. B, Summary graph of SEP fluorescence as in A measured 35 min after cLTP induction and normalized to control, in neurons expressing SEP-GluA1 and either shNT or shKCC2 exposed to Ser3 peptide, LIMKi, IPA-3, or the RhoA inhibitor Rhosin (30 μm). ***p < 0.005. n = 7–29 neurons from the same cultures in each condition. C, Summary graph of average spine volume (normalized to control before cLTP induction) measured 30 min after cLTP induction, from the same experiments as above, either in control neurons (shKCC2; black symbols) or after 4 h exposure to LIMK inhibitors (Ser 3 peptide or LIMKi; green symbols) or 24 h exposure to IPA-3 (red symbols). *p < 0.05. **p < 0.01. ***p < 0.005. D, Scheme representing how cofilin plays a central role in regulating actin dynamics as it catalyzes depolymerization of F-actin into G-actin. Cofilin activity is inhibited by phosphorylation of its Ser3 residue by LIMK. Several signaling cascades converge to activate LIMK in neurons. These include the RhoA/ROCK as well as the Rac1/PAK pathways. βPIX, a guanine nucleotide exchange factor (GEF) involved in Rac1 activation, has been shown to interact with KCC2 (Fig. 7D,E) (Llano et al., 2015). We propose that this interaction may hinder recruitment of βPIX and its scaffolding molecule GIT1 and, consequently, activation of Rac1/PAK at rest. Disrupting this interaction may then favor Rac1/PAK activation, increase LIMK activity, and thereby inactivate cofilin through Ser3 phosphorylation, resulting in enhanced actin polymerization. This in turn prevents LTP-induced membrane traffic of GluA1-containing AMPA receptors and increase in spine volume. The LIMK inhibitor LIMKi as well as the dominant-negative peptide Ser 3 pept. (gray) both favor actin depolymerization by antagonizing LIMK-mediated cofilin phosphorylation. The PAK inhibitor IPA-3 acts upstream to inhibit Rac1-mediated activation of LIMK. Rhosin specifically inhibits RhoA. In this scheme: orange represents kinases; blue represents Rho GTPases; purple represents GEF. Adapted from Cingolani and Goda (2008).
Electrophysiology.
Cultured neurons were recorded at 31°C under superfusion with a HEPES-buffered, artificial CSF (H-ACSF) containing (in mm) the following: 125 NaCl, 20 d-glucose, 10 HEPES, 4 MgCl2, 2 KCl, 1 CaCl2, pH 7.4. For miniature EPSC (mEPSC) recordings, neurons were recorded in whole-cell configuration with borosilicate glass micropipettes filled with a solution containing (in mm) the following: 110 CsMeSO4, 20 CsCl, 10 HEPES, 10 EGTA, 4 MgATP, and 0.4 Na3GTP, pH 7.4. Cells were held at −70 mV, and mEPSCs were isolated by adding TTX (1 μm) and bicuculline methochloride (20 μm) to the extracellular solution. Currents were recorded with a Multiclamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 20 kHz. Access and input resistance were regularly monitored with −5 mV voltage steps. mEPSCs were detected and analyzed offline using Detectivent software.
Chemical LTP (cLTP) was induced as described previously (Kopec et al., 2006) by switching extracellular solution for 16 min to a nominally Mg-free solution containing (in mm) the following: 125 NaCl, 20 d-glucose, 10 HEPES, 2 KCl, 5 CaCl2, pH 7.4, supplemented with 50 μm forskolin, 0.1 μm rolipram, and 20 μm bicuculline. Forskolin and rolipram stock solutions were prepared in anhydrous DMSO, leading to a final DMSO concentration of 0.2%. Hence, control experiments involved a 16 min application of a solution containing an equivalent DMSO concentration.
For ex vivo field EPSP (fEPSP) recordings, 3-week-old male Wistar rats were anesthetized by intraperitoneal injection of ketamine/xylazine (of 75/10 mg/kg). Rats were head-fixed into a stereotaxic apparatus, and holes were drilled bilaterally in the skull above the dorsal hippocampus (from bregma: −3.3 mm anteroposterior, ±2.3 mm mediolateral, −3.0 mm dorsoventral); 1 μl of a concentrated lentiviral solution (diluted at 1:10 from a titer of 375–440 ng/ml p24, as detected by ELISA) was injected in each site. After 2 weeks, acute hippocampal slices were prepared. Rats were anesthetized and perfused with ice-cold cutting solution containing the following (in mm): 110 choline chloride, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 25 glucose, 11.6 ascorbic acid, and 3.1 pyruvic acid. The brain was then rapidly removed, and 450-μm-thick parasagital slices were prepared with a vibratome (Microm, Thermo Fisher). Slices were then transferred and allowed to recover for 1 h in an interface chamber filled with bicarbonate-buffered ACSF (B-ACSF) preheated at 37°C and oxygenated with 5% CO2 in O2, containing the following (in mm): 124 NaCl, 1 NaH2PO4, 26.2 NaHCO3, 2.5 KCl, 11 glucose, 1.6 CaCl2, 1.2 MgCl2. For recordings, slices were transferred in a submerged recording chamber and superperfused with B-ACSF after a cut was made between the CA3 and CA1 areas. A recording borosilicate glass pipette (2–4 mΩ) filled with B-ACSF was inserted in the molecular layer of a densely infected dentate gyrus area, and a tungsten bipolar electrode (0.5 mΩ) was used to stimulate the perforant pathway. fEPSPs were recorded in the presence of the GABAA receptor antagonist bicuculline methochloride (20 μm), using a Multiclamp 700B amplifier (Molecular Devices) low-pass filtered at 5 kHz, and digitized at 20 kHz. fEPSP slopes were analyzed offline using Clampfit software (Molecular Devices). Briefly, baseline potential was set to zero, and recordings were low-pass filtered at 1 kHz using a Bessel filter. The initial slope of the fEPSP was then automatically measured using a 1 ms time-window manually positioned at the onset of the fEPSP. Data were acquired and analyzed blind to the experimental condition.
Immunocytochemistry and fluorescence image acquisition.
Cultures were fixed in 4% PFA and permeabilized with 0.2% Triton X-100 in PBS. KCC2 immunostaining was performed using rat KCC2 antibody (1:400, Sigma-Aldrich) and Cy3-coupled goat anti-rabbit (1:400); 4.1N (1:500, BD Biosciences) immunostainings were performed after methanol fixation and permeabilization (10 min at −20°C) and revealed with Cy3-coupled donkey anti-mouse antibodies (1:400). GFP immunostainings were performed using GFP (1:1000, Millipore Bioscience Research Reagents) and Alexa-488-coupled goat anti-chicken secondary antibodies (1:400). Rac1 GEF and GAP immunostaining was performed after 5 min fixation in 4% PFA by overnight incubation at 4°C with rabbit βPIX (1:100, Millipore) and monoclonal G protein-coupled receptor kinase-interacting protein 1 (GIT1, 1:100, clone N39B-8, Anticorpsenligne) antibodies, as in Smith et al. (2014). All secondary antibodies were from Jackson ImmunoResearch Laboratories. Alexa-456-coupled phalloidin (1:250, Invitrogen) was sometimes added, in the absence of serum, to stain F-actin.
Neurons were then imaged on a Leica DM6000 upright microscope using 40× (1.25 NA, for KCC2 immunofluorescence) or 100× (1.40 NA, for all other experiments) objectives. Images were acquired with a 12-bit cooled CCD camera (Micromax or Coolsnap fx, Roper Scientific) operated with MetaMorph (Molecular Devices). For quantification, cultures were stained simultaneously and images were acquired using the same exposure time. Mean or integrated fluorescence intensities were then quantified using ImageJ, blind to the experimental conditions. Normalization was performed for each culture by dividing mean fluorescence intensity from each cell by the average of the mean fluorescence intensity of all cells of the control group.
Immunohistochemistry and fluorescence image acquisition.
P40 rats were anesthetized by intraperitoneal injection of ketamine/xylazine and perfused transcardially with 4% PFA in PBS. Brains were then removed, postfixed overnight, and stored at −20°C in 30% ethylene glycol and 30% glycerol in PBS. Parasagital sections (40 μm thick) were obtained using a cryotome. Brain slices were preincubated 1 h with 0.5% Triton and 10% goat serum in PBS and incubated overnight at 4°C with the primary antibodies: GFP (chicken, 1:1000, Millipore Bioscience Research Reagents), KCC2 (rabbit, 1:400, Millipore). Slices were then incubated 1 h with secondary antibodies (goat CY3-coupled and donkey FITC-coupled directed against rabbit and chicken antibodies, respectively) and mounted with Mowiol/Dabco (25 mg/ml). Stacks of 10–40 sections (0.2 μm apart) were acquired using an upright confocal microscope (Leica TCS SP5, 40×/1.25 NA objective).
