

Abstract
Imprinted genes are dosage sensitive, and their dysregulated expression is linked to disorders of growth and proliferation, including fetal and postnatal growth restriction. Common sequelae of growth disorders include neurodevelopmental defects, some of which are indirectly related to placental insufficiency. However, several growth-associated imprinted genes are also expressed in the embryonic CNS, in which their aberrant expression may more directly affect neurodevelopment. To test whether growth-associated genes influence neural lineage progression, we focused on the maternally imprinted gene Zac1. In humans, either loss or gain of ZAC1 expression is associated with reduced growth rates and intellectual disability. To test whether increased Zac1 expression directly perturbs neurodevelopment, we misexpressed Zac1 in murine neocortical progenitors. The effects were striking: Zac1 delayed the transition of apical radial glial cells to basal intermediate neuronal progenitors and postponed their subsequent differentiation into neurons. Zac1 misexpression also blocked neuronal migration, with Zac1-overexpressing neurons pausing more frequently and forming fewer neurite branches during the period when locomoting neurons undergo dynamic morphological transitions. Similar, albeit less striking, neuronal migration and morphological defects were observed on Zac1 knockdown, indicating that Zac1 levels must be regulated precisely. Finally, Zac1 controlled neuronal migration by regulating Pac1 transcription, a receptor for the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP). Pac1 and Zac1 loss- and gain-of-function presented as phenocopies, and overexpression of Pac1 rescued the Zac1 knockdown neuronal migration phenotype. Thus, dysregulated Zac1 expression has striking consequences on neocortical development, suggesting that misexpression of this transcription factor in the brain in certain growth disorders may contribute to neurocognitive deficits.
SIGNIFICANCE STATEMENT Altered expression of imprinted genes is linked to cognitive dysfunction and neuropsychological disorders, such as Angelman and Prader–Willi syndromes, and autism spectrum disorder. Mouse models have also revealed the importance of imprinting for brain development, with chimeras generated with parthenogenetic (two maternal chromosomes) or androgenetic (two paternal chromosomes) cells displaying altered brain sizes and cellular defects. Despite these striking phenotypes, only a handful of imprinted genes are known or suspected to regulate brain development (e.g., Dlk1, Peg3, Ube3a, necdin, and Grb10). Herein we show that the maternally imprinted gene Zac1 is a critical regulator of neocortical development. Our studies are relevant because loss of 6q24 maternal imprinting in humans results in elevated ZAC1 expression, which has been associated with neurocognitive defects.
Keywords: neocortex, neuronal migration, Pac1, progenitor maturation, Zac1
Introduction
Development of a functional nervous system requires that appropriate numbers of the correct types of neurons first differentiate and then migrate to their proper destinations in which they establish specific synaptic connections. Long-term cognitive and behavioral deficits can arise when neurogenesis, neuronal migration, or circuit formation are disrupted. Infants with intrauterine growth restriction (IUGR), defined as birth weights below the 10th percentile for gestational age (Peleg et al., 1998), have an increased risk of long-term neurological disabilities (Geva et al., 2006a,b; Fattal-Valevski et al., 2009). Although IUGR-linked neurodevelopmental defects can be a secondary consequence of reduced nutrient/oxygen levels during pregnancy from placental insufficiency, several genes associated with IUGR are also expressed in the embryonic CNS, in which their dysregulated expression may more directly influence nervous system development. Included in this category are imprinted genes, which are expressed in a parent-of-origin-specific manner, and are emerging as key regulators of both intrauterine growth and brain development (Wilkinson et al., 2007; Diplas et al., 2009).
Zac1, also known as pleiomorphic adenoma gene like 1 (Plagl1), is a maternally imprinted gene that encodes a seven-C2H2 zinc finger protein (Abdollahi, 2007). Human ZAC1 is located on chromosome 6q24-25, a locus silenced in multiple carcinomas, including head and neck, ovarian, and pituitary tumors (Abdollahi, 2007). The ZAC1 maternal imprint is established during oogenesis by methylation of an imprinting control region (ICR), which silences transcription from a maternal P1 promoter (Arima and Wake, 2006). Loss of 6q24 maternal imprinting, resulting in biallelic expression, occurs in 70% of infants with transient neonatal diabetes mellitus (TNDM), a disorder associated with growth retardation (Temple and Shield, 2002; Azzi et al., 2014). In contrast, ICR hypermethylation reduces ZAC1 expression in ovarian tumor cells (Kamikihara et al., 2005). Reduced ZAC1 expression is also associated with growth restriction, developmental delay, and intellectual disability (e.g., Decipher identification numbers 248227 and 294593).
In mouse models, Zac1 regulates embryonic growth (Varrault et al., 2006), as well as keratinocyte (Basyuk et al., 2005), heart (Czubryt et al., 2010; Yuasa et al., 2010), pancreatic islet (Anderson et al., 2009), cerebellar (Chung et al., 2011), and retinal (Ma et al., 2007a,b) development. We identified Zac1 in a subtractive screen designed to identify new regulators of neocortical neurogenesis (Mattar et al., 2004). Here, we asked whether altered Zac1 expression in the embryonic neocortex, the seat of higher-order cognitive functioning, could give rise to morphological defects that may result in neurocognitive deficits (Geva et al., 2006a,b; Fattal-Valevski et al., 2009). Misexpression of Zac1 in neocortical progenitors inhibited progenitor maturation, while delaying neuronal differentiation and migration. The effects of Zac1 on neuronal migration were in part mediated by Pac1 (pituitary adenylate cyclase-activating polypeptide type I receptor), a Zac1 transcriptional target (Ciani et al., 1999; Rodríguez-Henche et al., 2002) that controls neocortical progenitor proliferation (Suh et al., 2001; Yan et al., 2013). We have thus identified a novel Zac1–Pac1 regulatory pathway that controls progenitor maturation, neuronal differentiation, and migration in the developing neocortex.
Materials and Methods
Animals.
Embryos were staged using the morning of the vaginal plug as embryonic day 0.5 (E0.5). CD1 mice (Charles River Laboratories) were used for in utero electroporation experiments. Zac1 null mutant embryos were obtained by crossing Zac1+/− males with C57BL/6 wild-type females. The resulting Zac1+m/− embryos, which obtained their wild-type allele from the dam, were the equivalent of Zac1 null mutants because of imprinting of the maternal Zac1 allele. Genotyping Zac1 mutant and wild-type alleles was performed as described previously (Ma et al., 2007b).
Constructs used for in utero electroporation.
For gain-of-function experiments, Zac1 and Pac1 were cloned into pCIG2 (Hand et al., 2005), a bicistronic expression vector containing a β-actin promoter/CMV enhancer and an internal ribosome entry site (IRES)–EGFP cassette (Hand et al., 2005). For knockdown experiments, shRNAs were obtained from ORIGENE: HuSH shRNA TG502444 Mus musculus Plagl1 (Zac1) in pGFP–V-RS; HuSH shRNA TG500044 M. musculus Adcyap1r1 in pGFP–V-RS. To identify which of the four shRNAs was most effective, NIH-3T3 cells were transfected with pCIG2–Zac1 or pCIG2–Pac1 either alone or together with individual shRNAs, and Western blots were performed 24 h later (as in the study by Li et al., 2012). The scrambled shRNA was from ORIGENE (TR30013). EGFP–CentII (Tanaka et al., 2004) and pEF/Myc/ER/GFP vectors (Invitrogen) were modified to incorporate RFP and mCherry reporters, as described previously (Shim et al., 2008).
In utero electroporation.
In utero electroporation was performed as described previously (Dixit et al., 2011). Briefly, endotoxin-free DNA was prepared according to the instructions of the manufacturer (Qiagen) and injected at 1.5 μg/μl into the telencephalic vesicles of embryos in time-staged pregnant females anesthetized under inhalable isoflurane (5 L/min) using a Femtojet microinjector apparatus (VWR CanLab) and three-axis coarse manipulator (Carl Zeiss). This was followed by seven 50 V pulses at 750 ms intervals applied by tweezer-style electrodes (5 mm for E12.5 and 7 mm for E14.5; Protech International) using a BTX square wave electroporator (VWR CanLab). The uterus was replaced in the body cavity, the peritoneum was sutured, the skin stapled, and normal embryonic development proceeded until the time of harvesting.
RNA extraction, cDNA synthesis, and quantitative real-time PCR.
RNA was extracted from E18.5 wild-type and Zac1 mutant cortices and from microdissected E13.5–E14.5 cortical tissue electroporated with pCIG2 or pCIG2–Zac1 using an RNeasy Mini kit (Qiagen) according to the instructions of the manufacturer. First-strand cDNA was synthesized using the Quantitect Reverse Transcription kit (Qiagen) according to the instructions of the manufacturer. Real-time qRT-PCR was performed using an Opticon 2 DNA engine (Bio-Rad Laboratories) using a Quantifast SYBR Green kit (Qiagen). For every primer pair, three different cDNA dilutions were tested with the following cycle conditions: one cycle of 95°C for 4 min, 40 cycles of 95°C for 1 min, 55–67°C for 1 min, 72°C for 1 min 30 s, and one cycle of 72°C for 10 min. The annealing temperature was optimized for each primer pair using a temperature gradient, selecting conditions that yielded >95% amplification efficiencies. Normalization was achieved using hypoxanthine phosphoribosyl-transferase 1 (Hprt) and Beta-2 microglobulin (B2m) as reference genes: Pac1 forward, TACTCCAGATGTGGTTCCAGGC; Pac1 reverse, AGTGAGGTCCGTGGGGTTTATC (66°C annealing); B2M forward, CCTGGTCTTTCTGGTGCTTGTC; B2M reverse, CAGTATGTTCGGCTTCCCATTC (63°C annealing); HPRT forward, AGCTACTGTAATGATCAGTCAACG; HPRT reverse, AGAGGTCCTTTTCACCAGCA (58.3°C annealing); Zac1 forward, AATGTGGCAAGTCCTTCGTCAC; and Zac1 reverse, TGGTTCTTCAGGTGGTCCTTCC (67°C annealing).
Tissue processing and immunolabeling.
Dissected brains were fixed overnight at 4°C in 4% paraformaldehyde (PFA)/1× PBS. Brains were rinsed three times for 10 min in 1× PBS and transferred to 20% sucrose/1× PBS overnight at 4°C. Cryopreserved brains were then embedded in O.C.T. (Tissue-Tek; Sakura Finetek) and stored at −80°C before cutting 10 μm cryosections. For immunolabeling, sections were blocked 1 h in 10% normal goat serum in 1× TBST (Tris-buffered saline: 25 mm Tris, 0.14 m NaCl, and 0.1% Triton X-100) at room temperature. Primary antibodies were diluted in blocking solution and incubated on slides overnight at 4°C. Slides were washed three times for 10 min in TBST before incubating in secondary antibody diluted in TBST for 1 h at room temperature. Nuclei were counterstained in 4′,6-diamidino-2-phenylindole (DAPI; Santa Cruz Biotechnology) diluted 1:10,000 in 1× PBS for 5 min and then destained with three PBS washes for 5 min before mounting in AquaPolymount (Polysciences). Primary antibodies included the following: mouse anti-BrdU (1:200; Roche Diagnostics), rabbit anti-GFP (1:500; Millipore Bioscience Research Reagents), rabbit anti-Pax6 (1:500; Covance), rabbit anti-Cux1 (anti-CDP; 1:500; Santa Cruz Biotechnology), mouse anti-Tuj1 (Neuronal class III β-tubulin; 1:500; Covance), rabbit anti-Tbr1 (1:3000; Millipore Bioscience Research Reagents), rabbit anti-Tbr2 (1:500; Abcam), rabbit anti-phospho-histone H3 (pHH3; 1:1000; Millipore Biotechnology), mouse anti-neuronal-specific nuclear protein (NeuN; 1:500; Millipore Bioscience Research Reagents), goat anti-Beta3 (1:300; Santa Cruz Biotechnology), rat anti-Ctip2 (1:100; Abcam), rabbit anti-Ki67 (1:200; Abcam), and rabbit anti-Zac1 (1:1000; Spengler et al., 1997). Secondary antibodies were conjugated to Alexa Fluor 488 (1:500; Invitrogen) or Cy3 (1:500; Jackson ImmunoResearch).
