

Abstract
The chromosome 15q13.3 microdeletion is a pathogenic copy number variation conferring epilepsy, intellectual disability, schizophrenia, and autism spectrum disorder (ASD). We generated mice carrying a deletion of 1.2 Mb homologous to the 15q13.3 microdeletion in human patients. Here, we report that mice with a heterozygous deletion on a C57BL/6 background (D/+ mice) demonstrated phenotypes including enlarged/heavier brains (macrocephaly) with enlarged lateral ventricles, decreased social interactions, increased repetitive grooming behavior, reduced ultrasonic vocalizations, decreased auditory-evoked gamma band EEG, and reduced event-related potentials. D/+ mice had normal body weight, activity levels, sensory gating, and cognitive abilities and no signs of epilepsy/seizures. Our results demonstrate that D/+ mice represent ASD-related phenotypes associated with 15q13.3 microdeletion syndrome. Further investigations using this chromosome-engineered mouse model may uncover the common mechanism(s) underlying ASD and other neurodevelopmental/psychiatric disorders representing the 15q13.3 microdeletion syndrome, including epilepsy, intellectual disability, and schizophrenia.
SIGNIFICANCE STATEMENT Recently discovered pathologic copy number variations (CNVs) from patients with neurodevelopmental/psychiatric disorders show very strong penetrance and thus are excellent candidates for mouse models of disease that can mirror the human genetic conditions with high fidelity. A 15q13.3 microdeletion in humans results in a range of neurodevelopmental/psychiatric disorders, including epilepsy, intellectual disability, schizophrenia, and autism spectrum disorder (ASD). The disorders conferred by a 15q13.3 microdeletion also have overlapping genetic architectures and comorbidity in other patient populations such as those with epilepsy and schizophrenia/psychosis, as well as schizophrenia and ASD. We generated mice carrying a deletion of 1.2 Mb homologous to the 15q13.3 microdeletion in human patients, which allowed us to investigate the potential causes of neurodevelopmental/psychiatric disorders associated with the CNV.
Keywords: 15q13.3, autism, CNV, epilepsy, intellectual disability, schizophrenia
Introduction
Contemporary approaches to neurogenetics have enabled the discovery of a substantial number of susceptibility genes for neurodevelopmental/psychiatric disorders and significant efforts have been made to understand the role of these genes and their encoded proteins in the etiology and pathophysiology of the illnesses. The identification of these genes in patient populations has guided the generation of genetically engineered mouse models with targeted mutations, which have been used for two main purposes: (1) to investigate the function/role of the susceptibility gene and (2) to make disease models to further the understanding of disease mechanisms. Simple, single-gene knock-out or transgenic overexpression strategies are an acceptable approach to making mutant mice for the former purpose, but are not ideal for the latter because most of the human disease single susceptibility genetic variances are noncoding single nucleotide polymorphisms and have weak penetration. The profound changes in gene expression levels often achieved in mutant mice by knock-out/transgenic strategies are rarely observed in human patients. Rather, neurodevelopmental/psychiatric disorders likely occur due to epistatic interactions among multiple gene variants that, in turn, cause the resultant pathology (Kleinman et al., 2011).
Recently discovered pathologic copy number variations (CNVs) from patients with neurodevelopmental/psychiatric disorders show very strong penetrance and thus are excellent candidates for mouse models of disease that can mirror the human genetic conditions with high fidelity (Glessner et al., 2009; St Clair, 2009; Tam et al., 2009; Mefford et al., 2010). A 15q13.3 microdeletion in humans results in a range of neurodevelopmental/psychiatric disorders, including autism spectrum disorder (ASD) (Ben-Shachar et al., 2009; Miller et al., 2009; Pagnamenta et al., 2009), schizophrenia (International Schizophrenia Consortium, 2008; Stefansson et al., 2008; Vassos et al., 2010; Levinson et al., 2011; Vacic et al., 2011), epilepsy (Sharp et al., 2008; Dibbens et al., 2009; Helbig et al., 2009; Muhle et al., 2011), and intellectual disability (Sharp et al., 2008; Ben-Shachar et al., 2009; Shinawi et al., 2009). The disorders conferred by a 15q13.3 microdeletion also have overlapping genetic architectures and comorbidity in other patient populations, such as those with epilepsy (Bolton et al., 2011), schizophrenia/psychosis (Clarke et al., 2012), and schizophrenia and ASD (Carroll and Owen, 2009).
The exceptionally low incidence of 15q13.3 microdeletion and other CNVs results in clinical studies with very small sample sizes, which limits the ability to study their contributions to the etiology of resultant syndromes. For these reasons, mouse models of CNVs are extremely valuable tools for investigating the potential causes of neurodevelopmental/psychiatric disorders associated with CNVs. We report here our phenotypic characterization of D/+ mice and our observations that these mice represent ASD-related phenotypes associated with 15q13.3 microdeletion syndrome.
Materials and Methods
Procedures
All experimental procedures were performed in accordance with guidelines published in the National Research Council's Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Astellas Research Institute of America LLC, Institutional Animal Care and Use Committee or by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Generation of 15q13.3/7qC microdeletion mutant mice
We generated mice carrying a deletion encompassing whole genes deleted in human 15q13.3 microdeletion patients (Fan1, Mtmr10, Trpm1, Klf13, Otud7a and Chrna7) using FLP recombinase target (FRT) chromosome engineering. A proximal FRT site was targeted into the gene Chrna7 (downstream of the last exon 10) in mouse C57BL/6N (C57BL/6NTac) embryonic stem (ES) cells. One of the targeted cell lines (clone A3) was used to insert the second, complementary cassette into the gene Fan1 (upstream of the first exon 1), which is located ∼1.2 Mb telomeric to Chrna7. We obtained four cell lines in which both the Chrna7 and Fan1 genes were replaced (clones B4, B5, B8, and B10). Recombination between the two FRT sites in these cell lines was induced by transient expression of Flp recombinase. Aliquots of cells were collected and assayed by PCR using primers located in flanking regions of the deletion (forward 5′-GGTCCGCTCCGATCTGTATCCACT-3′ and reverse 5′-GCTTGGAGGTCATCTAACATCTAAAGGGT-3′). PCR detection of the 1.2 Mb deletion in cells from clones B5 and B10 indicated successful intrachromosomal cis-recombination between the two FRT sites. Cells from these two transfections were plated and one single clone (B10/B8) with 1.2 Mb deletion was selected by PCR. We used this cell line to generate chimaeras that transmitted the deletion to their progeny. After confirmation of germline transmission, we expanded the mutant mouse colony using in vitro fertilization (IVF) with a C57BL/6N (C57BL/6NTac) background at Taconic. These IVF-derived mutant mice and their WT littermates were used for gene expression and large-cohort five basic behavioral analyses of locomotor activity, social interaction, anxiety-related behavior, working memory, and long-term memory. Then, the mutant mice were transferred to Hilltop Lab Animals and maintained as a mutant mouse line harboring heterozygous a 1.2 Mb deletion with a C57BL/6N (C57BL/6NHla) background. These mutant mice and WT littermates were used for subsequent behavioral and EEG analyses and brain morphological studies.
Measurement of gene dosage and gene expression in brain
Table 1 shows primers used in the present study. For gene dosage analysis, genomic DNA was isolated from tails using DNeasy Blood and Tissue Kit (Qiagen) and analyzed using quantitative PCR. For gene expression analysis, total RNA was isolated from brain regions and analyzed using quantitative RT-PCR as described previously (Chen et al., 2012). Quantitative PCR was conducted using SYBR Green PCR Master Mix in a ViiA real-time PCR machine (Life Technologies). All amplification data were normalized with mean value of WT group (defined as two copies) for gene dosage analysis and normalized with glyceraldehyde-3-phophate dehydrogenase (Gapdh) for gene expression analysis.
Table 1.