GluA-SEP fluorescence and spine tracking.
Neurons expressing GluA1/2-SEP and mCherry were maintained at 37°C in a thermostatted chamber and superfused with H-ACSF preheated at 37°C. Time-lapse confocal images of neurons were acquired using an inverted spinning-disc microscope (Leica DMI4000, Yokogawa CS20 spinning Nipkow disk, 100×/1.4 NA objective). At every time point, neurons were successively illuminated by 491 nm/561 nm light from an Ar/Kr laser. Emitted light was collected using corresponding emission filters (525-39 and 607-36 nm) and a cooled EM-CCD camera (512 × 512, 16 μm pixel size). Importantly, laser intensity, time of illumination, and acquisition parameters of the camera remained identical to allow comparison between experiments and bleach correction. An X, Y, Z motorized platform (Märzhäuser) was used to perform acquisition from multiple neurons and z-stacks (6 μm stack, 0.2 μm steps).
Confocal stacks were analyzed with Neuronstudio (Dumitriu et al., 2011), blind to experimental conditions, to quantify changes in spine head volume overtime using the mCherry channel. Only spines that could be accurately followed over the entire experiments were measured. Spine volumes from each cell were then averaged to test statistical significance. SEP fluorescence was measured on registered maximal projection in z of confocal stacks. ROIs were automatically drawn around the entire dendrites using thresholded mCherry signal, and mean SEP fluorescence intensity was calculated. Fluorescence intensity values were then background-subtracted and bleach-corrected. Normalization of SEP intensity was performed for each cell by dividing the mean fluorescence intensity by the average of fluorescence intensities of the three time points before cLTP induction.
In experiments using NH4 Cl application to collapse transmembrane pH gradients (Ashby et al., 2006) (see Fig. 3), rapid alkalization of intracellular compartments due to passive NH3 diffusion through membranes was followed by a delayed, rebound acidification, likely reflecting KCC2-mediated ammonium transport (Tietz et al., 1999). Accordingly, this rebound acidification was not observed in neurons with suppressed KCC2 expression, whereas the initial alkalization was unaffected (data not shown). We therefore always measured SEP fluorescence before the onset of the rebound acidification (<7 min).
Figure 3.

Suppression of activity-driven, membrane insertion of AMPARs in neurons lacking KCC2. A, Neurons were transfected with GluA1-SEP. Left, Summary graph of mean SEP fluorescence (normalized to control) at pH 5.5 (n = 11) and upon NH4Cl application (n = 26 cells). ***p < 0.005. SEP fluorescence was detected in neurons imaged at pH 7.4 (Aa) but was near-completely quenched at pH 5.5 (Ab). Upon collapse of pH gradients using 50 mm NH4Cl at pH 7.4 (Ac), additional fluorescence was detected reflecting an intracellular pool of SEP-GluA1. Right, Spinning disc confocal fluorescence micrographs of SEP-GluA1-expressing hippocampal neurons at time points a–c. Scale bar, 2 μm. B, Overlay of mCherry (red) and SEP (pseudocolors) fluorescence micrographs of dendritic sections of neurons before (Control) and 35 min after cLTP induction. Filled arrowheads indicate cLTP-induced SEP fluorescence spots. Open arrowheads indicate spines with unchanged SEP fluorescence. KCC2 suppression by RNA interference, but not blockade by the antagonist VU0240551 (6 μm), abolished cLTP-induced increase in SEP fluorescence. Scale bar, 2 μm. C, SEP fluorescence 35 min after cLTP induction normalized to control in neurons expressing SEP-GluA1 or SEP-GluA2 and either nontarget (shNT) or KCC2-directed (shKCC2) shRNA, KCC2 shRNA together with shRNA-proof recombinant KCC2 (rescue) or neurons exposed to KCC2 antagonist VU0240551 (6 μm) or vehicle only. **p < 0.01. ***p < 0.005. n = 18–29 neurons in each condition. D, Left, SEP fluorescence before cLTP induction in neurons expressing SEP-GluA1 and either nontarget (n = 47) or KCC2-directed (n = 56) shRNA, normalized to the mean SEP fluorescence in shNT-expressing neurons. *p < 0.05. Right, Change in SEP fluorescence (p of control) upon 5 min perfusion of NH4Cl solution, pH 7.4, to reveal intracellular SEP-GluA1 pool (shNT, n = 23; shKCC2, n = 28). ***p < 0.005.
In experiments shown in Figure 4, αCaMKII1–290 expression was induced by 24 h application of doxycycline (2 μg/ml). Neurons were then maintained in H-ACSF in a thermostatted chamber for acquisition using an upright confocal microscope (Leica TCS SP5, 40×/0.8 NA, water-immersion objective). GluA1-SEP and mCherry were successively excited by 488 nm (Ar laser) and 561 nm (HeNe laser), and emitted fluorescence was filtered by 500–600 nm and 570–700 nm (Leica AOBS). Pinhole was set as fully open (600 μm, 10 μm thickness) to collect fluorescence originating from the entire dendritic section. Laser power and scanning parameters were maintained identical to allow quantitative measurements and comparison of SEP fluorescence between conditions. All mCherry-positive neurons were imaged, and GluA1-SEP-negative neurons were discarded during the analysis process.
STED microscopy.
Neurons expressing LifeAct-Venus and mCherry were fixed and mounted in mowiol. The STED microscope consisted of an inverted Leica TCS SP5 STED CW microscope with a 100×/1.4 NA objective, 488 nm Ar laser, 594 HeNe laser, and 561 nm 10 mW DPSS laser for depletion, nonresonant galvanometer mirror (used at 400 Hz, 1024 × 1024, 28.7 nm pixel size), Leica AOBS for emission filters, and GaAsP hybrid detectors (HyD). Focusing onto dendritic spines was performed using the mCherry signal, and a single confocal slice was acquired for each dendritic section. Diffraction limited- and STED-resolution images of LifeAct-Venus were successively acquired. STED images were then deconvolved using special Leica CW-STED plugin of Huygens (SVI) and recommended parameters from Huygens. A custom plugin on ImageJ was used to outline individual spines and measure the FWHM of LifeAct-labeled spine necks based on a Gaussian fit (accuracy of the fit was evaluated both by eye and by r2 measurements, blind to experimental conditions). Several spine head parameters were also computed: mean and variance of LifeAct-Venus fluorescence intensity and geometric center of the spine. Spine heads were then realigned using a line originating from the attachment point of the spine neck to the head (manually determined) and passing through the geometric center.
For spatial autocorrelation analysis of LifeAct-Venus signal (see Fig. 5), data from isolated spine heads were first normalized. The degree of clustering in each dataset was assessed using Moran's index analysis (Moran, 1948). This index ranges values from −1 (complete dispersion) to 1 (complete clustering), with 0 representing random distribution. For each pixel i, local Moran's index (Ii) was computed as follows:
 |
where zi is the value of the pixel i and W represents the matrix of neighboring weights. Equal weights were attributed to all pixels within a 2-pixel distance from pixel i, all other weights being set to 0. To compute a statistical significance threshold for I, a bootstrap analysis was performed for each spine head. Data were resampled randomly 1000 times, and local Moran's indexes were computed for each pixel in the random distributions. For each pixel, Moran's index was compared with the 1000 values derived from bootstrap analysis to compute a local p value. Statistical significance of positive spatial autocorrelation was considered for pixels with p values <0.05. The proportion of high-intensity clustered pixels per spine was then computed as the proportion of pixels with significant autocorrelation index and intensity above the mean, and was used for statistical comparison between experimental groups.
Figure 5.

Enhanced F-actin content in dendritic spines upon KCC2 suppression. A, C, Wide-field fluorescent micrographs of representative dendritic sections of neurons expressing plasma-membrane (A) or cytosolic (C) EGFP and either nontarget (shNT) or KCC2-directed shRNA (shKCC2). Neurons were immunostained for GFP (green) and 4.1N (red, in A) or stained with Alexa-546-phalloidin (red, in C). Scale bar, 1 μm. B, Quantification of immunostainings as in A showing 4.1N mean immunofluorescence intensity per pixel either from dendritic spines only (left) or from entire dendritic sections (right), normalized to internal controls (gray bars) for each experimental condition (black bars). n = 41 (shNT), n = 37 (shKCC2), n = 54 (GFP), and n = 43 (KCC2-CTD) cells, respectively. n.s., Nonsignificant difference. D, Normalized fluorescence intensity of phalloidin staining in dendritic spines from neurons expressing shNT (1022 spines from 36 cells), shKCC2 (861 spines, from 29 cells), EGFP (538 spines from 15 cells), or KCC2-CTD (591 spines from 18 cells) from two independent experiments. ***p < 0.005. E, Normalized fluorescence intensity of phalloidin staining in dendritic spines from shNT-expressing neurons plotted against spine area. No significant correlation was observed between these variables (linear regression r2 = 0.02; p > 0.05, Spearman rank order correlation test).
