

Abstract
Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH) is an infantile SMA variant with additional manifestations, particularly severe microcephaly. We previously identified a nonsense mutation in Vaccinia-related kinase 1 (VRK1), R358X, as a cause of SMA-PCH. VRK1-R358X is a rare founder mutation in Ashkenazi Jews, and additional mutations in patients of different origins have recently been identified. VRK1 is a nuclear serine/threonine protein kinase known to play multiple roles in cellular proliferation, cell cycle regulation, and carcinogenesis. However, VRK1 was not known to have neuronal functions before its identification as a gene mutated in SMA-PCH. Here we show that VRK1-R358X homozygosity results in lack of VRK1 protein, and demonstrate a role for VRK1 in neuronal migration and neuronal stem cell proliferation. Using shRNA in utero electroporation in mice, we show that Vrk1 knockdown significantly impairs cortical neuronal migration, and affects the cell cycle of neuronal progenitors. Expression of wild-type human VRK1 rescues both proliferation and migration phenotypes. However, kinase-dead human VRK1 rescues only the migration impairment, suggesting the role of VRK1 in neuronal migration is partly noncatalytic. Furthermore, we found that VRK1 deficiency in human and mouse leads to downregulation of amyloid-β precursor protein (APP), a known neuronal migration gene. APP overexpression rescues the phenotype caused by Vrk1 knockdown, suggesting that VRK1 affects neuronal migration through an APP-dependent mechanism.
Keywords: APP, neuronal migration, SMA-PCH (spinal muscular atrophy pontocerebellar hypoplasia), VRK1
Introduction
The spinal muscular atrophies (SMAs) are heterogeneous disorders characterized by degenerative loss of spinal cord anterior horn cells, leading to muscle weakness and atrophy (Wee et al., 2010). SMA-PCH [or pontocerebellar hypoplasia type 1A (PCH1A) MIM (Mendelian Inheritance in Man) #607596], is an infantile SMA variant with additional manifestations, including severe congenital microcephaly, mental retardation, and cerebellar signs (Barth, 1993; Rudnik-Schöneborn et al., 2003). We identified a vaccinia-related kinase 1 (VRK1) nonsense mutation (R358X) as a cause of SMA-PCH in a consanguineous Ashkenazi Jewish family and other cases have since been reported (Gonzaga-Jauregui et al., 2013). VRK1-R358X is a rare founder allele: carrier frequency in Ashkenazi Jews is ∼1/250 (Renbaum et al., 2009). Other VRK1 mutations have been reported: homozygosity for VRK1-R133C in Iranian siblings with SMA-PCH (Najmabadi et al., 2011), and compound heterozygosity for VRK1 V236M and R89Q in two sisters with microcephaly and complex motor and sensory axonal neuropathy (Gonzaga-Jauregui et al., 2013).
VRK1 is a serine-threonine kinase involved in cell cycle regulation (Valbuena et al., 2011), and is ubiquitously expressed, including in fetal and adult brain and cerebellum (Nezu et al., 1997). Its subcellular localization is mainly nuclear, with a small fraction found in the cytoplasm and membrane compartments (Valbuena et al., 2007). VRK1 function has been mostly studied in the context of cellular proliferation and tumorigenesis (Santos et al., 2006; Valbuena et al., 2011). In cell cycle regulation, VRK1 phosphorylates several proteins, including p53, BAF (barrier-to-autointegration factor), histone H3, and CREB (Valbuena et al., 2011).
Restriction of the SMA-PCH phenotype to the nervous system suggested that VRK1 has previously unrecognized neuronal functions. Brain MRI findings from a newly diagnosed Israeli SMA-PCH patient, together with expression array data, implicated VRK1 in neuronal migration. Using in utero electroporation (IUE) knockdown of Vrk1 in mouse embryo brains, we provide evidence that, in addition to affecting neuronal progenitor proliferation, VRK1 is indeed important for neuronal migration, and that its role in migration is largely independent of its kinase activity. Finally, we show that amyloid-β precursor protein (APP), known to be involved in neuronal migration, acts downstream of VRK1 in this process.