PCR primers used in the present study
| Magel2 | Forward | CGGGAGTCCTGATGCTACA |
| Reverse | GGAGGAGGATGAGCCAAAG | |
| Mkrn3 | Forward | GAAGCAAATCCTCTGCAGGT |
| Reverse | GACAGTTATCGCCCTCCTTG | |
| Peg12 | Forward | CAGCTCGGCCTTGAGGTAT |
| Reverse | GCTGCGGGATGCTTACTTT | |
| Chrna7 | Forward | CCAGAGGAGGCTGTACAAGG |
| Reverse | TCTCCAGCGGGTTGTAGTTC | |
| Otud7a | Forward | TGGGCAGCACTTCTACATGA |
| Reverse | CTGACAGGACTGCGTCCATA | |
| Klf13 | Forward | GACAGCGCCTCGCTTTTC |
| Reverse | CTGGTTGAGGTCCGCTAGG | |
| Trpm1 | Forward | CCTACGACACCAAGCCAGAT |
| Reverse | CCAGTCCTTCACCATGAGGT | |
| Mtmr10 | Forward | GGGGCAAGTTGATATGCAGT |
| Reverse | AATGGCATTGGGTCATCTGT | |
| Fan1 | Forward | TGCACCACCTGCTAAACTTG |
| Reverse | GACTCATCAAGGTGCCGAAT | |
| Mphosph10 | Forward | TCGTCGACAGACTCTGGAAC |
| Reverse | TTGGTGGCTTTCCTGATTTC | |
| Mcee | Forward | TGGAACTGCTTCATCCACTG |
| Reverse | CCTCCAGCCTTGTTCTTCTG | |
| Apba2 | Forward | TGCCACAACCATAGTCCTGA |
| Reverse | GCCCTCGTCATAGTCCTCCT |
Behavioral analysis
Open field locomotor activity.
Using the Ethovision XT version 7 software program (Noldus Information Technology) to track movement in infrared light, total horizontal distance traveled over a 30 min period in a 50 cm L × 50 cm W × 50 cm H transparent Plexiglas chamber (Med Associates) was measured.
Social interaction.
Ovariectomized BALB/c female mice (Hilltop Labs) at 4 months of age were used to examine social investigative behaviors in the male's home cage (20 × 30 × 13 cm) for a duration of 5 min. Investigation times were counted when the male's head was in contact with the female. Trials were videotaped and manually scored with investigators blinded to genotype.
Anxiety-related behavior (elevated zero-maze).
The elevated zero maze consists of a circular runway with two open quadrants (25 cm × 5 cm) and two enclosed quadrants of the same size with 15 cm high opaque walls. The maze is elevated to a height of 55 cm above the floor. Each mouse was placed in the maze facing one of the closed quadrants. Mouse behavior was recorded during a 5 min session using Noldus Ethovision video-tracking software. The time spent in open and enclosed quadrants, as well as total distance traveled, was recorded.
Working memory (Y-maze).
Working memory deficits during a spontaneous delayed alternation task were assessed using an automated Y-maze (Med Associates) with three electronically controlled gray guillotine doors and infrared detection beams. The task began when the animal was randomly placed in any 37 cm arm with all doors closed for 30 s, after which it was free to choose to enter any arm (start arm). One minute later, the animal could choose to enter any arm (choice arm). Once a choice had been made, all doors were closed for 30 s, after which it could choose any arm (final arm). There were five trials in total with an intertrial interval (ITI) of 1 min. Maximum time allowed to complete this task was 15 min. A perfect score was attained when the animal did not reenter an arm previously chosen within a particular trial. Percentage correct was calculated by averaging all five trials. All arms of the maze were unbaited.
Long-term memory (fear conditioning).
Fear conditioning was assessed using an automated infrared video fear conditioning system. The conditioning chambers (Med Associates) were monitored by CCD infrared-sensitive video cameras. The Med Associates software analyzes the number of freezing bouts, bout duration, and percentage freezing in determined time intervals. Stimuli for conditioning were as follows: unconditioned stimulus (US; foot shock, 0.7 mA, 2 s) and conditioned stimulus (CS; 3K Hz, 80db, 30 s coterminating with US). Mice were in the chambers for 60 s before the first CS presentation. Mice were trained with five trials with a 60 s ITI.
Sensitivity to pilocarpine.
Mice (n = 5/dose) were administered a single injection of pilocarpine (200, 230, 250, 300, or 350 mg/kg, i.p., for WT; 30, 50, 70, or 100 mg/kg, i.p,. for mutants). The doses were determined from pilot studies and literature review. The lowest dose of pilocarpine required to elicit a seizure was recorded (Shin et al., 2013).
Visual paired discrimination and reversal learning.
Using methods similar to those described previously (Brigman et al., 2010; Romberg et al., 2013), a visual pairwise discrimination was assessed using an automated touchscreen system (Camden Instruments and Lafayette Instruments). Mice were required to learn to discriminate black and white, brightness-matched visual stimuli on a video screen. A trial began with the presentation of two stimuli on the screen: one was programmed as being correct (S+) and one as being incorrect (S−). Whether the S+ was on the right or left was determined pseudorandomly. The mouse had to touch the correct stimulus to elicit the tone/reward tray light and reward delivery (strawberry-flavored milk). If the mouse touched the incorrect stimulus, no reward was delivered and a time-out of 5 s followed before the mouse was given the opportunity to attempt a correction trial. Correction trials (representation of the stimulus array in the same L-R configuration) ensued until the correct stimulus was chosen. Mice were trained in daily sessions consisting of a maximum of 30 trials in 60 min (20 s ITI) until they performed at a response rate of 80% correct discrimination for 2 consecutive days. Reversal learning training immediately followed the initial discrimination task. The same criteria were applied to the reversal learning task, during which mice had to respond correctly to the previously nonrewarded S− stimulus. Data were analyzed for the number of days and number of trials required to reach criterion in each task.
Three-chamber sociability test.
General sociability was assessed in a three-chamber social approach apparatus using a modification of the protocol described by Crawley (2007). Each chamber in the apparatus is 50 cm L × 32 cm W × 40 cm H. There are social, neutral and nonsocial chambers with cups 7 cm in diameter in the social and nonsocial chambers, which can house one mouse. Mice were placed in the apparatus and confined to the center chamber for 10 min. They were then acclimated to the entire apparatus for an additional 10 min. Immediately after the acclimation period, a novel mouse (C57BL/6 male, 3–4 months old) was placed in one of the cups with the opposite cup remaining empty. The placement of the novel mouse was switched for each experiment to avoid any potential side bias to the chambers. Time spent in the social and nonsocial chambers was recorded for 10 min by Noldus Ethovision.
Self-grooming behavior.
Mice were placed in a novel test cage with no bedding or food for 20 min, acclimated for 10 min, and then time spent grooming was recorded for the second 10 min segment.
Ultrasonic vocalization analysis.
Male and female mouse pups 6 d of age were removed from their mothers and littermates in the home cage and placed on a small platform inside a sound attenuating chamber (Med Associates). An ultrasonic microphone (Avisoft Ultrasound Gate) was suspended 10 cm above the mouse platform. Ultrasonic vocalization spectrograms were recorded using the Avisoft SAS Lab Pro software and exported into Excel. Median and maximum amplitude (in decibels) for each vocalization emitted during the 5 min recording interval was averaged for every individual pup.
Olfaction test.
Before testing, animals were presented with small pieces of Kellogg's Pop-Tarts, which were used as nonsocial odor, in the home cage for 2 d. The olfaction test was composed of three consecutive sessions and performed under dim light. Each mouse was habituated in an empty cage with a cotton swab hanging from the lid for 10 min. In the first session, each mouse was presented with cotton swabs absorbed with water for 2 min 3 consecutive times. Immediately after the first session, the cotton swab was scented with Pop-Tart extract and presented for 2 min 3 consecutive times. The Pop-Tart extract was prepared by soaking pieces of Pop-Tarts in water and collecting supernatants. The third session used the cotton swabs that were scented with soiled cage bedding from a different mouse cage (social odor). The amount of time a mouse spent sniffing the cotton swabs during each presentation was measured.
EEG and event-related potentials (ERPs)
Electrode implantation occurred under 1% isoflurane anesthesia. A lead electrode was lowered into the right hippocampal region 1.8 mm posterior, 2.65 mm lateral, and 2.75 mm dorsal to bregma. Reference and ground electrodes were placed on the surface of the ipsilateral cortex 0.8 mm posterior and 0.2 mm anterior to bregma, respectively, at the same lateral coordinate as the lead electrode. Ethyl cyanoacrylate (Loctite; Henkel) and dental cement (Ortho Jet; Lang Dental) were used to secure the electrodes to the skull.
Auditory stimuli were generated by Micro1401 hardware and Spike2 version 6.0 software (Cambridge Electronic Design). EEGs were recorded during a series of repeated white noise bursts (12 ms duration, 85 decibels). Mice were initially exposed to 200 white noise bursts with an interstimulus interval (ISI) of 1 s, followed by an identical number of white noise bursts with a 2 s ISI, followed by a 4 s ISI, then by an 8 s ISI. One week later, EEGs were assessed during a series of paired-click white noise bursts (12 ms duration, 85 decibels, 500 ms ISI); 8.5 s separated each click pair and a total of 300 pairs were presented.