Biochemical assays.
Hippocampal cultures were infected at 15–17 DIV using lentiviral vectors expressing shRNAs and eGFP using 500 ng/ml of purified virus solution as above. One week later, cultures were processed for cLTP (see below) or exposed to vehicle only (DMSO 0.2%) for 2–16 min. Immediately after cLTP induction, cultures were washed twice in ice-cold PBS and lysed in lysis buffer (25 mm Tris-HCl, pH 7.4, 250 mm NaCl, 50 mm NaF, 5 mm PPi, 5 mm EDTA, 5 mm EGTA, 1 mm sodium orthovanadate, 1% Triton-X, and protease inhibitor mixture; Roche). After centrifugation at 14.000 × g for 15 min., supernatants were mixed with Laemmli sample buffer and boiled at 95°C for 5 min. Samples were subjected to standard SDS-PAGE and transferred to nitrocellulose membranes. Blots were probed with primary antibodies against cofilin (Abcam), cofilin phospho-Ser3 (Abcam), GluA1 (Millipore), GluA1 phospho-Ser845 (Millipore), KCC2 (Millipore), Tuj1 (R&D Systems), and detected with fluorescent secondary antibodies (DyLight 800) using Odyssey infrared imaging system (LI-COR Bioscience). For quantification, all data were normalized to internal Tuj1 control signals.
For coimmunoprecipitation, neurons were homogenized in modified RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 0.1% SDS, 0.5% DOC, and protease inhibitors; Roche). Lysed cells were kept on ice for 20 min and then centrifuged at 14,000 × g for 10 min. Lysates were then incubated with KCC2 (Millipore) or βPIX IgG (Millipore, 2 μg) (Smith et al., 2014) overnight at 4°C. The pulldown was performed using Protein G Magnetic beads (Invitrogen) for 2 h at 4°C. The beads were washed 3 times in RIPA buffer and one time in RIPA buffer without detergents. Bound proteins were eluted in SDS sample and separated by SDS-PAGE.
Rac1 and RhoA activity was measured using effector pulldown assays. Briefly, neurons infected with lentiviruses expressing shRNA sequences were washed with PBS, lysed with ice-cold lysis buffer (25 mm Tris pH 7.5, 1% NP40, 100 mm NaCl, 10 mm MgCl2, 5% glycerol, 5 mm NaF, 1 mm PMSF, 1 μl/ml protease inhibitor mixture; Sigma) and clarified by centrifugation at 1600 × g for 5 min at 4°C; 300 μg of lysate proteins was incubated with 10 or 50 μg of PAK-PBD or Rhotekin-RBD beads (Cytoskeleton), respectively, for 1 h at 4°C. Bead pellets were washed twice with 25 mm Tris, pH 7.5, 0.5% NP40, 40 mm NaCl, 30 mm MgCl2, 1 mm DTT, 1 mm PMSF, 1 μl/ml protease inhibitor mixture (Sigma) before addition of 5× Laemmli buffer. Fractions were analyzed by Western blotting. Proteins were separated on a 12% SDS polyacrylamide gel and transferred onto a nitrocellulose or PVDF membrane (GE Healthcare). Blots were probed with mouse monoclonal anti-Rac1 (1:1000, BD Transduction Laboratory) or anti-RhoA (1:200, Santa Cruz Biotechnology), as described previously (Causeret et al., 2004; Backer et al., 2007). Band densities were quantified by Fusion software (Vilber Lourmat). The relative densities of pulled down activated Rac1 and RhoA were normalized to the maximal Rac1 or RhoA in the same samples incubated with 200 μm GTP-γS for 15 min at room temperature, according to the manufacturer's instructions.
Biochemical assays were repeated at least 3–4 times on independent cultures, and data were compared using Student's t test, after testing normality and equal variance of the data with SigmaPlot 12.5 (SPSS). In all other experiments, data were compared using nonparametric paired (Wilcoxon) or unpaired (Mann–Whitney) tests. Differences were considered significant for p values <0.05.
Results
Suppressing KCC2 expression, but not function, precludes LTP in hippocampal neurons
We first investigated the impact of KCC2 expression on LTP in hippocampal slices using in vivo RNA interference. Juvenile rats (P21) were stereotaxically injected in the dorsal dentate gyrus with lentiviruses expressing previously validated either nontarget or KCC2-directed small hairpin RNA sequences (Gauvain et al., 2011). Suppression of KCC2 expression in transduced neurons was confirmed by immunohistofluorescence imaging 2 weeks after infection (Fig. 1A,B). An fEPSP was evoked in the molecular layer of densely infected areas of the dentate gyrus by extracellular stimulation of perforant path inputs in the presence of the GABAA receptor antagonist bicuculline (20 μm). High-frequency stimulation (4 × 1 s at 100 Hz) induced a robust and long-lasting (>60 min) increase in the initial slope of the fEPSP with no detectable post-tetanic potentiation, in both uninfected slices (data not shown) as well as slices infected with nontarget shRNA lentiviruses (220.4 ± 40.9% of control, n = 8 slices, p < 0.01; Fig. 1C). In slices infected with KCC2-directed shRNA lentiviruses, however, this form of LTP was suppressed (103.5 ± 12.8%, n = 10 slices, p = 1.0; Fig. 1D).
To explore the molecular mechanisms involved in LTP hindrance upon KCC2 suppression, we next used a classical, cLTP paradigm in primary hippocampal neurons. This paradigm consisted of a 16 min application of rolipram/forskolin/bicuculline in the absence of external Mg (Otmakhov et al., 2004; Kopec et al., 2006) to induce a persistent (>80 min) potentiation in the efficacy of excitatory synapses (Fig. 2A,B). This potentiation was detected as a 30% increase in mEPSC amplitude compared with neurons exposed to vehicle only (26.8 ± 1.6 vs 20.6 ± 1.6 pA, p = 0.01) with no significant change in their mean frequency (34.7 ± 5.5 vs 29.6 ± 5.6 Hz, n = 19 and n = 16 cells, respectively, p = 0.48), suggesting that its locus of expression was primarily postsynaptic (Otmakhov et al., 2004; Kopec et al., 2006).
Figure 2.
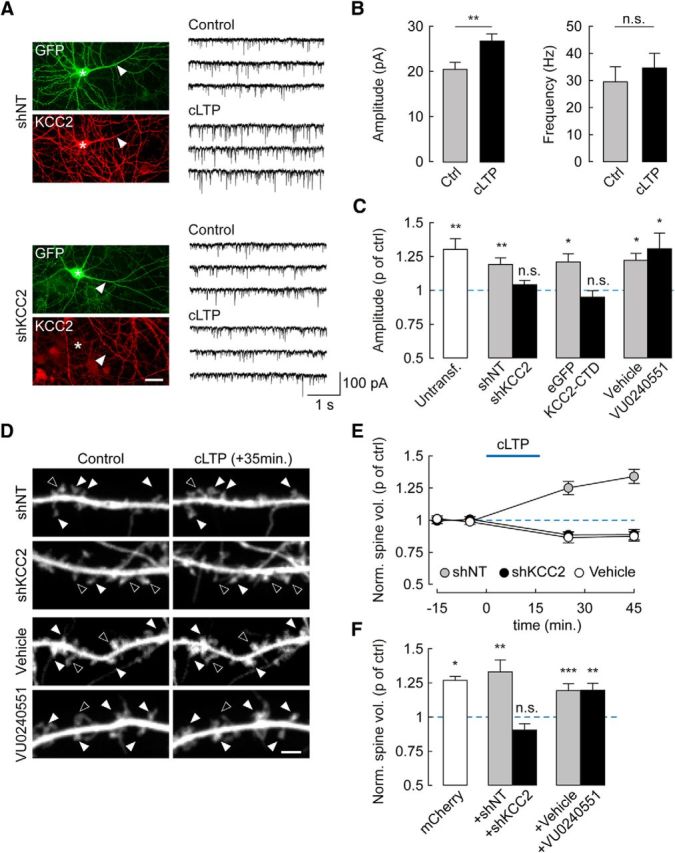
KCC2 suppression precludes cLTP expression through an ion transport-independent mechanism. A, Left, Wide-field fluorescence micrographs showing GFP fluorescence and KCC2 immunolabeling in hippocampal neurons transfected with nontarget (shNT, top) or KCC2-specific (shKCC2, bottom) shRNA. Stars and arrows represent the soma and primary dendrite of transfected cells, respectively. Scale bar, 20 μm. Right, 12 s recordings of mEPSCs from neurons either after cLTP protocol (see Materials and Methods, cLTP) or vehicle application only (DMSO 0.6 μl/ml, Control). B, Summary data from 16 (Ctrl) and 19 (cLTP) untransfected neurons showing that cLTP specifically increases mEPSC amplitude (p < 0.01) but not frequency (p = 0.5). C, Summary data showing change in mEPSC amplitude (proportion of control) upon cLTP in untransfected neurons (p < 0.01), or neurons expressing either nontarget (p < 0.01) or KCC2-specific shRNA (p = 0.2), eGFP alone (p < 0.05), or eGFP with KCC2 carboxy-terminal domain (KCC2-CTD, p = 0.4), and neurons exposed to the KCC2 antagonist VU0240551 (6 μm, p < 0.05) or vehicle only (DMSO, p < 0.05). n = 11–18 in each condition. D, Spinning disc confocal fluorescence micrographs of dendritic sections of neurons expressing mCherry and either shNT or shKCC2, or exposed for 72 h to VU0240551 or vehicle only. Images represent maximal projections of confocal stacks acquired 15 min before and 35 min after cLTP induction. Filled arrowheads indicate spines that increased in volume during cLTP. Open arrowheads indicate spines that decreased in volume or remained unchanged. Scale bar, 2 μm. E, Time course of changes in spine volume upon cLTP in neurons expressing shNT (n = 11), shKCC2 (n = 9), and neurons expressing mCherry only and exposed for 16 min to vehicle only (vehicle, n = 6). F, Summary graph of average spine volume (normalized to control before cLTP induction) measured 30 min after cLTP induction in neurons expressing mCherry alone (n = 7), or together with shNT (n = 11), shKCC2 (n = 9), or after 72 h exposure to VU0240551 (n = 14) or vehicle only (n = 20). *p < 0.05. **p < 0.01. ***p < 0.005. n.s., Nonsignificant difference.
