

Abstract
The nucleotide-binding and oligomerization domain (NOD) is an innate pattern recognition receptor that recognizes pathogen- and damage-associated molecular patterns. The 29-kDa amino-terminal fibronectin fragment (29-kDa FN-f) is a matrix degradation product found in the synovial fluids of patients with osteoarthritis (OA). We investigated whether NOD2 was involved in 29-kDa FN-f-induced pro-catabolic gene expression in human chondrocytes. The expression of mRNA and protein was measured using quantitative real-time polymerase chain reaction (qrt-PCR) and Western blot analysis. Small interfering RNAs were used for knockdown of NOD2 and toll-like receptor 2 (TLR-2). An immunoprecipitation assay was performed to examine protein interactions. The NOD2 levels in human OA cartilage were much higher than in normal cartilage. NOD1 and NOD2 expression, as well as pro-inflammatory cytokines, including interleukin-1beta (IL-1β) and tumor necrosis factor-alpha (TNF-α), were upregulated by 29-kDa FN-f in human chondrocytes. NOD2 silencing showed that NOD2 was involved in the 29-kDa FN-f-induced expression of TLR-2. Expressions of IL-6, IL-8, matrix metalloproteinase (MMP)-1, -3, and -13 were also suppressed by TLR-2 knockdown. Furthermore, NOD2 and TLR-2 knockdown data demonstrated that both NOD2 and TLR-2 modulated the expressions of their adaptors, receptor-interacting protein 2 (RIP2) and myeloid differentiation 88, in 29-kDa FN-f-treated chondrocytes. 29-kDa FN-f enhanced the interaction of NOD2, RIP2 and transforming growth factor beta-activated kinase 1 (TAK1), an indispensable signaling intermediate in the TLR-2 signaling pathway, and activated nuclear factor-κB (NF-κB), subsequently leading to increased expressions of pro-inflammatory cytokines and cartilage-degrading enzymes. These results demonstrate that 29-kDa FN-f modulated pro-catabolic responses via cross-regulation of NOD2 and TLR-2 signaling pathways.
Keywords: Fibronectin fragments, IL-6, IL-8, Matrix metalloproteinase, NOD2, Osteoarthritis
INTRODUCTION
Osteoarthritis (OA) is characterized by the degeneration of articular cartilage, synovial inflammation, and joint pain (1). Degradation of the extracellular matrix (ECM) is a crucial event leading to joint destruction in OA, rheumatoid arthritis (RA), and septic arthritis (2). Chondrocytes express catabolic mediators in response to proinflammatory cytokines, chemokines, adipokines, and mechanical loading (3). Fibronectin (FN) fragments result from the breakdown of FN, an ECM glycoprotein of cartilage, and in arthritis synovial fluid, up to 50% of the FN is fragmented into molecular sizes of 29 to 200 kDa (4). The most potent fragment, 29-kDa FN-f, stimulates cartilage ECM degradation by increasing the expression of nitric oxide (NO) and of matrix metalloproteinase (MMP)-1, -3, and -13 (5, 6). Innate pattern recognition receptors (PRRs), including toll-like receptors (TLRs) and nucleotide-binding and oligomerization domain (NOD) receptors, recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (7, 8). In response to various stresses, endogenous ligands, including fibronectin, heparin sulfate, and high mobility group box1 (9), are released by injured tissues or activated cells and activate signals of innate and adaptive immunity (10). Previously, we reported that 29-kDa FN-f induced catabolic responses, including MMPs expression in articular chondrocyte cultures by activation of NF-kappaB (NF-κB) and p38 through an MyD88-dependent TLR-2 signaling pathway (11). Thus, there is a possibility that activation of DAMP signaling forms a vicious cycle in OA pathogenesis by causing matrix damage and generation of matrix degradation products, which result in more DAMP signaling. A recent study provided additional evidence that PPRs are involved in OA development (12). Polymorphisms of the promoter TLR-3 and TLR-9 genes were associated with knee OA in a Chinese population (13, 14). We previously demonstrated that TLR-2, -3, -4, and -5 were significantly upregulated in OA cartilage compared to normal cartilage (11), and that TLR-2 and TLR-4 ligands strongly induced catabolic responses in chondrocytes (3). In addition to cartilage degeneration, TLR activation has implications in the development of synovitis in OA, since TLRs 1–7 and 9 have been detected in synovial membranes in OA and in vitro synovial fibroblasts have been shown to respond to many microbial TLR agonists (15).