Raw EEG data were analyzed and processed using EEGLAB (Schwartz Center for Computational Neuroscience) to create a time-frequency measure for gamma power. Single-trial epochs between 199 and 399 ms relative to tone onset were extracted from the continuous EEG recording. For each epoch, power was calculated by the EEGLAB MatLab toolbox using Morlet wavelets in 116 logarithmically spaced frequency bins between 4 and 120 Hz, with wavelet cycle numbers ranging from 2 to 10 (Delorme and Makeig, 2004). Gamma power was defined as between 30 to 80 Hz and was quantified as the average from 0 to 60 ms after the stimulus.
Baseline EEG was averaged over each ISI duration or across all 300 paired clicks. The waveform average was then baseline corrected by removing the baseline ERP response. The amplitude of each ERP waveform component was quantified as the maximum positive deflection between 15 and 30 ms (for P20) or the maximum negative deflection occurring between 25 and 55 ms (for N40). For the paired-click experiment, each component was calculated after both the S1 and S2 periods.
Brain analysis
MRI was performed using a Varian 7T Horizontal Bore Small Animal MRI system. After standard MRI preparation (optimization of shimming, pulse power calibration, line width determination, and scout images to locate the tumor), a scout T2-weighted (T2w) fast spin-echo multislice sequence was used to image the entire brain to optimize coverage for the subsequent high-resolution scan. The acquisition parameters were as follows: repetition time = 4 s, 8 echoes, echo-spacing = 14.2 ms, k-space centered on the fourth echo, and 2 averages. A 15-mm-square field of view was used with an image matrix of 128 × 128 and 17 contiguous, 1-mm-thick transaxial slices that covered the entire brain volume. Immediately after the T2w scan, a higher-resolution spin-echo multislice sequence was acquired to visualize the entire brain volume and finer structures throughout. The acquisition parameters were as follows: repetition time = 3.5 s, 2 averages; a 15-mm-square field of view was used with an image matrix of 256 × 256 and 60 contiguous, 0.3-mm-thick transaxial slices that covered the entire brain. During the MRI procedures, anesthesia was maintained using a 1–2% isoflurane-in-air mixture. Body temperature was maintained using a thermally regulated water heating blanket. The images were reconstructed using a custom written script in Matlab (The MathWorks). For morphological brain analysis, Nissl staining, Golgi staining, and brain slice preparation were performed as described previously (Matsumoto et al., 2008).
Statistics
Differences between D/+ mice and WT littermates were assessed with a Student's t test (two-tailed). EEG data were analyzed using repeated-measures ANOVA with genotype (D/+ and WT) and ISI as variables. Data are presented as mean ± SEM. Statistical analyses were conducted with Prism 5.0 (GraphPad Software).
Results
Generation of mouse model for human 15q13.3 microdeletion syndrome
To mimic human genetic conditions, we studied heterozygous mutants harboring one chromosome deletion but retaining one intact chromosome (D/+ mice). We used C57BL/6 ES cells for the chromosome engineering to avoid the additional burden of backcrossing to C57BL/6, a standard genetic background in CNS research. Given that a region of mouse chromosome 7qC is highly syntenic to the human 15q13.3 microdeletion region in gene content and order, we have been able to generate mice carrying a deletion encompassing the identical six genes deleted in human 15q13.3 microdeletion patients (Fan1, Mtmr10, Trpm1, Klf13, Otud7a, and Chrna7) using Flp-FRT chromosome engineering methods (Fig. 1A). Proximal and distal FRT sites were targeted into the gene Chrna7 and the gene Fan1 in mouse C57BL/6 ES cells, respectively. Recombination between the two FRT sites in double-targeted cells was induced by transient expression of Flp recombinase. Cells harboring the expected 1.2 Mb deletion by successful cis-recombination between the two FRT sites were used to generate chimaeras that transmitted the deletion to their progeny. To mirror the human patient genetic conditions, we studied heterozygous mutant mice on a C57BL/6 background harboring one intact chromosome and one 1.2 Mb deletion (D/+ mice).
Figure 1.
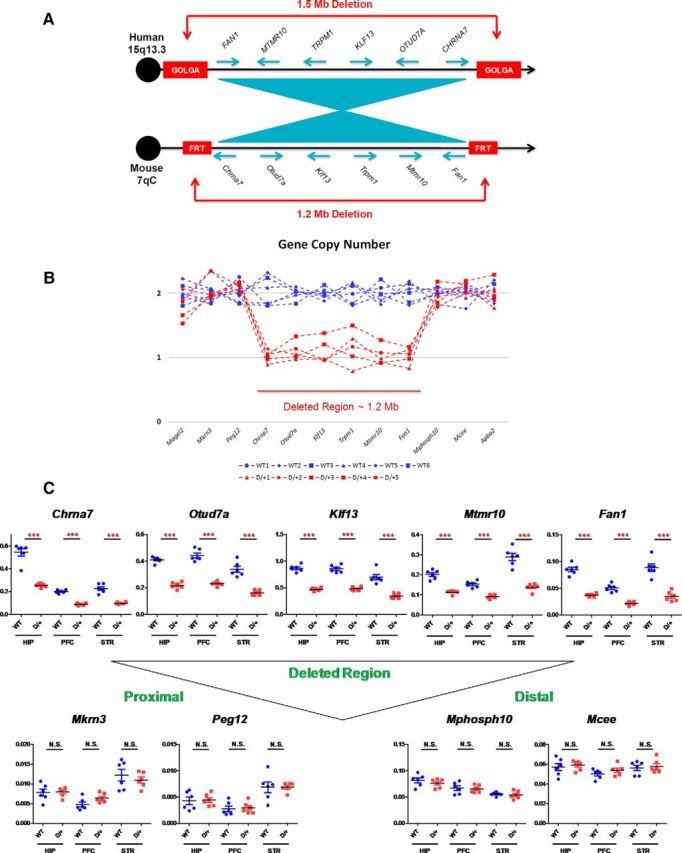
Generation of 15q13.3 microdeletion mouse model (D/+ mice). A, Gene content and order in the region commonly deleted in 15q13.3 microdeletion patients compared with that in a region of mouse chromosome 7. Note that the relative order of genes is conserved but with inverted centromere–telomere orientation. Blue arrows indicate the transcriptional orientation of genes. In humans, commonly observed 1.5 Mb deletion in 15q13.3 region arises through nonallelic homologous recombination between duplication blocks, which harbor copies of the GOLGA gene family. In mice, artificial FRT sites were inserted into regions corresponding to human GOLGA-containing duplication blocks to induce cis-recombination mediated 1.2 Mb deletion. B, Gene copy number in the deleted region and flanking regions. C, Gene expression levels in the hippocampus (HIP), prefrontal cortex (PFC), and striatum (STR). Levels of gene expression were normalized by Gapdh levels. ***p < 0.001; N.S., Nonsignificant.
The deletion allele was transmitted at the expected Mendelian ratios from both male and female mice. Mice with either a paternally or maternally inherited deletion bred normally and were fertile and apparently healthy. We used adult (4–7 months old) male D/+ mice with a paternally inherited deletion and compared them to their adult male WT littermates in the present study, unless otherwise specified.
Gene expression levels in multiple brain regions correspond to 15q13.3 dosage in 15q13.3 microdeletion mouse model
To validate the D/+ mouse model molecularly, we analyzed gene copy numbers and gene expression levels in the brain and determined whether the expression levels corresponded to gene dosage. Gene copy numbers within the deleted region were decreased by 50%, as expected (Fig. 1B). Using quantitative PCR, we determined mRNA expression levels in three brain regions (hippocampus, prefrontal cortex, and striatum). We could not detect reliable levels of mRNA expression of Trpm1, presumably due to its extremely low abundance in the three brain regions tested. The other five genes in the deleted chromosome (Fan1, Mtmr10, Klf13, Otud7a, and Chrna7) were expressed in all brain regions and their mRNA expression levels were decreased in D/+ mouse brain (Fig. 1C). Levels of mRNA expression from genes in flanking regions (proximal two genes, Mkrn3 and Peg12, and distal two genes, Mphosph10 and Mcee) were analyzed in the same three brain regions. There were no differences observed in mRNA levels of these genes flanking the deleted chromosome region between WT and D/+ mice (Fig. 1C).
15q13.3 microdeletion mouse model did not show phenotypic diversity
Clinical manifestation of the 15q13.3 microdeletion syndrome in human patients is heterogeneous, with diagnoses including the overlapping neurodevelopmental and psychiatric disorders of epilepsy, intellectual disability, schizophrenia, and ASD. Our initial observations involved a relatively large cohort of adult D/+ mice and WT littermates to determine whether there were phenotypic subgroups within the population of mutant mice that might mimic the clinical diversity of the human patients.