Suppressing KCC2 expression for 7–10 d by RNA interference completely abolished cLTP in mature hippocampal neurons (Fig. 2A–C). Thus, mEPSC amplitude was not significantly different after a cLTP paradigm compared with control (19.2 ± 0.6 vs 18.4 ± 0.5 pA, n = 13 cells each, p = 0.34). In contrast, expression of a nontarget shRNA sequence did not affect cLTP expression (24.1 ± 1.0 vs 20.2 ± 0.8 pA, n = 12 and n = 13 cells, respectively, p = 0.006). This effect of KCC2 suppression was independent of the loss of ion transport function because chronic application of the specific antagonist VU0240551 (Delpire et al., 2009; Gauvain et al., 2011) (6 μm) had no effect on cLTP expression (30.0 ± 2.6 vs 23.0 ± 1.7 pA, n = 12 cells each, p < 0.05; Fig. 2C). In contrast, specifically preventing KCC2 interactions with intracellular partners, using overexpression of a dominant-negative peptide (KCC2-CTD) (Li et al., 2007; Gauvain et al., 2011; Puskarjov et al., 2014), completely abolished cLTP (20.2 ± 1.0 vs 21.3 ± 0.9 pA, n = 11 and n = 18 cells, respectively, p = 0.44).
LTP at hippocampal synapses is associated with a concomitant and persistent increase in spine volume (e.g., Kopec et al., 2006, 2007; Harvey and Svoboda, 2007). We monitored spine volume in cLTP experiments using time-lapse, spinning-disc confocal imaging of mCherry-expressing neurons (Fig. 2D–F). In neurons expressing mCherry only, cLTP was associated with a significant increase in spine volume that persisted >30 min after induction (to 27.8 ± 3.0% of control at 30 min after induction, n = 143 spines from 7 cells, p < 0.05; Fig. 2F). Similarly, neurons expressing nontarget shRNA showed a 33.4 ± 8.9% increase in spine volume 30 min after cLTP induction. In contrast, no significant change in spine volume was detected in neurons expressing KCC2-directed shRNA (−9.5 ± 4.7% of control, n = 9 cells, p = 0.1; Fig. 2D–F), similar to shNT-expressing neurons exposed to vehicle only (DMSO 0.2%, −12.6 ± 3.7% of control, n = 6 cells, p = 0.06; Fig. 2E). Again, this effect did not involve KCC2 function because a 72 h exposure to the specific antagonist VU0240551 did not prevent a cLTP-induced increase in spine volume (to 19.7 ± 5.1% of control, n = 576 spines from 20 cells, p < 0.005 Fig. 2F) to a similar extent as in neurons exposed to vehicle only (n = 20 and n = 15 cells, respectively, p = 0.6). Our results show that KCC2 is strictly required for both synaptic and structural LTP at hippocampal excitatory synapses, through a mechanism likely requiring intracellular protein interactions but independent of ion transport.
KCC2 is required for activity-driven GluA1 exocytosis
LTP in hippocampal pyramidal neurons relies in part on activity-driven membrane insertion of AMPARs (Malinow and Malenka, 2002; Poncer, 2003; Huganir and Nicoll, 2013). We therefore asked whether activity-driven AMPAR exocytosis might be compromised in the absence of KCC2. We used SEP-tagged GluA1 to specifically monitor the membrane-inserted pool of GluA1-containing AMPARs (Ashby et al., 2006; Kopec et al., 2006; Lin et al., 2009; Makino and Malinow, 2011) (Fig. 3A). Application of a pH 5.5 external solution led to complete quenching of SEP-GluA1 (to 12.7 ± 3.1% of control, n = 11 cells, p < 0.001), whereas a collapse of transmembrane proton gradients by ammonium increased SEP-GluA1 fluorescence (to 125.0 ± 13.3% of control, n = 26 cells, p < 0.001), likely revealing intracellular receptor pools (Fig. 3A). SEP-GluA1 fluorescence was rapidly and persistently increased upon cLTP induction in hippocampal neurons, indicating membrane insertion of new SEP-GluA1-containing AMPARs (23.0 ± 5.1% of control after 35 min, n = 19 cells, p < 0.001; Fig. 3B,C). This effect was not observed upon application of vehicle only (DMSO, −2.1 ± 3.7% of control, n = 5 cells, p = 0.3) and was specific to SEP-GluA1 but not SEP-GluA2-containing receptors (2.3 ± 3.2% of control, n = 18 cells, p = 0.33).
Using this assay, we then examined how SEP-GluA1 trafficking may be affected by altering KCC2 expression or function. Chronic suppression of KCC2 expression by RNA interference prevented cLTP-induced membrane insertion of SEP-GluA1 containing receptors (3.2 ± 1.7% of control, n = 21 cells, p = 0.44; Fig. 3B,C). This effect was specific to KCC2 suppression as it could be rescued by overexpressing an shRNA-proof recombinant KCC2 (17.4 ± 4.5% of control, n = 20 cells, p < 0.001; Fig. 3C; see Materials and Methods). However, it was not mimicked by chronically blocking KCC2 function using the specific antagonist VU0240551 (Fig. 3C), again suggesting that KCC2 expression, but not function, is required for cLTP-induced AMPAR membrane traffic. Importantly, SEP-GluA1 steady-state fluorescence was reduced in neurons expressing KCC2-specific compared with nontarget shRNA (−24.8 ± 9.5%, n = 28 and n = 23 cells, respectively, p < 0.01). However, its relative increase upon application of NH4Cl was similar in both conditions (20.6 ± 2.2%, n = 28 cells, p < 0.001 and 22.1 ± 4.0%, n = 23 cells, p < 0.001, respectively; Fig. 3D). These results suggest that altered GluA1 membrane insertion in the absence of KCC2 may not reflect a loss of intracellular GluA1, but rather a specific disruption of its activity-driven exocytosis.
During LTP in hippocampal neurons, αCaMKII activation induces membrane insertion of GluA1-containing AMPARs (Hayashi et al., 2000; Esteban et al., 2003), which requires PKA phosphorylation of GluA1 Ser845 (Esteban et al., 2003). A defect in either αCaMKII activation or GluA1 phosphorylation on Ser845 in neurons lacking KCC2 may then preclude activity-dependent AMPAR traffic. To bypass αCaMKII activation, we overexpressed a constitutively active αCaMKII to directly promote GluA1 exocytosis. The catalytic domain of αCaMKII (CaMKII1–290) (Hayashi et al., 2000; Poncer et al., 2002; Esteban et al., 2003) fused to mCherry was expressed under a doxycycline-inducible promoter to allow for a precise control over the timing of its expression (Fig. 4A). We first verified that mCherry-CaMKII1–290 transgene expression was strictly restricted to neurons exposed to doxycycline, as shown by the lack of mCherry expression in neurons transfected with this construct for 8–10 d (Fig. 4B).