BrdU and 5-ethynyl-2′-deoxyuridine labeling.
For birthdating and proliferation studies, 100 μg/g body weight BrdU (Sigma) was injected intraperitoneally at E14.5. For BrdU immunolabeling, sections were treated with 2N HCl for 25 min at 37°C before processing (Britz et al., 2006). Cell proliferation was assayed via 5-ethynyl-2′-deoxyuridine (EdU) staining, using the Click-iT EdU Alexa Fluor 594 kit (Invitrogen). Two hundred microliters 1 μg/μl EdU dissolved in PBS was injected subcutaneously into pregnant dams 30 min before they were killed. For gain-of-function studies, sections were first stained with αGFP and postfixed for 1 h in 4% PFA/1× PBS at 4°C. Slides were the rinsed three times for 10 min in 5% bovine serum albumin/1× PBS and then stained for EdU as per the instructions of the manufacturer. Slides were rinsed three times for 5 min in 1× PBS, counterstained with DAPI, and mounted using Aqua Polymount.
Biphoton time-lapse video microscopy.
E15.5 cortices were dissected and electroporated with pCIG2 (control) or pCIG2–Zac1 expression constructs. Briefly, 1.5 μl of plasmid at 1–2 μg/μl was mixed with Fast Green (0.01 mg/ml; Sigma) and injected into the lateral ventricles of whole heads using a Hamilton syringe. Gold electrodes (Genetrode BTX model 514; Harvard Apparatus) were used to deliver five pulse of 30 V, 50 ms on/1000 ms off. The anode was oriented dorsally and the cathode ventrally. Cortices were then sliced and maintained in culture for 4 d (37°C, 7.5% CO2) as 150 μm organotypic slices. Biphoton time-lapse video microscopy was performed on 24 recorded positions in both hemispheres of 12 brain slices (two from each brain, n = 3 for each construct) over 3 d beginning 30 h after electroporation. One image was taken per hour on 100 μm with 20 z slices. A total of 195 control neurons (pCIG2) and 133 Zac1-transfected neurons were traced. Migration parameters extracted for each neuron included time of departure and arrival (number of hours after electroporation), migration duration (T, in hours), distance (D, in micrometers), velocity (micrometers per hours, D/T), and number and duration of pauses during saltatory locomotion. A pause was defined as nonsignificant movement (<3.5 μm) during at least 2 consecutive hours. Migration parameters were calculated for each neuron on a fragmented track. The neocortex thickness was subdivided into 20 bins parallel to the ventricular border, and the values were calculated for movements made in each bin. As neurons can begin and finish their track in different bins, only bins with at least half of the population making a part of their tracks in a bin were taken into account.
RNA in situ hybridization.
RNA in situ hybridization was performed as described previously (Touahri et al., 2015). The Zac1 digoxygenin-labeled riboprobe was generated as described previously (Alam et al., 2005).
Imaging, tracing, quantitation, and statistics.
Images were captured with a QImaging RETIGA 2000R or QImaging RETIGA EX digital camera and a Leica DMRXA2 optical microscope using OpenLab5 software (Improvision). Confocal images were acquired using a Nikon C1si Spectral confocal microscope. Using these images, neurons were traced with the paint tool in Photoshop CS6 (64 bit; Adobe Systems). Cell counts were performed on a minimum of three embryos per genotype or treatment group and a minimum of three cortical sections from each embryo. Statistical significance was calculated using a Student's t test when comparing two values, and three or more values were compared using a two-way ANOVA with a Bonferroni's correction unless indicated. Graphs and statistics were generated using GraphPad Prism Software (GraphPad Software). Error bars represent SEM. p values were denoted as follows: *p < 0.05, **p < 0.01, and ***p < 0.005.
Results
Overexpression of Zac1 in neocortical progenitors perturbs late-born neuronal migration
Previous analyses revealed that Zac1 is expressed in a regionalized manner in the developing nervous system, including in the telencephalon, the anlage of the neocortex (Alam et al., 2005). To better understand how Zac1 might function during neocortical development, we assessed its expression in this region of the embryonic neural tube in more detail. At E12.5, Zac1 transcripts were detected at high levels in dorsal telencephalic (neocortical) progenitor cells in the ventricular zone (VZ) and at lower levels in the ventral telencephalic VZ (Fig. 1A,A′). At E15.5, Zac1 continued to be expressed in neocortical VZ progenitors, as well as in deep layers of the developing cortical plate (CP; Fig. 1B,B′). A similar spatiotemporal distribution was observed using Zac1-specific antisera; at E12.5 (Fig. 1C,C′) and E14.5 (Fig. 1D,D′), Zac1 protein was detected in most neocortical VZ progenitors but not in postmitotic Tuj1+ neurons. By E16.5 (Fig. 1E,E′,E″) and at E18.5 (Fig. 1F,F′,F″), Zac1 protein continued to be expressed widely in the neocortical VZ and could now be detected in a small number of subventricular zone (SVZ) cells and deep layer neurons. Thus, Zac1 is expressed primarily in neocortical VZ progenitors and in a smaller number of SVZ progenitors and deep layer postmitotic neurons.
Figure 1.

Zac1 overexpression perturbs cell migration during later stages of corticogenesis. A, B, Zac1 transcript distribution in E12.5 (A, A′) and E15.5 (B, B′) telencephalon. A′ and B′ are high-magnification images of A and B, respectively. C–F, Distribution of Zac1 (red, C–F″) and Tuj1 (green, C, D) protein in the E12.5 (C, C′), E14.5 (D, D′), E16.5 (E–E″), and E18.5 (F–F″) neocortex. C′–F′ are higher-magnification images of C–F, respectively. Arrowheads in E and F mark CP expression. Comparison of E12.5–E18.5 (G–I) and E14.5–E18.5 (J–L) electroporations of pCIG2 control (G, J) and pCIG2-Zac1 (H, K) analyzed for the distribution of GFP+ cells/zone (I, L).
To mimic the upregulation of Zac1 expression associated with loss of the maternal imprint in TNDM, a bicistronic pCIG2–Zac1 expression vector containing an IRES–EGFP cassette or an empty vector pCIG2 control were introduced into E12.5 and E14.5 neocortical progenitors via in utero electroporation. The positions of GFP-expressing (GFP+) electroporated cells were then assessed at E18.5. Control and Zac1 E12.5–E18.5 electroporations looked similar, with most GFP+ electroporated cells concentrated in deep neocortical layers (n = 3; p > 0.05 comparing all layers; Fig. 1G–I), in accordance with the E12.5 birthdate of layer VI neurons (Caviness, 1982; Caviness et al., 1995). In contrast, a striking migratory block was observed in E14.5–E18.5 Zac1 electroporations, with Zac1-overexpressing cells aggregating in the intermediate zones (IZs; n = 6; p < 0.01) instead of migrating into upper (n = 6; p < 0.005) layers of the CP (Fig. 1J–L). Thus, overexpression of Zac1 strongly perturbs cellular migration at later stages of neocortical development, either because overexpressing cells fail to differentiate and/or because Zac1 impairs neuronal migration, which we addressed further below.
Zac1 misexpression delays progenitor cell maturation and neuronal differentiation
To further dissect the effects of Zac1 overexpression on neocortical progenitors, we focused on E14.5 electroporations, when Zac1-induced migratory defects were most profound. At E14.5, Pax6+ radial glial cell (RGC) progenitors in the VZ give rise to Tbr2+ intermediate neuronal progenitors (INPs) in the SVZ, which divide once or twice before differentiating into Tbr1+ neurons (Noctor et al., 2004; Fig. 2A). To test whether Zac1 overexpression perturbed the RGC-to-INP transition, we performed shorter E14.5–E15.5 electroporations. By 24 h after electroporation, the vast majority of pCIG2-transfected GFP+ progenitors migrated to the upper VZ/SVZ, in transition to becoming an INP (Fig. 2B,D). In contrast, more Zac1-transfected cells remained in the lower VZ (n = 3; p < 0.005), and many fewer cells reached the SVZ (n = 3; p < 0.005; Fig. 2C,D), consistent with a possible block in the RGC-to-INP transition.
Figure 2.
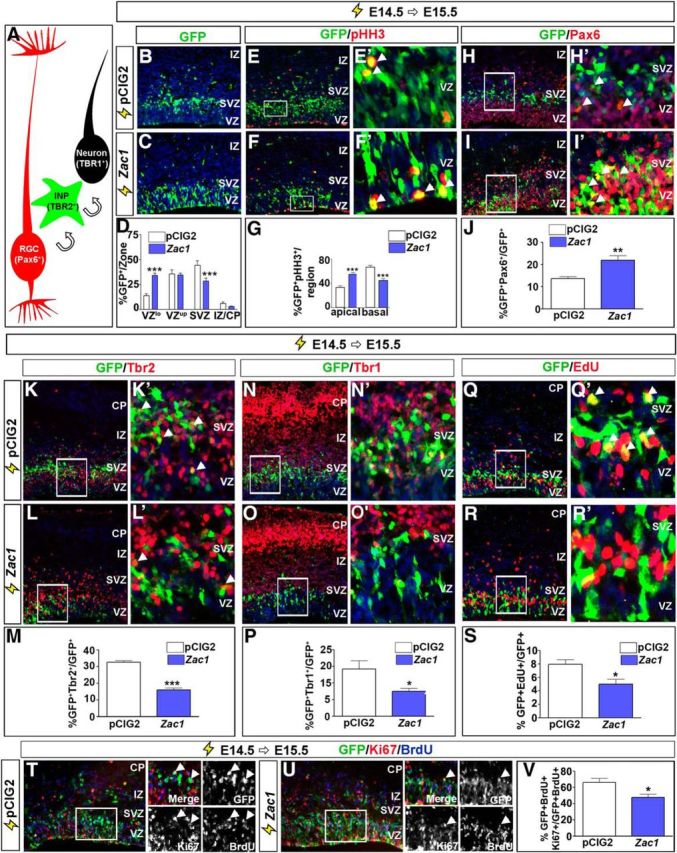
Zac1 overexpression delays progenitor cell maturation and neuronal differentiation. A, Schematic illustration of cells transitioning from Pax6+ RGCs to Tbr2+ INPs to Tbr1+ differentiated neurons. B–S, E14.5–E15.5 electroporations of pCIG2 control (B, E, H, K, N, Q) and pCIG2–Zac1 (C, F, I, L, O, R) costained for GFP and pHH3 (E, E′, F, F′), GFP and Pax6 (H, H′, I, I′), GFP and Tbr2 (K, K′, L, L′), GFP and Tbr1 (N, N′, O, O′), and GFP and EdU (Q, Q′, R, R′). E′, F′, H′, I′, K′, L′, N′, O′, R′, and Q′ are high-magnification images of boxed regions in E, F, H, I, K, L, N, O, R, and Q, respectively. Arrowheads in E′, F′, H′, I′, K′, L′, and Q′ mark double-positive cells. Quantitation of GFP+ cells/zone (D), percentage pHH3+GFP+ mitotic cells in apical and basal regions of the cortex (G), percentage Pax6+GFP+/GFP+ cells (J), percentage Tbr2+GFP+/GFP+ cells (M), percentage Tbr1+GFP+/GFP+ cells (P), and percentage EdU+ GFP+/GFP+ cells (S) after the electroporation of pCIG2 (white bars, n = 3) and pCIG2–Zac1 (blue bars, n = 3). T–V, E14.5–E15.5 electroporations of pCIG2 control (T) and pCIG2-Zac1 (U) costained for GFP (green), Ki67 (blue), and BrdU (red) after a 24 h BrdU pulse. Quantitation of percentage Ki67+BrdU+GFP+/GFP+BrdU+ cells (V). Arrowheads in T and U point to BrdU+ proliferating cells that have been electroporated (GFP+) and have remained in the cell cycle (Ki67+). DAPI labeling is in blue for B–R′.
