

Abstract
Emerging evidence has indicated that the pathogenesis of neuropathic pain is mediated by spinal neural plasticity in the dorsal horn, which provides insight for analgesic therapy. Here, we report that the abundance of tumor necrosis factor receptor-associated factor 2 and NcK-interacting kinase (TNIK), a kinase that is presumed to regulate neural plasticity, was specifically enhanced in ipsilateral dorsal horn neurons after spinal nerve ligation (SNL; left L5 and L6). Spinal TNIK-associated allodynia is mediated by downstream TNIK–GluR1 coupling and the subsequent phosphorylation-dependent trafficking of GluR1 toward the plasma membrane in dorsal horn neurons. Tumor necrosis factor receptor-associated factor 2 (TRAF2), which is regulated by spinal F-box protein 3 (Fbxo3)-dependent F-box and leucine-rich repeat protein 2 (Fbxl2) ubiquitination, contributes to SNL-induced allodynia by modifying TNIK/GluR1 phosphorylation-associated GluR1 trafficking. Although exhibiting no effect on Fbxo3/Fbxl2/TRAF2 signaling, focal knockdown of spinal TNIK expression prevented SNL-induced allodynia by attenuating TNIK/GluR1 phosphorylation-dependent subcellular GluR1 redistribution. In contrast, intrathecal administration of BC-1215 (N1,N2-Bis[[4-(2-pyridinyl)phenyl]methyl]-1,2-ethanediamine) (a novel Fbxo3 inhibitor) prevented SNL-induced Fbxl2 ubiquitination and subsequent TFAF2 de-ubiquitination to ameliorate behavioral allodynia via antagonizing TRAF2/TNIK/GluR1 signaling. By targeting spinal Fbxo3-dependent Fbxl2 ubiquitination and the subsequent TRAF2/TNIK/GluR1 cascade, spinal application of a TNF-α-neutralizing antibody ameliorated SNL-induced allodynia, and, conversely, intrathecal TNF-α injection into naive rats induced allodynia via a spinal Fbxo3/Fbxl2-dependent modification of the TRAF2/TNIK/GluR1 cascade. Together, our results suggest that spinal TNF-α contributes to the development of neuropathic pain by upregulating TRAF2/TNIK/GluR1 signaling via Fbxo3-dependent Fbxl2 ubiquitination and degradation. Thus, we propose a potential medical treatment strategy for neuropathic pain by targeting the F-box protein or TNIK.
SIGNIFICANCE STATEMENT TNF-α participates in neuropathic pain development by facilitating the spinal TRAF2-dependent TNIK–GluR1 association, which drives GluR1-containing AMPA receptor trafficking toward the plasma membrane. In addition, F-box protein 3 modifies this pathway by inhibiting F-box and leucine-rich repeat protein 2-mediated TRAF2 ubiquitination, suggesting that protein ubiquitination contributes crucially to the development of neuropathic pain. These results provide a novel therapeutic strategy for pain relief.
Keywords: Fbxl2, Fbxo3, GluR1, neuropathic, TNIK, ubiquitination
Introduction
Emerging evidence has linked tumor necrosis factor receptor-associated factors 2- and NcK-interacting kinase (TNIK), a member of the germinal center kinase family (Fu et al., 1999), to neural plasticity. Knockdown of TNIK expression in cultured neurons decreases the amount of membrane-bound GluR1-containing AMPA receptors (AMPARs) (Hussain et al., 2010) and alters the synchrony of neuronal activity (MacLaren et al., 2011). Similarly, introducing a synthesized peptide inhibiting TNIK activity results in decreased cell-surface GluR1–AMPAR and AMPAR-dependent currents in rat hippocampal cultures (Q. Wang et al., 2011), indicating that TNIK contributes to neural plasticity by regulating the subcellular redistribution of GluR1–AMPARs. Similar to plastic changes in brain areas, studies investigating visceral (Galan et al., 2004; Peng et al., 2011), inflammatory (Choi et al., 2010; Peng et al., 2012b), and neuropathic (Garry and Fleetwood-Walker, 2004) pain revealed that subcellular GluR1–AMPAR trafficking underlies the spinal plasticity that is essential for the development of allodynia and/or hyperalgesia (Tao, 2012). Importantly, our previous studies demonstrated that spinal serine/threonine kinase (Peng et al., 2012b) and scaffold protein (Peng et al., 2011) both drive GluR1–AMPAR redistribution in dorsal horn neurons to induce spinal plasticity, mediating pain hypersensitivity. Because TNIK has both serine/threonine kinase and scaffold domains, we hypothesized that TNIK is a crucial mediator of neuropathic pain via its effects on GluR1–AMPARs trafficking.
Tumor necrosis factor-α (TNF-α), a proinflammatory cytokine released in response to various neural insults or injury (Merrill and Benveniste, 1996), has been proposed to be a key initiator for pain hypersensitivity (Wagner and Myers, 1996; Sorkin and Doom, 2000; Zimmermann, 2001). Tumor necrosis factor receptor-associated factors (TRAFs) are adaptor molecules that couple with TNF-α. Previous studies have revealed that TRAFs convey signals from cell-surface TNF-α receptors to downstream signaling cascades (Y. Wang et al., 2010) and hence contribute to the development of neuropathic pain (Lu et al., 2014). Among the well characterized TRAF1–TRAF7 proteins (Inoue et al., 2000; Xu et al., 2004), TRAF2 acts as a crucial mediator of TNF-α-associated inflammatory signaling (Hong et al., 2007). Strikingly, TNIK was identified originally as a kinase that interacts with TRAF2 and Nck (Fu et al., 1999), and a recent study revealed that TNIK functions as a downstream molecule of TRAF2 (Schürch et al., 2012). Thus, these several observations prompted us to test whether spinal TNF-α contributes to pain-associated neural plasticity by regulating the TRFA2/TNIK cascade and its downstream GluR1–AMPAR trafficking in the spinal cord.
By ubiquitinating and subsequently degrading TRAF proteins, F-box and leucine-rich repeat protein 2 (Fbxl2), an E3-ubiquitin ligase subunit, crucially inhibits TRAF function (Chen et al., 2013). Recently, studies have shown that F-box protein 3 (Fbxo3), another E3-ubiquitin ligase subunit, potently stimulates cytokine secretion from monocytes by destabilizing the sentinel TRAF inhibitor Fbxl2 (Chen et al., 2013; Mallampalli et al., 2013). Conversely, administration of an Fbxo3-targeting inhibitor, which prevents Skp–Cullin 1–Fbxo3-catalyzed Fbxl2 ubiquitination (Chen et al., 2013), sufficiently ameliorated cytokine-driven inflammation in H1N1 influenza-induced lung injury, dextran sulfate sodium-induced colitis, and tissue plasminogen activator-induced edema (Mallampalli et al., 2013). Considering that these studies indicate a cascade of Fbxo3-dependent activation of TRAF via destabilization of Fbxl2, in this study, we determined whether this signaling contributes to the development of neuropathic pain by affecting the spinal TRAF2/TNIK/GluR1–AMPAR trafficking pathway and whether TNF-α contributes to neuropathic pain by activating spinal Fbxo3.
To the best of our knowledge, our results provide, for the first time, a mechanistic basis supporting the role of spinal TNIK in the development of neuropathic pain. This pathway involves activity-dependent TNIK expression triggering subcellular GluR1–AMPAR trafficking in dorsal horn neurons, which is activated by the TNF-α cooperative action of TRAF2 via TNF-α/Fbxo3/Fbxl2/TRAF2 signaling. Our findings provide a new strategy for the development of potential medical interventions for neuropathic pain relief.
Materials and Methods
Animals.
Adult male Sprague Dawley rats (180–230 g) were used throughout this study. All rats were housed in groups of three to four in per cage at a temperature of 22 ± 1°C with a 12 h light/dark cycle and were fed food and water ad libitum. All of the animal procedures in this study were conducted in accordance with the guidelines of the International Association for the Study of Pain (Zimmermann, 1983) and were reviewed and approved by the Institutional Review Board of Taipei Medical University (Taipei, Taiwan).
Anesthesia.
For surgical procedures, animals were first anesthetized using 5% isoflurane delivered in a mix of oxygen (50%) and air (50%) for the rapid induction of anesthesia, and then the concentration of isoflurane was reduced to 2% to maintain stable anesthesia. During surgery, the respiratory rate and the color of the plantar paw surface were observed closely to monitor adequate anesthesia.
Spinal nerve ligation.
We surgically induced neuropathic pain in rats using a previously described method (Peng et al., 2012a). Under isoflurane anesthesia (induction 5%, maintenance 2%), a small incision on the left at L4–S1 was performed. The paraspinal muscle was removed from the L5 and L6 transverse processes, and these transverse process components were removed to expose the parallel-lying L5 and L6 spinal nerves. The left L5 and L6 spinal nerves were then isolated carefully and ligated with 6-0 silk sutures 2–5 mm distal to the dorsal root ganglia. Next, the wound and surrounding skin were sutured with 5-0 silk. The procedures for the sham operation were identical to the nerve ligation group, with the exception that the silk sutures were left unligated. Immediately after surgery, animals were housed individually in a separate cage until they completely recover from anesthesia (for minutes but <1 h) to avoid possible injury caused by other rats.
Intrathecal catheter.
After anesthesia, the occipital crest of the skull was exposed, and the atlanto-occipital membrane was incised at the midline with the tip of an 18-gauge needle. A polyethylene-10 catheter was inserted through the slit and passed caudally to the dorsal arachnoid space at the L4–L5 level. Once in place, 20–30 μl of saline was injected through the catheter to clear the tip of any possible blood or tissue. The length of the external catheter was trimmed to ∼2 cm that was enough to grasp the end for injection or infusion, and a small length of 28 gauge stainless-steel wire was used to plug the catheter. Animals were allowed to recover for 3 d after implantation. Rats with neurological deficits after surgery were then killed and excluded from the statistical analyses (Peng et al., 2012a).
Behavioral studies.
To assess mechanical sensitivity, rats were placed individually in an opaque plastic cylinder that was placed on a wire mesh, and the animals were allowed to habituate for 30 min to allow acclimatization to their new environment. Next, von Frey monofilaments (Stoelting) were applied to the plantar surface of the hindpaws of different groups to measure the paw-withdrawal threshold. In addition, the animals' tactile thresholds before the operation were set at 15 g (Peng et al., 2012a). At 1 d before sham operation or spinal nerve ligation (SNL), tactile threshold was measured in animals using von Frey hair. Because Chung et al. (2004) and our previous studies (Lin et al., 2015a,b) have showed the mean withdrawal threshold of rats are ∼15 g, rats that displayed thresholds <15 g (which could be more sensitive to painful stimulation) and exhibited no withdrawal when the hair of 15 g was applied (which could be insensitive to painful stimulation) were excluded from additional experiments. Before various biochemical and immunohistochemical analyses, rats' tactile threshold was measured at the time point of either sham operation or SNL to confirm that SNL animals displayed allodynia but sham-operated animals did not. In some animals, the motor function was assessed using an accelerating rotarod apparatus (LE8500; Ugo Basile). For acclimatization, the animals were subjected to three training trials with 304 h intervals on 2 independent days. In the training sessions, the rod was set to accelerate from 3 to 30 rpm over a 180 s period. In the test session, the performance times of the rats were recorded up to a cutoff time of 180 s. Three measurements performed with 5 min intervals were averaged for each test (Hori et al., 2010).