Materials and Methods
Microarrays.
cDNA extracted from lymphoblastoid cell lines of a VRK1-R358X homozygous patient and two normal controls was analyzed using the HuGene_ST_1.0 array (Affymetrix). Array data were analyzed using RMA (probeset signals, >20) after quality assessment. Relative expression levels were analyzed by ANOVA (Partek Genomics Suite, Partek). Pathway analysis was performed using Ingenuity Pathway Analysis (Ingenuity Systems).
Quantitative real-time PCR analysis.
Quantitative real-time PCR was performed using Power SYBR master mix (Applied Biosystems). Threshold cycle (Ct) values were normalized to GAPDH and calculated as 2−ΔCt.
Plasmids.
Vrk1-shRNA lentiviral plasmids were purchased from Open BioSystems (F10 shRNA, TRCN0000023783). Control shRNA was previously described (Sapir et al., 2008). pCAGGS-GFP [BCCM/LMBP (Belgian Co-ordinated Collections of Micro-organisms/Laboratory of Molecular Biology Plasmids)] was used to label shRNA-transfected neurons. Human WT-VRK1 and kinase-dead VRK1-K179E cDNAs (Sanz-García et al., 2011) were subcloned into pCAGGS and pNeuroD (Lee et al., 1995). Fluorescent ubiquitination-based cell cycle indicator (FUCCI) cell cycle reporters (Sakaue-Sawano et al., 2008) were subcloned into pCAGGS. pCAX-FLAG-APP and pBABE-puro were purchased from Addgene.
Cell lines, tissue culture, and transfections.
Human lymphoblastoid cells were propagated in RPMI (Roswell Park Memorial Institute) medium, human primary fibroblasts, HEK293T, and mouse 3T3 cells, in DMEM all supplemented with 10% fetal calf serum (FCS) antibiotics and glutamine. Transfections were performed using jetPEI (Polyplus Transfection) in six-well dishes in triplicate with shRNA constructs or control vectors (3 μg) and pBABE-puro (1.5 μg). Forty-eight hours after transfection, the growth medium was supplemented with puromycin (3 μg/ml). Lysates were prepared after 3 additional days.
Western blotting and immunofluorescence.
Western blotting and immunofluorescence staining were performed as described previously (Sanz-García et al., 2011) using polyclonal anti-hVRK1 (Sigma-Aldrich HPA000660; antigen sequence includes residues 5–141), anti-GAPDH (Abcam), anti-mVRK1 (kindly provided by Prof. Kyong-Tai Kim, Pohang University of Science and Technology, Pohang, South Korea), and monoclonal anti-APP (Millipore MAB348; antigen sequence includes residues 66–81) along with anti-mouse (Jackson Immunoresearch) or anti-rabbit (Santa Cruz Biotechnology) HRP-conjugated secondary antibodies. Quantifications were performed using ImageJ on two independent Western blots. In all Western blots GAPDH was used as a loading control. APP was detected by immunofluorescence using anti-APP and Cy-3-conjugated secondary antibody (Jackson Immunoresearch) and was photographed using a Zeiss confocal microscope.
In utero electroporation.
Neuronal migration was analyzed by IUE on ICR pregnant dams, as described previously (Sapir et al., 2008), in ≥3 independent animals of either sex per treatment. Fluorescence intensity (presented in relation to the total number of cells) was quantified using ImageJ. Cell cycle analysis was performed by IUE in ≥3 independent animals of either sex using mVenus-hGeminin (1.5 μg/μl) and mCherry-hCdt1 (0.5 μg/μl) FUCCI reporters (Sakaue-Sawano et al., 2008). Cells were counted in separate channels using Imaris. Animal protocols were approved by the Weizmann Institute Institutional Animal Care and Use Committee.