NOD1 and NOD2, members of the leucine-rich repeat containing receptor (NLR) family, sense specific bacterial peptidoglycans and modulate expression of inflammatory genes through activation of the NF-kB signaling pathway (16). NOD2 plays an important role in activation of the innate immune response to muramyl dipeptide (MDP) found in all gram-positive and gram-negative bacteria, whereas NOD1 recognizes γ-D-glutamyl-meso-diaminopimelic acid which is contained in the proteoglycan of all gram-negative bacteria (17). NOD2 recruits the adaptor protein receptor interacting-protein 2 (RIP2/RICK2) and stimulates a TLR-2 downstream molecule, TNF receptor-associated factor 6, which drives the activation of NF-κB and mitogen-activated protein kinase (MAPK) pathways (16, 18, 19). In particular, NOD2 mutation and polymorphism lead to various diseases, including Crohn’s disease, Blau syndrome, and early onset sarcoidosis (17). We aimed to reveal the role of NOD2 in 29-kDa FN-f-induced pro-catabolic responses in primary chondrocytes. Tight regulation of innate immune receptor cross-signaling would thus be necessary to evade hyper-activation of pro-inflammatory and pro-catabolic responses.
In this study, we investigated whether 29-kDa FN-f modulated pro-catabolic responses through the NOD2 signaling pathway in articular chondrocytes. In addition, crosstalk between TLR-2 and NOD2 signaling pathways was explored.
RESULTS
NOD2 expression was elevated in OA cartilage tissue and 29-kDa FN-f-treated chondrocytes
To investigate the role of NOD2 in OA pathogenesis, the differences of NOD2 expression in OA and normal cartilage were compared using quantitative real-time reverse transcription polymerase chain reactions (qRT-PCR) and immunohistochemical (IHC) analyses. NOD2 expression at the mRNA level was significantly increased in OA cartilage compared to normal cartilage (Fig. 1A). IHC analyses also showed that OA cartilages showed strong expression of NOD2 in the chondrocytes in all zones, including the superficial, middle, and deep zones of the articular cartilage, together with the loss of cartilage matrix, compared to normal cartilage (Fig. 1B).
Fig. 1.

NOD2 expression increased in osteoarthritis (OA) cartilage and 29-kDa FN-f-treated chondrocytes. (A) Relative expression of NOD2 in human normal and OA cartilage. Relative expression of NOD2 in normal and OA cartilage was measured using SYBR Green-based real-time polymerase chain reaction (qPCR). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an endogenous control. *P < 0.05 vs. normal cartilage. Data are presented as the mean ± standard deviation (SD) of data from duplicate experiments using cartilages from different donors (normal cartilage, n = 7 and OA cartilage, n= 25). (B) IHC staining for NOD2 in normal and OA cartilage. NOD2 expression was determined by IHC using an antibody against NOD2. Sections were counterstained with methyl green. Data are representative of results from three normal and OA cartilages. Scale bars = 100 μm for 100× and 200× magnifications. The arrows indicate NOD2-positive chondrocytes. (C) 29-kDa FN-f and inflammatory cytokines, including (D) IL-1β and (E) TNF-α, positively increased NOD1 and NOD2 expression. Chondrocytes were exposed to 29-kDa FN-f, IL-1β, or TNF-α for 6, 24, and 48 h and mRNA levels of NOD1 and NOD2 were measured by qPCR. Data are presented as the mean ± SD of duplicate data from more than five different donors. *P < 0.05, ***P < 0.005, and ****P < 0.001 vs. untreated control. ns, not significant.