Western blotting analyses.
Rats were anesthetized deeply, and the L4–L5 spinal cords were removed immediately. The dissected sample was homogenized in 25 mm Tris-HCl, 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS with a complete protease inhibitor mixture (Roche). After 1 h of incubation on ice, the lysates were centrifuged (14,000 rpm, 20 min, 4°C), and the supernatant was collected for Western blotting (WB) analysis. The protein concentrations were determined using a bicinchoninic acid assay. Briefly, the supernatant was subjected to separation using acrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes. After blocking with nonfat milk (5%), the membranes were incubated with primary antibodies: mouse anti-TNIK (1:500; Santa Cruz Biotechnology), rabbit anti-GluR1 antibody (1:1000; Millipore), rabbit anti-phosphorylated GluR1 (pGluR1) antibody (pS845; 1:1000; Invitrogen), mouse anti-TRAF2 antibody (1:500; Santa Cruz Biotechnology), goat anti-fbxl2 antibody (1:250; Santa Cruz Biotechnology), rabbit anti-fbxo3 antibody (1:500; Santa Cruz Biotechnology), and mouse anti-ubiquitin antibody (1:1000; Santa Cruz Biotechnology). The membranes were then washed and incubated with an HRP-conjugated secondary antibody (1:8000; Jackson ImmunoResearch). The blots were visualized using an enhanced chemiluminescence detection kit (ECL Plus; Millipore), and densitometric analysis of the Western blot membranes was performed using Science Lab 2003 (Fuji). The relative values of each protein were normalized against either GAPDH (mouse, 1:4000; Genetex) or N-cadherin (mouse, 1:2000; Thermo Fisher Scientific) as the loading control.
Coprecipitation studies.
Extractions of dorsal horn samples were incubated with a rabbit polyclonal antibody against total GluR1 (tGluR1) overnight at 4°C. The 1:1 protein agarose suspension (Millipore) slurry was added to the protein immunocomplex, and the mixture was incubated at 4°C for 2–3 h. Agarose beads were washed once with 1% (v/v) Triton X-100 in immunoprecipitation (IP) buffer [50 mm Tris-Cl, pH 7.4, 5 mm EDTA, and 0.02% (w/v) sodium azide], twice with 1% (v/v) Triton X-100 in IP buffer plus 300 mm NaCl, and three times with IP buffer only. Bound proteins were eluted with SDS-PAGE sample buffer at 95°C. The proteins were then separated using SDS-PAGE, electrophoretically transferred onto polyvinylidene difluoride membranes, and detected using a mouse anti-TNIK antibody (1:500; Santa Cruz Biotechnology), rabbit anti- GluR1 antibody (1:1000; Millipore), rabbit anti-pGluR1 antibody (pS845; 1:1000; Invitrogen), and mouse anti-TRAF2 antibody (1:500; Santa Cruz Technology).
Immunofluorescence.
Rats were anesthetized deeply and perfused transcardially with PBS, followed by ice-cold 4% paraformaldehyde/PBS. The L4–L5 segment of the lumbar spinal cord was removed, postfixed in the same fixative (4°C for 4 h), and placed in a 30% sucrose solution for 24 h at 4°C. Transverse spinal cord sections (30 μm) were incubated for 24 h at 4°C with primary antibody [TNIK, mouse, 1:200 (Santa Cruz Biotechnology); pGluR1, rabbit, 1:1000 (pS845; Invitrogen); or TRAF2, mouse, 1:500 (Santa Cruz Biotechnology)]. Identification of cell types was performed using the following markers: rabbit anti-neuronal nuclear antigen (NeuN; a neuronal marker, 1:500; Abcam), rabbit anti-glial fibrillary acidic protein (GFAP; a marker of astroglial cells; 1:1000; Abcam), and rabbit anti-integrin αM (OX-42; a marker of microglia; 1:500; Biobyt). Next, the sections were incubated (1 h, 37°C) with secondary antibodies conjugated to Alexa Fluor 488 (1:1500) and Alexa Fluor 594 (1:1500; Invitrogen). Mix-n-Stain (Biotium) direct immunofluorescence staining was performed to eliminate the need for the secondary antibody incubation and wash steps and to allow the use of multiple primary antibodies from the same species for multicolor detection or for staining of animal tissues with antibodies raised in the same species without secondary antibody cross-reactivity. The spinal sections were subsequently rinsed in PBS, and coverslips were applied. When these fluorescent markers were excited, they were detected easily by a camera-coupled device (X-plorer; Diagnostic Instruments) through a fluorescent microscope (DM2500; Leica). Seven sections from the neuropathic or sham-operated spinal cord were used for cell counting in each rat. Cell counting was performed under a microscope at 200× magnification (Lin et al., 2015b).
Intrathecal injection of small-interfering RNA.
The 19 nt duplexes of the small-interfering RNAs (siRNAs) for TNIK were 5′-CAUAUUCAGGGUAAUAUCA-3′, and the missense nucleotides were 5′-UGAUAUUACCCUGAAUAUG-3′. The missense or siRNA with a polyethyleneimine (10 μl, 25 kDa; Sigma)-based gene-delivery system was injected intrathecally into the dorsal subarachnoid space (L4–L5) of animals daily for 4 d through the implanted catheter.
Drug application.
BC-1215 (N1,N2-Bis[[4-(2-pyridinyl)phenyl]methyl]-1,2-ethanediamine) (a novel inhibitor of Fbxo3 activity; 10, 30, and 100 nm, 10 μl; Merck), TNF-α-neutralizing antibody (10, 30, and 100 ng, 10 μl; R&D Systems), or TNF-α (1 pm, 10 μl; Sigma) was administered intrathecally by bolus injection. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX; a glutamatergic AMPAR antagonist; 10 μm, 10 μl; Sigma-Aldrich) was administered intrathecally daily for 4 d from day 3 to day 6 after SNL or by bolus injection (1, 3, and 10 μm, 10 μl). A vehicle solution of equal volume to that of the tested agents was dispensed to serve as a control.
Data analysis.
All of the data in this study were analyzed using SigmaPlot 10.0 (Systat Software) or Prism 6.0 (GraphPad Software) and are expressed as the mean ± SEM. Statistical comparison was performed by one-way ANOVA (using Tukey's tests for post hoc analysis), two-way ANOVA (using Tukey's tests for post hoc analysis), or paired Student's t test when appropriate. Significance was set at p < 0.05. In experiments exploring the effects of treatments on SNL-induced protein expression (WB), interaction (IP), and location [immunohistochemistry (IHC)], one-way ANOVA was first used to compare the statistical difference among groups. In case the difference was significant, Tukey's test was used to examine the difference between sham-operated and SNL animals. If the value of SNL is statistically different from the sham-operated animals, then Tukey's test was used to compare SNL animals with SNL animals that received various treatments.
Results
SNL induces pain-associated dorsal horn TNIK expression
As the first to address whether spinal TNIK plays a role in neuropathic pain, TNIK expression in the dorsal horn (L4–L5) after SNL was examined. SNL increased significantly the abundance of TNIK in the ipsilateral, but not the contralateral, dorsal horn at days 3, 7, 14, and 21 after surgery (0.42 ± 0.03, 0.58 ± 0.05, 0.52 ± 0.03, and 0.48 ± 0.05, respectively; n = 6; Fig. 1A). SNL-enhanced TNIK expression was aligned temporally with tactile allodynia, as shown by significant decreases in the withdrawal threshold of the ipsilateral hindpaw at days 3, 7, 14, and 21 after surgery (1.28 ± 0.48, 0.34 ± 0.13, 0.23 ± 0.08, and 0.28 ± 0.07 g, respectively; n = 7; Fig. 1B). At day 7 after surgery, a time point in which SNL provoked maximal allodynia and TNIK expression, IHC analysis of the spinal cord slices (L4–L5) showed that SNL increased TNIK immunofluorescence in the ipsilateral dorsal horn (159.86 ± 21.03 neurons; n = 7; Fig. 1C). Double-labeled staining revealed TNIK colocalization with neuronal (NeuN), but not microglial (OX-42) or astrocytic (GFAP), markers. Together, these results suggest that SNL enhances TNIK expression in ipsilateral dorsal horn neurons associated with the development of neuropathic pain.
Figure 1.

SNL upregulates TNIK expression in dorsal horn neurons. A, Representative WB and statistical analyses (normalized to GAPDH) revealing SNL-increased TNIK expression in the ipsilateral (I and IPSI), but not contralateral (C and CONTRA), dorsal horn. Two-way ANOVA with repeated measures over time: treatment, F(3,20) = 45.8, p = 0.002; time, F(4,80) = 4.672, p < 0.001; and treatment × time, F(12,80) = 5.322, p < 0.001. **p < 0.01 versus Sham IPSI; ##p < 0.01 versus SNL day 1. n = 6. IB, Immunoblotting. B, Time course of SNL-reduced withdrawal threshold of the hindpaw (von Frey test). Two-way ANOVA with repeated measures over time: treatment, F(3,24) = 56.39, p < 0.001; time, F(5,120) = 2.912, p = 0.016; and treatment × time, F(15,120) = 4.282, p < 0.001. **p < 0.01 versus Sham IPSI; ##p < 0.01 versus day 1. n = 7. C, Images and statistical analysis at day 7 after surgery revealing SNL-increased TNIK immunofluorescence (red) in the ipsilateral dorsal horn (SNL 7D), which is colocalized with neuronal (NeuN, green), but not microglial (OX-42, green) or astrocytic (GFAP, green), markers. One-way ANOVA, post hoc Tukey's test: F(3,24) = 24.68, p < 0.001. **p < 0.01 versus Sham IPSI; ##p < 0.01 versus SNL CONTRA. n = 7. Scale bar, 50 μm.