A distinguishing feature of RGC progenitors is their cell cycle-dependent interkinetic nuclear movements, with nuclei in G2/M-phase of the cell cycle dividing at the apical surface, whereas INP mitoses occur in basal positions. To determine whether Zac1 influenced the apical-to-basal mitotic transition, we examined the expression of pHH3, a G2/M-phase marker (Fig. 2E–G). In E14.5–E15.5 transfections, of the Zac1-transfected (GFP+) cells that coexpressed pHH3, most divided in apical regions of the VZ (n = 3; p < 0.005), whereas fewer divided basally compared with control pCIG2 transfections (n = 3; p < 0.005; Fig. 2G). These data are consistent with the idea that Zac1 maintains an RGC identity while blocking the transition to an INP fate. To provide additional support for this conclusion, we examined the expression of Pax6 and Tbr2, which are expressed specifically in, and are essential determinants of, RGC and INP progenitor cell fates, respectively (Englund et al., 2005; Sessa et al., 2008). At 24 h after E14.5 electroporation, significantly more Zac1-misexpressing cells versus control-transfected cells expressed Pax6 (n = 3; p < 0.01; Fig. 2H–J). Concomitantly, fewer Zac1-transfected cells expressed Tbr2 (n = 3; p < 0.005; Fig. 2K–M). Thus, Zac1 misexpression blocks the maturation of cortical progenitors from an apical Pax6+ RGC identity to a basal Tbr2+ INP fate.
The delay in progenitor cell maturation associated with Zac1 overexpression suggested that this transcription factor may also block neuronal differentiation. To assess the effects of Zac1 on neuronal differentiation, we examined the expression of Tbr1 (Fig. 2N–P), a T-box transcription factor that is expressed at high and low levels, respectively, in deep and upper layer cortical neurons (Englund et al., 2005). In E14.5–E15.5 electroporations, the number of Zac1-transfected cells that expressed Tbr1 was reduced compared with control transfections (n = 3; p < 0.05; Fig. 2P). These data suggest that Zac1 does indeed block neuronal differentiation in the neocortex. However, these results were somewhat unexpected, because Zac1 promotes cell-cycle exit when misexpressed in cell lines (Spengler et al., 1997; Schmidt-Edelkraut et al., 2014) or in the retina (Ma et al., 2007b), and exit from the cell cycle is a hallmark feature of neuronal differentiation. To test whether Zac1 influenced the proliferative capacity of E14.5 cortical progenitor cells, we performed a 30 min pulse label with the thymidine analog EdU (Fig. 2Q–S). In E14.5–E15.5 electroporations of Zac1, fewer GFP+EdU+/GFP+ proliferating S-phase progenitors were detected compared with pCIG2 control transfections (n = 3; p < 0.05). To confirm that Zac1-misexpressing cells exited the cell cycle at a higher frequency, we administered BrdU immediately after electroporation of pCIG2 or Zac1 at E14.5. At E15.5, 24 h after electroporation, embryos were harvested and quantified based on the number of electroporated GFP+ cells that incorporated BrdU while also expressing Ki67 (Fig. 2T–V). This value gave us a measure of the number of GFP+ cells that were proliferating at the time of electroporation and also remained in the cell cycle 24 h later. The number of cells that remained in the cell cycle 24 h after electroporation was reduced when Zac1 was overexpressed (n = 3) compared with pCIG2 (n = 3; p < 0.05; Fig. 2V). Thus, Zac1 promotes cell-cycle exit in cortical progenitors, although it does not initiate the expression of neuronal differentiation markers such as Tbr1.
Zac1 overexpression reduces the expression of neuronal differentiation markers
To test whether Zac1 overexpression blocked as opposed to delayed the expression of neuronal differentiation markers, we extended the time after which E14.5 electroporated brains were analyzed to E18.5 (Fig. 3A–P). Four days after electroporation, NeuN (neuronal nuclear antigen), which is a late neuronal marker, was expressed in comparatively fewer Zac1- versus control-transfected cells (n = 3; p < 0.005; Fig. 3A–C), particularly in the germinal zone (GZ; n = 3; p < 0.01; Fig. 3D). Notably, the overall number of GFP+NeuN+ cells was low even in control transfections because NeuN was not expressed at high levels in upper layers of the neocortex. Thus, we also examined the effects of Zac1 misexpression on the differentiation of deep layer (Ctip2) and upper layer (Cux1 and Beta3) neurons using cell type-specific markers. Ctip2 is expressed in layer V neurons, most of which differentiate before E14.5. Accordingly, <6.0 ± 0.8% of GFP+ neurons in pCIG2 control electroporations expressed Ctip2 (Fig. 3E,G), and even fewer GFP+Ctip2+ neurons were observed during Zac1 overexpression (n = 3; p < 0.01), particularly in deep layers of the CP (n = 3; p < 0.005; Fig. 3H). Consistent with the idea that E14.5 progenitors preferentially differentiate into upper layer neurons, many more control transfected cells expressed Cux1 (Fig. 3I,K) and Beta3 (Fig. 3M,O), markers for upper layers II–IV and II–V, respectively. Zac1 misexpression reduced the number of progenitors that differentiated into both Cux1+ (n = 3; p < 0.005; Fig. 3I–K) and Beta3+ (n = 3; p < 0.05; Fig. 3M–O) neurons, particularly in upper neocortical layers [Cux1, n = 3, p < 0.005 (Fig. 3L); Beta3, n = 3, p < 0.005 (Fig. 3P)]. More Cux1+ and Beta3+ neurons were also observed in the IZ during Zac1 misexpression [n = 3 for both; p < 0.005 for Cux1 (Fig. 3L) and p < 0.005 for Beta3 (Fig. 3P)], suggesting that some neurons differentiate when Zac1 is overexpressed, but these neurons fail to migrate to their correct position in the CP.
Figure 3.

Zac1 overexpression blocks neuronal differentiation. A–P, E14.5–E18.5 electroporations of pCIG2 control (A, E, I, M) and pCIG2–Zac1 (B, F, J, N) analyzed for the expression of GFP and NeuN (A, B), GFP and Ctip2 (E, F), GFP and Cux1 (I, J), and GFP and Beta3 (M, N). Insets to the right are high-magnification images of boxed regions in the IZ and CP in A, B, E, F, I, J, M, and N, and arrowheads mark double-positive cells. Quantitation of percentage NeuN+GFP+/GFP+ cells in total (C) and per zone (D), Ctip2+ GFP+/GFP+ cells in total (G) and per zone (H), Cux1+ GFP+/GFP+ cells in total (K) and per zone (L), and Beta3+ GFP+/GFP+ cells in total (O) and per zone (P) after the electroporation of pCIG2 (white bars, n = 3) and pCIG2–Zac1 (blue bars, n = 3).
Together, these data suggest that there is a block in neuronal differentiation in a subset of Zac1-overexpressing progenitors, whereas many of the neurons that differentiate fail to migrate to their correct location in the CP.
Zac1-overexpressing cells exhibit decreased migratory velocities and increased pause time
Defects in the migration of Zac1-overexpressing neurons were evident 96 h after transfection of E14.5 cortical progenitors (Fig. 1J–L). To better assess the effects of Zac1 misexpression on the migratory behavior of differentiating neurons, we used time-lapse biphoton laser scanning microscopy to image transfected cells in real time after ex utero electroporation of E15.5 cortical slices. Recordings were initiated 30 h after electroporation and were continued over 3 d with one image taken per hour. In total, 133 control (n = 3) and 195 (n = 3) Zac1-misexpressing cells were traced through 71 positions along the radial cortical axis, with video microscopy ending at 101 h after electroporation (Fig. 4A,B). Newly born neurons generated at E15.5 were expected to exit the GZ within 48 h (Langevin et al., 2007), but a large proportion (63.7% by 72 h) of Zac1-electroporated cells did not exit the GZ until 72 h after transfection compared with controls (66.3% by 60 h; Fig. 4C,D). Thus, on average, the peak departure time, defined as the time when neurons left the GZ and entered the IZ (Fig. 4C), occurred significantly later in Zac1-misexpressing cells compared with controls (p < 0.005; Fig. 4D,E). As migration proceeded, a large subset of control cells (32.8%) managed to arrive at the CP within 56 h after transfection, whereas most Zac1-overexpressing cells (53.1%) took in excess of 80 h to complete this phase of migration (Fig. 4F,G). Accordingly, the peak arrival time of Zac1-misexpressing cells in the CP was delayed (p < 0.005; Fig. 4H), and the overall distance migrated was reduced from 79.6 ± 1.7 μm for control cells to 62.8 ± 1.2 μm for Zac1-transfected cells (p < 0.005; Fig. 4I,J). Zac1 overexpression also affected migration velocity, with Zac1-transfected cells (43.9%) averaging a velocity of ∼7.5 ± 0.3 μm/h compared with control cells, which migrated on average at 9.3 ± 0.3 μm/h (p < 0.005; Fig. 4K,L).
Figure 4.
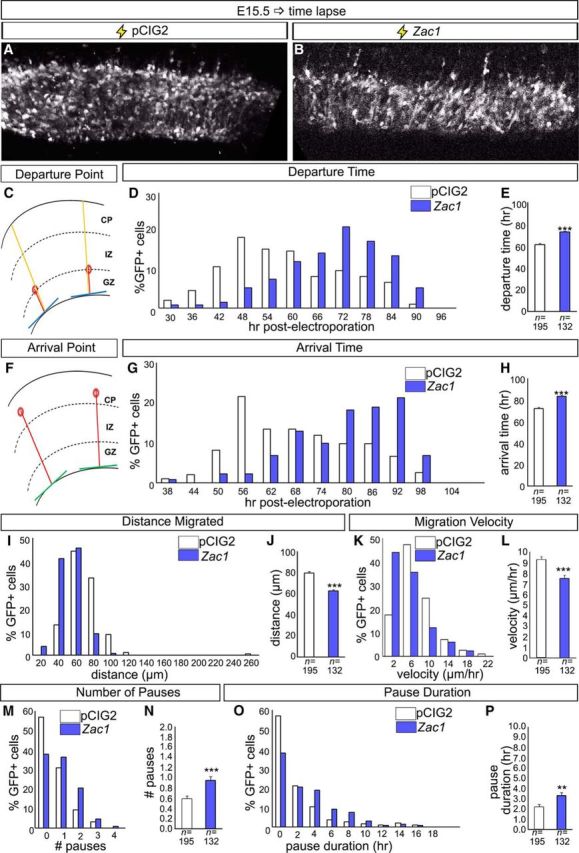
Altered migratory properties of Zac1 overexpressing cortical cells. A–P, Biphoton time-lapse microscopy of E15.5 cortical slice cultures electroporated with pCIG2 and pCIG2–Zac1. A, B, Photomicrographs of GFP+ cells imaged 30 h after electroporation of pCIG2 (A) and pCIG2–Zac1 (B). C–E, Measurement of departure time defined as hours after transfection when GFP+ cells left the GZ and entered the IZ (C). Departure times for 195 pCIG2 (white bars) and 133 pCIG2–Zac1 (blue bars) transfected cells were recorded individually (D) and averaged (E). F–H, Measurement of arrival time defined as hours after transfection when GFP+ entered the CP (F). Arrival times for 195 pCIG2 (white bars) and 133 pCIG2–Zac1 (blue bars) transfected cells were recorded individually (G) and averaged (H). I, J, Measurement of total distance (micrometers) migrated for 195 pCIG2 (white bars) and 133 pCIG2–Zac1 (blue bars) transfected cells recorded individually (I) and averaged (J). K, L, Measurement of migration velocity (micrometers per hours) migrated for 195 pCIG2 (white bars) and 133 pCIG2–Zac1 (blue bars) transfected cells recorded individually (K) and averaged (L). M, N, Quantitation of pauses in migration defined as any movement <3.5 μm over 2 consecutive hours of recording for 195 pCIG2 (white bars) and 133 pCIG2–Zac1 (blue bars) individual transfected cells (M) and averaged (N). O, P, Quantitation of pause duration in hours for 195 pCIG2 (white bars) and 133 pCIG2–Zac1 (blue bars) individual transfected cells (O) and averaged (P).