We assessed five basic behaviors in D/+ mice and their WT littermates: locomotor activity in an open field, social interaction in a home cage free interaction task, anxiety-related behavior on an elevated zero maze, working memory in a Y-maze delayed spontaneous alternation task, and long-term memory in a contextual fear conditioning task (Fig. 2). Compared with WT littermates, D/+ mice showed significantly less social interaction duration with a novel mouse in the free interaction task (D/+: 65.0 ± 3.6 s, n = 30; WT: 86.5 ± 3.5 s, n = 30; t(58) = −4.29, p < 0.0001), significantly more time in the open sectors of the zero maze (D/+: 36.7 ± 2.8 s, n = 30; WT: 23.8 ± 2.9 s, n = 29; t(57) = 3.16, p < 0.01), a trend toward hypoactivity in the open field (D/+: 810.6 ± 24.6 cm, n = 28; WT: 872.2 ± 20.2 cm, n = 28; t(54) = 1.93, p = 0.059), no differences in percentage correct alternation in their Y-maze behavior (D/+: 49.3 ± 3.4%, n = 30; WT: 53.0 ± 2.1%, n = 30; t(58) = 0.92, p = 0.36), and no differences in the freezing time in the contextual fear conditioning task between the two groups (D/+: 42.0 ± 2.6%, n = 30; WT: 42.9 ± 3.1%, n = 30; t(58) = 0.22, p = 0.83).
Figure 2.

Basic behavioral phenotypes in D/+ mice based on basic behavioral assessment, open field locomotor (activity), free interaction (social interaction), zero-maze (anxiety), delayed Y-maze (working memory), and fear-conditioning (contextual memory). *p < 0.05; **p < 0.01; ***p < 0.001; N.S., Nonsignificant.
Overall, in the five behavior assessments, we did not see any indication of subgroups or clinically analogous phenotypic diversity (pleiotropy) in D/+ mice.
15q13.3 microdeletion mouse model showed behavioral phenotypes related to ASD
After we confirmed that there is no obvious diversity in D/+ mouse phenotypes, we assessed behavioral phenotypes using a regular size cohort.
Home cage behavior was videotaped for 2 consecutive days for D/+ mice (n = 24) and WT littermates (n = 12). There were no spontaneous seizures observed in either D/+ or WT mice. An absence of an epilepsy-related phenotype in D/+ mice was corroborated by no significant differences in the sensitivity to pilocarpine-induced seizures between mutant and WT mice. We observed an apparent resistance to seizure induction in D/+ mice compared with WT littermates (Fig. 3A).
Figure 3.
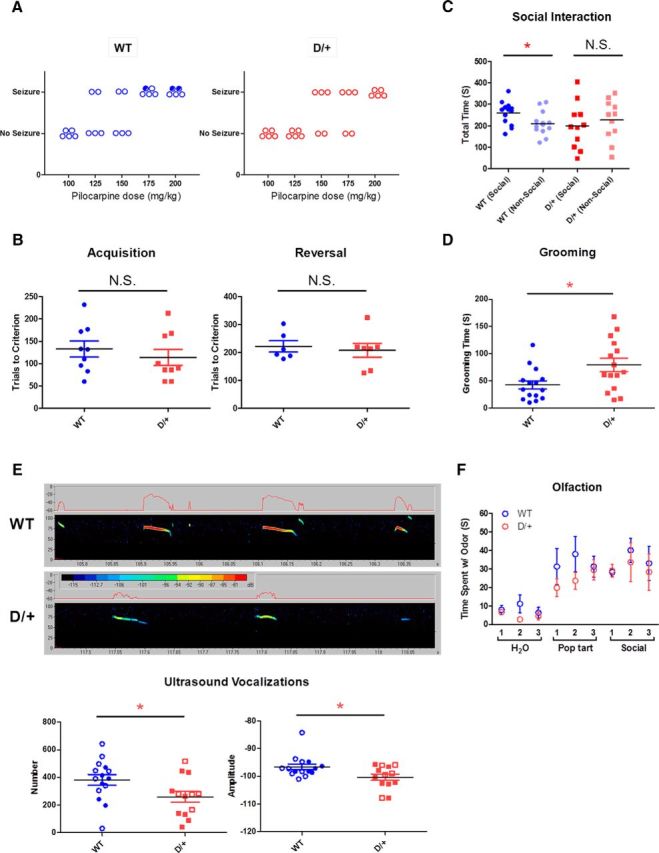
Autism spectrum disorder (ASD)-related behavioral phenotypes in D/+ mice. A, Seizure threshold with pilocarpine. The minimum dose required to elicit convulsive seizures in some of the mice was 150 and 125 mg/kg for D/+ mice and WT littermates, respectively. Open circles represent mice that survived the treatment; closed circles represent mice that died within 90 min of pilocarpine injection. B, Visual pairwise discrimination task. Both the acquisition and the reversal of the discrimination task are shown. N.S., Nonsignificant. C, Sociability deficits in D/+ mice in the three-chamber apparatus. D, Elevated self-grooming durations in D/+ mice. E Fewer and lower-amplitude USVs in D/+ mice. Closed circles and squares represent males; open circles and squares represent females. Representative USVs from D/+ mice and WT littermates are shown. *p < 0.05; N.S., Nonsignificant. F, Normal olfaction in D/+ mice. The mice were examined for their sensitivity to odor. The mice were presented with three types of scents (water, Pop-Tarts, and social odor) three times for each scent.
Because D/+ mice did not show working and contextual memory deficits, we further examined the cognitive abilities of the mice using a visual pairwise discrimination task. This task uses a touchscreen method in which the mice were trained to discriminate between two visual stimuli projected onto a touch-sensitive computer screen to receive a liquid reward. There was no significant difference in the acquisition of the discrimination task between D/+ and WT littermates (D/+: 114.0 ± 17.9 trials to criteria, n = 9; WT: 132.8 ± 17.9 trials to criteria, n = 9; t(16) = 0.74, p = 0.47) or in the reversal task (D/+: 207.9 ± 24.9 trials to criteria, n = 7; WT: 222.0 ± 20.1 trials to criteria, n = 6; t(11) = 0.43, p = 0.67) (Fig. 3B).
Children with 15q13.3 microdeletion syndrome are frequently diagnosed with ASD (Ben-Shachar et al., 2009) and D/+ mice displayed behavioral traits more similar to ASD-related features than other disorders linked to 15q13.3 microdeletion (social withdrawal and risk-taking behavior without hyperactivity, seizure, or cognitive impairment). Therefore, we tested D/+ mice using additional tasks that are sensitive to the core phenotypes related to ASD. The triad of behavioral symptoms diagnostic of ASD are abnormal social interactions, high levels of stereotyped or repetitive behaviors, and communication deficits (American Psychiatric Association and American Psychiatric Association Task Force on DSM-IV, 1994).
The three-chamber sociability test has been used widely to characterize the social behavior of mouse models of ASD (Crawley, 2007). We retested the sociability of D/+ mice and observed that they did not show a preference for the social chamber over for the nonsocial chamber of the apparatus during the three chamber test, whereas their WT littermates did show a preference (D/+ social: 199.6 ± 32.3 s, n = 11; D/+ nonsocial: 226.8 ± 28.7 s, n = 11; t(20) = 0.63, p = 0.54; WT social: 259.8 ± 15.1 s, n = 13; WT nonsocial: 210.7 ± 15.5 s, n = 13, t(24) = −2.27, p < 0.05; Fig. 3C). We assessed D/+ and WT mice for stereotyped repetitive behaviors in a novel environment. D/+ mice showed significantly increased durations of self-grooming (repetitive) behaviors compared with WT mice (D/+: 79.1 ± 12.2 s, n = 15; WT: 42.5 ± 7.6 s, n = 15; t(28) = 2.54, p < 0.05; Fig. 3D). Ultrasonic vocalizations (USVs) emitted by pups when they are separated from their mother and littermates are a well characterized method for assessing social communication between mice. USVs were recorded for 5 min from male and female D/+ and WT pups on postnatal day 6. D/+ mice showed significantly fewer (D/+: 259.0 ± 40.2, n = 13; WT: 381.5 ± 38.6, n = 15; t(26) = 2.19, p < 0.05) and less loud (D/+: −100.3 ± 1.2 dB, n = 13; WT: −96.6 ± 1.0 dB, n = 15; t(26) = 2.45, p < 0.05) USVs (Fig. 3E). To rule out any potential impairment in olfaction that may confound the outcome of the social interaction or other assays, we tested the animal's responses to detect various odors by presenting cotton swabs scented with nonsocial and social scents. Both D/+ mice (n = 9) and WT littermates (n = 8) spent similar time interacting with the cotton swabs, suggesting that D/+ mice display normal olfactory responses (Fig. 3F).