Knockdown of TNIK expression alleviates SNL-induced allodynia
To further confirm the role of TNIK in neuropathic pain hypersensitivity, we examined whether the lack of spinal TNIK affects the development of SNL-induced allodynia. An intrathecal injection of TNIK mRNA-targeting siRNA (1, 3, and 5 μg, 10 μl, once daily for 4 d), but not missense siRNA (5 μg, 10 μl) or polyethylenimine (a transfection reagent, 10 μl), decreased the abundance of TNIK in dorsal horn samples (0.35 ± 0.06, 0.15 ± 0.03, and 0.11 ± 0.02, respectively; n = 6; Fig. 2A) in a dose-dependent manner, indicating that spinal TNIK expression was sufficiently knocked down by specific siRNAs. In the rotarod performance test, which is used frequently to evaluate rodent motor coordination, we found no significant difference between naive and polyethylenimine-, missense siRNA (5 μg, 10 μl)-, or TNIK mRNA-targeting siRNA (5 μg, 10 μl)-treated animals (Fig. 2B), indicating that neither our procedures nor spinal TNIK knockdown resulted in motor deficits in rats. Moreover, although it exhibited no effect on the withdrawal threshold in the sham-operated animals (Fig. 2C), administration of TNIK mRNA-targeting siRNA (5 μg, 10 μl) partially ameliorated SNL-induced behavioral allodynia by significantly increasing the withdrawal threshold at days 5 and 7 after surgery (5.91 ± 1.12 and 6.86 ± 0.86 g, respectively; n = 7; Fig. 2D). Together, the data obtained from the siRNA-treated rats suggested that spinal TNIK contributes significantly to SNL-associated pain hypersensitivity.
Figure 2.

Knockdown of spinal TNIK expression relieves SNL-induced allodynia. A, Representative WB and statistical analyses (normalized to GAPDH) demonstrating intrathecal administration with TNIK mRNA-targeting siRNA (TNIK RNAi; 1, 3, and 5 μg, 10 μl), but not missense siRNA (TNIK MS; 5 μg, 10 μl) or polyethylenimine (PEI; 10 μl), dose dependently decreased spinal TNIK expression. One-way ANOVA, post hoc Tukey's test: F(6,35) = 12.90, p < 0.001. **p < 0.01 versus Naive. n = 6. IB, Immunoblotting; it, implantation of an intrathecal catheter. B, Intrathecal TNIK mRNA-targeting siRNA (TNIK RNAi; 5 μg, 10 μl) resulted in no motor deficits in the animals (rotarod test). Two-way ANOVA with repeated measures over time: treatment, F(3,24) = 0.482, p = 0.698; time, F(4,96) = 0.379, p = 0.823; treatment × time, F(12,96) = 0.565, p = 0.865. n = 7. C, D, Although it exhibited no effect on that of sham-operated animals, intrathecal TNIK mRNA-targeting siRNA (5 μg, 10 μl) increased the withdrawal threshold of SNL animals at days 5 and 7 after surgery (von Frey test). In sham-operated animals, two-way ANOVA with repeated measures over time: treatment, F(3,24) = 0.059, p = 0.981; time, F(4,96) = 0.572, p = 0.683; treatment × time, F(12,96) = 0.091, p > 0.999. n = 7; In SNL animals, two-way ANOVA with repeated measures over time: treatment, F(3,24) = 21.26, p < 0.001; time, F(4,96) = 598.8, p < 0.001; treatment × time, F(12,96) = 11.19, p < 0.001. **p < 0.01 versus SNL. n = 7.
SNL consecutively provoked spinal GluR1 coupling with TNIK, phosphorylation, and trafficking
TNIK, as a serine/threonine kinase and a scaffold domain, has been shown recently to regulate cell-surface GluR1–AMPAR trafficking (Hussain et al., 2010). Our laboratory has shown previously that both serine/threonine kinases (Peng et al., 2012b) and postsynaptic scaffolding proteins (Peng et al., 2011) drive the phosphorylation-dependent subcellular trafficking of spinal GluR1–AMPARs to underlie the pain-associated central sensitization. Thus, we hypothesized that SNL-enhanced TNIK expression provokes spinal GluR1 phosphorylation and subsequent trafficking. We found that SNL significantly increased the abundance of GluR1, but not tGluR1, in the ipsilateral dorsal horn samples at days 3 and 7 after the operation (0.58 ± 0.05 and 0.62 ± 0.04, respectively; n = 6; Fig. 3A). These time points are presumed to reflect the induction (W. Wang et al., 2011) and maintenance (Lee et al., 2011) phases of neuropathic pain, respectively. Furthermore, SNL also increased the amount of membrane-bound GluR1 [GluR1(m)] in the ipsilateral dorsal horn sample at the identical time points (0.69 ± 0.05 and 0.70 ± 0.05, respectively; n = 6), suggesting that the phosphorylation-dependent subcellular trafficking of spinal GluR1–AMPARs plays a crucial role in SNL-induced neuropathic pain. Strikingly, we found that daily administration of TNIK mRNA-targeting siRNA (5 μg, 10 μl) significantly and quantitatively reversed SNL-enhanced abundances of pGluR1 (Fig. 3B; from 0.74 ± 0.06 to 0.44 ± 0.07, n = 6) and GluR1(m) (Fig. 3C; from 0.71 ± 0.07 to 0.47 ± 0.04, n = 6) in the ipsilateral dorsal horn at day 7 after surgery. Based on these observations, we next assayed the interaction between tGluR1, pGluR1, and TNIK in the ipsilateral dorsal horn at day 7 after surgery and the effect of TNIK knockdown on their interactions. Compared with the sham-operated group, SNL increased the abundance of tGluR1-bound TNIK and pGluR1 in the tGluR1 antibody-bound precipitates [IP: tGluR1, ∼1.5- and 2.2-fold above the 7 d sham group (Sham 7D), respectively], which were both attenuated by daily administration of TNIK mRNA-targeting siRNA [Fig. 3D; 5 μg, 10 μl; ∼0.2 and 0.4-fold of 7 d SNL group (SNL 7D), respectively]. To further determine the upstream and downstream relationship between TNIK and GluR1, we intrathecally injected the selective AMPAR antagonist CNQX daily for 4 d (from day 3 to day 6 after SNL) to allodynic animals. We found that administration with neither CNQX (10 μm, 10 μl) nor a vehicle solution (10 μl) affected SNL-upregulated spinal TNIK expression (Fig. 3E). Nevertheless, a bolus of CNQX (1, 3, and 10 μm, 10 μl), but not a vehicle solution (10 μl), dose dependently ameliorated SNL-induced behavioral allodynia, as shown by the significant increase in the withdrawal threshold of the ipsilateral hindpaw at 1–4 h after injection (Fig. 3F; 1 μm, 3.29 ± 0.64, 3.14 ± 0.59, 2.42 ± 0.43, and 1.69 ± 0.41 g; 3 μm, 2.77 ± 0.44, 4.86 ± 0.59, 4.86 ± 0.86, and 1.91 ± 0.39 g; 10 μm, 5.14 ± 0.74, 7.43 ± 0.72, 5.14 ± 0.74, and 3.14 ± 0.40 g). In addition, SNL significantly enhanced the number of TNIK-positive, pGluR1-positive, and TNIK/pGluR1 double-labeled neurons at day 7 after surgery in the ipsilateral dorsal horn, which were all reversed by knockdown of spinal TNIK expression (Fig. 4A; 5 μg, 10 μl; from 154.0 ± 18.37 to 61.14 ± 19.28, from 122.28 ± 21.82 to 40.57 ± 11.86, and from 65.14 ± 11.51 to 22.14 ± 6.34 neurons, respectively; n = 7). Together, these results indicated SNL upregulated TNIK expression, which results in TNIK–GluR1 coupling and subsequent phosphorylation-dependent GluR1–AMPAR trafficking toward the plasma membrane in dorsal horn neurons.
Figure 3.
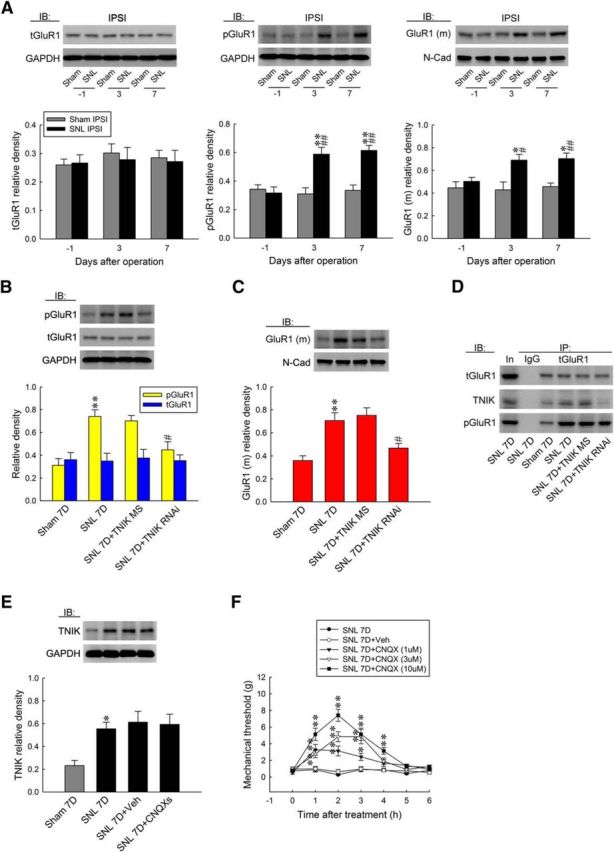
SNL provokes spinal TNIK–GluR1 coupling and subsequent phosphorylation-dependent GluR1–AMPAR trafficking. A, Representative WB and statistical analyses (normalized to GAPDH or N-cadherin) demonstrating SNL-increased pGluR1 and GluR1(m), but not tGluR1 expression, in the ipsilateral (IPSI) dorsal horn. One-way ANOVA, post hoc Tukey's test: pGluR1, F(2,15) = 16.03, p < 0.001; GluR1(m), F(2,15) = 5.965, p = 0.012; tGluR1, F(2,15) = 0.024, p = 0.976. *p < 0.05, **p < 0.01 versus Sham IPSI. #p < 0.05, ##p < 0.01 versus SNL day 1. n = 6. IB, Immunoblotting. B, C, Intrathecal TNIK mRNA-targeting siRNA (SNL 7D+TNIK RNAi, 5 μg, 10 μl), but not missense small-interfering RNA (TNIK MS, 10 μl), reduced SNL-enhanced pGluR1 and GluR1(m) expression. However, both conditions exhibited no effect on tGluR1 expression. One-way ANOVA, post hoc Tukey's test; pGluR1, F(3,20) = 10.36, p < 0.001; GluR1(m), F(3,20) = 11.46, p < 0.001; tGluR1, F(3,20) = 0.285, p = 0.836. **p < 0.01 versus Sham 7D. #p < 0.05 versus SNL 7D. n = 6. D, SNL increased the levels of tGluR1–TNIK and pGluR1, which were both reduced by intrathecal administration with TNIK mRNA-targeting siRNA. E, Administration with neither CNQX (daily for 4 d from day 3 to day 6 after SNL, 10 μm, 10 μl) nor vehicle (Veh; 10 μl) affected SNL-upregulated spinal TNIK expression. One-way ANOVA, post hoc Tukey's test: F(3,20) = 5.747, p = 0.005 F, Intrathecal administration with CNQX (SNL 7D+CNQX; 1, 3, and 10 nm, 10 μl), but not the vehicle solution (SNL 7D+Veh, 10 μl), increased the withdrawal threshold of the ipsilateral hindpaw. Two-way ANOVA with repeated measures over time: treatment, F(4,30) = 93.76, p < 0.001; time, F(6,180) = 34.15, p < 0.001; treatment × time, F(24,180) = 8.539, p < 0.001. *p < 0.05, **p < 0.01 versus SNL 7D. n = 7.