We investigated whether NOD1 and NOD2 expressions could be regulated by 29-kDa FN-f and pro-inflammatory cytokines, including IL-1β and TNF-α, in primary human chondrocytes. Both NOD1 and NOD2 expressions were significantly increased 24 h and 48 h following treatment with 29-kDa FN-f compared to untreated cells (Fig. 1C). Increased expressions of both receptors were also observed in IL-1β- and TNF-α-treated cells (Fig. 1D and E). Taken together, these results demonstrate that 29-kDa FN-f modulated the expression of PRR, including NOD1 and NOD2.
29-kDa FN-f induced pro-catabolic gene expressions via NOD2 in primary human chondrocytes
To examine whether NOD2 and/or TLR-2 signaling pathways were involved in 29-kDa FN-f-induced expression of inflammatory genes, chondrocytes were transfected with control siRNA, si-NOD2, or si-TLR-2, followed by treatment with 29-kDa FN-f. QRT-PCR analysis showed that 29-kDa FN-f highly induced the expression of the IL-6 and IL-8 pro-inflammatory cytokines, and the proteoglycan degrading enzymes MMP-1, -3, and -13, whereas a single knockdown of NOD2 or TLR-2 significantly suppressed the 29-kDa FN-f-induced expression of IL-6, IL-8, and MMPs (Fig. 2A). In addition, double knockdown of NOD2 and TLR-2 exhibited greater reductions in the expressions of IL-6, IL-8, MMP-1, and MMP-3 than a single knockdown of NOD2 or TLR-2 (Fig. 2A). These data demonstrate that NOD2 and TLR-2 signaling pathways, which are responsible for 29-kDa FN-f-induced expression of pro-catabolic genes, controlled the expressions both independently and synergistically.
Fig. 2.

29-kDa FN-f increased pro-catabolic factor expressions through synergistic activation of NOD2 and TLR-2. (A) NOD2 and TLR-2 silencing inhibited 29-kDa FN-f-induced IL-6, IL-8, MMP-1, -3, and -13 expressions. Chondrocytes were transfected with control small interfering (si) RNA (si-con), si-NOD2 RNA (si-NOD2), or TLR-2 RNA (si-TLR-2), and 48 h later chondrocytes were treated with 29-kDa FN-f for 24 h. mRNA levels were measured using qPCR. Data represent the mean ± SD of duplicate data from more than five different donors. GAPDH served as an endogenous control. *P < 0.05 and ****P < 0.001 vs. si-contransfected cells. #P < 0.05, ##P < 0.01, ###P < 0.005, and ####P < 0.001 vs. 29-kDa FN-f+si-NOD2 and siTLR-2-transfected cells. ns, not significant. (B) The reciprocal regulation of NOD2 and TLR-2 on their mRNA expressions. mRNA expression of NOD2 and TLR-2 was measured in 29-kDa FN-f+si-NOD2- or 29-kDa FN-f+si-TLR-2-transfected chondrocytes using SYBR Green-based real-time PCR assay. GAPDH was used as an endogenous control. Data represent the mean ± SD of duplicate data from more than five different donors. ***P < 0.005 and ****P < 0.001 vs. si-NOD2- or si-TLR-2-transfected cells.
Next, we investigated whether NOD2 and TLR-2 affected reciprocal expression of the genes. NOD2 knockdown significantly suppressed 29-kDa FN-f-induced expressions of TLR-2, while TLR-2 knockdown suppressed NOD2 expression induced by 29-kDa FN-f (Fig. 2B). Our data demonstrate that NOD2 and TLR-2 reciprocally regulated 29-kDa FN-f-stimulated expression of each other, and suggested that they may synergistically work together in 29-kDa FN-f-induced pro-catabolic gene expression.