Results
Brain MRI in SMA-PCH reveals a simplified gyral pattern, consistent with impaired neuronal migration
SMA-PCH patients from the original family we investigated (Renbaum et al., 2009) are deceased, and brain MRIs were unavailable. An unrelated Ashkenazi Jewish VRK1-R358X homozygous boy presented at birth with a head circumference of 30.5 cm (−3 SD) and developed progressive postnatal microcephaly; at age 30 months, head circumference was 39.5 cm (−8 SD). Brain MRI at age 1 year demonstrated a simplified gyral pattern and hypoplasia of the posterior vermis without brainstem involvement (Fig. 1). EMG at 2.5 years showed markedly reduced bilateral common peroneal and posterior tibial compound muscle action potentials with normal distant latencies and velocities, and normal sensory studies. Upper limb studies were normal. At age 4.5 years, muscle tone was generally increased with increased tendon reflexes and sustained clonus. Muscle mass of the distal lower limbs was decreased. His general developmental quotient was 56, based on the following sub-scores: cognitive, 75; language understanding, 65; expression, 32; gross motor, 32; fine motor, 36; social, 43.
Figure 1.
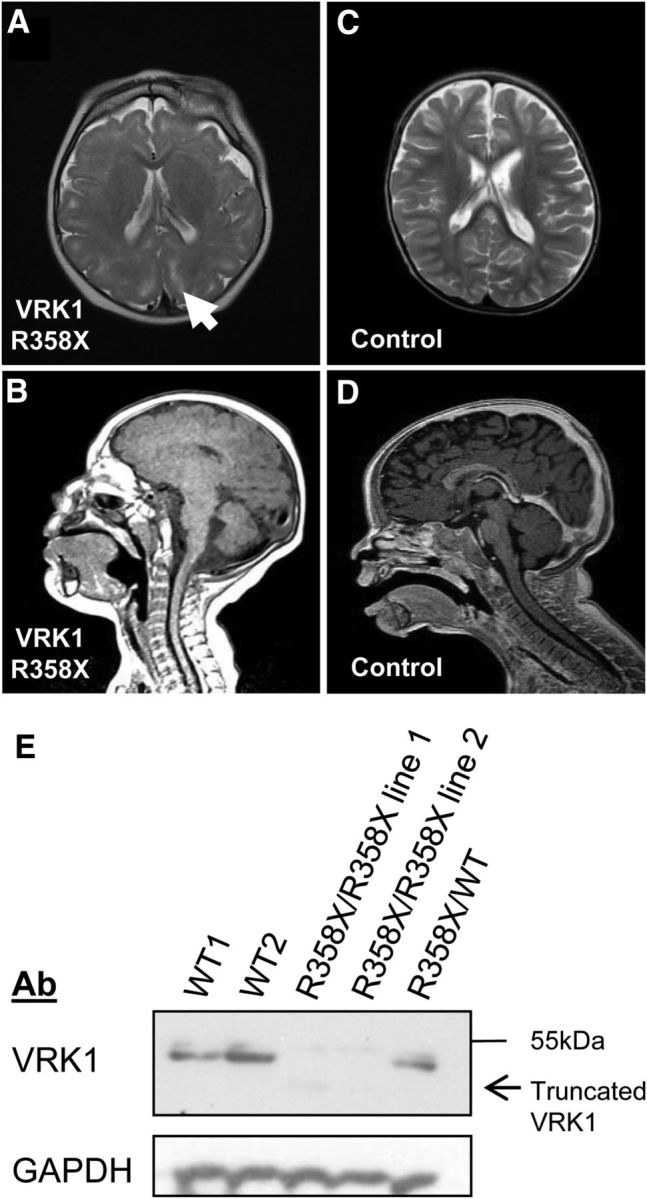
Brain MRI and VRK1 expression in a VRK1-R358X homozygote. A-D, Brain MRI (age 1 year). A, Axial T2-weighted image shows simplified gyral pattern and enhancement of the perivascular spaces in the subcortical zones (arrow). B, Midsagittal T1-weighted image shows posterior vermis hypoplasia. C–D, Images from an age-matched, healthy child. E, VRK1-R358X is a null mutation. Representative Western blot of endogenous VRK1 and GAPDH in B-cell lysates from two healthy individuals (WT1, WT2), two VRK1-R358X homozygous cell lines, and a VRK1-R358X carrier. In homozygous VRK1-R358X cell lines, mean VRK1 levels are reduced by 18.5-fold (vs WT cell lines) and there is a barely detectable ∼41 kDa band (consistent with truncated mutant VRK1).