Figure 4.

Knockdown of spinal TNIK expression reduces SNL-enhanced TNIK, pGluR1, and TNIK/pGluR1 colocalization in the dorsal horn. A, Images and statistical analyses at day 7 after surgery, SNL increased TNIK (red), pGluR1 (green), and TNIK/pGluR1 double-labeled (yellow) immunofluorescence in the ipsilateral dorsal horn (SNL 7D), which were all reduced by intrathecal administration with TNIK mRNA-targeting siRNA (SNL 7D+TNIK RNAi, 5 μg, 10 μl). One-way ANOVA, post hoc Tukey's test: TNIK, F(2,18) = 11.60, p < 0.001; pGluR1, F(2,18) = 27.18, p < 0.001; TNIK/pGluR1, F(2,18) = 32.45, p < 0.001. **p < 0.01 versus Sham 7D. ##p < 0.01 versus SNL 7D. n = 7. Scale bar, 50 μm.
SNL provoked spinal TRAF2/TNIK/pGluR1-dependent GluR1 trafficking
TNIK was identified originally as a TRAF2- and Nck-interacting kinase (Fu et al., 1999), and a recent study revealed that TNIK acts as a downstream molecule of TRAF2 (Schürch et al., 2012). Thus, we investigated whether spinal TRAF2 contributes to the development of neuropathic pain via TNIK-dependent GluR1–AMPAR trafficking. WB analyses revealed that SNL increased the amount of TRAF2 in the ipsilateral dorsal horn at days 3 and 7 after surgery (Fig. 5A; 0.51 ± 0.06 and 0.59 ± 0.07, respectively; n = 6). At day 7 after surgery, administration with neither TNIK mRNA-targeting siRNA (5 μg, 10 μl) nor missense siRNA (5 μg, 10 μl) affected SNL-upregulated spinal TRAF2 expression (Fig. 5B). Coprecipitation analyses showed that SNL increased the amount of tGluR1-bound TRAF2, TNIK, and pGluR1 (IP: tGluR1, ∼2.4-, ∼1.8- and 2.1-fold above Sham 7D, respectively), which were all attenuated by the knockdown of spinal TNIK expression (Fig. 5C; ∼0.6, ∼0.2, and 0.5-fold of SNL 7D, respectively). Moreover, SNL significantly increased the number of TRAF2-, TNIK-, and pGluR1-positive neurons and TRAF2/TNIK/pGluR1 triple-labeled neurons in the ipsilateral dorsal horn (111.42 ± 13.60, 130.43 ± 17.15, 132.57 ± 13.34, and 74.14 ± 9.23, respectively; n = 7). Although knockdown of spinal TNIK expression did not affect the number of TRAF2-positive neurons, it did significantly reduce the number of SNL-enhanced TNIK-positive, pGluR1-positive, and TRAF2/TNIK/pGluR1 triple-labeled neurons (Fig. 5D; from 130.43 ± 17.15 to 61.00 ± 10.77, from 132.57 ± 13.34 to 65.71 ± 8.43, and from 74.14 ± 9.23 to 29.14 ± 5.62 neurons, respectively; n = 7). Together, these results indicate a crucial role for spinal TRAF2 in mediating neuropathic pain via downstream TNIK-dependent GluR1–AMPAR phosphorylation and trafficking.
Figure 5.
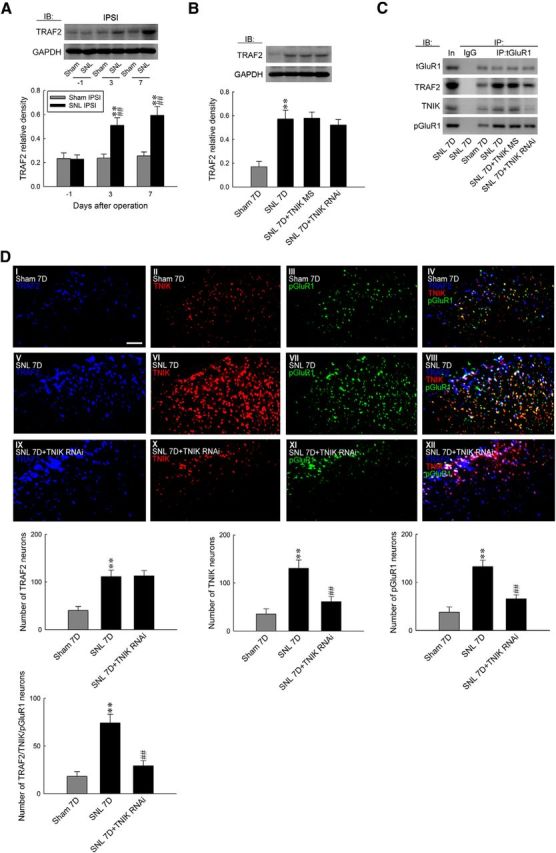
SNL induces spinal TRAF2/TNIK/pGluR1-dependent GluR1–AMPAR trafficking. A, Representative WB and statistical analyses (normalized to GAPDH) demonstrating SNL increased TRAF2 expression in the ipsilateral (IPSI) dorsal horn. One-way ANOVA, post hoc Tukey's test: F(2,15) = 14.97, p < 0.001. **p < 0.01 versus Sham IPSI. ##p < 0.01 versus SNL day 1. n = 6. B, Administration with neither TNIK mRNA-targeting siRNA (SNL 7D+TNIK RNAi, 5 μg, 10 μl) nor missense siRNA (SNL 7D+TNIK MS, 5 μg, 10 μl) affected SNL-enhanced spinal TRAF2 expression at day 7 after surgery (normalized to GAPDH). One-way ANOVA, post hoc Tukey's test: F(3,20) = 12.83, p < 0.001. **p < 0.01 versus Sham 7D. n = 6. C, SNL increased spinal tGluR1–TRAF2/TNIK/pGluR1 coprecipitations, which were reduced with TNIK mRNA-targeting siRNA treatment. D, Images and statistical analyses demonstrated that SNL increased TRAF2 (blue), TNIK (red), pGluR1 (green), and TRAF2/TNIK/pGluR1 triple-labeled (white) immunofluorescence in the ipsilateral dorsal horn. TNIK mRNA-targeting siRNA treatment reduced SNL-enhanced TNIK (red), pGluR1 (green), and TRAF2/TNIK/pGluR1 triple-labeled (white), but not TRAF2 (blue), immunofluorescence. One-way ANOVA, post hoc Tukey's test: TRAF2, F(2,18) = 14.08, p < 0.001; TNIK, F(2,18) = 13.87, p < 0.001; pGluR1, F(2,18) = 19.62, p < 0.001; TRAF2/TNIK/pGluR1, F(2,18) = 18.58, p < 0.001. **p < 0.01 versus Sham 7D. ##p < 0.01 versus SNL 7D. n = 7. Scale bar, 50 μm.
BC-1215 relieves allodynia by preventing ubiquitination-dependent Fbxl2/TRAF2/TNIK/pGluR1 signaling
Two F-box proteins, Fbxo3 and Fbxl2, were demonstrated recently to regulate inflammation-associated TRAF adaptor stability (Chen et al., 2013). Thus, we investigated the potential contribution of these proteins in neuropathic pain development by assaying spinal Fbxl2 and Fbxo3 expression in response to SNL. Interestingly, we observed that SNL significantly increased Fbxo3 but decreased Fbxl2 abundance in the ipsilateral dorsal horn sample at days 3 and 7 after surgery (Fig. 6A; Fbxo3, 0.53 ± 0.04 and 0.60 ± 0.07, respectively, n = 6; Fbxl2, 0.23 ± 0.04 and 0.17 ± 0.02, respectively, n = 6). After observing SNL-enhanced Fbxo3 expression, we determined whether the intrathecal application of BC-1215, a novel inhibitor of Fbxo3 activity, could modify SNL-induced pain behavior. The results of the von Frey test demonstrated that a bolus of BC-1215 (10, 30, and 100 nm, 10 μl), but not vehicle solution (10 μl), dose dependently ameliorated SNL-induced behavioral allodynia, as shown by the significant increase in the withdrawal threshold of the ipsilateral hindpaw at 2–6 h after injection (Fig. 6B; 10 nm BC-1215, 0.78 ± 0.27, 1.97 ± 0.41, 2.40 ± 0.43, 1.63 ± 0.18, and 1.05 ± 0.29 g; 30 nm BC-1215, 1.60 ± 0.15, 3.73 ± 1.00, 3.71 ± 0.80, 2.86 ± 0.40, and 2.29 ± 0.47 g; 100 nm BC-1215, 2.34 ± 0.45, 6.57 ± 0.72, 6.29 ± 1.11, 4.29 ± 0.81, and 2.71 ± 0.47 g, respectively). Nevertheless, application of neither the vehicle solution nor BC-1215 (100 nm, 10 μl) affected the withdrawal threshold of the contralateral hindpaw at 3 h after injection, a time point at which BC-1215 exhibited its maximal analgesic effect on the ipsilateral side (Fig. 6C). Based on these results and considering that Fbxo3 catalyzes the ubiquitination of Fbxl2 (Chen et al., 2013), which ubiquitinates and inhibits TRAFs (Mallampalli et al., 2013), we speculated that BC-1215 exerted its analgesic effect by preventing Fbxo3-dependent Fbxl2 ubiquitination, thereby enhancing TRAF2 ubiquitination and degradation. At day 7 after surgery, SNL predictably increased Fbxl2 ubiquitination in the ipsilateral dorsal horn, which was markedly reversed by an intrathecal BC-1215 injection (Fig. 6D; 100 nm, 10 μl). Moreover, treatment of SNL animals with BC-1215 significantly increased TRAF2 ubiquitination compared with the untreated SNL group. These results suggest that SNL-associated Fbxo3 expression participates in neuropathic pain development by ubiquitinating Fbxl2, thereby decreasing the Fbxl2-dependent ubiquitination of spinal TRAF2. When examining the effect of BC-1215 (100 nm, 10 μl) on protein expression, we observed that it reversed SNL-enhanced TRAF2 (from 0.59 ± 0.04 to 0.26 ± 0.02; n = 6), TNIK (from 0.55 ± 0.03 to 0.35 ± 0.03; n = 6), pGluR1 (from 0.75 ± 0.05 to 0.41 ± 0.03; n = 6), and GluR1(m) expression (from 0.78 ± 0.03 to 0.42 ± 0.04; n = 6) and SNL-decreased Fbxl2 expression (from 0.17 ± 0.01 to 0.38 ± 0.02; n = 6) without affecting the abundance of tGluR1 or Fbxo3 (Fig. 6E,F). Importantly, administration of neither TNIK mRNA-targeting siRNA (5 μg, 10 μl, daily for 4 d) nor missense siRNA (10 μl) affected the SNL-upregulated Fbxo3 or SNL-downregulated Fbxl2 expression (Fig. 6G). Moreover, SNL enhanced the abundance of tGluR1-bound TRAF2, TNIK, and pGluR1 (IP: tGluR1, ∼1.9-, ∼2.5-, and 2.1-fold above Sham 7D, respectively), which were all reduced by a spinal injection of BC-1215 (Fig. 6H; ∼0.5, ∼0.3, and 0.6-fold of SNL 7D, respectively). Furthermore, immunofluorescence studies revealed that a bolus injection of BC-1215 significantly reversed the number of SNL-enhanced TRAF2-positive (from 186.85 ± 14.18 to 49.42 ± 9.65 neurons; n = 7), TNIK-positive (from 150.28 ± 18.93 to 49.28 ± 12.03 neurons; n = 7), pGluR1-positive (from 175.85 ± 19.42 to 64.00 ± 10.81 neurons; n = 7), and TRAF2/TNIK/pGluR1 triple-labeled (from 55.28 ± 6.88 to 15.71 ± 5.75 neurons; n = 7) neurons in the ipsilateral dorsal horn (Fig. 7A). Together, these data suggest that SNL-induced Fbxo3 expression can activate TRAF2/TNIK/pGluR1 signaling and the subsequent GluR1 trafficking pathway via ubiquitin-dependent degradation of Fbxl2.