TLR-2 and NOD2 reciprocally modulated their downstream adaptor expression
To explore the mechanism of 29-kDa FN-f-induced expressions of pro-inflammatory factors via NOD2, we analyzed the effect of 29-kDa FN-f on the protein expression of NOD2 and its downstream adaptor RIP2. 29-kDa FN-f increased both NOD2 and RIP2 expressions and elevated TLR-2 expression (Fig. 3A), showing that 29-kDa FN-f positively regulated the expression of the NOD2 receptor and its downstream molecule. We next examined whether the increased levels of NOD2 or TLR-2 influenced the expression of either one’s adaptor. Human articular chondrocytes were transfected with si-NOD2 or si-TLR-2, then stimulated with 29-kDa FN-f for 24 h. 29-kDa FN-f greatly increased TLR-2 expression, whereas NOD2 silencing significantly suppressed the expression, as well as the expressions of RIP2 and MyD88 (Fig. 3B). In addition, we observed similar results for TLR-2 knockdown, which significantly suppressed the 29-kDa FN-f-stimulated expressions of NOD2 and the downstream molecule, RIP2 (Fig. 3C). These results demonstrate that TLR-2 and NOD2 reciprocally influenced their signaling pathways through modulation of the expression of their respective receptors and adapters in response to 29-kDa FN-f.
Fig. 3.
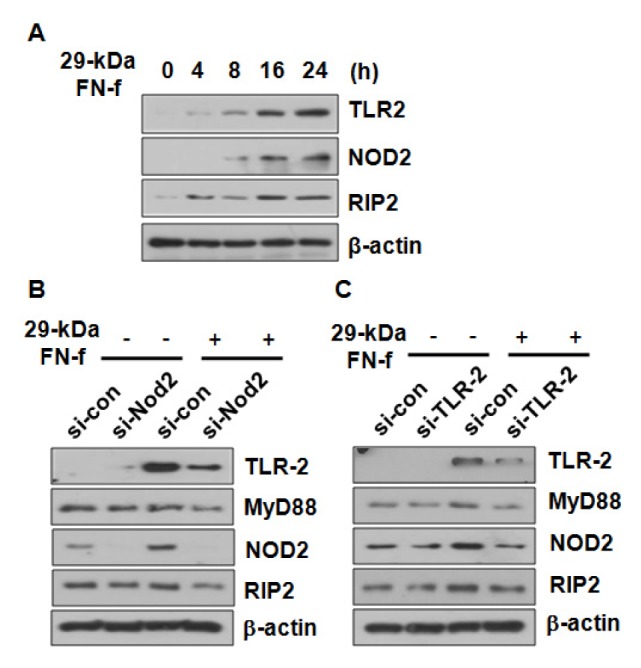
NOD2 and TLR-2 cross-regulated the expression of their respective adaptor molecules. (A) 29-kDa FN-f increased the protein levels of TLR-2 and NOD2 in a time-dependent manner. Chondrocytes were incubated with 29-kDa FN-f for 4, 8, 16, and 24 h. (B, C) NOD2 and TLR-2 receptors were involved in expression of the adaptor molecules of NOD2 and TLR-2. Chondrocytes were transfected with control siRNA and (B) si-NOD2 or (C) si-TLR-2. Forty-eight hours later, chondrocytes were stimulated with 29-kDa FN-f for 24 h. The protein levels were measured by immunoblot analyses. β-actin served as a loading control.
29-kDa FN-f activated the NF-kB signaling pathway through enhanced formation of NOD2, RIP2, and TAK1 complex
29-kDa FN-f-mediated activation of the NF-κB and p38 MAPK signaling pathways was associated with TLR-2 in human chondrocytes (11). To investigate the involvement of NOD2 in the 29-kDa FN-f-mediated signaling pathway, control siRNA or si-NOD2-transfected chondrocytes were stimulated with 29-kDa FN-f for 15 to 60 min. Immunoblot analyses showed that higher phosphorylation of IκBα was observed 15 minutes after 29-kDa FN-f treatment than in untreated cells, while NOD2 knockdown significantly inhibited the phosphorylation (Fig. 4). In contrast, the phosphorylation of JNK, ERK1/2, and p38 MAPK was not altered by NOD2 knockdown (Fig. 4). The results demonstrate that 29-kDa FN-f-induced inflammatory factor expression was dependent on the NOD2-dependent NF-κB signaling pathway.
Fig. 4.