The microcephaly observed in SMA-PCH patients is consistent with VRK1's established role in cell proliferation (Valbuena et al., 2011) (although this has not been studied in neurons). However, the simplified gyral pattern observed in this and other cases of VRK1 mutations (Gonzaga-Jauregui et al., 2013) suggested a possible role for VRK1 in neuronal migration.
VRK1-R358X is a null mutation
We have previously shown that the R358X transcript undergoes nonsense-mediated decay, and that overexpressed VRK1-R358X does not properly localize to the nucleus (Sanz-García et al., 2011). Consistent with these observations, expression arrays in lymphoblastoid cell lines from the VRK1-R358X homozygote showed 6.2-fold reduced VRK1 expression, compared with VRK1 wild-type (WT) controls (confirmed by real-time qPCR; data not shown). VRK1 protein is barely detectable in VRK1-R358X homozygous cells (Fig. 1E). Together, these results demonstrate that VRK1-R358X is essentially a null mutation.
Global mRNA expression in VRK1 homozygous cells reveals altered Reelin signaling
Comparison of global gene expression from VRK1-R358X homozygote and VRK1 WT control B-cells revealed 414 genes upregulated or downregulated by ≥2-fold (p < .05), and 55 genes up-regulated or down-regulated by >5-fold (p < 0.01; full data accessible at Gene Expression Omnibus). Pathway analysis showed that the Reelin signaling was the pathway most significantly associated with expression differences in VRK1-mutant cells (p < 0.002; Table 1). Reelin signaling is required for proper positioning of neurons within the laminated telencephalon (Rice and Curran, 2001; Frotscher et al., 2009), and also plays a role in neuronal migration in the spinal cord (Yip et al., 2009). The association of altered Reelin pathway expression and the simplified gyral patterns observed in brain MRIs of affected individuals (Fig. 1A–D; Gonzaga-Jauregui et al., 2013), strengthened the putative role for VRK1 in Reelin signaling and neuronal migration.
Table 1.
Altered expression of Reelin signaling-associated genes in a VRK1 R358X mutant cell line
| Gene symbol | Global expression array fold changea | Quantitative real-time PCR fold change (±SD)b |
|---|---|---|
| APP | −38.2* | −333.4 (±52.3)* |
| PIK3CG | −2.6** | −2.3 (±0.3)** |
| CNR1 | 2.0** | 9.8 (±6.5)c |
| APBB1 | 2.1* | 77.3 (±36.5)* |
| FYN | 2.3* | 6.9 (±0.5)** |
| ARHGEF3 | 4.1* | 7.1 (±2.4)* |
aVRK1 R358X cell line versus mean of two different control cell lines.
bVRK1 R358X cell line versus mean of three different control cell lines.
cNot significant.
*p < 0.05,
**p < 0.001.
Vrk1 downregulation inhibits neuronal migration in the cerebral cortex of mouse embryos
Since VRK1-R358X is a null mutation (Fig. 1E), we used IUE of Vrk1 shRNA to knockdown Vrk1 and study the role of this protein in developing mouse brains. Among five Vrk1-shRNAs analyzed in mouse 3T3 cells, F10 shRNA was chosen for further experiments since it caused the greatest reduction of Vrk1 transcript, with a mean 8.6-fold reduction of Vrk1 protein (Fig. 2A). F10 shRNA was specific for murine transcripts: human VRK1 mRNA was resistant to F10 transfection into human HEK293 cells (Fig. 2B). For neuronal migration studies, F10 shRNA was introduced with GFP into the lateral brain ventricles of mouse embryos at embryonic day (E)14 and brains were examined on E18. The location of GFP-stained neurons, which labeled Vrk1-shRNA-treated cells, was quantified by measuring the fluorescence intensity in eight arbitrary bins across the width of the cerebral cortex (Fig. 2C′–H′). In control brains (IUE of control shRNA), 32.7 ± 2.8% of GFP-positive neurons were localized to the cortical plate (CP; Fig. 2C,C′, bins 1, 2), whereas 23 ± 3.1% were in the ventricular (VZ) and subventricular zones (SVZ; Fig. 2C,C′, bins 7, 8). Vrk1 knockdown resulted in strong inhibition of neuronal migration: only 17.1 ± 3.5% of GFP-positive neurons in F10-treated brains localized to the CP (bins 1 and 2) with 42.1 ± 7.9% in the SVZ/VZ (Fig. 2C,C′,D,D′, bins 7, 8). Differences between control shRNA and F10 Vrk1-shRNA-treated brains were significant in each of the 8 bins (Fig. 2D′). Similar results were obtained with an additional Vrk1-shRNA (data not shown).