Figure 6.
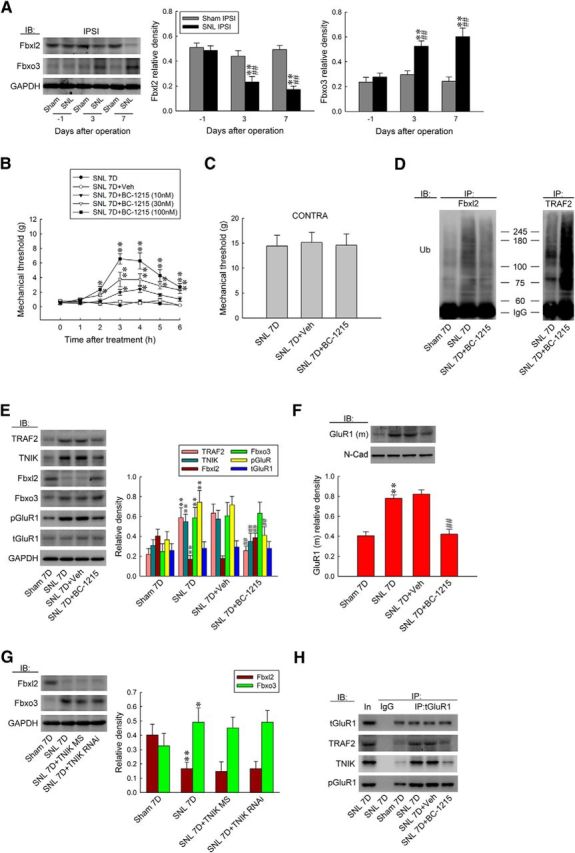
BC-1215 relieves allodynia by preventing ubiquitination-dependent spinal Fbxl2/TRAF2/TNIK/pGluR1 signaling. A, Representative WB and statistical analyses (normalized to GAPDH) demonstrating SNL decreased Fbxl2 but increased Fbxo3 abundance in the ipsilateral (IPSI) dorsal horn. One-way ANOVA, post hoc Tukey's test: Fbxl2, F(2,15) = 20.63, p < 0.001; Fbxo3, F(2,15) = 11.21, p = 0.001. **p < 0.01 versus Sham. ##p < 0.01 versus SNL day 1. n = 6. B, C, Intrathecal administration with BC-1215 (SNL 7D+BC-1215, 10, 30, and 100 nm, 10 μl), but not the vehicle solution (SNL 7D+Veh, 10 μl), increased the withdrawal threshold of the ipsilateral hindpaw. Nevertheless, both these treatments failed to affect the contralateral hindpaw at 3 h after injection (BC-1215 100 nm; von Frey test). Ipsilateral hindpaw, two-way ANOVA with repeated measures over time: treatment, F(4,30) = 46.98, p < 0.001; time, F(6,180) = 22.40, p < 0.001; treatment × time, F(24,180) = 6.460, p < 0.001. *p < 0.05, **p < 0.01 versus SNL 7D. n = 7. Contralateral hindpaw, one-way ANOVA, post hoc Tukey's test: F(2,18) = 0.031, p = 0.970. D, At day 7 after surgery, SNL increased Fbxl2 ubiquitination in the ipsilateral dorsal horn, which was reversed markedly by spinal injection of BC-1215, which increased TRAF2 ubiquitination. E, F, Without affecting the abundances of tGluR1 and Fbxo3, BC-1215 reversed the SNL-increased TRAF2, TNIK, pGluR1, and GluR1(m) expressions and the SNL-decreased Fbxl2 expression. One-way ANOVA, post hoc Tukey's test: TRAF2, F(3,20) = 48.08, p < 0.001; TNIK, F(3,20) = 18.75, p < 0.001; Fbxl2, F(3,20) = 43.52, p < 0.001; Fbxo3, F(3,20) = 16.80, p < 0.001; pGluR1, F(3,20) = 26.66, p < 0.001; tGluR1, F(3,20) = 0.260, p = 0.854; GluR1(m), F(3,20) = 28.80, p < 0.001. **p < 0.01 versus Sham 7D. ##p < 0.01 versus SNL 7D. n = 6. G, Administration with neither TNIK mRNA-targeting siRNA (SNL 7D+TNIK RNAi, 5 μg, 10 μl) nor missense siRNA (SNL 7D+TNIK MS, 5 μg, 10 μl) affected SNL-upregulated Fbxo3 and SNL-downregulated Fbxl2 expression. One-way ANOVA, post hoc Tukey's test: Fbxl2, F(3,20) = 23.94, p < 0.001; Fbxo3, F(3,20) = 4.992, p = 0.010. *p < 0.05, **p < 0.01 versus Sham 7D. n = 6. H, SNL-enhanced tGluR1-TRAF2/TNIK/pGluR1 precipitations were reduced by spinal injection of BC-1215.
Figure 7.
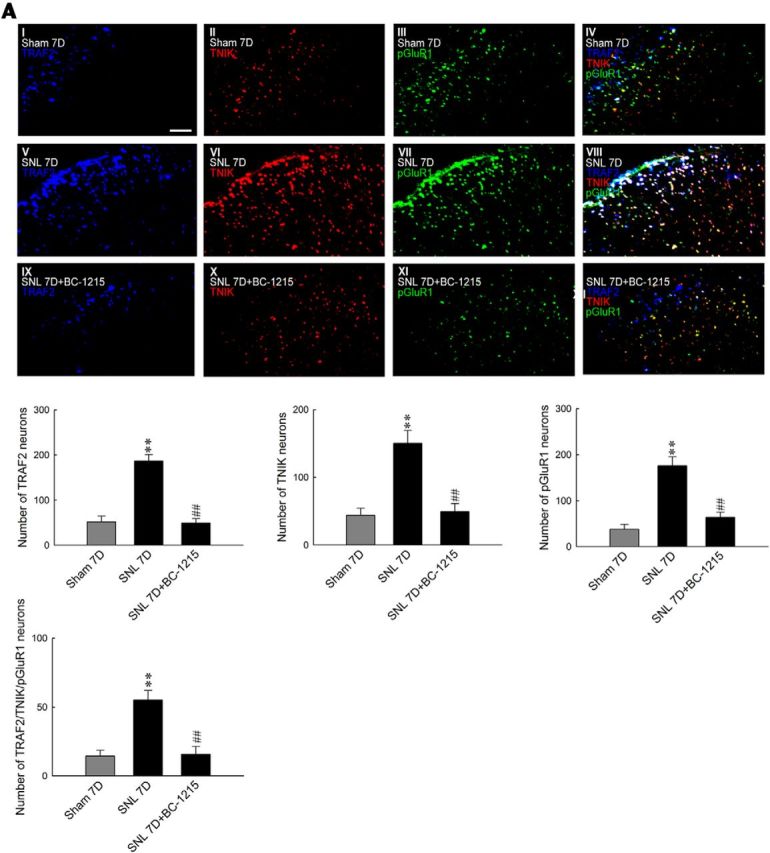
BC-1215 reduces SNL-enhanced TRAF2, TNIK, pGluR1, and triple-labeled immunofluorescence. A, Images and statistical analyses showing SNL enhanced TRAF2 (blue), TNIK (red), pGluR1 (green), and TRAF2/TNIK/pGluR1 triple-labeled (white) immunofluorescence in the ipsilateral dorsal horn at 7 d after surgery, which were all reduced by a spinal BC-1215 injection (SNL 7D+BC-1215, 100 nm, 10 μl). One-way ANOVA, post hoc Tukey's test: TRAF2, F(2,18) = 40.67, p < 0.001; TINK, F(2,18) = 17.53, p < 0.001; pGluR1, F(2,18) = 26.50, p < 0.001; TRAF2/TNIK/pGluR1, F(2,18) = 29.46, p < 0.001. **p < 0.01 versus Sham 7D. ##p < 0.01 versus SNL 7D. n = 7. Scale bar, 50 μm.