29-kDa FN-f activated the NF-κB signaling pathway through the NOD2/RIP2/TAK1 signaling pathway. (A) NOD2 silencing inhibited the 29-kDa FN-f-induced NF-kB activation. Human chondrocytes were transfected with control small interfering RNA (si-con) or si-NOD2, then stimulated with 29-kDa FN-f (300 nM) for 15, 30, and 60 min. The levels of p-ERK/ERK, p-JNK/JNK, p-p38/p38, and p-IκBα/IκBα were determined by immunoblot analyses. Immunoblot data are representative of three independent experiments from different donors. (B) 29-kDa FN-f increased the formation of the NOD2/RIP2/TAK1 complex. 29-kDa FN-f-stimulated chondrocytes were lysed with RIPA buffer and the cell extracts were immunoprecipitated with an antibody against RIP2. The proteins were analyzed by immunoblot analyses using antibodies against NOD2, RIP2, and TAK1. Immunoprecipitation with IgG served as a negative control.
Activated NOD2 binds to its adaptor, RIP2, and recruits transforming growth factor β-activated kinase 1 (TAK1) to the complex, subsequently leading to NF-κB activation (20). We investigated whether 29-kDa FN-f influenced formation of the NOD2/RIP2/TAK1 complex. Primary chondrocytes were transfected with si-NOD2 and treated with 29-kDa FN-f for 24 h. Cell lysates were immunoprecipitated (IP) with an antibody against RIP2. 29-kDa FN-f induced the expressions of NOD2, RIP2, and TAK1, but NOD2 silencing lowered their expressions (Fig. 4B). IP data revealed that 29-kDa FN-f increased formation of the NOD2/RIP2/TAK1 complex, whereas NOD2 silencing inhibited it due to decreased expression of NOD2, RIP2, and TAK1, indicating that 29-kDa FN-f activated the NOD2/RIP2/TAK1 pathway (Fig. 4B). Therefore, these results suggest that 29-kDa FN-f activated the pro-catabolic NF-κB signaling pathway via the NOD2 signaling pathway.
DISCUSSION
In this study, we examined whether NOD2 expression was elevated in OA cartilage compared to normal cartilage. Consistent with previous reports, 29-kDa FN-f significantly induced pro-inflammatory gene expressions, including IL-6 and IL-8, and proteoglycan degrading enzymes, including MMP-1, -3, and -13, through synergistic action of the NOD2 and TLR-2 signaling pathways. In addition, NOD2 and TLR-2 reciprocally modulated the 29-kDa FN-f-stimulated expression of their respective adaptors, RIP2 and MyD88. Further, 29-kDa FN-f activated NF-κB through NOD2 signaling and promoted the formation of NOD2/RIP2/TAK1, subsequently increasing expressions of pro-catabolic factors.
Synovial fluids contain various molecules released from damaged joint tissue. In particular, molecules derived from the cartilage extracellular matrix, including fragments of aggrecan, hyaluronan, collagen, and fibronectin (FN), have been found in the cartilage and synovial fluid from patients with OA and RA. For example, FN-fs in OA synovial fluids were present at concentrations of more than 1 μM, together with increased proteolytic enzymes (21). Aggrecan fragments (150–250 kDa high-molecular and 32-amino acids) were also identified in the synovial fluid from OA patients (22). A recent report showed that the naturally-occurring aggrecan 32-mer fragment triggered catabolic factor expression via TLR-2 and activation of NF-κB (23). Another report showed that the aggrecan 32-mer excited dorsal root ganglion nociceptive neurons, both in culture and in intact explants, which was mediated through TLR-2 (24), suggesting that DAMPs induce the cartilage catabolic process, as well as pain, in OA. In addition, promoter polymorphism of the TLR-9 gene was associated with end-stage knee OA, while that of TLR-3 was associated with susceptibility to knee OA in a Chinese Han population (13, 14). Thus, the elucidation of signaling mechanisms controlling the DAMP response is gaining importance in understating the pathogenesis of OA.