Doxycycline-induced overexpression of CaMKII1–290 for 24 h led to a significant increase in SEP-GluA1 fluorescence in neurons expressing nontarget shRNA (28.3 ± 8.8% of control, n = 105 and n = 121 cells, respectively, p = 0.006; Fig. 4C,D). However, this effect was abolished in neurons in which KCC2 expression was suppressed (9.3 ± 12.1% of control, n = 84 and n = 90 cells, respectively, p = 0.8; Fig. 4C,D). In contrast, in hippocampal neurons transduced with shRNA-expressing lentiviruses, we observed no difference in GluA1 Ser845 phosphorylation in neurons expressing KCC2-specific compared with nontarget shRNA, either at rest (1.7 ± 4.5%, n = 3 experiments, p = 0.9) or upon cLTP (758 ± 131% vs 687 ± 246% of control, respectively, p = 1.0; Fig. 4E,F). These results demonstrate that KCC2 is required for activity-dependent GluA1 membrane delivery in hippocampal neurons and acts via mechanisms downstream of αCaMKII activation and pKA-dependent GluA1 phosphorylation on Ser845.
Altered spine actin polymerization upon KCC2 suppression
How may KCC2 gate activity-driven AMPAR membrane insertion? KCC2 has been shown to interact with spine cytoskeleton through the FERM-domain protein 4.1N (Li et al., 2007) and GluA1 binding to 4.1N is required for its activity-dependent membrane insertion (Lin et al., 2009). We therefore tested whether the loss of KCC2 may affect 4.1N expression in hippocampal neurons. However, suppressing KCC2 or overexpressing KCC2-CTD had no apparent effect on 4.1N expression as detected by immunofluorescence, as measured either in the dendritic spines and shafts together (97.2 ± 3.4% and 104.3 ± 5.3% of control intensity, p = 0.7 and p = 0.6, respectively) or specifically in dendritic spines (97.7 ± 4.0% and 110.7 ± 5.2% of control, n = 37 (41 control) and n = 43 (54 control) cells, p = 0.6 and p = 0.1, respectively; Figure 5A,B). Therefore, altered 4.1N expression and/or clustering seem unlikely to be responsible for the defect in GluA1 membrane traffic upon KCC2 suppression.
Both activity-driven AMPAR trafficking and spine enlargement rely on remodeling of actin cytoskeleton. Whereas transient actin depolymerization by cofilin may be permissive for AMPAR membrane insertion (Gu et al., 2010), delayed increase in F-actin spine content may be required for spine enlargement (Okamoto et al., 2004). We previously reported increased lateral diffusion of several transmembrane actin-binding proteins in dendritic spines upon KCC2 suppression (Gauvain et al., 2011), suggestive of submembrane cytoskeleton alterations. This hypothesis was tested by using phalloidin staining of F-actin in dendritic spines, revealing a 40% increase of fluorescence in neurons with suppressed KCC2 expression (1.42 ± 0.12 of control, n = 29 and n = 36 cells, respectively, p < 0.005; Fig. 5C,D). This effect was even more pronounced in neurons expressing KCC2-CTD (1.84 ± 0.22 of control, n = 18 and n = 15 cells, respectively, p < 0.001) and was likely independent of changes in spine volume because no significant correlation was detected between phalloidin staining and spine size (Fig. 5E). To examine the subspine spatial distribution of actin, we next used STED microscopy of the synthetic, actin-binding peptide LifeAct-Venus (Riedl et al., 2008; Urban et al., 2011). Using this approach, we could resolve individual actin bundles within spine necks and heads (Fig. 6A). The FWHM of LifeAct-labeled spine necks in neurons transfected with nontarget shRNA was 93.7 ± 1.9 nm, (n = 128 spines from 19 neurons, Fig. 6B), consistent with previous data in hippocampal pyramidal neurons (Urban et al., 2011). In neurons with suppressed KCC2 expression, however, the FWHM of LifeAct-labeled spine necks was significantly increased by 20.3% (112.8 ± 5.6 nm, n = 60 spines from 18 neurons, p = 0.001; Fig. 6C), suggestive of an increased recruitment of actin bundles in spine necks. We next asked whether suppression of KCC2 might lead to a redistribution of F-actin within spines or a global increase in F-actin content. LifeAct-Venus fluorescence was heterogeneously distributed within individual spines with some spines showing intense, calyx-shaped bundles and others more widely distributed signal (Fig. 6D,E). Overall, however, we could not detect a significant difference in the coefficient of variation of LifeAct-Venus fluorescence between neurons expressing nontarget or KCC2-directed shRNA (0.57 ± 0.02 vs 0.60 ± 0.02, n = 149 and 85 spines, from 19 and 20 neurons, respectively, p = 0.13; Fig. 6E–G). To compare the spatial distribution of F-actin within spines, we performed spatial autocorrelation analysis of LifeAct-Venus signal using Moran's index (Moran, 1948) (see Materials and Methods; Fig. 6F). Again, we found no significant difference in the proportion of high-intensity clustered pixels (Moran's index p value <0.05) between neurons expressing nontarget or KCC2-directed shRNA (23.0 ± 0.3% vs 23.1 ± 0.4%, same dataset as above, p = 1.0; Fig. 6F–H). Together, our results show that suppression of KCC2 results in a net increase in F-actin accumulation within dendritic spines with no substantial subspine redistribution.
Figure 6.

Subspine distribution of F-actin is unchanged upon KCC2 suppression. A, Representative fluorescence micrographs of a dendritic section of a neuron expressing mCherry and LifeAct-Venus (pseudocolors), imaged in confocal (left) and CW-STED microscopy with or without deconvolution (right). B, Gaussian fit of the pixel intensity profile across spine neck following the blue line shown in A. C, Summary graph showing FWHM of Gaussian fit of LifeAct-Venus signals across spine necks of neurons expressing nontarget or KCC2-directed shRNA (n = 128 and n = 60 spines from 19 and 18 neurons, respectively). ***p < 0.001. D, Representative, pseudocolored fluorescence STED micrographs of 6 individual spines from 12 different neurons expressing either nontarget or KCC2-directed shRNA. Scale bar, 500 nm. E, Pseudocolored averages of 149 and 85 individual spine images from 19 and 20 neurons, respectively, as in D, aligned as described in Materials and Methods to show the mean distribution of LifeAct-Venus fluorescence within spine heads. Scale bar, 500 nm. F, Schematic representation of spine heads (yellow) isolated from the micrographs shown in D with high-intensity clustered pixels (orange), as derived from spatial autocorrelation analysis using Moran's index (for details, see Materials and Methods). G, H, Summary graphs of the mean coefficient of variation (CV) of LifeAct-Venus fluorescence (G) and the mean percentage of high-intensity clustered pixels (H) in spine heads of neurons expressing nontarget or KCC2-directed shRNA (same dataset as in E). n.s., Nonsignificant difference.
Rac1- and LIMK-mediated reduction of cofilin activity accounts for the lack of activity-driven GluA1 traffic and LTP upon KCC2 suppression
We asked whether increased actin polymerization upon KCC2 suppression might be responsible for the defect in LTP expression in hippocampal neurons. Transient actin depolymerization has been shown to be required for activity-driven AMPAR membrane traffic and spine enlargement (Ouyang et al., 2005; Gu et al., 2010). This reflects a transient activation (i.e., dephosphorylation) of the actin-severing enzyme cofilin upon LTP induction. We therefore compared cofilin phosphorylation in hippocampal neurons transduced with lentiviruses expressing nontarget or KCC2-specific shRNA and observed a nearly twofold increase in phospho-cofilin/total cofilin ratio upon KCC2 suppression under basal conditions (85.8 ± 14.3%, n = 3 independent experiments, p < 0.02; Fig. 7A,B). However, the lack of KCC2 did not prevent cLTP-induced decrease in cofilin phosphorylation (−79.7 ± 9.4% of control after 16 min of cLTP induction, p < 0.001), to a similar extent as in neurons expressing nontarget shRNA (p = 0.4).
Two major signaling pathways involving the Rho GTPases RhoA and Rac1 converge on LIM kinase to control cofilin phosphorylation and actin dynamics (Cingolani and Goda, 2008) (Fig. 8D). We tested whether enhanced cofilin phosphorylation may result from activation of either GTPase. Pulldown experiments revealed a specific increase in active (GTP-bound) Rac1 in neurons with suppressed KCC2 expression. Thus, we observed a near fivefold increase in activated/total Rac1 ratio in neurons transduced with KCC2-directed compared with with nontarget shRNA (471 ± 131% of shNT ratio, n = 4 independent experiments, p < 0.01; Figure 7C,D) with no significant change in RhoA activity (92 ± 19% of shNT ratio, n = 3 independent experiments, p = 0.5).