Locomotion is a saltatory, discontinuous process whereby neurons undergo periods of active movement interspersed by pauses (Nadarajah et al., 2001, 2003). Zac1-misexpressing cells paused more often (25.0% of cells paused two or more times) during migration when compared with the migratory progress of control cells (12.3% paused two or more times; p < 0.005; Fig. 4M,N), and the length of their pauses was longer compared with control transfected cells (p < 0.01; Fig. 4O,P). The motility index, defined as the migration capacity of each cell without considering pauses, was also reduced in Zac1-misexpressing cells (Zac1, 11.1 ± 0.3 μm/h vs pCIG2, 11.9 ± 0.2 μm/h).
Together, these data indicate that Zac1 overexpression in neocortical progenitors reduces migratory velocity and increases pause time and frequency.
Zac1-overexpressing neurons exhibit aberrant morphologies
Cortical neurons undergo a series of morphological transitions as they differentiate and migrate, the perturbation of which can block radial migration. To examine whether Zac1 misexpression influenced the morphology of migrating neurons, we used spectral confocal microscopy to image transfected neurons after E14.5–E18.5 electroporations of pCIG2 and Zac1 (Fig. 5A–I). Cortical neurons born at E14.5 use glial-guided locomotion to migrate, with their leading process contacting RGCs, which serve as glial guides. As differentiating neurons exit the GZ, they initially stall in the upper SVZ and IZ, in which they acquire a transient multipolar morphology that is associated with the dynamic extension and retraction of neurites (Tabata and Nakajima, 2003; Noctor et al., 2004). This is followed by the acquisition of a motile, bipolar morphology, with neurons extending a leading process toward the pial surface and a smaller lagging process oriented toward the ventricle (Nadarajah et al., 2001; Noctor et al., 2004). Because the waiting or pause period was increased after Zac1 misexpression, we questioned whether the multipolar-to-bipolar transition was disrupted. We first traced 82 pCIG2-transfected and 137 Zac1-transfected Tuj1+ neurons in the IZ (n = 3). In both pCIG2 and Zac1 electroporations, many Tuj1+ neurons in the IZ had a multipolar phenotype (34.4 ± 6.7% for pCIG2 and 36.4 ± 5.5% for Zac1), but the vast majority of neurons had transited to typical unipolar or bipolar neuronal morphologies (65.6 ± 6.7% for pCIG2 and 53.6 ± 4.6% for Zac1), with processes extending toward the apical (ventricular) and basal (pial) surfaces (Fig. 5A,A′,E,F). However, although most pCIG2-transfected neurons extended neurites (99.8 ± 0.2%), 10.4 ± 2.6% of Zac1-overexpressing neurons in the IZ lacked any detectable processes, instead acquiring an amorphous cell shape (p < 0.01; Fig. 5B,B′,G). Thus, Zac1 overexpression perturbs the ability of cortical neurons to extend processes in the IZ (Fig. 5G).
Figure 5.

Zac1 overexpression alters the morphology of migrating neurons. A–D, E14.5–E18.5 electroporations of pCIG2 (A, C; white bars) and pCIG2–Zac1 (B, D; blue bars). GFP+Tuj1+ neurons (A–D) were traced (A′–D′) in the IZ (A′, B′) and CP (C′, D′) from pCIG2 (n = 82 in IZ; n = 101 in CP) and pCIG2–Zac1 (n = 137 in IZ; n = 121 in CP) electroporations. E–I, Quantitation of percentage multipolar neurons in the IZ (E), percentage unipolar/bipolar neurons in the IZ (F), percentage neurons with neurites in the IZ (G), number of branches per neuron in the CP (H), and average number of branches in the CP (I). J, Schematic illustration of glial guided locomotion; saltatory movements begin with the centrosome, which is in front of the nucleus, and sends out microtubules to form a fork/cage around the nucleus (i). The leading process of the migrating neuron dilates and the centrosome enters (ii). Other organelles, such as the Golgi apparatus and ER, enter the dilated leading process (iii). Finally, microtubules attached to the centrosome pull the nucleus into the dilation (iv). K–N, E14.5–E18.5 coelectroporations of pCIG2 (K, K′, M, M′) or pCIG2–Zac1 (L, L′, N, N′) with RFP–CENT2 (K, K′, L, L′) or pEF/Myc/ER/mCherry (M, M′, N, N′). GFP+ cells were traced in K′–N′ to highlight the position of the organelles within the transfected cells. ns, Not significant.
Once locomoting neurons reach their destination in the CP, their leading process extends multiple branches that attach to the pial surface, providing traction for the rapid pulling of neurons into their final laminar position in a process known as somal translocation (Nadarajah et al., 2001). To determine whether Zac1 misexpression perturbed these late morphological changes, we examined the morphology of neurons in upper layer II/III of the CP, tracing 101 pCIG2-transfected and 121 Zac1-transfected Tuj1+ neurons. In E14.5–E18.5 pCIG2 control electroporations, almost all (98.9%) of the GFP+Tuj1+ neurons had two or more secondary branches extending out of the leading process (Fig. 5C,C′,H,I). In contrast, when Zac1 was overexpressed, only 44.9% of GFP+Tuj1+ neurons elaborated branches in the CP (Fig. 5D,D′,H,I). Thus, Zac1 overexpression prevents neurite branching, most notably in the CP, likely interfering with the final somal translocation of migrating neurons.
Concomitant with the dynamic changes in neurite branching patterns, intracellular organelles also undergo active movements in migrating neurons. The centrosome, which is located basal to the nucleus in a migrating neuron, first translocates into a swelling within the leading process, followed by the endoplasmic reticulum (ER). The centrosome then pulls the nuclear cage upward and the saltatory migratory movements are repeated (Fig. 5J). To examine whether organelle movements were disrupted during Zac1 misexpression, we labeled the centrosome and ER by coelectroporating RFP–CENT2 (White et al., 2000) and pEF/Myc/ER/mCherry (Shim et al., 2008), respectively. In control pCIG2-transfected neurons, the centrosome (Fig. 5K,K′) and ER membranes (Fig. 5M,M′) were located on the basal side of the nucleus and were clearly in the process of translocating into the leading process. In contrast, in Zac1-overexpressing neurons, especially those with an amorphous shape, the centrosome (Fig. 5L,L′) and ER membranes (Fig. 5N,N′) remained on the apical side of the nucleus. These data suggest that organelle movements are perturbed in neurons that overexpress Zac1, likely contributing to the aberrant morphological transitions and migratory patterns of these neurons.
Neuronal migration is perturbed in Zac1 mutant neocortices
ZAC1 is a critical developmental gene in humans, because both the increase and decrease in ZAC1 expression in humans is associated with intellectual disability and smaller size for gestational age (e.g., Decipher identification numbers 248227 and 251465; Temple and Shield, 2002; Azzi et al., 2014). Growth restriction is also observed in Zac1 mutant mice (Varrault et al., 2006). To determine whether the loss of Zac1 expression also influenced neocortical development, we examined Zac1 mutant mice. Zac1 is a maternally imprinted gene (Piras et al., 2000; Smith et al., 2002). Consequently, crosses between Zac1+/− males and wild-type C57BL/6 females yield Zac1+m/− heterozygotes with a silenced, maternal wild-type allele; these embryos are effectively null for Zac1 and are hereafter designated as Zac1 mutants. We first examined Zac1 mutant neocortices at E14.5 to determine whether proliferation and progenitor cell dynamics were altered. After a 30 min exposure to BrdU, similar numbers of progenitors were labeled in E14.5 Zac1 mutant and wild-type cortices (Fig. 6A–D,I). In addition, there were no differences in the numbers of Pax6+ RGCs (Fig. 6A,B,E) and Tbr2+ INPs (Fig. 6C,D,G) in E14.5 Zac1 mutants or in the numbers of progenitors that coexpressed Pax6/BrdU (Fig. 6F) or Tbr2/BrdU (Fig. 6H). To provide additional support for the lack of an effect of the Zac1 mutation on progenitor cell maturation, we also analyzed progenitor populations at E15.5, 24 h after BrdU injection at E14.5. The ratios of Pax6/BrdU (Fig. 6J,K,N) and Tbr2/BrdU (Fig. 6L,M,O) coexpression, as well as the total BrdU counts (Fig. 6P), were not significantly different in E15.5 wild-type and Zac1 mutants. Thus, the loss of Zac1 does not alter the transition of cortical progenitors from Pax6+BrdU+ RGCs to Tbr2+Brdu+ INPs. So although Zac1 is sufficient to block the RGC-to-INP transition and promote cell-cycle exit, it is not required for these events.
Figure 6.
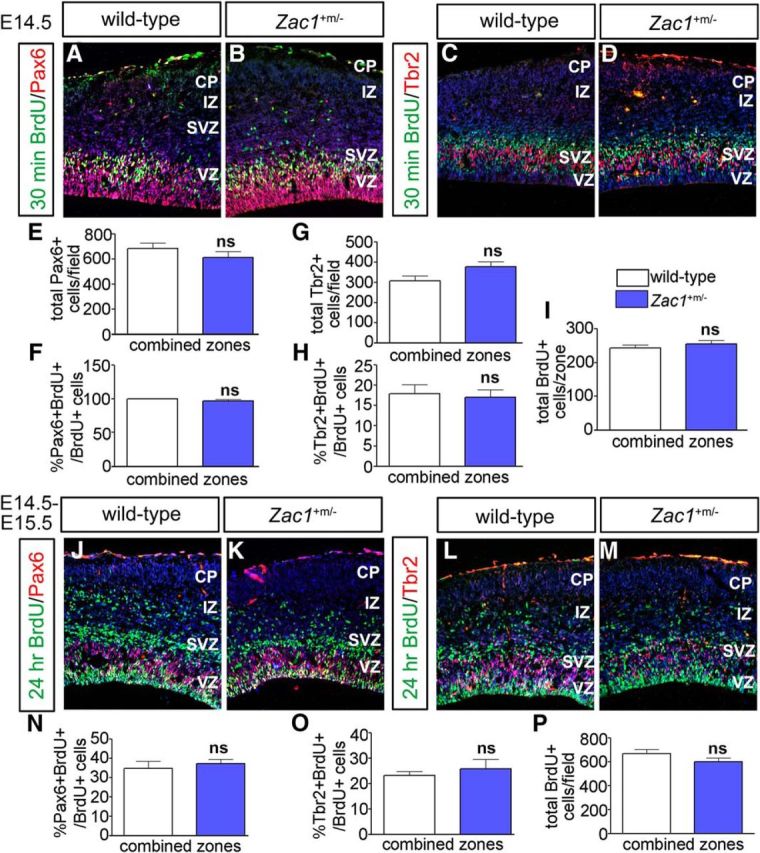
Loss of Zac1 does not alter progenitor cell dynamics. A–I, Analysis of Pax6/BrdU (A, B) and Tbr2/BrdU (C, D) coexpression in E14.5 wild-type and Zac1 mutant (B) cortices after a 30 min BrdU pulse. DAPI labeling is blue counterstain. Quantitation of total number of Pax6+ cells (E), percentage Pax6+BrdU+/BrdU+ cells (F), total number of Tbr2+ cells (G), percentage Tbr2+BrdU+/BrdU+ cells (H), and total BrdU+ cells (I) in wild-type (n = 3; white bars) and Zac1 mutants (n = 3; blue bars). J–P, Analysis of Pax6/BrdU (J, K) and Tbr2/Brdu (L, M) coexpression in E15.5 wild-type (L) and Zac1 mutant (M) cortices after a 24 h BrdU pulse. DAPI labeling is blue counterstain. Quantitation of the percentage Pax6+BrdU+/BrdU+ cells (N), percentage Tbr2+BrdU+/BrdU+ cells (O), and total BrdU+ cells (P) in wild types (n = 3; white bars) and Zac1 mutants (n = 3; blue bars).