Cell surface TLRs sense intact bacterial peptidoglycans (PGN), whereas intracellular NOD2 recognizes degradation products resulting after the uptake of PGN and its release into the cytosol (25). NOD1 and NOD2 mRNA expressions have been demonstrated in the synovial tissues from RA and OA patients, and NOD2-deficient mice showed decreases in joint inflammation and cytokine production in a streptococcal cell wall (SCW)-induced arthritis model (26). A previous report showed that Ag-induced arthritis developed in NOD2 and RIPK2-deficient mice, and impaired NOD2/RIP2 signaling pathways in joints led to reduced production of proinflammatory cytokines and chemokines, including TNF, IL-1β, and CXCL1 (27). TLR-2 and NOD2 receptor expressions were enhanced by IL-32γ and Pam3Cys/MDP stimulation, and AdIL-32γ transduction, followed by the injection of SCW, displayed aggravated joint inflammation and cartilage destruction in mice (28). TLR-2/NOD2 activation significantly induced cartilage-degrading mediators, including MMP-1, -3, inducible nitric oxide synthase, TLR-2, and NOD2, in fibroblast-like synoviocytes (28). Several studies have reported cross-regulation between NOD2 and TLR-2 signaling. IL-12 expression was increased by a variety of TLR ligands in NOD2-deficient splenic macrophages compared to wild type cells, indicating that NOD2 is a negative regulator of the TLR-2 response (29). On the other hand, NOD2 and TLR-2 were required for PGN-induced cytokine production and had a synergistic influence on murine peritoneal macrophages (29, 30). In our in vitro study, we demonstrated that 29-kDa FN-f modulated IL-6 and IL-8 expressions, as well as MMPs, through the coordinated actions of NOD2 and TLR-2, indicating a close synergistic relationship between the NOD2 and TLR-2 signaling pathways in the 29-kDa FN-f-induced catabolic responses.
IL-1β induces expression of proteoglycan-degrading enzymes and functions synergistically with other cytokines and chemokines, such as IL-6 and IL-8, subsequently worsening inflammation (31, 32). In addition, cartilage proteoglycan loss was inhibited in the knee joints of IL-6-deficient mice compared to wild type, suggesting that IL-6 aggravates cartilage destruction (33). NOD2 activation produced IL-6 and IL-8 in RA synovial fibroblasts by synergistic action with TLRs, indicating that up-regulation of proinflammatory cytokines was produced by the increased expression of NOD2 from TLR-2 activation (34). A previous study using human B lymphocytes showed that the NOD2 ligands enhanced TLR-induced B cell activation (35). Thus, the intracellular cytoplasmic sensor, NOD2, and membrane-bound TLR-2 may recognize a variety of pathogens and stimuli due to different localization, whereas upon recognition of PAMPs, the two receptors converge into common pathways, i.e. NF-κB and MAPK, leading to inflammatory immune responses and host defense (36). Furthermore, NOD2 may respond to PAMPs that are delivered into the cytoplasm without detection by extracellular TLRs. Therefore, these receptors may function synergistically through tight regulation of the crosstalk between their signaling pathways.
Taken together, our findings underline the important role of NOD2 signaling in the 29-kDa FN-f-induced catabolic responses in articular chondrocytes. NOD2 and TLR-2 pathways contribute to the independent triggering of 29-kDa FN-f-induced catabolic gene expressions and, at least in part, cooperate through managing the complex formation of NOD2/RIP2 and TAK1, a downstream effector of TLR-2, suggesting that TLR-2 and NOD2 cross-regulation pathways may be useful targets to prevent the development and progression of arthritis.
MATERIALS AND METHODS
Materials, IHC analysis, transfection with siRNA, qRT-PCR, immunoblot analysis, and statistical analyses are described in the Supplementary Materials.
Immunoprecipitation (IP) assays
Proteins were extracted from chondrocytes with RIPA lysis buffer and supernatants were obtained by centrifugation at 13,000 × g for 10 min at 4°C. Supernatants containing equal amounts of proteins were pre-incubated with a protein A agarose bead slurry for 4 h at 4°C on a rotating shaker, then incubated with specific antibodies against RIP2 or IgG in the presence of protein A agarose beads at 4°C overnight with gentle rotation. Proteins eluted in 2x SDS sample buffer were separated on SDS-PAGE and subjected to immunoblot analyses as described above. A portion of the protein extracts was used as an input control.