Figure 2.
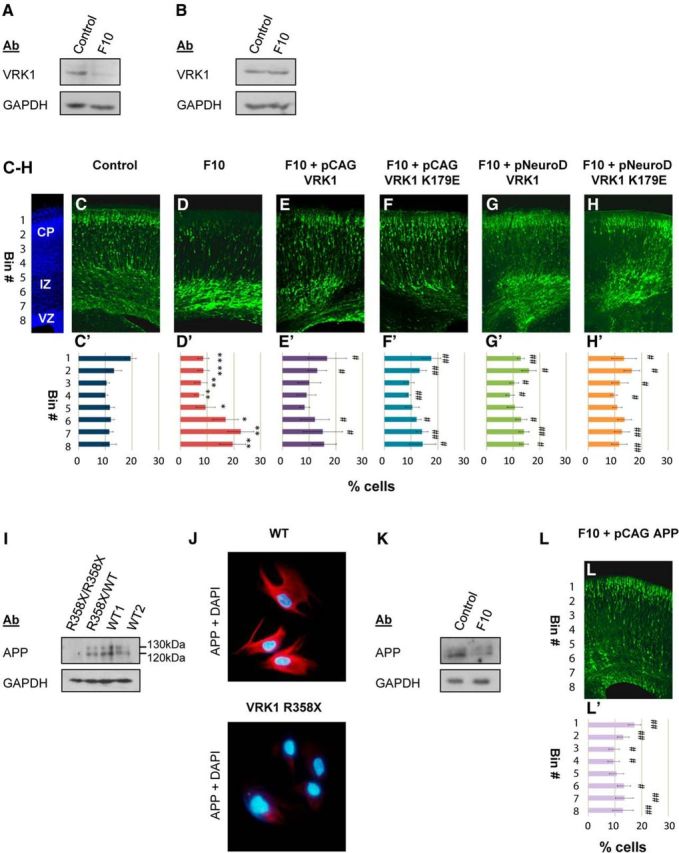
Knockdown of Vrk1 affects neuronal migration through an APP-dependent mechanism. A, Representative Western blot of lysates from murine 3T3 cells transfected with F10 or control shRNA shows F10 shRNA reduces Vrk1 protein. B, Representative Western blot of lysates from HEK293 cells transfected with F10 or control shRNA shows human VRK1 is resistant to F10 shRNA. C–H, Representative mouse brain slices following IUE of control shRNA (C; N = 5 brains), F10 shRNA (D; N = 6), F10 shRNA with various human VRK1 rescue vectors: CAG-driven WT-VRK1 (E; N = 3), CAG-VRK1–K179E (F; N = 3), NeuroD-driven WT-VRK1 (G; N = 3), NeuroD-VRK1–K179E (H; N = 3). C′–H′, Quantification of GFP signal detected in eight arbitrary bins spanning the width of the cortex, from ≥3 independent brains per treatment. Error bars indicate standard deviation. D′, F10 versus control shRNA (*p < 0.05, **p < 0.001). E′–H′, F10 shRNA with the rescue vector indicated versus F10 shRNA only (#p < 0.05, ##p < 0.001). CP, cortical plate; IV, intermediate zone; VZ,ventricular zone. I, Representative Western blot of APP in lysates of B-cell lines homozygous, heterozygote, or WT for VRK1-R358X. In the VRK1-R358X homozygote, levels of both APP isoforms (120 and 130 kDa) are reduced, by 6.4-fold and 23-fold respectively. J, WT and VRK1-R358X homozygous primary fibroblasts stained with anti-APP (red)/DAPI (blue). K, Representative Western blot of lysates from GFP-positive cells of mouse embryo brains after IUE with control shRNA or F10 shRNA. L, L′, IUE of F10 shRNA with CAG-driven human APP. L, Representative brain slice. L′, Eight-bin analysis of GFP-stained cells, compared with F10 shRNA alone (#p < 0.05, ##p < 0.001; N = 4 brains).