TNF-α-neutralizing antibody relieves allodynia via attenuation of Fbxo3/Fbxl2/TRAF2/TNIK/pGluR1-dependent GluR1 trafficking
TNF-α released after nerve injury has been proposed to be an initiator of neuropathic pain (Zimmermann, 2001), and blockade of TNF-α release reduces the pain behavior (Schäfers et al., 2003). To characterize the role of TNF-α in neuropathic pain, we first examined whether antagonizing spinal TNF-α affects SNL-induced allodynia. At day 7 after SNL, intrathecal injections of a TNF-α-neutralizing antibody (SNL 7D + TNF-α Ab; 10, 30, and 100 ng, 10 μl), but not a nonspecific IgG (100 ng, 10 μl), dose dependently increased the withdrawal threshold of the ipsilateral hindpaw at 1–6 h after injection (Fig. 8A; 10 ng, 1.91 ± 0.39, 4.00 ± 0.62, 3.14 ± 0.40, 2.25 ± 0.47, 2.11 ± 0.33, and 1.22 ± 0.29 g; 30 ng, 3.57 ± 0.61, 6.00 ± 0.87, 5.14 ± 0.86, 4.57 ± 0.84, 2.43 ± 0.43, and 2.06 ± 0.36 g; 100 ng, 4.29 ± 0.81, 7.43 ± 0.84, 6.57 ± 0.72, 5.14 ± 0.73, 4.86 ± 0.96, and 3.57 ± 0.87 g). Moreover, the neutralizing antibody injection (100 ng) did not affect the response of the contralateral hindpaw at 2 h after injection, a time point at which the antibody displays its maximal analgesic effect (Fig. 8B). Considering that TRAF2 is a central mediator in TNF-α signaling (Locksley et al., 2001; Zhu et al., 2010), we next examined whether spinal TNF-α participates in neuropathic pain development via the TRAF2/TNIK/pGluR1 signaling cascade. The WB results demonstrated that TNF-α-neutralizing antibody (100 ng, 10 μl) significantly reversed SNL-upregulated TRAF2 (from 0.55 ± 0.03 to 0.20 ± 0.02; n = 6), TNIK (from 0.64 ± 0.03 to 0.36 ± 0.03; n = 6), pGluR1 (from 0.65 ± 0.03 to 0.39 ± 0.03; n = 6), and GluR1(m) expression (from 0.71 ± 0.05 to 0.48 ± 0.05; n = 6) in the ipsilateral dorsal horn at day 7 after surgery but had no effect on the level of tGluR1 (Fig. 8C,D). Unexpectedly, we found that SNL-associated decreases in Fbxl2 (from 0.63 ± 0.03 to 0.47 ± 0.03; n = 6) and increases in Fbxo3 levels (from 0.15 ± 0.02 to 0.38 ± 0.19; n = 6) were also reversed by TNF-α-neutralizing antibody treatment. Together, these results indicate that the development of neuropathic pain involves TNF-α and downstream spinal Fbxo3/Fbxl2/TRAF2/TNIK/pGluR1-dependent GluR1–AMPAR trafficking.
Figure 8.
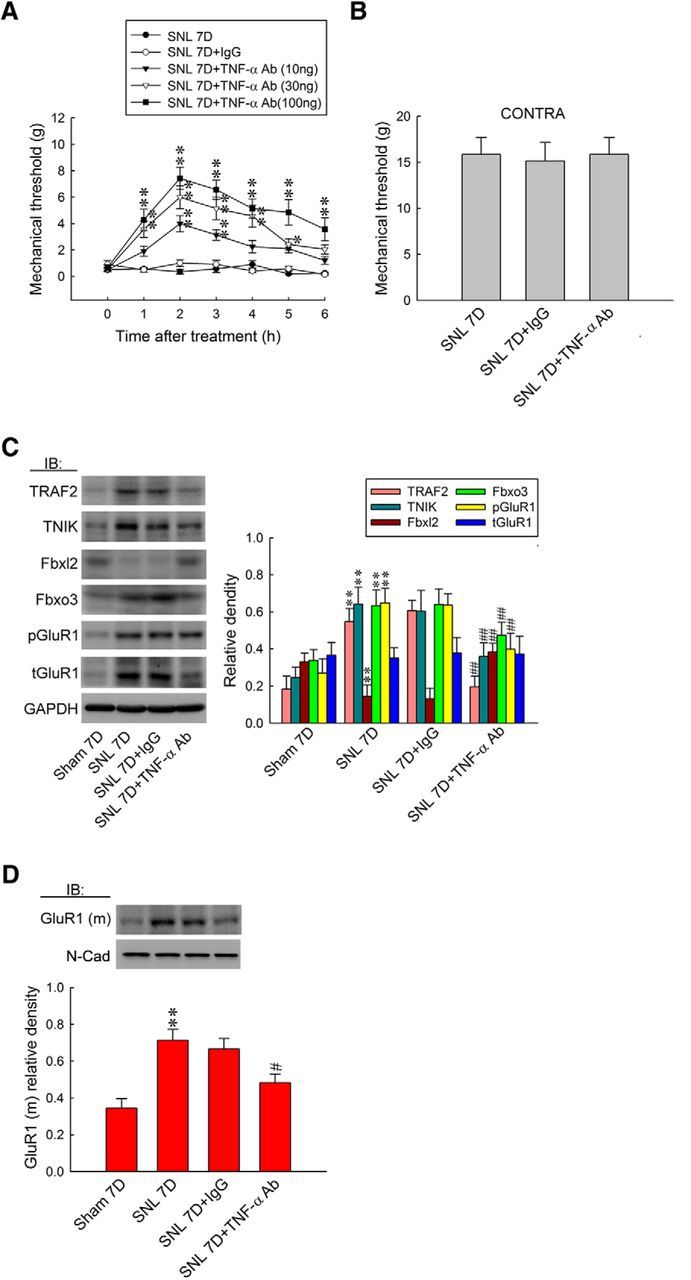
TNF-α-neutralizing antibody relieves allodynia by attenuating spinal Fbxo3/Fbxl2/TRAF2/TNIK/pGluR1-dependent GluR1–AMPAR trafficking. A, B, Intrathecal administration with an TNF-α-neutralizing antibody (SNL 7D+TNF-α Ab; 10, 30, and 100 ng, 10 μl), but not nonspecific IgG (SNL 7D+IgG; 100 ng, 10 μl), dose dependently increased the withdrawal threshold of the ipsilateral hindpaw. In addition, neither treatment affected the contralateral hindpaw at 2 h after injection (TNF-α-neutralizing antibody, 100 ng; von Frey test). Ipsilateral hindpaw, two-way ANOVA with repeated measures over time: treatment, F(4,30) = 64.39, p < 0.001; time, F(6,180) = 24.44, p < 0.001; treatment × time, F(24,180) = 4.657, p < 0.001. *p < 0.05, **p < 0.01 versus SNL 7D. n = 7. Contralateral hindpaw, one-way ANOVA, post hoc Tukey's test: F(2,18) = 0.047, p = 0.954. C, D, Although it had no effect on tGluR1, administrating with TNF-α-neutralizing antibody reversed SNL-upregulated TRAF2, TNIK, Fbxo3, pGluR1, GluR1(m), and SNL-downregulated Fbxl2 (normalized to GAPDH or N-cadherin) expressions. One-way ANOVA, post hoc Tukey's test: TRAF2, F(3,20) = 75.39, p < 0.001; TNIK, F(3,20) = 29.62, p < 0.001; Fbxl2, F(3,20) = 34.78, p < 0.001; Fbxo3, F(3,20) = 22.30, p < 0.001; pGluR1, F(3,20) = 24.55, p < 0.001; tGluR1, F(3,20) = 0.086, p = 0.966; GluR1(m), F(3,20) = 10.10, p < 0.001. **p < 0.01 versus Sham 7D. ##p < 0.01 versus SNL 7D. n = 6.
TNF-α induces allodynia via Fbxo3/Fbxl2/TRAF2/TNIK/pGluR1-dependent GluR1 trafficking
To further confirm the role of TNF-α in the pain-associated spinal Fbxo3/Fbxl2/TRAF2/TNIK/pGluR1-dependent GluR1–AMPAR trafficking pathway, recombinant rat TNF-α was administered intrathecally to naive rats. The results of the von Frey test showed that the intrathecal application of TNF-α (1 pm, 10 μl) decreased significantly the withdrawal threshold at 2 h after injection (Fig. 9A; 0.97 ± 0.30 g, n = 7). The TNF-α-decreased mechanical threshold was significantly prevented by once daily pretreatment with TNIK mRNA-targeting siRNAs (5 μg, 10 μl) for 4 d before TNF-α application (from 0.97 ± 0.30 to 6.57 ± 1.13 g; n = 7) and BC-1215 (100 nm, 10 μl) 2 h before TNF-α application (from 0.97 ± 0.30 to 7.43 ± 0.84 g; n = 7) but not by missense siRNA (5 μg, 10 μl) or the vehicle solution (10 μl). When examining protein expression, we observed that a spinal TNF-α injection significantly decreased the levels of Fbxl2 (0.25 ± 0.03; n = 6) but increased the levels of Fbxo3 (0.68 ± 0.03; n = 6), TRAF2 (0.57 ± 0.04; n = 6), TNIK (0.61 ± 0.03; n = 6), pGluR1 (0.71 ± 0.04; n = 6), and GluR1(m) (0.78 ± 0.03; n = 6), whereas the level of tGluR1 remained stable in the dorsal horn (Fig. 9B,C). Daily administration of TNIK mRNA-targeting siRNAs significantly prevented TNF-α-dependent increases in TNIK (from 0.61 ± 0.03 to 0.32 ± 0.02; n = 6), pGluR1 (from 0.71 ± 0.04 to 0.35 ± 0.02; n = 6), and GluR1(m) (from 0.78 ± 0.03 to 0.42 ± 0.03; n = 6) and exhibited no effect on the levels of Fbxl2, Fbxo3, TRAF2, or tGluR1. Intrathecal pretreatment of BC-1215 (100 nm, 10 μl) also prevented increases in the expression of Fbxo3 (from 0.68 ± 0.03 to 0.32 ± 0.02; n = 6), TRAF2 (from 0.57 ± 0.04 to 0.27 ± 0.04; n = 6), TNIK (from 0.61 ± 0.03 to 0.36 ± 0.03; n = 6), pGluR1 (from 0.71 ± 0.04 to 0.43 ± 0.02; n = 6), and GluR1(m) (from 0.78 ± 0.03 to 0.37 ± 0.04; n = 6) and prevented decreases in Fbxl2 expression (from 0.25 ± 0.03 to 0.43 ± 0.04; n = 6) after TNF-α injection but had no effect on the abundance of tGluR1. Moreover, daily TNIK mRNA-targeting siRNAs and a bolus of BC-1215 prevented the TNF-α-enhanced levels of tGluR1-bound TRAF2, TNIK, and pGluR1 (Fig. 9D; TNIK mRNA-targeting siRNAs, ∼0.5, ∼0.2, and 0.7-fold of SNL 7D, respectively; BC-1215, ∼0.6, ∼0.4, and 0.2-fold of SNL 7D, respectively). These results provide a pharmacological basis that supports the crucial role of TNF-α in SNL-induced neuropathic pain, potentially via an Fbxo3/Fbxl2-dependent modification of the TRAF2/TNIK/GluR1-trafficking cascade.
Figure 9.