15q13.3 microdeletion mouse model showed altered gamma-band EEG and ERPs
EEG was recorded in adult male D/+ mice (n = 10) and WT littermates (n = 9). One D/+ mouse was removed from the sensory gating experiment due to poor quality of ERP response. A main effect of genotype was observed on evoked gamma (F(1,17) = 7.2, p < 0.05) and intertrial coherence (ITC; F(1,17) = 6.7, p < 0.05), with D/+ mice showing reductions in both measures relative to WT mice (Fig. 4A). Likewise, significant main effects were observed for ISI on both measures (evoked F(3,51) = 17.9, p < 0.05; ITC F(3,51) = 19.7, p < 0.05), but this did not differ according to genotype, as indicated by the lack of interaction between genotype and ISI (evoked F(3,51) = 2.1, p > 0.05; ITC F(3,51) = 1.3, p > 0.05). In contrast, no main effects were found for genotype on induced gamma (F(1,17) = 0.46, p > 0.05) (Fig. 4A) or baseline gamma (F(1,17) = 3.23, p > 0.05), nor was the interaction between ISI and genotype significant on either measure (induced F(3,51) = 0.9, p > 0.05; baseline F(3,51) = 1.3, p > 0.05). A significant effect was observed for ISI on induced (F(3,51) = 13.4, p < 0.05), but not baseline gamma (F(3,51) = 0.95, p > 0.05).
Figure 4.

Gamma EEG response and ERPs in D/+ mice. A, Top, Evoked gamma response to white noise auditory stimuli as a function of ISI in WT and D/+ mice. A, Middle, Gamma band ITC as a function of ISI in WT and D/+ mice. Bottom, Induced gamma response to auditory stimuli as a function of ISI in WT and D/+ mice. Gamma = 30–80 Hz for all measures. B, Top, Maximum amplitude of the P20 component of the ERP (15–30 ms) as a function of ISI in WT and D/+ mice and maximum amplitude of the N40 component of the ERP (25 to 60 ms) as a function of ISI in WT and D/+ mice. Bottom, Sensory gating in WT and D/+ mice. *p < 0.05; N.S., Nonsignificant.
D/+ mice showed a reduction in amplitude of the P20 component of the auditory ERP, as indicated by a significant main effect of genotype (F(1,17) = 5.1, p < 0.05; Fig. 4B). In contrast, no main effect of genotype was observed for the later N40 component (F(1,16) = 0.63, p > 0.05; Fig. 4B). Both D/+ and WT mice showed significant sensory gating during assessment on a paired-click paradigm, as defined as a significant main effect of stimulus type (S1 > S2; P20, F(1,16) = 5.2, p < 0.05; N40, F(1,16) = 16.9, p < 0.05; Fig. 4B). No significant interactions were seen between genotype and stimulus type (P20, F(1,16) = 0.015, p > 0.05; N40, F(1,16) = 0.66, p > 0.05), suggesting that the reduction in S2 over S1 was not sensitive to the 15q13.3 deletion.
Enlarged brains and lateral ventricles were observed in the 15q13.3 microdeletion mouse model
We measured body and brain weights of adult D/+ mice and WT littermates (male, ∼4 months old; Fig. 5A). There was no difference in body weight (D/+: 32.7 ± 0.5 g, n = 30; WT: 31.9 ± 0.5 g, n = 30; t(58) = 1.06, p = 0.84), but there was a highly significant difference in brain weight, with D/+ mice having heavier brains than WT mice (D/+: 0.476 ± 0.003 g, n = 30; WT: 0.456 ± 0.003 g, n = 30; t(58) = 4.77, p < 0.0001). We then determined whether brain weight differences emerged at birth. There were no differences in either body weight (D/+: 5.65 ± 0.20 g, n = 11; WT: 5.67 ± 0.13 g, n = 14; t(23) = 0.11, p = 0.91) or brain weight (D/+: 0.334 ± 0.004 g, n = 11; WT: 0.336 ± 0.003 g, n = 14; t(23) = 0.34, p = 0.74) at postnatal day 10 (both male and female pups mixed; Fig. 5B). However, we observed significant differences in brain weight (D/+: 0.446 ± 0.005 g, n = 5; WT: 0.420 ± 0.003 g, n = 7; t(10) = 4.63, p < 0.001), but not body weight (D/+: 24.0 ± 0.4 g, n = 5; WT: 23.4 ± 0.7 g, n = 7; t(10) = 0.65, p = 0.53), between male D/+ and WT mice at 2 months of age (Fig. 5B).
Figure 5.
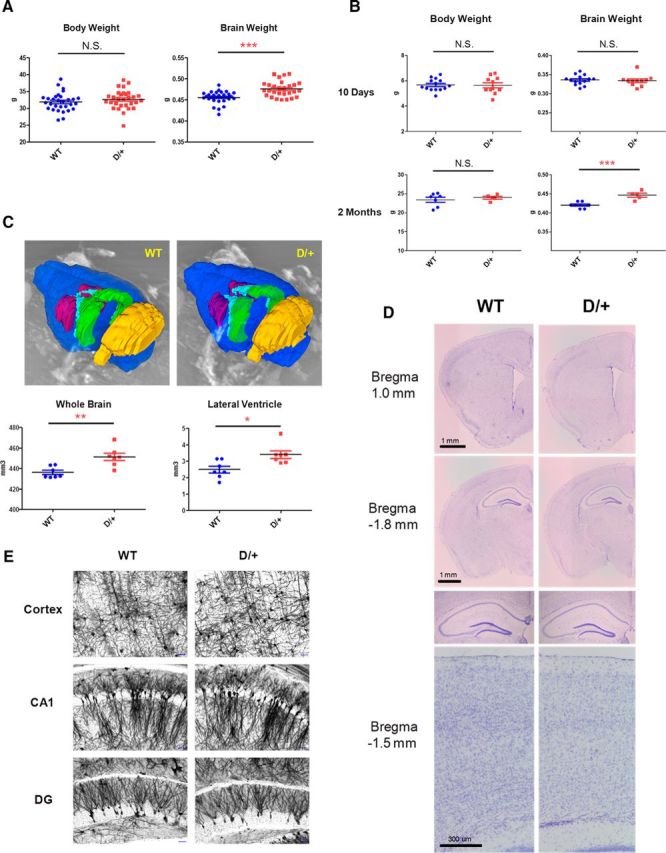
Enlarged brain and lateral ventricles in D/+ mice. A, Body and brain weights of adult D/+ mice and WT littermates. B, Body and brain weights of D/+ mice and their WT littermates at postnatal day 10 and at 2 months. C, Enlarged whole-brain volume and lateral ventricle volume in D/+ mice. Representative MRIs from D/+ mice and WT littermates are shown. Blue, whole brain; purple, striatum; light blue, lateral ventricle; green, hippocampus; yellow, cerebellum. D, Representative brain images of coronal sections from D/+ mice and WT littermates. Sections were Nissl stained. Note that no gross malformations in D/+ mice are seen. E, Representative neuron images from D/+ mice and WT littermates. Sections were Golgi stained. Note that no apparent differences in arborization between D/+ mice and WT littermates are seen. Scale bars, 50 μm. *p < 0.05; **p < 0.01; ***p < 0.001; N.S., Nonsignificant.
Using MRI, we confirmed that the whole-brain volume was significantly larger in 2-month-old male D/+ mice compared with WT littermates (D/+: 451 ± 4 mm3, n = 7; WT: 436 ± 2 mm3, n = 7; t(12) = 3.64, p < 0.01). We also found that the lateral ventricles were enlarged in D/+ mice (D/+: 3.40 ± 0.23 mm3, n = 7; WT: 2.50 ± 0.20 mm3, n = 7; t(12) = 2.97, p < 0.05; Fig. 5C) compared with WT. There were no differences measured in the volume of hippocampus (D/+: 16.86 ± 0.33 mm3, n = 7; WT: 16.70 ± 0.51 mm3, n = 7; t(12) = 0.26, p = 0.80), striatum (D/+: 14.83 ± 0.49 mm3, n = 7; WT: 15.06 ± 0.53 mm3, n = 7; t(12) = 0.32, p = 0.76), or cerebellum (D/+: 51.38 ± 0.75 mm3, n = 7; WT: 51.14 ± 1.02 mm3, n = 7; t(12) = 0.19, p = 0.85) between the two groups.