Rescue experiments with human VRK1 expression vectors resistant to the murine-specific F10 shRNA vector allowed us to address several issues. Vrk1 knockdown specificity at E14 was addressed by rescue with a constitutive (CAG promoter) human VRK1 construct (Fig. 2E). The necessity for VRK1 kinase activity in migrating neurons was examined using a constitutively expressed kinase-dead VRK1 (K179E; Fig. 2F; Sanz-García et al., 2011). Furthermore, using the postmitotic NeuroD promoter (Lee et al., 1995) allowed us to probe the need for VRK1 protein (with or without kinase activity) in migrating cells and not in stem cell progenitors (Fig. 2G–H). Expression of either WT-VRK1 or VRK1-K179E, driven by either a constitutive or postmitotic promoter, resulted in significant, partial rescue of the abnormal migration phenotype (Fig. 2E–H). When VRK1 rescue vectors were added to Vrk1 shRNA, the proportion of GFP stained cells in the CP (bins 1 and 2) increased to 29.8 ± 8% with pCAG WT-VRK1 (Fig. 2E′), 28.9 ± 3.3% with pNeuroD WT-VRK1 (Fig. 2G′), 30.8 ± 5% with pCAG VRK1-K179E (Fig. 2F′), and 29.8 ± 6.3% with pNeuroD VRK1-K179E (Fig. 2H′). There were also fewer GFP-positive cells in the SVZ/VZ: 30.7 ± 9% with pCAG WT-VRK1, 28.2 ± 3.1% with pNeuroD WT-VRK1, 28.4 ± 5.4% with pCAG VRK1-K179E, and 24.3 ± 5.5% with pNeuroD VRK1-K179E (Fig. 2E′–H′). Differences between F10 shRNA alone and the rescue treatments were significant in ≥4 bins for each treatment (Fig. 2E′–H′). Rescue with either postmitotic (NeuroD) or constitutive (pCAG) VRK1-driven expression was similar. Collectively these results suggest that VRK1 is important for migrating neurons independently of cell proliferation and that kinase activity may not be required for proper radial neuronal migration.
Amyloid-β precursor protein (APP) is regulated by Vrk1, and APP overexpression rescues the Vrk1 knockdown neuronal migration phenotype
APP was the most downregulated gene in the VRK1-R358X homozygous B-cell line. APP expression was shown to be reduced by 38.2-fold in the expression array (p < 0.000005), and by 333-fold using real-time qPCR (Table 1). APP protein expression was assessed by Western blot of lysates from the R358X homozygous and control B-cell lines. While two APP isoforms were clearly detected in two WT controls and a VRK1-R358X heterozygote, only one isoform (120 kDa) was weakly detected in the VRK1-R358X homozygous cells (a 6.4-fold reduction; Fig. 2I). In VRK1-R358X homozygous primary fibroblasts, immunostaining with anti-APP antibodies resulted in a barely detectable signal compared with WT fibroblasts (Fig. 2J).
APP is crucial for neuronal migration; APP knockdown in mouse embryo brains inhibits migration of neuronal precursor cells to the CP (Young-Pearse et al., 2007). We confirmed reduced APP expression in Vrk1-shRNA IUE brains (Fig. 2K). The GFP-positive areas of cortices in utero electroporated at E14 with Vrk1 F10 shRNA or control shRNA and a GFP vector were isolated at E16 and analyzed by Western blot. APP levels were reduced by threefold in the brain regions transfected with Vrk1-shRNA compared with controls (Fig. 2K).
We hypothesized that if APP levels decreased due to VRK1 mutation in human cells or following knockdown in murine brains, then perhaps addition of human APP would rescue the position of migrating neurons in mouse embryonic brains. Therefore, Vrk1-F10 shRNA and the GFP vector were in utero electroporated with or without a human APP expression vector. APP overexpression significantly rescued the localization of the GFP-positive neurons caused by Vrk1 knockdown (Fig. 2L,L′): GFP-positive cells in the CP (bins 1 and 2) increased to 30.5 ± 3.3% and only 26.5 ± 6.4% of the cells remained in the SVZ/VZ (bins 7 and 8) compared with 17.1 ± 3.5% in the CP and 42.1 ± 7.9% in the SVZ/VZ with F10 shRNA alone (Fig. 2D′). Differences between F10 shRNA alone or with APP rescue were significant in seven of eight bins. These results suggest that VRK1 affects neuronal migration via an APP-dependent mechanism.