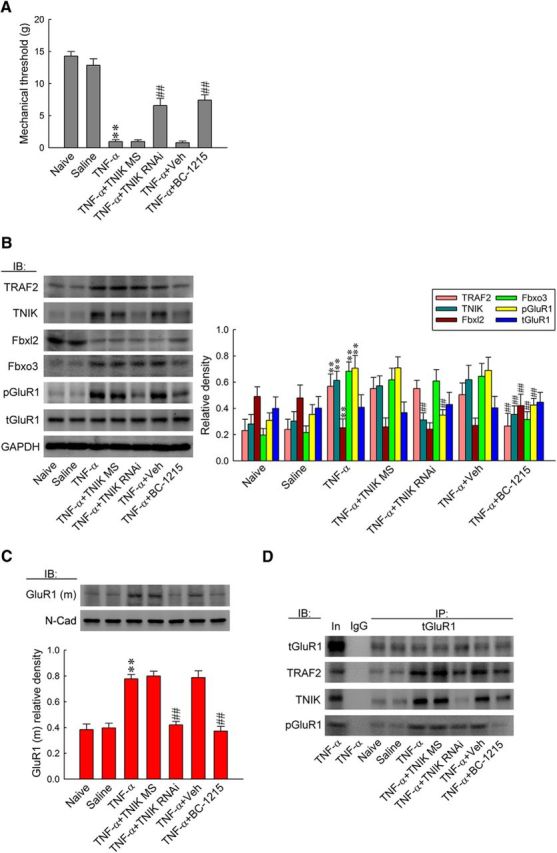
TNF-α induces allodynia via spinal Fbxo3/Fbxl2/TRAF2/TNIK/pGluR1-dependent GluR1–AMPAR trafficking. A, Statistical analysis of von Frey test showing intrathecal application of TNF-α (1 pm, 10 μl) significantly decreased the withdrawal threshold of the ipsilateral hindpaw at 2 h after injection, which was prevented by pretreatment with TNIK mRNA-targeting siRNA (5 μg, 10 μl, once daily for 4 d before TNF-α application) and BC-1215 (100 nm, 10 μl, 2 h before TNF-α application) but not by missense siRNA (5 μg, 10 μl) or the vehicle solution (10 μl). One-way ANOVA, post hoc Tukey's test: F(6,42) = 61.05, p < 0.001. **p < 0.01 versus Naive; ##p < 0.01 versus TNF-α. n = 7. B, C, Administration with TNIK mRNA-targeting siRNA prevented TNF-α-increased TNIK, pGluR1, and GluR1(m) expression but exhibited no effect on the levels of TRAF2, Fbxl2, Fbxo3, and tGluR1. Intrathecal pretreatment with BC-1215 prevented increases in TRAF2, TNIK, Fbxo3, pGluR1, and GluR1(m) expressions and decreases in Fbxl2 expression caused by TNF-α injection. However, it had no effect on tGluR1 expression. One-way ANOVA, post hoc Tukey's test: TRAF2, F(6,35) = 21.42, p < 0.001; TNIK, F(6,35) = 23.9, p < 0.001; Fbxl2, F(6,35) = 14.28, p < 0.001; Fbxo3, F(6,35) = 51.42, p < 0.001; pGluR1, F(6,35) = 37.03, p < 0.001; tGluR1, F(6,35) = 0.422, p = 0.859; GluR1(m), F(6,35) = 29.21, p < 0.001. **p < 0.01 versus Sham 7D; ##p < 0.01 versus SNL 7D. n = 6. D, Bolus of BC-1215 prevented TNF-α-enhanced tGluR1-TRAF2/TNIK/pGluR1 coprecipitation. Daily TNIK mRNA-targeting siRNA administration exhibited similar effects with the exception that it failed to affect TNF-α-enhanced tGluR1-TRAF2 coprecipitation.
Discussion
Although TNIK has emerged as a potential candidate for regulating the network activity of glutamatergic synapses in the CNS (Hussain et al., 2010; Q. Wang et al., 2011; Coba et al., 2012), several pivotal questions regarding its function remain unanswered. In the present study, we found that neuropathic injury-provoked behavioral allodynia was accompanied by enhanced dorsal horn TNIK expression, TNIK–GluR1 coupling, and GluR1 phosphorylation and trafficking toward the cytoplasmic membrane, all of which were prevented by focal knockdown of spinal TNIK expression. Our results provide molecular, histological, and genetic evidence supporting the role of spinal TNIK in the development of neuropathic pain via modification of phosphorylation-dependent glutamatergic GluR1–AMPAR trafficking. Our results are consistent with a study linking TNIK to subcellular GluR1–AMPAR redistribution by demonstrating that treatment with a synthetic peptide dramatically reduced TNIK expression and the amount of GluR1(m) in hippocampal neurons (Q. Wang et al., 2011). Our previous data showed that spinal serum- and glucocorticoid-induced protein kinase 1, a serine/threonine kinase, drives the subcellular redistribution of spinal GluR1–AMPARs, which underlies central sensitization-mediating inflammatory pain (Peng et al., 2012b). In addition, TNIK is a member of the germinal center serine/threonine kinase family (Fu et al., 1999). Thus, our results suggest that spinal serine/threonine kinases are involved crucially in the regulation of the machinery contributing to pain sensitization, potentially via modification of phosphorylation-dependent GluR1–AMPAR trafficking. To the best of our knowledge, our results are the first demonstration of the involvement of TNIK in the spinal plasticity underlying pain hypersensitivity caused by neuropathic injury.
In the present study, SNL induced Fbxo3 expression, Fbxl2 ubiquitination and downregulation, and TRAF2 expression in the dorsal horn. Conversely, intrathecal application of an Fbxo3 inhibitor attenuated allodynia as well as spinal Fbxl2 ubiquitination and degradation, which were accompanied by an increase in TRAF2 ubiquitination. Studies investigating cytokine-driven inflammation have linked Fbxo3-dependent Fbxl2 ubiquitination to TRAF2 expression through the modification of TRAF2 ubiquitination and degradation because transfection of functional Fbxo3 ubiquitinates and therefore destabilizes Fbxl2 and that Fbxo3 depletion reduces ubiquitination and increases the half-life of Fbxl2 in cultured cells (Mallampalli et al., 2013). Moreover, ectopic expression of Fbxl2 in lung epithelial cells resulted in TRAF protein ubiquitination and degradation (Chen et al., 2013). Thus, our findings are consistent with and demonstrate that Fbxo3-dependent Fbxl2 ubiquitination crucially regulates spinal TRAF2 expression by altering its ubiquitination-induced degradation. These findings further extend the contribution of spinal Fbxo3/Fbxl2/TRAF2 signaling to neuropathic injury-induced spinal neural plasticity underlying pain hypersensitivity. Interestingly, in this study, intrathecal administration of TNF-α induced behavioral allodynia and increased the levels of Fbxo3 and TRAF2 but decreased the levels of Fbxl2 in the dorsal horn. In addition to ameliorating SNL-associated allodynia, application of a TNF-α-neutralizing antibody efficiently prevented SNL-upregulated Fbxo3 and TRAF2 expression and SNL-downregulated Fbxl2 expression. Although the possibility that TNF-α independently modifies these protein levels cannot be excluded, it is less likely because Fbxo3 is a well known catalyst of the ubiquitination-dependent degradation of the sentinel TRAF inhibitor Fbxl2, which regulates TRAF function (Chen et al., 2013). Moreover, these findings suggest that spinal Fbxo3/Fbxl2/TRAF2 signaling potentially functions downstream to mediate TNF-α-induced allodynia. In addition, although the focal knockdown of spinal TNIK expression sufficiently prevented TNF-α-enhanced spinal TNIK, phosphorylation and cell-surface GluR1 expression, and tGluR1-TNIK/pGluR1 coprecipitation, it failed to affect TNF-α-increased Fbxo3 levels and TRAF2 and TNF-α-decreased Fbxl2 levels in the dorsal horn, suggesting that TNIK/pGluR1-dependent GluR1–AMPAR trafficking acts downstream of TNF-α-activated Fbxo3/Fbl2/TRAF2 signaling in dorsal horn neurons. Together, these findings revealed that pain hypersensitivity caused by neuropathic injury could be attributed to spinal TNF-α-induced Fbox3 and subsequent Fbxl2 ubiquitination, which promotes membrane GluR1–AMPAR trafficking via the downstream TRAF2/TNIK/GluR1 signaling cascade. Although the mechanism remains unclear, to the best of our knowledge, this is the first demonstration that TNF-α upregulates the expression of Fbxo3, which catalyzes the ubiquitination-dependent degradation of Fbxl2 to crucially regulate TRAF function. Because TRAF2 is well recognized as a potential mediator of TNF-α signaling (Hong et al., 2007), whether a microbial infection and/or neuropathic injury could activate the TNF-α/Fbxo3/Fbxl2/TRAF2 signaling cascade in parallel with traditional TNF-α/TRAF2 signaling to enhance cytokine signaling and/or pain hypersensitivity requires additional investigation.
In the present study, both SNL and a spinal TNF-α injection enhanced the level of Fbox3 in dorsal horn neurons. Nevertheless, intrathecal administration of BC-1215, a reagent that was designed originally to prevent Skp1–Cullin–Fbxo3 catalysis of Fbxl2 ubiquitination (Mallampalli et al., 2013), attenuated TNF-enhanced spinal Fbxo3 expression but had no effect on SNL-induced spinal Fbxo3 expression. Although the detailed mechanism underlying such a discrepancy remains unclear (Mallampalli et al., 2013) and considering that we assayed the levels of Fbox3 at day 7 after SNL and at 2 h after TNF-α injection, we speculated that BC-1215 failed to reverse SNL-enhanced Fbox3 levels because it has no effect on Fbxo3 protein expression. In contrast, BC-1215 administered as early as 2 h after a TNF-α injection inhibited the activation of Fbxo3, which is potentially crucial for subsequent Fbxo3 expression. Nevertheless, the machinery that is potentially involved and the temporal profile of Fbxo3 expression warrant additional experiments.
Over the past few years, investigations of pain pathology have revealed that E3-ubiquitin ligase-mediated protein ubiquitination contributes to chronic pain by regulating the levels of synaptic proteins and subsequently modifying synaptic efficacy (Ehlers, 2003; Ossipov et al., 2007; Yao et al., 2007). Our results revealed that SNL-induced allodynia involves spinal TRAF2, and we identified that Fbxo3 regulates TRAF2 expression by mediating ubiquitination-dependent Fbxl2 degradation. Agents that antagonize protein ubiquitination/degradation seem plausible to relieve pain. For example, MG-132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal), an ubiquitin inhibitor, reduces the degradation of ubiquitin-conjugated proteins and has been shown to attenuate pain and inflammation in rats suffering from arthritis (Ahmed et al., 2010, 2012). Nevertheless, our present data revealed that Fbxo3 modifies spinal TRAF2 expression by mediating Fbxl2 ubiquitination and that Fbxl2 also inhibits TRAF2 protein via the ubiquitination and degradation of TRAF2. Therefore, whether the application of a wide range of ubiquitination inhibitors might produce complicated effects in these ubiquitination pathways and whether the systemic reduction of protein ubiquitination, even restricted to the CNS, could result in unopposed effects needs be taken cautiously into consideration because specific ubiquitination defects have been linked to several neural developmental disorders (Kishino et al., 1997). Importantly, Chen et al. (2013) and Mallampalli et al. (2013) identified BC-1215 as a novel inhibitor of Fbxo3 activity. When evaluating its biological effect using in vivo preparations, they found that BC-1215 exerted robust therapeutic effects in cytokine-driven inflammation, including pseudomona-induced lung injury and cecum ligation/punctured-induced sepsis, by targeting Fbxo3/Fbxl2/TRAF-mediated cytokine release. On the basis of these findings, we tested whether the focal antagonism of Fbxo3 activity could ameliorate neuropathic pain development by thwarting the underlying spinal plasticity. As described previously, spinal BC-1215 application reversed the SNL-associated plastic changes in the dorsal horn, including Fbxl2 downregulation, as well as protein expression, phosphorylation, trafficking, coprecipitation, and colocalization in addition to alleviating behavioral allodynia, but did not affect Fbxo3 expression. Our findings revealed a potential therapeutic effect of BC-1215 in neuropathic pain development by limiting the underlying spinal plasticity and therefore providing a good starting point for the treatment of neuropathic pain by targeting the F box protein. Moreover, MG-132 was shown to inhibit the nuclear factor-κB cascade (Ahmed et al., 2010, 2012), and additional studies are required to determine whether the analgesic effect caused by MG-132 is attributable to an attenuation of the inflammation process. In summary, our results in this study will provide a basis for future studies investigating the role of protein ubiquitination in synaptic-dependent neuropathic pain and will provide insight toward novel therapeutic strategies in pain management.