KCC2 was recently reported to interact with the Rac1-specific guanine nucleotide exchange factor βPIX, both in adult mouse brain extracts and in heterologous cells, and to inhibit βPIX-induced Rac1 activation (Llano et al., 2015). βPIX is also known to interact with GIT1, and their recruitment to specific subcellular compartments together with downstream effectors may serve to locally regulate actin dynamics (Zhang et al., 2003). We confirmed KCC2 interaction with βPIX in primary hippocampal neurons using coimmunoprecipitation assays using antibodies against either protein (Fig. 7E). Suppressing KCC2 expression resulted in changes in both βPIX and GIT1 clustering, as detected in immunostaining experiments (Fig. 7F,G). Thus, the integrated intensity of βPIX and GIT1 clusters was increased by 55.0 ± 9.4% and 30.6 ± 7.2%, respectively, in the dendrites of neurons expressing KCC2-specific compared with with nontarget shRNA (p < 0.001 for both). Suppression of KCC2 also resulted in a 27.3 ± 5.0% increase in the density of βPIX clusters (p < 0.001, n = 110 and n = 104 cells, respectively). Together, our results suggest that KCC2 suppression may enhance actin polymerization through Rac1- and LIMK-mediated inhibition of cofilin, likely due to subcellular recruitment of βPIX-GIT1 complexes.
Enhanced cofilin phosphorylation at the time of cLTP induction may then occlude activity-driven AMPAR membrane insertion (Gu et al., 2010). Inhibiting steady-state cofilin phosphorylation in neurons lacking KCC2 might then be sufficient to rescue LTP expression. We tested this hypothesis by monitoring cLTP-induced SEP-GluA1 exocytosis in neurons pretreated with either LIMK or Rac1-PAK inhibitors. A peptide containing the first 16-amino acid sequence of cofilin (Ser3) fused to penetratin for cell internalization was first used to inhibit endogenous LIMK activity (Gu et al., 2010). A 4 h application of Ser3 peptide had no effect on cLTP-induced SEP-GluA1 membrane insertion in neurons transfected with nontarget shRNA (11.2 ± 1.8% vs 9.6 ± 1.9% of control, n = 13 and n = 17 cells, respectively, p = 0.6; Fig. 8A,B) but rescued it in neurons with suppressed KCC2 expression (8.6 ± 1.4% vs −1.6 ± 2.0% of control, n = 24 and n = 21 cells, respectively, p < 0.001). A similar rescue was obtained in experiments using the LIMK inhibitor LIMKi (Ross-Macdonald et al., 2008) (15.7 ± 2.9% of control, n = 20 cells, p < 0.001) or the PAK inhibitor IPA-3 (13.3 ± 3.3% of control, n = 29 and n = 22 cells, respectively, p < 0.001). In contrast, prior incubation with the RhoA-specific inhibitor Rhosin failed to rescue cLTP-induced SEP-GluA1 membrane insertion in neurons transfected with KCC2-specific shRNA (−0.2 ± 1.5% of control, n = 14 and n = 13 cells, respectively, p = 0.98; Fig. 8A,B).
Finally, because activity-induced GluA1 traffic gates spine enlargement during LTP (Kopec et al., 2007), we asked whether rescuing activity-driven AMPAR insertion by inhibiting LIMK or PAK upon KCC2 suppression may be sufficient to also restore structural LTP. In neurons treated with either Ser3 peptide or LIMKi, cLTP led to a similar increase in spine volume in neurons expressing KCC2-specific (17.3 ± 5.5% of control, n = 355 spines from 21 cells) and nontarget shRNA (19.2 ± 4.7% of control, n = 208 spines from 9 cells, p = 0.8; Fig. 8C). Similar results were obtained upon pretreatment with the PAK inhibitor IPA-3 (13.3 ± 2.3 vs 14.7 ± 2.9% of control spine volume; n = 29 and n = 23 cells, respectively, p = 0.9; Fig. 8C). Together, these data demonstrate that the loss of KCC2 expression affects actin polymerization through enhanced cofilin phosphorylation and thereby precludes both synaptic and structural LTP expression.
Discussion
Our results reveal an unexpected role of KCC2 in the regulation of glutamatergic signaling in hippocampal neurons. We show that KCC2 membrane expression, but not function, is required for activity-driven AMPAR membrane insertion and thereby gates the expression of LTP at glutamatergic synapses. This effect involves enhanced Rac-1- and LIMK-mediated cofilin phosphorylation resulting in increased F-actin mobilization in dendritic spines. Thus, preventing Rac1/PAK activation or cofilin phosphorylation upon KCC2 suppression rescues activity-driven AMPAR membrane insertion as well as structural LTP. KCC2-mediated control of actin polymerization may then represent a new modality of metaplasticity at hippocampal glutamatergic synapses.
LTP of excitatory synapses involves a cascade of biochemical events (Poncer, 2003), each of which could in principle be perturbed by KCC2 suppression. Our results, however, suggest that synaptic and structural LTP was primarily compromised due to altered actin dynamics. Direct activation of CaMKII failed to induce AMPAR membrane insertion, suggesting that LTP was likely not compromised due to a defect in induction mechanisms. In addition, cLTP-induced GluA1 Ser845 phosphorylation was unaffected in neurons lacking KCC2, suggesting that only the ultimate steps of activity-driven AMPAR traffic were affected. The reduced density of extrasynaptic, GluA1-containing AMPARs upon KCC2 suppression (Fig. 3D) may in principle contribute to hinder LTP expression by limiting their activity-dependent recruitment at synapses. However, our data are based on total GluA1-SEP fluorescence in dendritic sections and therefore suggest instead a defect in activity-dependent membrane insertion in neurons lacking KCC2. Phalloidin staining showed that suppressing KCC2 expression or overexpressing its C-terminal domain increases F-actin content in spines (Fig. 5). This may reflect increased aggregation of F-actin bundles within spines or increased actin polymerization. KCC2 interaction with the spectrin/actin-binding protein 4.1N (Li et al., 2007) suggests that KCC2 may be part of a complex anchoring submembrane actin cytoskeleton to plasma membrane. A similar role has been ascribed to other ion-transport proteins in various cell types (Baines et al., 2014). Accordingly, we previously observed that suppressing KCC2 increased membrane diffusion of GluA1-containing AMPARs and NCAM180 (Gauvain et al., 2011), both of which interact with actin. Conversely, suppressing 4.1N expression increased membrane diffusion of KCC2 (Chamma et al., 2013). However, we could not detect changes in 4.1N expression in spines upon KCC2 suppression, suggesting that KCC2 is not strictly required for 4.1N aggregation in spines. In addition, phalloidin staining of endogenous F-actin and STED imaging of LifeAct, respectively, reveal an increased F-actin content in dendritic spines with no significant difference in its intraspine distribution (Fig. 6). Although more subtle changes in F-actin interactions with plasma membrane may occur upon KCC2 suppression, our observations are consistent with increased actin polymerization rather than a major reorganization of actin cytoskeleton within dendritic spines.
Consistent with this conclusion, we observed a reduction in active cofilin in neurons lacking KCC2 (Fig. 7). Cofilin plays a major role in actin signaling (Cingolani and Goda, 2008; Bosch and Hayashi, 2012). Its active, dephosphorylated form promotes actin depolymerization. Our results show a near-complete recovery of AMPAR traffic and structural LTP by LIMK inhibitors. This strongly suggests that LIMK is hyperactivated in the absence of KCC2, resulting in enhanced actin polymerization. Importantly, we observed a twofold increase in cofilin phosphorylation upon suppression of KCC2 expression at rest. However, phosphorylation was still reduced during cLTP (Fig. 7A,B), suggesting that steady-state, but not activity-driven, regulation of cofilin was specifically affected by KCC2 suppression. How may KCC2 influence LIMK activity? Among the numerous molecular interactions KCC2 may be engaged in, a recent report shows that KCC2 directly interacts with the Rac1 GEF βPIX, thereby inhibiting β-PIX-induced Rac1 activation (Llano et al., 2015). In addition, KCC2 interaction with β-PIX prevented Rac1 activation in heterologous cells. Our data support direct interaction between KCC2 and βPIX and further demonstrate (1) subcellular redistribution of both βPIX and its partner GIT1 in dendrites and (2) enhanced Rac1, but not RhoA, activity upon KCC2 suppression. Subcellular recruitment of βPIX and GIT1 with their downstream effectors mediates local Rac1 activation in neurons (Zhang et al., 2003). Because F-actin content in dendritic spines is similarly increased in neurons lacking KCC2 or overexpressing its C-terminal domain, it seems unlikely that KCC2 may solely compete with βPIX binding to other proteins. Instead, because membrane-inserted KCC2 usually surrounds but is excluded from the PSD (Gulyás et al., 2001; Chamma et al., 2013), it may act to trap βPIX at distance from PSD partners (Park et al., 2003), such as GIT1 and downstream effectors (Zhang et al., 2003), thereby spatially restricting βPIX-driven Rac1/PAK activation. Together, our results therefore strongly support that loss of KCC2 interaction with βPIX results in Rac1 activation, leading to LIMK activation and inhibition of cofilin. Finally, pharmacological blockade of the Rac1-PAK pathway, but not RhoA, fully restored activity-driven GluA1 membrane traffic and increased spine volume during cLTP. These observations demonstrate that LTP hindrance upon KCC2 suppression primarily relies on the loss of KCC2-βPIX interaction and subsequent hyperactivation of the Rac1/PAK/LIMK pathway.