We next examined whether the loss of Zac1 influenced neocortical neuronal migration. The majority of Zac1 mutant pups die within 24 h after birth (Varrault et al., 2006; Ma et al., 2007b), precluding us from examining mature laminar patterns in Zac1 mutants at postnatal day 7, when migration is normally complete. Nevertheless, we were able to analyze the initial partitioning of neurons into upper and deep layers of the cortex at E18.5 by BrdU birthdating. BrdU was administered at E14.5, when upper layer II–IV neurons are generated (Caviness, 1982; Caviness et al., 1995). The laminar positions of darkly labeled nuclei, corresponding to neurons derived from progenitors that underwent their last round of cell division immediately after labeling, were assessed at E18.5 (Fig. 7A–C). Cortical sections were subdivided into 13 10-μm bins, which were assigned to the VZ, IZ, or deep or upper CP layers based on differences in the size and distribution of DAPI+ nuclei, and pairwise comparisons were made between wild-type and Zac1 mutants. In wild-type cortices, the majority of labeled neurons were found in bins in upper layers II–IV (Fig. 7A–A″,C). In contrast, in Zac1 mutants, fewer darkly stained nuclei were present in the upper-most cortical layers (n = 3; p < 0.05, t tests to compare bins; Fig. 7B–B″,C). Instead, a subset of the postmitotic cells labeled at E14.5 in Zac1 mutants accumulated aberrantly in the upper GZ (n = 3; p < 0.05) and IZ (n = 3; p < 0.001; Fig. 7B–B″,C). Because progenitor cell maturation and neuronal differentiation were not notably different in Zac1 mutants, these data suggested that some neurons born at E14.5 fail to migrate into the upper CP in the absence of Zac1, instead aggregating in deep positions in the GZ/IZ.
Figure 7.

Aberrant distribution of laminar markers in Zac1 mutant cortices. A–C, E14.5–E18.5 BrdU birthdating in wild-type (A–A″) and Zac1 mutant (B–B″) cortices. Distribution of BrdU-labeled cortical neurons divided into 13 bins corresponding to upper CP layers (bins 10–13), deep CP layers (bins 5–9), IZ (bins 3–4), and GZ (bins 1–2) in wild-type (white bars; n = 3) and Zac1 mutant (blue bars; n = 3) cortices (C). D–N, E18.5 wild-type (D, D′, H, H′, L, L′) and Zac1 mutant (E, E′, I, I′, M, M′) cortices immunostained for Beta3 (D, D′, E, E′), Cux1 (H, H′, I, I′), and Ctip2 (L, L′, M, M′). DAPI labeling is blue counterstain. Quantitation of percentage Beta3+/DAPI+ cells in total (F) and in each layer (G), percentage Cux1+/DAPI+ cells in total (J) and in each layer (K), and percentage Ctip2+/DAPI+ cells in total (N) and in each layer (O) for wild types (n = 3; white bars) and Zac1 mutants (n = 3; blue bars).
To confirm that the aberrantly aggregating cells in Zac1 mutants were indeed neurons, we also examined the expression of layer-specific markers in E18.5 cortices. The total number of neurons expressing two upper layer markers—Beta3 (Fig. 7D–F), a basic-helix–loop–helix transcription factor expressed in layers II–V (Kim et al., 2002), and Cux1 (Fig. 7H–J), a Cut-like homeobox 1 transcription factor expressed in layers II–IV—were the same in E18.5 wild-type and Zac1 mutant cortices. However, the distribution of Beta3 (Fig. 7G) and Cux1+ (Fig. 7K) neurons was altered in E18.5 Zac1 mutants, with more of these neurons aberrantly aggregating in the GZ and IZ (n = 3; p < 0.05 for both Beta3 and Cux1 using t tests to compare bins; Fig. 7G,K). Cux1 was also found to be located ectopically in the deep regions of Zac1 mutants (n = 3; p < 0.05; Fig. 6K). In contrast, Ctip2 (Bcl11b), a layer V-specific transcription factor (Arlotta et al., 2005), was expressed in similar numbers of neurons and in a similar distribution throughout the cortical layers in both E18.5 wild-type and Zac1 mutant cortices (Fig. 7L–O).
Thus, a small subset of late-born Zac1 mutant neurons fail to migrate to their appropriate upper layers based on birthdating and laminar markers. Notably, these defects were overcome by P4, when cell counts revealed no differences in the number or distribution of upper layer neurons in the Zac1 mutants that survived (data not shown). Hence, there is a delay, rather than a block, in upper layer neuronal migration in Zac1 mutant neocortices.
Zac1 mutant neocortical neurons have aberrant morphologies
To further substantiate the requirement for Zac1 in regulating the migration of upper layer neurons and to examine underlying causes, we performed two electroporation assays. First, we performed knockdown experiments. A highly efficient Zac1–shRNA construct [Zac1–sh(3); hereafter designated Zac1–shRNA] was identified by transfecting NIH-3T3 cells with four shRNA constructs carrying different Zac1 target sequences (Fig. 8A). We then electroporated Zac1–shRNA or a scrambled shRNA control construct into E14.5 cortices and examined the distribution of electroporated cells at E18.5. Knockdown of Zac1 had a striking effect on cell migration, with the vast majority of GFP+ electroporated cells failing to migrate out of the GZ (n = 3; p < 0.005) and not reaching the IZ (n = 3; p < 0.005) and upper layers (n = 3; p < 0.005) of the neocortex (Fig. 8B–D). To confirm that Zac1–shRNA did not have off-target effects, we repeated the E14.5–E18.5 electroporations of sh-scrambled and sh-Zac1 constructs in Zac1 mutant cortices; neither shRNA construct blocked neuronal migration in Zac1 mutants (Fig. 8E,F), indicating that Zac1–shRNA-induced migration errors are not off-target effects or an electroporation artifact. To assess whether a subset of neurons failed to migrate in Zac1 mutant cortices, we used a second electroporation assay, introducing pCIG2 into E14.5 Zac1 mutants and wild-type littermates and analyzing the distribution of GFP+ cells at E18.5. In Zac1 mutants, significantly more GFP+ cells were found in deep cortical layers compared with wild-type controls (n = 5 for Zac1 mutants and n = 6 for wild-type littermates; p < 0.05; Fig. 8G–I). Thus, there are subtle migratory defects in Zac1 genetic mutants.
Figure 8.
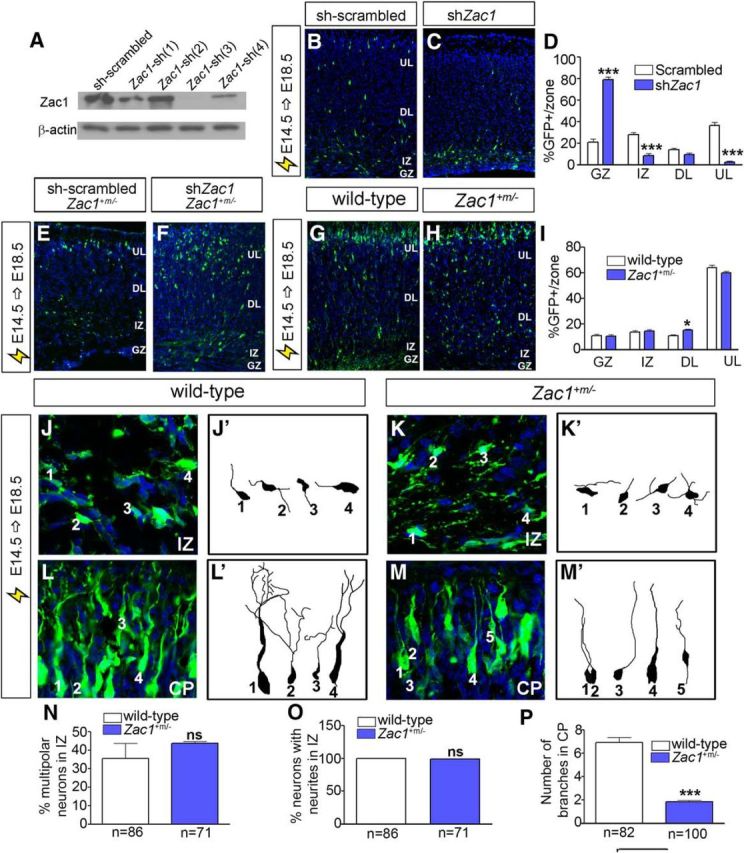
Aberrant morphology of migrating neurons in Zac1 mutant cortices. A, Western blot analysis of Zac1 and β-actin protein levels in NIH-3T3 cells cotransfected with pCIG2–Zac1 along with different shRNA constructs. B–D, E14.5–E18.5 electroporations of sh-scrambled control (B) and shZac1 vectors (C) in wild-type CD1 timed pregnant females. Quantitation of percentage GFP+ cells/layer for sh-scrambled (n = 3; white bars) and shZac1 (n = 3; blue bars) (D). E, F, E14.5–E18.5 electroporations of Zac1 mutant cortices with sh-scrambled and sh-Zac1 constructs. G–I, E14.5–E18.5 electroporation of pCIG2 in wild-type (G) and Zac1 mutant (H) cortices. Quantitation of percentage GFP+ cells in each layer for wild-type (n = 3; white bars) and Zac1 mutant (n = 3; blue bars) cortices (I). J–P, E14.5–E18.5 electroporation of pCIG2 in wild-type (J, L) and Zac1 mutant (K, M) cortices, with images taken in the IZ (J, K) and CP (L, M). GFP+Tuj1+ neurons in wild-type IZ (J′) and CP (L′) and in Zac1+m/− IZ (K′) and CP (M′) were traced. Quantitation of percentage multipolar neurons in IZ of wild-type (n = 86; white bars) and Zac1 mutant (n = 71; blue bars) cortices (N). Quantitation of percentage neurons with neurites in the IZ of wild-type (n = 86; white bars) and Zac1 mutant (n = 71; blue bars) cortices (O). Quantitation of the number of branches in the CP of wild-type (n = 82; white bars) and Zac1 mutant (n = 100; blue bars) cortices (P). DL, Deep layer; UL, upper layer.
Next, to study the underlying cause of the migration defects in Zac1 mutants in more detail, we examined the morphologies of pCIG2-transfected GFP+Tuj1+ neurons in the IZ and upper CP, comparing the with littermate controls. Within the IZ, there were no differences in the number of GFP+Tuj1+ multipolar neurons (Fig. 8J,J′,K,K′,N) or neurite-bearing neurons (Fig. 8L,L′,M,M′,O) in wild-type (n = 86) and Zac1 mutant cortices (n = 71). However, many fewer branches were seen in the pCIG2-transfected GFP+Tuj1+ neurons in Zac1 mutant (n = 100; p < 0.005; Fig. 8M,M″,P) versus wild-type embryos (n = 82; Fig. 8L,L″,P) in the CP.
Combined, these data suggest that neuronal migration is perturbed when Zac1 is knocked down and to a lesser extent when it is knocked out. However, there was a similar reduction in the branching of upper layer neurons in both genetic null mice and in transient knockdown experiments, suggesting that Zac1 is absolutely required for this branching process, with no compensatory mechanisms in place. Given that similar branching defects were observed whether Zac1 was overexpressed or underexpressed, we can conclude that Zac1 is a dosage-sensitive gene, similar to other imprinted genes.