At 2 months of age, macroscopic analysis of the male D/+ mouse brains using Nissl staining revealed no gross structural abnormalities such as laminar formation of cerebral cortices or position of major brain nuclei (Fig. 5D). Microscopically, Golgi staining revealed no apparent abnormalities at the level of dendrite branching or arborization in their cerebral cortex and hippocampus (Fig. 5E).
Discussion
The present study demonstrates that a deletion of mouse chromosome 7qC syntenic to the region that is deleted in patients with 15q13.3 microdeletion syndrome is similarly pathogenic in mice. The 15q13.3 microdeletion in humans results in a range of neurodevelopmental/psychiatric disorders including schizophrenia (International Schizophrenia Consortium, 2008; Stefansson et al., 2008; Vassos et al., 2010; Levinson et al., 2011; Vacic et al., 2011), epilepsy (Sharp et al., 2008; Dibbens et al., 2009; Helbig et al., 2009; Muhle et al., 2011), intellectual disability (Sharp et al., 2008; Ben-Shachar et al., 2009; Shinawi et al., 2009), and ASD (Ben-Shachar et al., 2009; Miller et al., 2009; Pagnamenta et al., 2009). The 15q13.3 microdeletion mouse model generated here displayed phenotypes largely relevant to ASD but potentially representing other neurodevelopmental/psychiatric disorders observed due to 15q13.3 microdeletion. Considering the epidemiological and genetic findings of comorbidity and shared genetic susceptibility between ASD and epilepsy (Tuchman and Rapin, 2002; Tuchman et al., 2010; Bolton et al., 2011), intellectual disabilities (Matson and Shoemaker, 2009; Mefford et al., 2012), and schizophrenia (Carroll and Owen, 2009; Guilmatre et al., 2009; King and Lord, 2011), this CNV mutant mouse model should be a valuable tool with which to investigate the mechanism(s) underlying the variable penetrance of phenotypes in 15q13.3 microdeletion syndrome.
15q13.3 microdeletion mouse model demonstrated phenotypes related to ASD
We found that D/+ mice demonstrated phenotypes relevant to the three core behavioral features of ASD: impairment in social interactions, restricted-repetitive behaviors, and deficits in communication (Chadman et al., 2012).
The deficits in reciprocal social interaction reported here in D/+ mice were assessed by both a free social interaction task in the subject's home cage with a novel partner mouse and by a three-chamber sociability test (Crawley, 2007). Longer bouts of self-grooming behaviors are the most commonly reported ASD-related phenotype in mouse models of the disorder (Provenzano et al., 2012; Jiang and Ehlers, 2013). We measured the number of ultrasonic vocalizations emitted by maternally isolated pups at 6 d of age and found that D/+ pups vocalized less frequently and less loudly than WT pups, which could be translated to human disorder by recent studies showing that infants at risk for ASD and prelanguage toddlers with ASD have abnormal cry and speech characteristics that are useful correlates of later developmental communication deficits in ASD (Esposito and Venuti, 2010; Sheinkopf et al., 2012; Sullivan et al., 2013). Although not usually used as a critical diagnostic criterion, increased risk-taking behavior is also a symptom of ASD and was mirrored by the behavior of D/+ mice in the elevated zero maze, where they displayed lower anxiety/higher risk-taking behavior (Cavalari and Romanczyk, 2012).
Reductions in time-locked gamma response, both in terms of power of response (evoked-gamma) and consistency of response across trials (ITC), were observed in D/+ mice. In contrast, these mice showed no change in non-time-locked induced gamma or baseline gamma response. There is evidence that time-locked gamma responses, including steady-state response and intertrial coherence, are reduced in subjects with ASD (Grice et al., 2001; Wilson et al., 2007; Rojas et al., 2008; Gandal et al., 2010; Sun et al., 2012). Studies explicitly addressing induced gamma in ASD are rare. Rojas et al. (2008) showed increased auditory induced gamma in ASD, in contrast to the reductions that they observed in evoked gamma. Reductions in both induced and evoked gamma have been reported in schizophrenia (Krishnan et al., 2009). Therefore, the gamma band disruptions observed in D/+ mice would seem consistent with the time-locked gamma changes observed in both autism and schizophrenia. Notwithstanding their relationship to a particular human disorder, the changes reported here in D/+ mice suggest substantial deficits in stimulus processing and sensory registration. D/+ mice showed a reduced P20 response in the ERP, as well as a more modest, nonsignificant reduction in N40. Reductions of ERP amplitudes are commonly reported in schizophrenia, but not autism, especially in regard to the later negative component. Interestingly, D/+ mice showed normal sensory gating of both the P20 and N40. Gating studies are mixed in schizophrenia, in which more recent data suggest that the amplitude of response, rather than gating, is abnormal. Several studies have failed to report altered P50 gating in autistic subjects, consistent with the data in D/+ mice (Kemner et al., 2002; Orekhova et al., 2008; Oranje et al., 2013) and suggesting that D/+ mice represent EEG phenotypes related to ASD in this regard.
We found the phenotype of increased brain weight and size in D/+ mice. This phenotype is consistent with the accumulating evidence demonstrating that individuals with ASD have enlarged brains (Hardan et al., 2001; Courchesne et al., 2007; Hazlett et al., 2011). Although a recent reexamination of previous studies raised the question of brain overgrowth as a generalized biomarker of ASD, the existence of a subgroup-specific brain enlargement in ASD patients was evident (Raznahan et al., 2013). Importantly, it was reported that 15q13.3 microdeletion patients diagnosed with ASD also had larger head circumferences (Pagnamenta et al., 2009). D/+ mice may represent features of a subset of ASD patients with brain overgrowth and would thus be a good experimental model with which to study dysregulation of brain growth and its impact on ASD-related behavioral phenotypes. Our MRI study revealed an enlargement of the entire brain of D/+ mice with an increase in the lateral ventricle volume, which is also a neuroanatomical feature that has been observed in patients with ASD (Hardan et al., 2001).
Mechanistic insight into various disorders related to 15q13.3 microdeletion syndrome
The 15q13.3 microdeletion region contains CHRNA7, which has generated a large amount of attention due to its association with both schizophrenia (Leonard et al., 1996; Hajós and Rogers, 2010) and autism (Martin-Ruiz et al., 2004; Ray et al., 2005). However, studies have revealed no array of behavioral phenotypes or brain morphological deficits in either Chrna7 heterozygous or homozygous knock-out mice that are relevant to these disorders except for a subtle cognitive deficit, which has been detected only in homozygous null knock-out mice (Orr-Urtreger et al., 1997; Young et al., 2007; Young et al., 2011). Therefore, we propose that a heterozygous deletion of Chrna7 is not sufficient by itself to evoke phenotypes in D/+ mice, but rather that haploinsufficiency of some other gene(s) within the deleted region (Fan1, Mtmr10, Trpm1, Klf13, or Otud7a) must be needed.
Recently, another 15q13.3 microdeletion mouse Df(h15q13)/+ was reported (Fejgin et al., 2014). The phenotype of the Df(h15q13)/+ mouse is both similar to and different from the D/+ mouse in several respects. Both mouse models had slightly lower locomotor activity under baseline conditions and both had no spontaneous seizure activity but reduced sensitivity to pharmacologically induced seizures (resistant to seizures). Both mice had normal working memory in the Y-maze and intact long-term memory in contextual fear conditioning, although the Df(h15q13)/+ mice also had deficits in long-term spatial memory. In addition, both mice had complex electrophysiological phenotypes, including decreased auditory-evoked gamma band EEG and reduced ERPs, with the Df(h15q13)/+ mice also having increased gamma-band power during their active state. Conversely, although the Df(h15q13)/+ mice had increased body weight and normal brain weight, the D/+ mice had normal body weight and increased brain weight. The pleiotropy of the two mouse models may represent the phenotypic diversity in human subjects harboring the 15q13.3 microdeletion.
Studies using genetically engineered mice have shown that homeostatic regulation sometimes leads to compensation in perturbed molecular pathways that can result in the absence or even inversion of expected phenotypes in the CNS (Kelly et al., 2007; Zhang et al., 2007) and that this regulation (e.g., brain size homeostasis) can be strongly modulated by the genetic background of mice (Houde et al., 2004). Therefore, it is tempting to speculate that the pleiotropy of the two 15q13.3 microdeletion mouse models, brain overgrowth in D/+ versus normal brain size in Df(h15q13)/+, may be caused by different compensation levels influenced by genetic architectures of these two mouse models. It is worth noting that there could be a subtle difference in the substrain of mice used (see Materials and Methods), although both 15q13.3 microdeletion mouse lines have been maintained on a C57BL/6 strain background. This homeostasis/compensation mechanism may also explain the reason that both 15q13.3 microdeletion mouse models showed an apparent seizure-resistant phenotype, which is opposite from the epilepsy phenotype anticipated from the 15q13.3 microdeletion, the most prevalent risk factor associated with ∼1% of patients with generalized epilepsy (Helbig et al., 2009). Because a diametric molecular and morphological relationship between ASD and schizophrenia has been postulated (Crespi et al., 2010), the possible homeostasis/compensation mechanism with diverse genetic variations in humans may explain the reason that ASD and schizophrenia are conferred by the same 15q13.3 microdeletion.