How may reduced cofilin activity prevent activity-driven AMPAR exocytosis and spine enlargement? Although LTP is associated with F-actin accumulation in dendritic spines (Bosch and Hayashi, 2012), a transient increase in both cofilin activity and density of actin barbed ends has been reported during cLTP induction (Gu et al., 2010). Preventing cofilin activation by dephosphorylation or stabilizing F-actin pharmacologically compromised AMPAR membrane insertion during cLTP. Therefore, transient, cofilin-mediated, actin depolymerization represents a permissive step for activity-driven AMPAR membrane insertion. The underlying mechanisms remain unclear but may involve hindering of vesicle fusion to plasma membrane by cortical actin, as described in non-neuronal cells (Gasman et al., 1999), or cytoskeleton entrapping of secretory vesicles preventing their traffic toward the membrane. Suppressing KCC2 expression also abolished cLTP-induced spine enlargement. This effect is unlikely to be due to the increase in spine volume induced by the loss of KCC2 function (Gauvain et al., 2011) because the KCC2 antagonist VU0241550 produced the same increase in volume as KCC2 suppression but did not prevent cLTP-induced spine enlargement (Fig. 2F). In neurons lacking KCC2, inhibiting PAK or LIMK rescued both activity-driven AMPAR insertion and spine enlargement (Fig. 8). This suggests the two processes may either be linked together or both independently linked to actin dynamics. Although not sufficient on its own, synaptic translocation of GluA1-containing AMPARs is permissive to cLTP-induced spine enlargement (Kopec et al., 2007). However, GluA1 synaptic translocation in the absence of actin polymerization fails to induce spine enlargement. These observations suggest spine enlargement during LTP requires both activity-driven GluA1 translocation and actin polymerization. Therefore, Rac1/PAK- and LIMK-mediated cofilin inactivation in neurons lacking KCC2 is likely to prevent spine enlargement during cLTP primarily by hindering GluA1 traffic.
KCC2 membrane expression and stability are tightly regulated by neuronal activity (Chamma et al., 2012; Kahle et al., 2013). In particular, Ca2+ influx through postsynaptic NMDARs rapidly reduces KCC2 function at least in part through calcium-dependent Ser940 dephosphorylation (Lee et al., 2011; Chamma et al., 2013) and calpain-mediated cleavage (Puskarjov et al., 2012; Chamma et al., 2013), leading to reduced membrane stability of the transporter. Phosphorylation of Tyr903 and Tyr1087 by Src-family tyrosine kinases on the other hand was suggested to enhance (Wake et al., 2007; Watanabe et al., 2009) or reduce (Lee et al., 2010) KCC2 membrane stability. Activation of muscarinic acetylcholine receptors enhances KCC2 tyrosine phosphorylation while promoting its endocytosis and degradation (Lee et al., 2010). Thus, both Ca2+ influx and tyrosine kinase activation may rapidly affect the membrane pool of KCC2. Our results predict that this may in turn influence the ability of excitatory synapses to undergo LTP. This raises the possibility that activity-dependent regulation of KCC2 membrane stability may not only tune GABA signaling to ongoing neuronal activity (Woodin et al., 2003; Fiumelli et al., 2005) but also act as a metaplastic switch at glutamatergic synapses. In this context, it is interesting to note that NMDA receptor activation before induction was shown to suppress LTP at hippocampal synapses (Huang et al., 1992). Although the underlying mechanisms were not fully identified, they may involve recruitment of protein phosphatase 1 (Kato et al., 1999), which mediates KCC2 membrane destabilization (Lee et al., 2011), consistent with a contribution of activity-dependent KCC2 internalization in gating LTP expression at excitatory synapses. More experiments are clearly needed to fully explore how activity-driven downregulation of KCC2 membrane stability may affect plasticity rules at glutamatergic synapses.
In addition to activity-induced changes at the post-translational level, KCC2 expression is also down-regulated in a variety of pathological conditions ranging from epilepsy (Rivera et al., 2004; Jin et al., 2005; Miles et al., 2012; Pallud et al., 2014), stroke (Jaenisch et al., 2010), to schizophrenia (Hyde et al., 2011) and autism (Tyzio et al., 2014). These conditions are associated with increased neuronal activity, which is often assumed to reflect reduced GABAergic signaling due to altered chloride extrusion. Therefore, agents acting to compensate for the loss of KCC2 function represent promising therapeutic options. For instance, antagonists of the NKCC1 chloride importer, such as bumetanide, effectively suppress interictal discharges in human epileptic cortex in vitro (Pallud et al., 2014). These strategies, however, are predicted not to compensate altered glutamatergic signaling induced by KCC2 suppression. Although the net impact of a reduced excitatory synaptic function (Gauvain et al., 2011) and long-term plasticity (this study) on network activity is somewhat difficult to predict, rhythmogenesis and mnesic performances are likely to be affected. In this respect, therapeutic strategies acting to restore KCC2 expression or membrane stability (Bos et al., 2013; Gagnon et al., 2013) rather than just chloride homeostasis may be more effective in fully compensating the effects of its suppression in neurological and psychiatric disorders.
Footnotes
This work was supported by Human Frontier Science Program Research Grant (RGP0022/2013) to J.C.P., Institut National de la Santé et de la Recherche Médicale, Ville de Paris Biomedical and Health Research Program to J.C.P., Fédération pour la Recherche sur le Cerveau to J.C.P., Fondation pour la Recherche Médicale (DEQ20140329539) to J.C.P., and PICPEN (Center for Psychiatry and Neuroscience, Paris). Q.C. was the recipient of doctoral fellowships from Université Pierre and Marie Curie and Fondation pour la Recherche Médicale. We thank Ingrid Chamma for the quantitative analysis of phalloidin staining data; Theano Irinopoulou for support with spinning disc and confocal imaging; David Geny at PICPEN for assistance with STED microscopy; Alain Prochiantz and David diGregorio for helpful discussions; and Eric J. Schwartz, Manuel Mameli, Kai Kaila, Kevin J. Staley, and Richard Miles for constructive comments on an earlier version of the manuscript.
The authors declare no competing financial interests.