SUPPLEMENTARY INFORMATION
ACKNOWLEDGEMENTS
This study was supported by a grant (HI16C0287) from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea, the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education (2016R1D1A 1B03932259 and 2017R1A2B2001881), and in part by Hallym University Research Fund.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Felson DT. Clinical practice. Osteoarthritis of the knee. N Engl J Med. 2006;354:841–848. doi: 10.1056/NEJMcp051726. [DOI] [PubMed] [Google Scholar]
- 2.Goldring MB. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43:1916–1926. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 3.Kim HA, Cho ML, Choi HY, et al. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006;54:2152–2163. doi: 10.1002/art.21951. [DOI] [PubMed] [Google Scholar]
- 4.Xie DL, Meyers R, Homandberg GA. Fibronectin fragments in osteoarthritic synovial fluid. J Rheumatol. 1992;19:1448–1452. [PubMed] [Google Scholar]
- 5.Homandberg GA, Hui F. Association of proteoglycan degradation with catabolic cytokine and stromelysin release from cartilage cultured with fibronectin fragments. Arch Biochem Biophys. 1996;334:325–331. doi: 10.1006/abbi.1996.0461. [DOI] [PubMed] [Google Scholar]
- 6.Pichika R, Homandberg GA. Fibronectin fragments elevate nitric oxide (NO) and inducible NO synthetase (iNOS) levels in bovine cartilage and iNOS inhibitors block fibronectin fragment mediated damage and promote repair. Inflamm Res. 2004;53:405–412. doi: 10.1007/s00011-004-1279-8. [DOI] [PubMed] [Google Scholar]
- 7.Wu J, Zhang Y, Xin Z, Wu X. The crosstalk between TLR2 and NOD2 in Aspergillus fumigatus keratitis. Mol Immunol. 2015;64:235–243. doi: 10.1016/j.molimm.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Ko R, Lee SY. Glycogen synthase kinase 3beta in Toll-like receptor signaling. BMB Rep. 2016;49:305–310. doi: 10.5483/BMBRep.2016.49.6.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basu M, Paichha M, Swain B, et al. Modulation of TLR2, TLR4, TLR5, NOD1 and NOD2 receptor gene expressions and their downstream signaling molecules following thermal stress in the Indian major carp catla (Catla catla) 3 Biotech. 2015;5:1021–1030. doi: 10.1007/s13205-015-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 11.Hwang HS, Park SJ, Cheon EJ, Lee MH, Kim HA. Fibronectin fragment-induced expression of matrix metalloproteinases is mediated by MyD88-dependent TLR-2 signaling pathway in human chondrocytes. Arthritis Res Ther. 2015;17:320. doi: 10.1186/s13075-015-0833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Jiang C, Zhu W, et al. NOD2 pathway via RIPK2 and TBK1 is involved in the aberrant catabolism induced by T-2 toxin in chondrocytes. Osteoarthritis Cartilage. 2015;23:1575–1585. doi: 10.1016/j.joca.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 13.Su SL, Yang HY, Lee CH, Huang GS, Salter DM, Lee HS. The (−1486T/C) promoter polymorphism of the TLR-9 gene is associated with end-stage knee osteoarthritis in a Chinese population. J Orthop Res. 2012;30:9–14. doi: 10.1002/jor.21494. [DOI] [PubMed] [Google Scholar]
- 14.Yang HY, Lee HS, Lee CH, et al. Association of a functional polymorphism in the promoter region of TLR-3 with osteoarthritis: a two-stage case-control study. J Orthop Res. 2013;31:680–685. doi: 10.1002/jor.22291. [DOI] [PubMed] [Google Scholar]