Vrk1 downregulation affects the cell cycle of neural progenitors in the mouse developing cortex
VRK1 is important for cell cycle regulation and mitosis (Valbuena et al., 2011), and the severe microcephaly observed in patients with VRK1 mutations could be related to impaired neuronal proliferation. The effect of Vrk1 knockdown on neuronal proliferation in the mouse developing cortex was examined using modified FUCCIs (Sakaue-Sawano et al., 2008). These short-lived fluorescent proteins allow visualization of cells in G1 (red), G1-to-S transition (yellow, simultaneous expression of the red and the green fluorescent proteins), and S,G2,M (green, Fig. 3E). FUCCI cell cycle reporter plasmids were introduced into E13 developing brains together with either control or Vrk1-F10 shRNA. Analysis at E15 revealed that Vrk1 knockdown was associated with an increased proportion of cells at the S, G2, or M phases: 32% versus 27% in controls (p = 0.0045; Fig. 3A,A′,B,B′). The combined proportion of cells in G1 and in G1-to-S transition was concomitantly significantly reduced, but separately, each of these cell cycle phases did not differ between Vrk1-F10 shRNA and control brains. We attempted to rescue Vrk1 knockdown by CAG-driven VRK1, either WT or kinase-dead. While addition of WT-VRK1 completely rescued the phenotype, the catalytically inactive protein did not. In Vrk1-F10 shRNA brains rescued with WT-VRK1, proportion of cells in the S, G2, or M phases was 26%, which was similar to the proportion in control brains (Fig. 3C,C′). In brains electroporated with Vrk1-F10 shRNA and VRK1-K179E, no rescue was observed, and 35% of cells were in the S, G2, or M phase (Fig. 3D,D′, green). These results suggest that VRK1 functions in cell cycle progression of neural progenitors, and that this role depends on its catalytic kinase activity, similar to other known functions of VRK1 in cellular proliferation (Valbuena et al., 2011).
Figure 3.
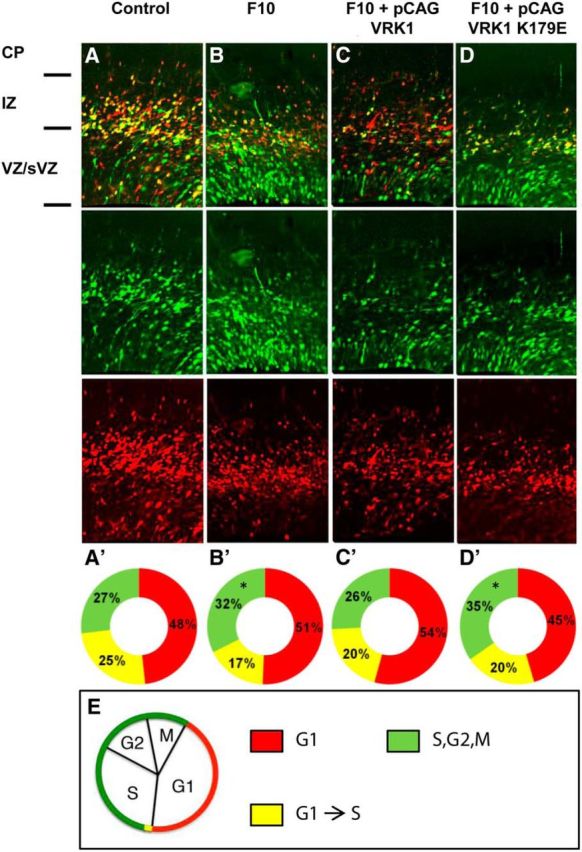
Vrk1 knockdown affects cell cycle in the developing mouse brain. A–D, Representative mouse brain slices after IUE with FUCCI cell cycle markers and control shRNA (A; N = 4 brains), F10 shRNA (B; N = 3), F10 and CAG-driven WT VRK1 (C; N = 3), F10 and CAG-driven VRK1–K179E (D; N = 3). Top, Merged image of: Middle, green channel, mVenus-hGeminin (S,G2,M)-positive cells and Bottom, red channel, mCherry-hCdt1(G1)-positive cells. A′–D′, The distribution of the proportion of cells expressing each of the FUCCI reporters (counted in separate channels) is represented in circular graphs. Red indicates mCherryCdt1+ (G1) cells. Green indicates mVenus-hGeminin+ (S/G2/M) cells. Yellow indicates that both markers are expressed (G1-to-S transition). Asterisks indicate significant changes compared with control (*p < 0.005). E, Schematic presentation showing the appearance of FUCCI chimeric markers during the cell cycle. CP, cortical plate; IZ, intermediate zone; VZ/sVZ, ventricular and subventricular zones.