References
- Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backer S, Hidalgo-Sánchez M, Offner N, Portales-Casamar E, Debant A, Fort P, Gauthier-Rouvière C, Bloch-Gallego E. Trio controls the mature organization of neuronal clusters in the hindbrain. J Neurosci. 2007;27:10323–10332. doi: 10.1523/JNEUROSCI.1102-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines AJ, Lu HC, Bennett PM. The Protein 4.1 family: hub proteins in animals for organizing membrane proteins. Biochim Biophys Acta. 2014;1838:605–619. doi: 10.1016/j.bbamem.2013.05.030. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Bos R, Sadlaoud K, Boulenguez P, Buttigieg D, Liabeuf S, Brocard C, Haase G, Bras H, Vinay L. Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci U S A. 2013;110:348–353. doi: 10.1073/pnas.1213680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Hayashi Y. Structural plasticity of dendritic spines. Curr Opin Neurobiol. 2012;22:383–388. doi: 10.1016/j.conb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Causeret F, Hidalgo-Sanchez M, Fort P, Backer S, Popoff MR, Gauthier-Rouvière C, Bloch-Gallego E. Distinct roles of Rac1/Cdc42 and Rho/Rock for axon outgrowth and nucleokinesis of precerebellar neurons toward netrin 1. Development. 2004;131:2841–2852. doi: 10.1242/dev.01162. [DOI] [PubMed] [Google Scholar]
- Chamma I, Chevy Q, Poncer JC, Lévi S. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci. 2012;6:5. doi: 10.3389/fncel.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamma I, Heubl M, Chevy Q, Renner M, Moutkine I, Eugène E, Poncer JC, Lévi S. Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J Neurosci. 2013;33:15488–15503. doi: 10.1523/JNEUROSCI.5889-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.3410/f.1010572.158257. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Delpire E, Days E, Lewis LM, Mi D, Kim K, Lindsley CW, Weaver CD. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci U S A. 2009;106:5383–5388. doi: 10.1073/pnas.0812756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu D, Rodriguez A, Morrison JH. High-throughput, detailed, cell-specific neuroanatomy of dendritic spines using microinjection and confocal microscopy. Nat Protoc. 2011;6:1391–1411. doi: 10.1038/nprot.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Briner A, Puskarjov M, Blaesse P, Belem BJ, Dayer AG, Kaila K, Martin JL, Vutskits L. An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb Cortex. 2013;23:378–388. doi: 10.1093/cercor/bhs027. [DOI] [PubMed] [Google Scholar]
- Gagnon M, Bergeron MJ, Lavertu G, Castonguay A, Tripathy S, Bonin RP, Perez-Sanchez J, Boudreau D, Wang B, Dumas L, Valade I, Bachand K, Jacob-Wagner M, Tardif C, Kianicka I, Isenring P, Attardo G, Coull JA, De Koninck Y. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med. 2013;19:1524–1528. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasman S, Chasserot-Golaz S, Popoff MR, Aunis D, Bader MF. Involvement of Rho GTPases in calcium-regulated exocytosis from adrenal chromaffin cells. J Cell Sci. 1999;112:4763–4771. doi: 10.1242/jcs.112.24.4763. [DOI] [PubMed] [Google Scholar]
- Gauvain G, Chamma I, Chevy Q, Cabezas C, Irinopoulou T, Bodrug N, Carnaud M, Lévi S, Poncer JC. The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc Natl Acad Sci U S A. 2011;108:15474–15479. doi: 10.1073/pnas.1107893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Sík A, Payne JA, Kaila K, Freund TF. The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur J Neurosci. 2001;13:2205–2217. doi: 10.1046/j.0953-816x.2001.01600.x. [DOI] [PubMed] [Google Scholar]
- Harrington WW, Britt C, Wilson J, Milliken N, Binz J, Lobe D, Oliver W, Lewis M, Ignar D. The effect of PPARalpha, PPARdelta, PPARgamma, and PPARpan agonists on body weight, body mass, and serum lipid profiles in diet-induced obese AKR/J Mice. PPAR Res. 2007;2007:97125. doi: 10.1155/2007/97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Huang YY, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255:730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, Straub RE, Ye T, Colantuoni C, Herman MM, Bigelow LB, Weinberger DR, Kleinman JE. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivakine EA, Acton BA, Mahadevan V, Ormond J, Tang M, Pressey JC, Huang MY, Ng D, Delpire E, Salter MW, Woodin MA, McInnes RR. Neto2 is a KCC2 interacting protein required for neuronal Cl- regulation in hippocampal neurons. Proc Natl Acad Sci U S A. 2013;110:3561–3566. doi: 10.1073/pnas.1212907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke. 2010;41:e151–e159. doi: 10.1161/STROKEAHA.109.570424. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4:490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Li ST, Zorumski CF. Modulation of long-term potentiation induction in the hippocampus by N-methyl-D-aspartate-mediated presynaptic inhibition. Neuroscience. 1999;92:1261–1272. doi: 10.1016/S0306-4522(99)00080-9. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Jurd R, Moss SJ. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci. 2010;45:173–179. doi: 10.1016/j.mcn.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABA(A) receptor-mediated currents. Nat Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci. 2009;12:879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano O, Smirnov S, Soni S, Golubtsov A, Guillemin I, Hotulainen P, Medina I, Nothwang HG, Rivera C, Ludwig A. KCC2 regulates actin dynamics in dendritic spines via interaction with beta-PIX. J Cell Biol. 2015;209:671–686. doi: 10.1083/jcb.201411008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- Mahadevan V, Pressey JC, Acton BA, Uvarov P, Huang MY, Chevrier J, Puchalski A, Li CM, Ivakine EA, Airaksinen MS, Delpire E, McInnes RR, Woodin MA. Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Rep. 2014;7:1762–1770. doi: 10.1016/j.celrep.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron. 2011;72:1001–1011. doi: 10.1016/j.neuron.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P, Pellegrino C. Current view on the functional regulation of the neuronal K(+)-Cl(−) cotransporter KCC2. Front Cell Neurosci. 2014;8:27. doi: 10.3389/fncel.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Blaesse P, Huberfeld G, Wittner L, Kaila K. Chloride homeostasis and GABA signaling in temporal lobe epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies. Bethesda, MD: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- Moran PA. The interpretation of statistical maps. J R Stat Soc Series B. 1948;10:243–251. [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–9331. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Wong M, Capani F, Rensing N, Lee CS, Liu Q, Neusch C, Martone ME, Wu JY, Yamada K, Ellisman MH, Choi DW. Transient decrease in F-actin may be necessary for translocation of proteins into dendritic spines. Eur J Neurosci. 2005;22:2995–3005. doi: 10.1111/j.1460-9568.2005.04521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallud J, Le Van Quyen M, Bielle F, Pellegrino C, Varlet P, Labussiere M, Cresto N, Dieme MJ, Baulac M, Duyckaerts C, Kourdougli N, Chazal G, Devaux B, Rivera C, Miles R, Capelle L, Huberfeld G. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci Transl Med. 2014;6:244ra289. doi: 10.1126/scitranslmed.3008065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Na M, Choi J, Kim S, Lee JR, Yoon J, Park D, Sheng M, Kim E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem. 2003;278:19220–19229. doi: 10.1074/jbc.M301052200. [DOI] [PubMed] [Google Scholar]
- Poncer JC. Hippocampal long term potentiation: silent synapses and beyond. J Physiol. 2003;97:415–422. doi: 10.1016/j.jphysparis.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Esteban JA, Malinow R. Multiple mechanisms for the potentiation of AMPA receptor-mediated transmission by alpha-Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2002;22:4406–4411. doi: 10.1523/JNEUROSCI.22-11-04406.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO Rep. 2014;15:723–729. doi: 10.1002/embr.201438749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho GJ, Kim DS, Son WJ, Cho SR, Kim JG, Choe SY. Influence of in vitro oxygen concentrations on preimplantation embryo development, gene expression and production of Hanwoo calves following embryo transfer. Mol Reprod Dev. 2007;74:486–496. doi: 10.1002/mrd.20502. [DOI] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. LifeAct: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Macdonald P, de Silva H, Guo Q, Xiao H, Hung CY, Penhallow B, Markwalder J, He L, Attar RM, Lin TA, Seitz S, Tilford C, Wardwell-Swanson J, Jackson D. Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol Cancer Ther. 2008;7:3490–3498. doi: 10.1158/1535-7163.MCT-08-0826. [DOI] [PubMed] [Google Scholar]
- Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Görlich A, Sassoè-Pognetto M, Banchaabouchi MA, Giustetto M, Triller A, Choquet D, Witke W. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010;29:1889–1902. doi: 10.1038/emboj.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107. doi: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Davenport EC, Wei J, Li X, Pathania M, Vaccaro V, Yan Z, Kittler JT. GIT1 and betaPIX are essential for GABA(A) receptor synaptic stability and inhibitory neurotransmission. Cell Rep. 2014;9:298–310. doi: 10.1016/j.celrep.2014.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietz EI, Kapur J, Macdonald RL. Functional GABAA receptor heterogeneity of acutely dissociated hippocampal CA1 pyramidal cells. J Neurophysiol. 1999;81:1575–1586. doi: 10.1152/jn.1999.81.4.1575. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, Khalilov I, Tsintsadze V, Brouchoud C, Chazal G, Lemonnier E, Lozovaya N, Burnashev N, Ben-Ari Y. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343:675–679. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- Urban NT, Willig KI, Hell SW, Nägerl UV. STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys J. 2011;101:1277–1284. doi: 10.1016/j.bpj.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Watanabe M, Moorhouse AJ, Kanematsu T, Horibe S, Matsukawa N, Asai K, Ojika K, Hirata M, Nabekura J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci. 2007;27:1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Wake H, Moorhouse AJ, Nabekura J. Clustering of neuronal K+-Cl− cotransporters in lipid rafts by tyrosine phosphorylation. J Biol Chem. 2009;284:27980–27988. doi: 10.1074/jbc.M109.043620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM. Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron. 2003;39:807–820. doi: 10.1016/S0896-6273(03)00507-5. [DOI] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Horwitz AF. Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol. 2003;161:131–142. doi: 10.1083/jcb.200211002. [DOI] [PMC free article] [PubMed] [Google Scholar]