Zac1 regulates neuronal migration via Pac1
Because Zac1 functions as a transcriptional activator or repressor (Varrault et al., 1998; Hoffmann et al., 2003), its effects on neuronal migration are likely mediated by downstream effectors. Several Zac1 transcriptional targets have been identified, including Pac1 (Ciani et al., 1999; Rodríguez-Henche et al., 2002), which encodes a receptor for the neuropeptide PACAP. We focused on Pac1 as a potential downstream effector of Zac1 because Pac1 is expressed at high levels in the neocortical VZ and to a lesser extent in the CP (Suh et al., 2001; Yan et al., 2013), similar to the Zac1 expression profile (Fig. 1A–F). Moreover, PACAP promotes cell-cycle exit in cortical progenitors after E13.5 (Suh et al., 2001), mimicking the effects we observed during Zac1 overexpression, although the authors did not examine whether Pac1 also influenced neuronal migration.
To determine whether Pac1 may be a downstream Zac1 effector in the developing neocortex, we first asked whether Zac1 regulated Pac1 expression levels in this region of the neural tube. Pac1 transcript levels were quantitated by qPCR in E18.5 microdissected wild-type (n = 4) and Zac1 mutant (n = 4) neocortices, revealing a 1.2-fold decrease in Zac1 mutants relative to wild type (Fig. 9A,B). Next, to test whether Zac1 was sufficient to induce Pac1 expression in neocortical cells, pCIG2 control (n = 6) and pCIG2-Zac1 (n = 6) expression vectors were electroporated into E13.5 cortices and GFP+ electroporated patches in the dorsal telencephalon were microdissected 24 h later (Fig. 9C,D). Zac1 was upregulated 4.2-fold in Zac1-transfected cortical cells compared with control transfections, resulting in a 3.3-fold increase in Pac1 transcript levels (Fig. 9D). Thus, Zac1 is required and sufficient to regulate Pac1 transcript levels in neocortical progenitors.
Figure 9.

Zac1 regulates neuronal migration by regulating Pac1 transcription in the developing neocortex. A–D, Schematic of the experimental design to test whether Zac1 regulates the expression of Pac1 in E15.5 Zac1+m/− cortices (A) and in E13.5–E14.5 Zac1 gain-of-function assays (C). Quantitation of qPCR data, showing reduced Pac1 transcript levels in Zac1+m/− cortices [n = 4 for both wild-type (white bars) and Zac1 mutant (blue bars); B] and increased Pac1 transcript levels after Zac1 misexpression [n = 6 for both pCIG2 (white bars) and pCIG2–Zac1 (blue bars); D]. E–G, E14.5–E18.5 electroporations of pCIG2 (E) and pCIG2-Pac1 (F). Quantitation of percentage GFP+ cells in each layer for pCIG2 control (n = 3; white bar) and pCIG2–Pac1 (n = 3; blue bar) (G). H–J, E14.5–E18.5 electroporations of sh-scrambled (H) and shPac1 (I). Quantitation of percentage GFP+ cells in each layer for pCIG2 control (n = 3; white bar) and shPac1 (n = 3; blue bar) (J). K–R, E14.5–E18.5 electroporations of pCIG2 (K, M) and pCIG2–Pac1 (L, N), showing coimmunolabeling of GFP (green) and Tuj1 (red). Blue is DAPI counterstain. Tracing of GFP+Tuj1+ neurons in the IZ from pCIG2 control (n = 82; K′) and pCIG2–Pac1 (n = 93; L′) electroporations. Quantitation of percentage multipolar cells (O), percentage cells with neurites (P), and percentage unipolar or bipolar neurons (Q) for pCIG2 control (n = 3; white bars) and pCIG2–Pac1 (n = 3; blue bars). Tracing of GFP+Tuj1+ neurons in the CP from pCIG2 control (n = 101; M′) and pCIG2–Pac1 (n = 23; N′) electroporations. Quantitation of average number of branches per neuron in the CP (R). S–V, E14.5–E18.5 electroporations of sh-scrambled (S) and shPac1 (T), showing coimmunolabeling of GFP (green) and Tuj1 (red). Blue is DAPI counterstain. Tracing of GFP+Tuj1+ neurons in the IZ from pCIG2 control (n = 82; S′) and shPac1 (n = 87; T′). Quantitation of percentage multipolar neurons (U) and percentage unipolar or bipolar neurons (V) for sh-scrambled (n = 3; white bars) and shZac1 (n = 3; blue bars).
If Zac1 acts via Pac1 to regulate neuronal migration, we predicted that we would obtain the same perturbation of neuronal migration when Pac1 was either overexpressed or knocked down. To test this hypothesis, we first electroporated pCIG2 control and pCIG2–Pac1 expression vectors into E14.5 neocortices and harvested the embryos at E18.5 (Fig. 9E,F). In control electroporations, most GFP+ cells had migrated to upper regions of the CP (Fig. 9E,G), whereas misexpression of Pac1 led to the accumulation of more GFP+ cells in the IZ (n = 3; p < 0.01) and fewer cells reached upper layers of the CP (n = 3; p < 0.005; Fig. 9F,G). Therefore, Pac1 misexpression phenocopies the migration defects observed when Zac1 is misexpressed in E14.5 cortical progenitors (Fig. 1J–L). Next, to investigate whether Pac1 loss-of-function phenocopied Zac1 loss-of-function, we knocked down Pac1 using shRNA, targeting E14.5 cortical progenitors and examining the positions of electroporated cells at E18.5. Relative to control electroporations (Fig. 9H,J), more GFP+ cells electroporated with shPac1 were ectopically located in the IZ (n = 3; p < 0.005) and fewer GFP+ cells reached upper layers of the CP (n = 3; p < 0.005; Fig. 9I,J). Both the loss and gain of Pac1 function thus perturbs neuronal migration, similar to the phenotypes observed when Zac1 levels are manipulated.
As a final comparative measure of Zac1 and Pac1 functions, we examined the morphologies of cortical neurons after the overexpression or knockdown of Pac1. Similar to Zac1, overexpression of Pac1 increased the number of GFP+Tuj1+ neurons that lacked neurites, with 9.6% of Pac1-transfected neurons acquiring an amorphous shape in the IZ (n = 93; p < 0.005; Fig. 9K,K′,L,L′,P). Also similar to Zac1, Pac1 did not affect the multipolar-to-unipolar/bipolar ratio of the neurons that did extend neurites (Fig. 9O,Q). However, of the few GFP+Tuj1+ neurons that did reach the CP after Pac1 overexpression, there was a reduction in the average number of neurite branches that were extended (n = 23; p < 0.005; Fig. 9R), similar to the Zac1 gain-of-function phenotype. Conversely, when Pac1 was knocked down by electroporating shPac1 into E14.5 cortical progenitors (Fig. 9S–V), more GFP+Tuj1+ neurons acquired a multipolar shape compared with control transfections (n = 87 for shPac1 vs n = 63 for sh-scrambled; p < 0.05; Fig. 9U), whereas fewer were bipolar (p < 0.05; Fig. 9V). Thus, Pac1 is required for the multipolar-to-bipolar transition of locomoting neurons (Fig. 9U,V).
Together, these data indicate that Pac1 transcription is regulated by Zac1 in the neocortex and suggest that Pac1 is necessary and sufficient downstream of Zac1 to control the migratory behavior and morphologies of neocortical neurons.
Pac1 partially rescues migration defects associated with Zac1 knockdown
To provide additional support for the idea that Pac1 is a downstream effector of Zac1 in the developing neocortex, we performed rescue experiments. For this purpose, we first conducted E14.5–E18.5 electroporations of pCIG2, pCIG2–Zac1, pCIG2–Pac1, sh-scrambled, shZac1, and shPac1 constructs, confirming that the gain or loss of both Zac1 and Pac1 perturbed migration (Fig. 10B–G) and providing a comparative baseline for coelectroporation experiments. To provide a single measure of migration that could be compared between single and double electroporations, we calculated a migration index, dividing the cortex into seven bins of equal size, with the top-most bin, in which cells had migrated the farthest, given a value of 7, and the lowest bin, in which cells had migrated the least, assigned a value of 1 (Fig. 10A). The percentage of GFP+ cells within each bin was then multiplied by the assigned bin value, and all numbers were added together. Using this strategy, the migration indices of pCIG2 (n = 4) and scrambled shRNA (n = 3) control transfections were 4.3 ± 0.2 and 5.0 ± 0.1%, respectively (Fig. 10 B,E,J). In contrast, migration indices for pCIG2–Zac1 (3.3 ± 1.5; n = 3; p < 0.001) and pCIG2–Pac1 (3.4 ± 0.1%; n = 3; p < 0.01; Fig. 10C,D,J) were considerably lower than for pCIG2, whereas shPac1 (2.8 ± 0.1%; n = 4; p < 0.001) and shZac1 (2.7 ± 0.1%; n = 3; p < 0.001; Fig. 10F,G,J) were considerably lower than the scrambled shRNA control, as expected. Thus, we were able to use this migration index to compare the migratory effects of several constructs at once.
Figure 10.

Zac1 regulates neuronal migration via Pac1 in the developing neocortex. A, Schematic representation of method used to calculate migration index. B–F, E14.5–E18.5 electroporations of pCIG2 (B), pCIG2–Zac1 (C), pCIG2–Pac1 (D), sh-scrambled (E), shZac1 (F), shPac1 (G), pCIG2–Zac1 plus shPac1 (H), and shZac1 plus pCIG2–Pac1 (I). J, Quantitation of migration indices for all electroporations. K, Summary of regulatory interactions between Zac1 and Pac1. Zac1 gain-of-function perturbs radial migration even when Pac1 is knocked down, suggesting that Zac1 controls the expression of other migratory factors. Zac1 knockdown no longer perturbs migration when Pac1 is overexpressed, suggesting that Pac1 is the most critical regulator of migration downstream of Zac1. L, Summary of the role of Zac1 in guiding neuronal migration in the developing neocortex. At the end of glial guided locomotion (steps i–iv), neurons detach from the radial glial scaffold and the leading process extends multiple branches that arborize to the pial surface (step v in pCIG2 control). In neurons in which Zac1 expression is deregulated, branching of the leading process does not occur at the end of terminal translocation (step v; + or − Zac1).
To determine whether Pac1 was an essential Zac1 effector, we first asked whether Zac1 perturbed neuronal migration when Pac1 was knocked down. The migration index for pCIG2–Zac1 plus shPac1 was 2.6 ± 0.1% (n = 6; Fig. 10H,J), even lower than that observed for the gain-of-function of Zac1 (p < 0.01). Thus, Zac1 gain-of-function perturbs radial migration even when Pac1 is knocked down, suggesting that Zac1 must control the expression of other migratory factors in addition to Pac1. We next asked the converse question: whether Zac1 is required to initiate Pac1 expression for normal migration to occur. Indeed, by knocking down Zac1 and adding back Pac1, a rescue of neuronal migration defects was observed, with a resulting migration index of 4.2 ± 0.1% that was not significantly different from values observed in control electroporations (n = 3; p > 0.05; Fig. 10I,J). Given that the Zac1 knockdown no longer perturbs migration when Pac1 is overexpressed, we suggest that Pac1 is indeed a critical downstream effector of Zac1. Together, these data support the notion that Zac1 modulates neuronal migration at least in part by regulating the expression of Pac1, although other downstream effectors are also likely involved.
Discussion
Zac1 is a maternally imprinted, dosage-sensitive gene, and an increase or decrease in its expression is associated with developmental growth restriction and intellectual deficits in humans. Therefore, we investigated whether alterations in Zac1 expression in the developing murine neocortex, which is the seat of higher-order cognitive functioning, would influence brain development. Striking defects in progenitor cell maturation, neuronal differentiation, neuronal morphology, and neuronal migration were observed during overexpression of Zac1 in neocortical progenitors. Defects in neuronal migration were also observed in Zac1 loss-of-function models, albeit to a lesser extent. Mechanistically, we attribute the ability of Zac1 to control neocortical neuronal migration to its regulation of Pac1, a receptor for the neuropeptide PACAP that is known to regulate neocortical progenitor proliferation (Suh et al., 2001). Thus, we have uncovered a novel Zac1–Pac1 regulatory circuit that plays an essential role in regulating progenitor proliferation, neuronal differentiation, and migration in the developing neocortex. Many of our conclusions are based on gain-of-function experiments, which can in some instances induce experimental artifacts. However, Zac1 misexpression upregulates Pac1 expression, a known Zac1 transcriptional target, and Zac1 and Pac1 have similar gain-of-function phenotypes, thus supporting the specificity of our gain-of-function data. Moreover, because our goal was to mimic the increase in Zac1 expression observed in human TNDM, it was necessary to use a gain-of-function approach.