- 15.Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51:249–257. doi: 10.1016/j.bone.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27:373–383. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borzutzky A, Fried A, Chou J, Bonilla FA, Kim S, Dedeoglu F. NOD2-associated diseases: Bridging innate immunity and autoinflammation. Clin Immunol. 2010;134:251–261. doi: 10.1016/j.clim.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Elia PP, Tolentino YF, Bernardazzi C, de Souza HS. The role of innate immunity receptors in the pathogenesis of inflammatory bowel disease. Mediators Inflamm. 20152015 doi: 10.1155/2015/936193. 936193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim H, Lee YD, Kim MK, et al. Extracellular S100A4 negatively regulates osteoblast function by activating the NF-kappaB pathway. BMB Rep. 2017;50:97–102. doi: 10.5483/BMBRep.2017.50.2.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyle JP, Parkhouse R, Monie TP. Insights into the molecular basis of the NOD2 signalling pathway. Open Biol. 2014;4 doi: 10.1098/rsob.140178. 140178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zack MD, Arner EC, Anglin CP, Alston JT, Malfait AM, Tortorella MD. Identification of fibronectin neoepitopes present in human osteoarthritic cartilage. Arthritis Rheum. 2006;54:2912–2922. doi: 10.1002/art.22045. [DOI] [PubMed] [Google Scholar]
- 22.Fosang AJ, Last K, Gardiner P, Jackson DC, Brown L. Development of a cleavage-site-specific monoclonal antibody for detecting metalloproteinase-derived aggrecan fragments: detection of fragments in human synovial fluids. Biochem J. 1995;310(Pt 1):337–343. doi: 10.1042/bj3100337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lees S, Golub SB, Last K, et al. Bioactivity in an Aggrecan 32-mer Fragment Is Mediated via Toll-like Receptor 2. Arthritis Rheumatol. 2015;67:1240–1249. doi: 10.1002/art.39063. [DOI] [PubMed] [Google Scholar]
- 24.Miller RE, Ishihara S, Tran PB, et al. An aggrecan fragment drives osteoarthritis pain through Toll-like receptor 2. JCI Insight. 2018;3:e95704. doi: 10.1172/jci.insight.95704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 26.Joosten LA, Heinhuis B, Abdollahi-Roodsaz S, et al. Differential function of the NACHT-LRR (NLR) members Nod1 and Nod2 in arthritis. Proc Natl Acad Sci U S A. 2008;105:9017–9022. doi: 10.1073/pnas.0710445105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vieira SM, Cunha TM, Franca RF, et al. Joint NOD2/RIPK2 signaling regulates IL-17 axis and contributes to the development of experimental arthritis. J Immunol. 2012;188:5116–5122. doi: 10.4049/jimmunol.1004190. [DOI] [PubMed] [Google Scholar]
- 28.Heinhuis B, Koenders MI, van de Loo FA, et al. IL-32gamma and Streptococcus pyogenes cell wall fragments synergise for IL-1-dependent destructive arthritis via upregulation of TLR-2 and NOD2. Ann Rheum Dis. 2010;69:1866–1872. doi: 10.1136/ard.2009.127399. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 30.Netea MG, Ferwerda G, de Jong DJ, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–6523. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- 31.Kim JR, Yoo JJ, Kim HA. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. Int J Mol Sci. 2018;19:674. doi: 10.3390/ijms19030674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7:33–42. doi: 10.1038/nrrheum.2010.196. [DOI] [PubMed] [Google Scholar]
- 33.van de Loo FA, Kuiper S, van Enckevort FH, Arntz OJ, van den Berg WB. Interleukin-6 reduces cartilage destruction during experimental arthritis. A study in interleukin-6-deficient mice. Am J Pathol. 1997;151:177–191. [PMC free article] [PubMed] [Google Scholar]
- 34.Ospelt C, Brentano F, Jungel A, et al. Expression, regulation, and signaling of the pattern-recognition receptor nucleotide-binding oligomerization domain 2 in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60:355–363. doi: 10.1002/art.24226. [DOI] [PubMed] [Google Scholar]
- 35.Petterson T, Jendholm J, Mansson A, Bjartell A, Riesbeck K, Cardell LO. Effects of NOD-like receptors in human B lymphocytes and crosstalk between NOD1/NOD2 and Toll-like receptors. J Leukoc Biol. 2011;89:177–187. doi: 10.1189/jlb.0210061. [DOI] [PubMed] [Google Scholar]
- 36.Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365–398. doi: 10.1146/annurev.pathol.4.110807.092239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.