Footnotes
This research was supported by the National Science Council, Taipei, Taiwan: NSC 102-2628-B-715-001 and 101-2320-B-715-001-MY3 (H.-Y.P.) and NSC 101-2320-B-039-013-MY3 (T.-B.L.); the Ministry of Science and Technology, Taipei, Taiwan: MOST 104-2320-B-019-MY3 and MOST104-2320-B-038-027-MY3 (T.-B.L.) and MOST 104-2320-B-715-004-MY3 (H.-Y.P.); the Mackay Memorial Hospital, Taipei, Taiwan: MMH-MM-10206 and MMH-MM-10302 (H.-Y.P.); the Mackay Medical College, New Taipei City, Taiwan: 1031B07 and 1041B06 (H.-Y.P.), and the Taipei Medical University, Taipei, Taiwan: TMU102-AE1-B06 (T.-B.L.), and the Saint Paul's Hospital, Taoyuan, Taiwan SPMRD-U1-6003 (T.-B.L.).
The authors declare no competing financial interests.
References
- Ahmed AS, Li J, Ahmed M, Hua L, Yakovleva T, Ossipov MH, Bakalkin G, Stark A. Attenuation of pain and inflammation in adjuvant-induced arthritis by the proteasome inhibitor MG132. Arthritis Rheum. 2010;62:2160–2169. doi: 10.1002/art.27492. [DOI] [PubMed] [Google Scholar]
- Ahmed AS, Li J, Erlandsson-Harris H, Stark A, Bakalkin G, Ahmed M. Suppression of pain and joint destruction by inhibition of the proteasome system in experimental osteoarthritis. Pain. 2012;153:18–26. doi: 10.1016/j.pain.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y, Zou C, Ellis B, Sciurba FC, Zhang Y, Mallampalli RK. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol. 2013;14:470–479. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JI, Svensson CI, Koehrn FJ, Bhuskute A, Sorkin LS. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain. 2010;149:243–253. doi: 10.1016/j.pain.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JM, Kim HK, Chung K. Segmental spinal nerve ligation model of neuropathic pain. Methods Mol Med. 2004;99:35–45. doi: 10.1385/1-59259-770-X:035. [DOI] [PubMed] [Google Scholar]
- Coba MP, Komiyama NH, Nithianantharajah J, Kopanitsa MV, Indersmitten T, Skene NG, Tuck EJ, Fricker DG, Elsegood KA, Stanford LE, Afinowi NO, Saksida LM, Bussey TJ, O'Dell TJ, Grant SG. TNiK is required for postsynaptic and nuclear signaling pathways and cognitive function. J Neurosci. 2012;32:13987–13999. doi: 10.1523/JNEUROSCI.2433-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Fu CA, Shen M, Huang BC, Lasaga J, Payan DG, Luo Y. TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton. J Biol Chem. 1999;274:30729–30737. doi: 10.1074/jbc.274.43.30729. [DOI] [PubMed] [Google Scholar]
- Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Garry EM, Fleetwood-Walker SM. A new view on how AMPA receptors and their interacting proteins mediate neuropathic pain. Pain. 2004;109:210–213. doi: 10.1016/j.pain.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Hong S, Lim S, Li AG, Lee C, Lee YS, Lee EK, Park SH, Wang XJ, Kim SJ. Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat Immunol. 2007;8:504–513. doi: 10.1038/ni1451. [DOI] [PubMed] [Google Scholar]
- Hori K, Ozaki N, Suzuki S, Sugiura Y. Upregulations of P2X(3) and ASIC3 involve in hyperalgesia induced by cisplatin administration in rats. Pain. 2010;149:393–405. doi: 10.1016/j.pain.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Hussain NK, Hsin H, Huganir RL, Sheng M. MINK and TNIK differentially act on Rap2-mediated signal transduction to regulate neuronal structure and AMPA receptor function. J Neurosci. 2010;30:14786–14794. doi: 10.1523/JNEUROSCI.4124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Ji, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, Yamamoto T. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. 2000;254:14–24. doi: 10.1006/excr.1999.4733. [DOI] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Lee CY, Perez FM, Wang W, Guan X, Zhao X, Fisher JL, Guan Y, Sweitzer SM, Raja SN, Tao YX. Dynamic temporal and spatial regulation ofmu opioid receptor expression in primary afferent neurons following spinal nerve injury. Eur J Pain. 2011;15:669–675. doi: 10.1016/j.ejpain.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TB, Hsieh MC, Lai CY, Cheng JK, Chau YP, Ruan T, Chen GD, Peng HY. Modulation of nerve injury-induced HDAC4 cytoplasmic retention contributes to neuropathic pain in rats. Anesthesiology. 2015a;123:199–212. doi: 10.1097/ALN.0000000000000663. [DOI] [PubMed] [Google Scholar]
- Lin TB, Lai CY, Hsieh MC, Jiang JL, Cheng JK, Chau YP, Ruan T, Chen GD, Peng HY. Neuropathic allodynia involves spinal neurexin-1β-dependent neuroligin-1/postsynaptic density-95/NR2B cascade in rats. Anesthesiology. 2015b;123:909–926. doi: 10.1097/ALN.0000000000000809. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/S0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Lu Y, Jiang BC, Cao DL, Zhang ZJ, Zhang X, Ji RR, Gao YJ. TRAF6 upregulation in spinal astrocytes maintains neuropathic pain by integrating TNF-α and IL-1β signaling. Pain. 2014;155:2618–2629. doi: 10.1016/j.pain.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren EJ, Charlesworth P, Coba MP, Grant SG. Knockdown of mental disorder susceptibility genes disrupts neuronal network physiology in vitro. Mol Cell Neurosci. 2011;47:93–99. doi: 10.1016/j.mcn.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallampalli RK, Coon TA, Glasser JR, Wang C, Dunn SR, Weathington NM, Zhao J, Zou C, Zhao Y, Chen BB. Targeting F box protein Fbxo3 to control cytokine-driven inflammation. J Immunol. 2013;191:5247–5255. doi: 10.4049/jimmunol.1300456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Bazov I, Gardell LR, Kowal J, Yakovleva T, Usynin I, Ekström TJ, Porreca F, Bakalkin G. Control of chronic pain by the ubiquitin proteasome system in the spinal cord. J Neurosci. 2007;27:8226–8237. doi: 10.1523/JNEUROSCI.5126-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng HY, Chang CH, Tsai SJ, Lai CY, Tung KC, Wu HC, Lin TB. Protein kinase A-dependent spinal α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionate-receptor trafficking mediates capsaicin-induced colon-urethra cross-organ reflex sensitization. Anesthesiology. 2011;114:70–83. doi: 10.1097/ALN.0b013e3181fe4204. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Lai CY, Hsieh MC, Lin TB. Spinal SIRPα1-SHP2 interaction regulates spinal nerve ligation-induced neuropathic pain via PSD-95-dependent NR2B activation in rats. Pain. 2012a;153:1042–1053. doi: 10.1016/j.pain.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Hsieh MC, Lai CY, Huang YP, Lin TB. Spinal SGK1/GRASP-1/Rab4 is involved in complete Freund's adjuvant-induced inflammatory pain via regulating dorsal horn GluR1-containing AMPA receptor trafficking in rats. Pain. 2012b;153:2380–2392. doi: 10.1016/j.pain.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Schäfers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. 2003;23:3028–3038. doi: 10.1523/JNEUROSCI.23-07-03028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürch C, Riether C, Matter MS, Tzankov A, Ochsenbein AF. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J Clin Invest. 2012;122:624–638. doi: 10.1172/JCI45977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin LS, Doom CM. Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripher Nerv Syst. 2000;5:96–100. doi: 10.1046/j.1529-8027.2000.00012.x. [DOI] [PubMed] [Google Scholar]
- Tao YX. AMPA receptor trafficking in inflammation-induced dorsal horn central sensitization. Neurosci Bull. 2012;28:111–120. doi: 10.1007/s12264-012-1204-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Myers RR. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. 1996;7:2897–2901. doi: 10.1097/00001756-199611250-00018. [DOI] [PubMed] [Google Scholar]
- Wang Q, Charych EI, Pulito VL, Lee JB, Graziane NM, Crozier RA, Revilla-Sanchez R, Kelly MP, Dunlop AJ, Murdoch H, Taylor N, Xie Y, Pausch M, Hayashi-Takagi A, Ishizuka K, Seshadri S, Bates B, Kariya K, Sawa A, Weinberg RJ, Moss SJ, Houslay MD, Yan Z, Brandon NJ. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol Psychiatry. 2011;16:1006–1023. doi: 10.1038/mp.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Atianjoh F, Gauda EB, Yaster M, Li Y, Tao YX. Increased expression of sodium channel subunit Nav1.1 in the injured dorsal root ganglion after peripheral nerve injury. Anat Rec (Hoboken) 2011;294:1406–1411. doi: 10.1002/ar.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang P, Liu Y, Cheng G. TRAF-mediated regulation of immune and inflammatory responses. Sci China Life Sci. 2010;53:159–168. doi: 10.1007/s11427-010-0050-3. [DOI] [PubMed] [Google Scholar]
- Xu LG, Li LY, Shu HB. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem. 2004;279:17278–17282. doi: 10.1074/jbc.C400063200. [DOI] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Yang X, Li XY, Liu QH, Tang XQ, Zhou SF, Kong QY, Axelsson J, Yu XQ. Suppression of tumor necrosis factor receptor associated factor (TRAF)-2 attenuates the proinflammatory and proliferative effect of aggregated IgG on rat renal mesangial cells. Cytokine. 2010;49:201–208. doi: 10.1016/j.cyto.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/S0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]