Zac1 and the regulation of cortical progenitor cell proliferation
We found that Zac1 misexpression reduces EdU incorporation in the neocortex and that fewer Zac1-overexpressing cells re-enter S-phase of the cell cycle. This is similar to our findings in the embryonic retina, in which Zac1 misexpression also reduced S-phase progenitors (Ma et al., 2007b). Zac1 similarly induces cell-cycle arrest in epithelial cell lines; conversely, Zac1 expression is lost in several carcinomas that display an increased proliferative potential (Abdollahi, 2007). It is currently unknown how Zac1 regulates cell-cycle exit, in either the developing CNS or tumorigenic cells, but it is thought to function independently of Kip-family cyclin dependent kinase inhibitors and retinoblastoma (Spengler et al., 1997). We found that Zac1 promotes Pac1 expression in cortical progenitors, whereas Pac1 transcript levels were also reduced in Zac1 mutant cortices, although to a lesser extent. PACAP has anti-proliferative properties in the developing neocortex, reducing the number of progenitors entering S-phase of the cell cycle at E13.5 and later (Suh et al., 2001). Therefore, the ability of Zac1 to reduce proliferation is likely related to its ability to increase Pac1 transcription. A similar model was proposed based on analyses of Suz12 null cortices, which also display a reduction in cortical proliferation (Miró et al., 2009). Suz12 encodes a component of a polycomb repressive complex 2 that regulates the expression of imprinted genes, such as Zac1. Accordingly, Zac1 expression levels are upregulated in Suz12 mutant cortices, as are the expression levels of Pac1 (Miró et al., 2009). Zac1 in turn has been shown to regulate the expression of a number of imprinted genes, including Igf2, Dlk1, and H19, all of which are associated with growth control and proliferation (Varrault et al., 2006). Future studies will be required to see whether these genes are also regulated by Zac1 in the neocortex, accounting for the reduced proliferative capacity of Zac1-overexpressing cells. However, based on our studies, we can conclude that a Zac1–Pac1 transcriptional pathway is a key regulator of progenitor cell proliferation in the developing neocortex.
Although we found that Zac1 can promote cell-cycle exit, it was shown recently that Zac1 can enhance the tumorigenicity of a glioma cell line, in contrast to its original identification as a tumor suppressor gene (Hide et al., 2009). This is perhaps not unexpected given that members of several gene families, including the Runx transcription factors (Cameron and Neil, 2004), Pten phosphatase (Groszer et al., 2001; Marino et al., 2002), and TGFβ signaling molecules (Bachman and Park, 2005), are lineage-specific oncogenes or tumor suppressors, depending on the time and tissue in which they are expressed. Zac1 function is thus clearly context specific.
Zac1 blocks progenitor cell maturation and neuronal differentiation
We found that Zac1 misexpression blocks the apical-to-basal transition of neocortical progenitors, leading to the sustained expression of Pax6 and a reduction in Tbr2 expression. Furthermore, neuronal differentiation was delayed, but not completely blocked, by the overexpression of Zac1 in neocortical progenitors. The ability of Zac1 to block neocortical cells as early apical progenitors is consistent with a recent study in a glioma cell line, in which Zac1 promoted the expression of nestin, a marker of apical radial glia, and blocked neuronal differentiation (Hide et al., 2009). However, it is not clear whether Zac1 is a direct transcriptional regulator of apical progenitor genes, such as Nestin or Pax6, or whether it alters the expression of other genes that block progenitor cell maturation and neuronal differentiation (e.g., Pac1). Interestingly, Zac1 and Pax6 expression have very similar expression domains in the embryonic telencephalon, consistent with a potential regulatory interaction (Alam et al., 2005). Interestingly, Pax6 expression was reduced in the embryonic pancreas of a transgenic mouse engineered to overexpress a locus associated with TNDM, which included ZAC1 (Ma et al., 2004), suggesting a repressive interaction between Zac1 and Pax6. Indeed, Zac1 is known to function both as a transcriptional activator and repressor (Huang and Stallcup, 2000; Hoffmann et al., 2003). Additional studies are required to explore potential regulatory interactions between Zac1 and Pax6.
Zac1 regulates neocortical neuronal migration via Pac1
We found that Zac1 misexpression at E12.5 did not perturb neuronal migration, whereas misexpression of Zac1 at E14.5 prevented neurons from migrating out of the GZ and IZ and into the CP. Other studies have identified two temporal phases of neuronal migration: (1) an early period of somal translocation that occurs before E14.5; and (2) a later period of glial-guided locomotion that primarily occurs after E14.5 (Nadarajah et al., 2001). Based on the timing of its effects, Zac1 overexpression influences the latter period of locomotion. Indeed, several of the changes that locomoting neurons undergo were perturbed when Zac1 was overexpressed, including an increase in the length and number of pauses, and reduced neuronal branching in the IZ, in which a multipolar shape is associated with the waiting period. Zac1-overexpressing neurons also displayed decreased branching patterns in the upper CP, in which the final movement of neurons into their laminar position depends on somal translocation, with the force required for movement generated by the branching of the primary dendrites and their attachment to the pial surface.
Analyses of Zac1 mutant cortices also revealed defects in neocortical neuronal migration, albeit less severe than in our gain-of-function models. Consistent with this observation, we previously identified a role for Zac1 in mediating neuronal migration in the developing retina (Ma et al., 2007b). In addition, in the Zac1 mutant cerebellum, fewer neurons are found in medial cerebellar nuclei, and fewer Golgi cells populate cerebellar lobule IX, possibly also reflecting a neuronal migration defect (Chung et al., 2011). Consistent with the idea that Zac1 may mediate its effects on neuronal migration through Pac1 signaling, PACAP reduces the rate of granule cell migration in culture (Falluel-Morel et al., 2005; Cameron et al., 2009). Together, our data suggest that neuronal migration may be regulated by different signaling pathways early and late in corticogenesis.
Several Zac1 transcriptional targets have been identified, including Tcf4 (Schmidt-Edelkraut et al., 2014), Pparg1 (Barz et al., 2006), Cdkn1a (Liu, 2011), Rasgfr1 (Hoffmann and Spengler, 2012), Glut4 (Czubryt et al., 2010), and Pac1 (Ciani et al., 1999; Rodríguez-Henche et al., 2002). We focused on Pac1 as a potential downstream effector of Zac1 for several reasons. First, the overexpression of Pac1 signaling molecules in humans has been associated with developmental brain disorders (Cameron et al., 2009). Second, previous time-lapse video microscopy studies have shown that PACAP1–38, a Pac1 agonist, delays the migration of cerebellar granule cells (Falluel-Morel et al., 2005; Cameron et al., 2009). In contrast, PACAP6–38, a Pac1 antagonist, has no effect (Falluel-Morel et al., 2005; Cameron et al., 2009), and the overall laminar organization of the cerebellum is normal in PACAP mutant mice, indicating that this neuropeptide is sufficient but not required to regulate the migration of cerebellar granule cells (Allais et al., 2007). In contrast, in this study, we observe neuronal migration defects after Pac1 gain- and loss-of-function in the neocortex, suggesting that precise levels of signaling through this receptor is required for normal migration of neocortical neurons. Notably, Pac1 perturbations phenocopied those observed when Zac1 expression was altered, and misexpression of Pac1 could rescue the Zac1 knockdown migratory phenotype, suggesting that these genes act in the same genetic pathway. Conversely, when Zac1 was overexpressed and Pac1 was knocked down, migratory defects were not rescued, suggesting that Zac1 induces the expression of other downstream genes that perturb migration. Future experiments will be required to identify the exact nature of these genes and the underlying regulatory interactions.
Together, these data increase our understanding of the pathways that regulate the morphogenetic changes associated with neuronal differentiation and migration in the neocortex.
Footnotes
This project was supported March of Dimes and Canadian Institutes of Health Research Grant 89994 (C.S.). L.A., L.M.L., S.L., and G.W. were supported by a Canadian Institutes of Health Research (CIHR)/Alberta Children's Hospital Research Institute Training Grant in Genetics, Child Health, and Development. R.D. was supported by a CIHR Canada Hope Fellowship. We thank Dawn Zinyk and Nicole Gruenig for animal maintenance and Natasha Klenin for technical assistance.
The authors declare no competing financial interests.