In summary, our findings with D/+ mice indicate that these animals represent ASD-related phenotypes associate with 15q13.3 microdeletion syndrome. This CNV mutant mouse model should be a valuable tool with which to investigate the mechanism(s) underlying the variable penetrance of phenotypes in the 15q13.3 microdeletion syndrome, including ASD, schizophrenia, epilepsy, and intellectual disability.
Footnotes
This work was supported by Astellas Pharma, Inc. S.J.S. receives grant support from Astellas Pharma, Inc. We thank members of Xenogen Bioscience/Taconic Farms, Inc., for mutant generation, Hilltop Lab Animals, Inc., for animal husbandry, and Molecular Imaging, Inc., for MRI analysis.
Q.C., M.A., N.M.W., S. Miyoshi, S. Miyake, K.T., H.I., and M.M. are full-time employees of Astellas Pharma, Inc. J.H.K., A.K.G., R.S., and C.L.H. were full-time employees of Astellas at the time our research was conducted, but are no longer affiliated with the company. During these studies, A.L. was an intern working at Astellas Research Institute of America. LLC from University of Illinois at Urbana–Champaign. S.J.S. receives grant support from Astellas Pharma, Inc. and Boehringer Ingelheim and is a consultant for NuPathe, Inc. The remaining authors declare no competing financial interests.
References
- American Psychiatric Association, American Psychiatric Association. Task Force on DSM-IV. Diagnostic and statistical manual of mental disorders: DSM-IV. Ed 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- Ben-Shachar S, Lanpher B, German JR, Qasaymeh M, Potocki L, Nagamani SC, Franco LM, Malphrus A, Bottenfield GW, Spence JE, Amato S, Rousseau JA, Moghaddam B, Skinner C, Skinner SA, Bernes S, Armstrong N, Shinawi M, Stankiewicz P, Patel A, Cheung SW, Lupski JR, Beaudet AL, Sahoo T. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 2009;46:382–388. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton PF, Carcani-Rathwell I, Hutton J, Goode S, Howlin P, Rutter M. Epilepsy in autism: features and correlates. Br J Psychiatry. 2011;198:289–294. doi: 10.1192/bjp.bp.109.076877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigman JL, Graybeal C, Holmes A. Predictably irrational: assaying cognitive inflexibility in mouse models of schizophrenia. Front Neurosci. 2010:4. doi: 10.3389/neuro.01.013.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll LS, Owen MJ. Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 2009;1:102. doi: 10.1186/gm102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalari RN, Romanczyk RG. Caregiver perspectives on unintentional injury risk in children with an autism spectrum disorder. J Pediatr Nurs. 2012;27:632–641. doi: 10.1016/j.pedn.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Chadman KK, Guariglia SR, Yoo JH. New directions in the treatment of autism spectrum disorders from animal model research. Expert Opin Drug Discov. 2012;7:407–416. doi: 10.1517/17460441.2012.678828. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kogan JH, Gross AK, Zhou Y, Walton NM, Shin R, Heusner CL, Miyake S, Tajinda K, Tamura K, Matsumoto M. SREB2/GPR85, a schizophrenia risk factor, negatively regulates hippocampal adult neurogenesis and neurogenesis-dependent learning and memory. Eur J Neurosci. 2012;36:2597–2608. doi: 10.1111/j.1460-9568.2012.08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MC, Tanskanen A, Huttunen MO, Clancy M, Cotter DR, Cannon M. Evidence for shared susceptibility to epilepsy and psychosis: a population-based family study. Biol Psychiatry. 2012;71:836–839. doi: 10.1016/j.biopsych.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007;17:448–459. doi: 10.1111/j.1750-3639.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi B, Stead P, Elliot M. Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia. Proc Natl Acad Sci U S A. 2010;107:1736–1741. doi: 10.1073/pnas.0906080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, Bellows S, Leu C, Trucks H, Obermeier T, Wittig M, Franke A, Caglayan H, Yapici Z, Yapici Z, Sander T, Eichler EE, Scheffer IE, Mulley JC, Berkovic SF. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. 2009;18:3626–3631. doi: 10.1093/hmg/ddp311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Venuti P. Understanding early communication signals in autism: a study of the perception of infants' cry. J Intellect Disabil Res. 2010;54:216–223. doi: 10.1111/j.1365-2788.2010.01252.x. [DOI] [PubMed] [Google Scholar]
- Fejgin K, Nielsen J, Birknow MR, Bastlund JF, Nielsen V, Lauridsen JB, Stefansson H, Steinberg S, Sorensen HB, Mortensen TE, Larsen PH, Klewe IV, Rasmussen SV, Stefansson K, Werge TM, Kallunki P, Christensen KV, Didriksen M. A mouse model that recapitulates cardinal features of the 15q13.3 microdeletion syndrome including schizophrenia- and epilepsy-related alterations. Biol Psychiatry. 2014;76:128–137. doi: 10.1016/j.biopsych.2013.08.014. [DOI] [PubMed] [Google Scholar]
- Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TP, Siegel SJ. Validating gamma oscillations and delayed auditory responses as translational biomarkers of autism. Biol Psychiatry. 2010;68:1100–1106. doi: 10.1016/j.biopsych.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice SJ, Spratling MW, Karmiloff-Smith A, Halit H, Csibra G, de Haan M, Johnson MH. Disordered visual processing and oscillatory brain activity in autism and Williams syndrome. Neuroreport. 2001;12:2697–2700. doi: 10.1097/00001756-200108280-00021. [DOI] [PubMed] [Google Scholar]
- Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, Layet V, Rosier A, Briault S, Bonnet-Brilhault F, Laumonnier F, Odent S, Le Vacon G, Joly-Helas G, David V, Bendavid C, Pinoit JM, Henry C, Impallomeni C, Germano E, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajós M, Rogers BN. Targeting alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia. Curr Pharm Des. 2010;16:538–554. doi: 10.2174/138161210790361434. [DOI] [PubMed] [Google Scholar]
- Hardan AY, Minshew NJ, Mallikarjuhn M, Keshavan MS. Brain volume in autism. J Child Neurol. 2001;16:421–424. doi: 10.1177/088307380101600607. [DOI] [PubMed] [Google Scholar]
- Hazlett HC, Poe MD, Gerig G, Styner M, Chappell C, Smith RG, Vachet C, Piven J. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch Gen Psychiatry. 2011;68:467–476. doi: 10.1001/archgenpsychiatry.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde C, Banks KG, Coulombe N, Rasper D, Grimm E, Roy S, Simpson EM, Nicholson DW. Caspase-7 expanded function and intrinsic expression level underlies strain-specific brain phenotype of caspase-3-null mice. J Neurosci. 2004;24:9977–9984. doi: 10.1523/JNEUROSCI.3356-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MP, Isiegas C, Cheung YF, Tokarczyk J, Yang X, Esposito MF, Rapoport DA, Fabian SA, Siegel SJ, Wand G, Houslay MD, Kanes SJ, Abel T. Constitutive activation of Galphas within forebrain neurons causes deficits in sensorimotor gating because of PKA-dependent decreases in cAMP. Neuropsychopharmacology. 2007;32:577–588. doi: 10.1038/sj.npp.1301099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemner C, Oranje B, Verbaten MN, van Engeland H. Normal P50 gating in children with autism. J Clin Psychiatry. 2002;63:214–217. doi: 10.4088/JCP.v63n0307. [DOI] [PubMed] [Google Scholar]
- King BH, Lord C. Is schizophrenia on the autism spectrum? Brain Res. 2011;1380:34–41. doi: 10.1016/j.brainres.2010.11.031. [DOI] [PubMed] [Google Scholar]
- Kleinman JE, Law AJ, Lipska BK, Hyde TM, Ellis JK, Harrison PJ, Weinberger DR. Genetic neuropathology of schizophrenia: new approaches to an old question and new uses for postmortem human brains. Biol Psychiatry. 2011;69:140–145. doi: 10.1016/j.biopsych.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan GP, Hetrick WP, Brenner CA, Shekhar A, Steffen AN, O'Donnell BF. Steady state and induced auditory gamma deficits in schizophrenia. Neuroimage. 2009;47:1711–1719. doi: 10.1016/j.neuroimage.2009.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard S, Adams C, Breese CR, Adler LE, Bickford P, Byerley W, Coon H, Griffith JM, Miller C, Myles-Worsley M, Nagamoto HT, Rollins Y, Stevens KE, Waldo M, Freedman R. Nicotinic receptor function in schizophrenia. Schizophr Bull. 1996;22:431–445. doi: 10.1093/schbul/22.3.431. [DOI] [PubMed] [Google Scholar]
- Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Kendler KS, Freedman R, Dudbridge F, Pe'er I, Hakonarson H, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Ruiz CM, Lee M, Perry RH, Baumann M, Court JA, Perry EK. Molecular analysis of nicotinic receptor expression in autism. Brain Res Mol Brain Res. 2004;123:81–90. doi: 10.1016/j.molbrainres.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Matson JL, Shoemaker M. Intellectual disability and its relationship to autism spectrum disorders. Res Dev Disabil. 2009;30:1107–1114. doi: 10.1016/j.ridd.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Straub RE, Marenco S, Nicodemus KK, Matsumoto S, Fujikawa A, Miyoshi S, Shobo M, Takahashi S, Yarimizu J, Yuri M, Hiramoto M, Morita S, Yokota H, Sasayama T, Terai K, Yoshino M, Miyake A, Callicott JH, Egan MF, et al. The evolutionarily conserved G protein-coupled receptor SREB2/GPR85 influences brain size, behavior, and vulnerability to schizophrenia. Proc Natl Acad Sci U S A. 2008;105:6133–6138. doi: 10.1073/pnas.0710717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, Gurnett CA, Schreiber S, Bassuk AG, Guipponi M, Stephani U, Helbig I, Eichler EE. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Batshaw ML, Hoffman EP. Genomics, intellectual disability, and autism. N Engl J Med. 2012;366:733–743. doi: 10.1056/NEJMra1114194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Shen Y, Weiss LA, Korn J, Anselm I, Bridgemohan C, Cox GF, Dickinson H, Gentile J, Harris DJ, Hegde V, Hundley R, Khwaja O, Kothare S, Luedke C, Nasir R, Poduri A, Prasad K, Raffalli P, Reinhard A, et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhle H, Mefford HC, Obermeier T, von Spiczak S, Eichler EE, Stephani U, Sander T, Helbig I. Absence seizures with intellectual disability as a phenotype of the 15q13.3 microdeletion syndrome. Epilepsia. 2011;52:e194–e198. doi: 10.1111/j.1528-1167.2011.03301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oranje B, Lahuis B, van Engeland H, Jan van der Gaag R, Kemner C. Sensory and sensorimotor gating in children with multiple complex developmental disorders (MCDD) and autism. Psychiatry Res. 2013;206:287–292. doi: 10.1016/j.psychres.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Orekhova EV, Stroganova TA, Prokofyev AO, Nygren G, Gillberg C, Elam M. Sensory gating in young children with autism: relation to age, IQ, and EEG gamma oscillations. Neurosci Lett. 2008;434:218–223. doi: 10.1016/j.neulet.2008.01.066. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnamenta AT, Wing K, Sadighi Akha E, Knight SJ, Bölte S, Schmötzer G, Duketis E, Poustka F, Klauck SM, Poustka A, Ragoussis J, Bailey AJ, Monaco AP. A 15q13.3 microdeletion segregating with autism. Eur J Hum Genet. 2009;17:687–692. doi: 10.1038/ejhg.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano G, Zunino G, Genovesi S, Sgadó P, Bozzi Y. Mutant mouse models of autism spectrum disorders. Dis Markers. 2012;33:225–239. doi: 10.1155/2012/916082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray MA, Graham AJ, Lee M, Perry RH, Court JA, Perry EK. Neuronal nicotinic acetylcholine receptor subunits in autism: an immunohistochemical investigation in the thalamus. Neurobiol Dis. 2005;19:366–377. doi: 10.1016/j.nbd.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Raznahan A, Wallace GL, Antezana L, Greenstein D, Lenroot R, Thurm A, Gozzi M, Spence S, Martin A, Swedo SE, Giedd JN. Compared to what? Early brain overgrowth in autism and the perils of population norms. Biol Psychiatry. 2013;74:563–575. doi: 10.1016/j.biopsych.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas DC, Maharajh K, Teale P, Rogers SJ. Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism. BMC Psychiatry. 2008;8:66. doi: 10.1186/1471-244X-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberg C, Horner AE, Bussey TJ, Saksida LM. A touch screen-automated cognitive test battery reveals impaired attention, memory abnormalities, and increased response inhibition in the TgCRND8 mouse model of Alzheimer's disease. Neurobiol Aging. 2013;34:731–744. doi: 10.1016/j.neurobiolaging.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, Broomer A, Casuga I, Wang Y, Xiao C, Barbacioru C, Gimelli G, Bernardina BD, Torniero C, Giorda R, Regan R, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinkopf SJ, Iverson JM, Rinaldi ML, Lester BM. Atypical cry acoustics in 6-month-old infants at risk for autism spectrum disorder. Autism Res. 2012;5:331–339. doi: 10.1002/aur.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin R, Kobayashi K, Hagihara H, Kogan JH, Miyake S, Tajinda K, Walton NM, Gross AK, Heusner CL, Chen Q, Tamura K, Miyakawa T, Matsumoto M. The immature dentate gyrus represents a shared phenotype of mouse models of epilepsy and psychiatric disease. Bipolar Disord. 2013;15:405–421. doi: 10.1111/bdi.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, Lanpher B, Nagl S, Herding HS, Nevinny-Stickel C, Immken LL, Patel GS, German JR, Beaudet AL, Stankiewicz P. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet. 2009;41:1269–1271. doi: 10.1038/ng.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Clair D. Copy number variation and schizophrenia. Schizophr Bull. 2009;35:9–12. doi: 10.1093/schbul/sbn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K, Sharda M, Greenson J, Dawson G, Singh NC. A novel method for assessing the development of speech motor function in toddlers with autism spectrum disorders. Front Integr Neurosci. 2013;7:17. doi: 10.3389/fnint.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Grützner C, Bölte, Wibral M, Tozman T, Schlitt S, Poustka F, Singer W, Freitag CM, Uhlhaas PJ. Impaired gamma-band activity during perceptual organization in adults with autism spectrum disorders: evidence for dysfunctional network activity in frontal-posterior cortices. J Neurosci. 2012;32:9563–9573. doi: 10.1523/JNEUROSCI.1073-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam GW, Redon R, Carter NP, Grant SG. The role of DNA copy number variation in schizophrenia. Biol Psychiatry. 2009;66:1005–1012. doi: 10.1016/j.biopsych.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Tuchman R, Rapin I. Epilepsy in autism. Lancet Neurol. 2002;1:352–358. doi: 10.1016/S1474-4422(02)00160-6. [DOI] [PubMed] [Google Scholar]
- Tuchman R, Cuccaro M, Alessandri M. Autism and epilepsy: historical perspective. Brain Dev. 2010;32:709–718. doi: 10.1016/j.braindev.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Vacic V, McCarthy S, Malhotra D, Murray F, Chou HH, Peoples A, Makarov V, Yoon S, Bhandari A, Corominas R, Iakoucheva LM, Krastoshevsky O, Krause V, Larach-Walters V, Welsh DK, Craig D, Kelsoe JR, Gershon ES, Leal SM, Dell Aquila M, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassos E, Collier DA, Holden S, Patch C, Rujescu D, St Clair D, Lewis CM. Penetrance for copy number variants associated with schizophrenia. Hum Mol Genet. 2010;19:3477–3481. doi: 10.1093/hmg/ddq259. [DOI] [PubMed] [Google Scholar]
- Wilson TW, Rojas DC, Reite ML, Teale PD, Rogers SJ. Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biol Psychiatry. 2007;62:192–197. doi: 10.1016/j.biopsych.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, Finlayson K, Sharkey J. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur Neuropsychopharmacol. 2007;17:145–155. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Young JW, Meves JM, Tarantino IS, Caldwell S, Geyer MA. Delayed procedural learning in alpha7-nicotinic acetylcholine receptor knock-out mice. Genes Brain Behav. 2011;10:720–733. doi: 10.1111/j.1601-183X.2011.00711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Duan RS, Chen Z, Quezada HC, Mix E, Winblad B, Zhu J. IL-18 deficiency aggravates kainic acid-induced hippocampal neurodegeneration in C57BL/6 mice due to an overcompensation by IL-12. Exp Neurol. 2007;205:64–73. doi: 10.1016/j.expneurol.2007.01.019. [DOI] [PubMed] [Google Scholar]