References
- Abdollahi A. LOT1 (ZAC1/PLAGL1) and its family members: mechanisms and functions. J Cell Physiol. 2007;210:16–25. doi: 10.1002/jcp.20835. [DOI] [PubMed] [Google Scholar]
- Alam S, Zinyk D, Ma L, Schuurmans C. Members of the Plag gene family are expressed in complementary and overlapping regions in the developing murine nervous system. Dev Dyn. 2005;234:772–782. doi: 10.1002/dvdy.20577. [DOI] [PubMed] [Google Scholar]
- Allais A, Burel D, Isaac ER, Gray SL, Basille M, Ravni A, Sherwood NM, Vaudry H, Gonzalez BJ. Altered cerebellar development in mice lacking pituitary adenylate cyclase-activating polypeptide. Eur J Neurosci. 2007;25:2604–2618. doi: 10.1111/j.1460-9568.2007.05535.x. [DOI] [PubMed] [Google Scholar]
- Anderson AA, Helmering J, Juan T, Li CM, McCormick J, Graham M, Baker DM, Damore MA, Véniant MM, Lloyd DJ. Pancreatic islet expression profiling in diabetes-prone C57BLKS/J mice reveals transcriptional differences contributed by DBA loci, including Plagl1 and Nnt. Pathogenetics. 2009;2:1. doi: 10.1186/1755-8417-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima T, Wake N. Establishment of the primary imprint of the HYMAI/PLAGL1 imprint control region during oogenesis. Cytogenet Genome Res. 2006;113:247–252. doi: 10.1159/000090839. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Azzi S, Sas TC, Koudou Y, Le Bouc Y, Souberbielle JC, Dargent-Molina P, Netchine I, Charles MA. Degree of methylation of ZAC1 (PLAGL1) is associated with prenatal and post-natal growth in healthy infants of the EDEN mother child cohort. Epigenetics. 2014;9:338–345. doi: 10.4161/epi.27387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman KE, Park BH. Duel nature of TGF-beta signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol. 2005;17:49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]
- Barz T, Hoffmann A, Panhuysen M, Spengler D. Peroxisome proliferator-activated receptor gamma is a Zac target gene mediating Zac antiproliferation. Cancer Res. 2006;66:11975–11982. doi: 10.1158/0008-5472.CAN-06-1529. [DOI] [PubMed] [Google Scholar]
- Basyuk E, Coulon V, Le Digarcher A, Coisy-Quivy M, Moles JP, Gandarillas A, Journot L. The candidate tumor suppressor gene ZAC is involved in keratinocyte differentiation and its expression is lost in basal cell carcinomas. Mol Cancer Res. 2005;3:483–492. doi: 10.1158/1541-7786.MCR-05-0019. [DOI] [PubMed] [Google Scholar]
- Britz O, Mattar P, Nguyen L, Langevin LM, Zimmer C, Alam S, Guillemot F, Schuurmans C. A role for proneural genes in the maturation of cortical progenitor cells. Cereb Cortex. 2006;16:i138–i151. doi: 10.1093/cercor/bhj168. [DOI] [PubMed] [Google Scholar]
- Cameron DB, Raoult E, Galas L, Jiang Y, Lee K, Hu T, Vaudry D, Komuro H. Role of PACAP in controlling granule cell migration. Cerebellum. 2009;8:433–440. doi: 10.1007/s12311-009-0121-9. [DOI] [PubMed] [Google Scholar]
- Cameron ER, Neil JC. The Runx genes: lineage-specific oncogenes and tumor suppressors. Oncogene. 2004;23:4308–4314. doi: 10.1038/sj.onc.1207130. [DOI] [PubMed] [Google Scholar]
- Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-O. [DOI] [PubMed] [Google Scholar]
- Chung SH, Marzban H, Aldinger K, Dixit R, Millen K, Schuurmans C, Hawkes R. Zac1 plays a key role in the development of specific neuronal subsets in the mouse cerebellum. Neural Dev. 2011;6:25. doi: 10.1186/1749-8104-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani E, Hoffmann A, Schmidt P, Journot L, Spengler D. Induction of the PAC1-R (PACAP-type I receptor) gene by p53 and Zac. Brain Res Mol Brain Res. 1999;69:290–294. doi: 10.1016/S0169-328X(99)00116-3. [DOI] [PubMed] [Google Scholar]
- Czubryt MP, Lamoureux L, Ramjiawan A, Abrenica B, Jangamreddy J, Swan K. Regulation of cardiomyocyte Glut4 expression by ZAC1. J Biol Chem. 2010;285:16942–16950. doi: 10.1074/jbc.M109.097246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diplas AI, Lambertini L, Lee MJ, Sperling R, Lee YL, Wetmur J, Chen J. Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics. 2009;4:235–240. doi: 10.4161/epi.9019. [DOI] [PubMed] [Google Scholar]
- Dixit R, Lu F, Cantrup R, Gruenig N, Langevin LM, Kurrasch DM, Schuurmans C. Efficient gene delivery into multiple CNS territories using in utero electroporation. J Vis Exp. 2011;pii:2957. doi: 10.3791/2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falluel-Morel A, Vaudry D, Aubert N, Galas L, Bernard M, Basille M, Fontaine M, Fournier A, Vaudry H, Gonzales BJ. Effects of PACAP and C2-ceramide on motility of cerebellar granule neurons: the fastest is not the farthest (in French) Med Sci. 2005;21:696–698. doi: 10.1051/medsci/2005218-9696. [DOI] [PubMed] [Google Scholar]
- Fattal-Valevski A, Toledano-Alhadef H, Leitner Y, Geva R, Eshel R, Harel S. Growth patterns in children with intrauterine growth retardation and their correlation to neurocognitive development. J Child Neurol. 2009;24:846–851. doi: 10.1177/0883073808331082. [DOI] [PubMed] [Google Scholar]
- Geva R, Eshel R, Leitner Y, Fattal-Valevski A, Harel S. Memory functions of children born with asymmetric intrauterine growth restriction. Brain Res. 2006a;1117:186–194. doi: 10.1016/j.brainres.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Geva R, Eshel R, Leitner Y, Valevski AF, Harel S. Neuropsychological outcome of children with intrauterine growth restriction: a 9-year prospective study. Pediatrics. 2006b;118:91–100. doi: 10.1542/peds.2005-2343. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron. 2005;48:45–62. doi: 10.1016/j.neuron.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Hide T, Takezaki T, Nakatani Y, Nakamura H, Kuratsu J, Kondo T. Sox11 prevents tumorigenesis of glioma-initiating cells by inducing neuronal differentiation. Cancer Res. 2009;69:7953–7959. doi: 10.1158/0008-5472.CAN-09-2006. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Spengler D. Transient neonatal diabetes mellitus gene Zac1 impairs insulin secretion in mice through Rasgrf1. Mol Cell Biol. 2012;32:2549–2560. doi: 10.1128/MCB.06637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Ciani E, Boeckardt J, Holsboer F, Journot L, Spengler D. Transcriptional activities of the zinc finger protein Zac are differentially controlled by DNA binding. Mol Cell Biol. 2003;23:988–1003. doi: 10.1128/MCB.23.3.988-1003.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Stallcup MR. Mouse Zac1, a transcriptional coactivator and repressor for nuclear receptors. Mol Cell Biol. 2000;20:1855–1867. doi: 10.1128/MCB.20.5.1855-1867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamikihara T, Arima T, Kato K, Matsuda T, Kato H, Douchi T, Nagata Y, Nakao M, Wake N. Epigenetic silencing of the imprinted gene ZAC by DNA methylation is an early event in the progression of human ovarian cancer. Int J Cancer. 2005;115:690–700. doi: 10.1002/ijc.20971. [DOI] [PubMed] [Google Scholar]
- Kim MH, Gunnersen J, Augustine C, Tan SS. Region-specific expression of the helix-loop-helix gene BETA3 in developing and adult brains. Mech Dev. 2002;114:125–128. doi: 10.1016/S0925-4773(02)00036-9. [DOI] [PubMed] [Google Scholar]
- Langevin LM, Mattar P, Scardigli R, Roussigné M, Logan C, Blader P, Schuurmans C. Validating in utero electroporation for the rapid analysis of gene regulatory elements in the murine telencephalon. Dev Dyn. 2007;236:1273–1286. doi: 10.1002/dvdy.21126. [DOI] [PubMed] [Google Scholar]
- Li S, Mattar P, Zinyk D, Singh K, Chaturvedi CP, Kovach C, Dixit R, Kurrasch DM, Ma YC, Chan JA, Wallace V, Dilworth FJ, Brand M, Schuurmans C. GSK3 temporally regulates neurogenin 2 proneural activity in the neocortex. J Neurosci. 2012;32:7791–7805. doi: 10.1523/JNEUROSCI.1309-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JS. Molecular genetics of neuronal migration disorders. Curr Neurol Neurosci Rep. 2011;11:171–178. doi: 10.1007/s11910-010-0176-5. [DOI] [PubMed] [Google Scholar]
- Ma D, Shield JP, Dean W, Leclerc I, Knauf C, Burcelin R Ré, Rutter GA, Kelsey G. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004;114:339–348. doi: 10.1172/JCI200419876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Hocking JC, Hehr CL, Schuurmans C, McFarlane S. Zac1 promotes a Muller glial cell fate and interferes with retinal ganglion cell differentiation in Xenopus retina. Dev Dyn. 2007a;236:192–202. doi: 10.1002/dvdy.21002. [DOI] [PubMed] [Google Scholar]
- Ma L, Cantrup R, Varrault A, Colak D, Klenin N, Götz M, McFarlane S, Journot L, Schuurmans C. Zac1 functions through TGFbetaII to negatively regulate cell number in the developing retina. Neural Dev. 2007b;2:11. doi: 10.1186/1749-8104-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, Camenisch I, Berns A, Brandner S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development. 2002;129:3513–3522. doi: 10.1242/dev.129.14.3513. [DOI] [PubMed] [Google Scholar]
- Mattar P, Britz O, Johannes C, Nieto M, Ma L, Rebeyka A, Klenin N, Polleux F, Guillemot F, Schuurmans C. A screen for downstream effectors of Neurogenin2 in the embryonic neocortex. Dev Biol. 2004;273:373–389. doi: 10.1016/j.ydbio.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Miró X, Zhou X, Boretius S, Michaelis T, Kubisch C, Alvarez-Bolado G, Gruss P. Haploinsufficiency of the murine polycomb gene Suz12 results in diverse malformations of the brain and neural tube. Dis Model Mech. 2009;2:412–418. doi: 10.1242/dmm.001602. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex. 2003;13:607–611. doi: 10.1093/cercor/13.6.607. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Peleg D, Kennedy CM, Hunter SK. Intrauterine growth restriction: identification and management. Am Fam Physician. 1998;58:453–460. 466–457. [PubMed] [Google Scholar]
- Piras G, El Kharroubi A, Kozlov S, Escalante-Alcalde D, Hernandez L, Copeland NG, Gilbert DJ, Jenkins NA, Stewart CL. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: identification by a subtractive screen of novel uniparental fibroblast lines. Mol Cell Biol. 2000;20:3308–3315. doi: 10.1128/MCB.20.9.3308-3315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Henche N, Jamen F, Leroy C, Bockaert J, Brabet P. Transcription of the mouse PAC1 receptor gene: cell-specific expression and regulation by Zac1. Biochim Biophys Acta. 2002;1576:157–162. doi: 10.1016/S0167-4781(02)00303-2. [DOI] [PubMed] [Google Scholar]
- Schmidt-Edelkraut U, Daniel G, Hoffmann A, Spengler D. Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol Cell Biol. 2014;34:1020–1030. doi: 10.1128/MCB.01195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron. 2008;60:56–69. doi: 10.1016/j.neuron.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim SY, Wang J, Asada N, Neumayer G, Tran HC, Ishiguro K, Sanada K, Nakatani Y, Nguyen MD. Protein 600 is a microtubule/endoplasmic reticulum-associated protein in CNS neurons. J Neurosci. 2008;28:3604–3614. doi: 10.1523/JNEUROSCI.5278-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Arnaud P, Konfortova G, Dean WL, Beechey CV, Kelsey G. The mouse Zac1 locus: basis for imprinting and comparison with human ZAC. Gene. 2002;292:101–112. doi: 10.1016/S0378-1119(02)00666-2. [DOI] [PubMed] [Google Scholar]
- Spengler D, Villalba M, Hoffmann A, Pantaloni C, Houssami S, Bockaert J, Journot L. Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J. 1997;16:2814–2825. doi: 10.1093/emboj/16.10.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh J, Lu N, Nicot A, Tatsuno I, DiCicco-Bloom E. PACAP is an anti-mitogenic signal in developing cerebral cortex. Nat Neurosci. 2001;4:123–124. doi: 10.1038/83936. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol. 2004;165:709–721. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet. 2002;39:872–875. doi: 10.1136/jmg.39.12.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touahri Y, Adnani L, Mattar P, Markham K, Klenin N, Schuurmans C. Non-isotopic RNA in situ hybridization on embryonic sections. Curr Protoc Neurosci. 2015;70:1.22.1–1.22.25. doi: 10.1002/0471142301.ns0122s70. [DOI] [PubMed] [Google Scholar]
- Varrault A, Ciani E, Apiou F, Bilanges B, Hoffmann A, Pantaloni C, Bockaert J, Spengler D, Journot L. hZAC encodes a zinc finger protein with antiproliferative properties and maps to a chromosomal region frequently lost in cancer. Proc Natl Acad Sci U S A. 1998;95:8835–8840. doi: 10.1073/pnas.95.15.8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, Pavlidis P, Journot L. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell. 2006;11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- White RA, Pan Z, Salisbury JL. GFP-centrin as a marker for centriole dynamics in living cells. Microsc Res Tech. 2000;49:451–457. doi: 10.1002/(SICI)1097-0029(20000601)49:5<451::AID-JEMT7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Wilkinson LS, Davies W, Isles AR. Genomic imprinting effects on brain development and function. Nat Rev Neurosci. 2007;8:832–843. doi: 10.1038/nrn2235. [DOI] [PubMed] [Google Scholar]
- Yan Y, Zhou X, Pan Z, Ma J, Waschek JA, DiCicco-Bloom E. Pro- and anti-mitogenic actions of pituitary adenylate cyclase-activating polypeptide in developing cerebral cortex: potential mediation by developmental switch of PAC1 receptor mRNA isoforms. J Neurosci. 2013;33:3865–3878. doi: 10.1523/JNEUROSCI.1062-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa S, Onizuka T, Shimoji K, Ohno Y, Kageyama T, Yoon SH, Egashira T, Seki T, Hashimoto H, Nishiyama T, Kaneda R, Murata M, Hattori F, Makino S, Sano M, Ogawa S, Prall OW, Harvey RP, Fukuda K. Zac1 is an essential transcription factor for cardiac morphogenesis. Circ Res. 2010;106:1083–1091. doi: 10.1161/CIRCRESAHA.109.214130. [DOI] [PubMed] [Google Scholar]