Discussion
This work provides experimental evidence that VRK1 plays an essential role in development of the cerebral cortex. Recessive VRK1 mutations cause SMA-PCH (Renbaum et al., 2009), a disease associated with severe congenital microcephaly, and limited to the nervous system. We hypothesized a role for VRK1 in neuronal migration based on brain MRIs of affected children (Fig. 1A–D; Gonzaga-Jauregui et al., 2013) and the observed effect of the mutation on expression of genes associated with the Reelin pathway. Our results also suggested that VRK1 R358X is a null allele, and furthermore, any residual mutant protein that may be produced will not enter the nucleus (Sanz-García et al., 2011). Therefore, we proposed to model the effects of the human disease in the developing mouse brain by knockdown experiments. Our results show that Vrk1 knockdown strongly impairs neuronal migration, and human VRK1 (resistant to the shRNA) significantly rescues migration deficits when expressed in neural progenitors or in postmitotic neurons. In rescue experiments with kinase-dead VRK1, we found that it can also significantly rescue the position of migrating neurons. These results provide evidence that VRK1 plays a critical role in postmitotic neuronal migration, which is at least partly independent of its kinase activity.
VRK1-R358X cells showed an extreme reduction in the mRNA and protein levels of APP. These results were recapitulated in mouse embryonic brains electroporated with Vrk1-shRNA. Moreover, human APP rescued the impaired neuronal migration phenotype caused by Vrk1 knockdown. Collectively, our results suggest that APP acts, at least partially, downstream of VRK1 in regulating neuronal migration. APP is a type-I transmembrane glycoprotein expressed throughout the developing and adult brain (De Strooper and Annaert, 2000). APP is best known for its role in Alzheimer's disease: amyloid-β protein (Aβ), whose deposition in the brain is a hallmark of this disease, is produced by proteolytic cleavage of APP, and germline APP mutations can cause inherited, early-onset Alzheimer's disease. In early development, APP is important for neuronal migration. APP knock-down inhibits entry of migrating neurons into the CP, whereas APP overexpression accelerates migration of cells past the CP boundary, suggesting a requirement for balanced APP levels for correct neuronal migration (Young-Pearse et al., 2007). Mice lacking all three APP family members (APP, APLP1, and APLP2) have a phenotype resembling human type II cobblestone lissencephaly (Herms et al., 2004). APP binds Dab1, the intracellular mediator of Reelin signaling (Homayouni et al., 1999). Dab1 overexpression rescued migration deficits observed with APP knockdown, suggesting that Dab1 acts downstream of APP (Young-Pearse et al., 2007). Collectively, these data strongly suggest that APP affects neuronal migration through the Reelin-signaling pathway. Beyond Dab1, APP has been shown to interact with Pancortins to regulate neuronal entry into the CP (Rice et al., 2012); and in the hawkmoth Manduca, proper neuronal migration depends on the interaction between the insect APP (APPL) and the heterotrimeric G-protein Goα, suggesting that APP and its orthologs regulate neuronal migration by functioning as unconventional Goα-coupled receptors (Ramaker et al., 2013). This is of particular interest, since the second most significantly affected pathway in the SMA-PCH patient is G-protein-coupled receptor signaling.
We found that VRK1 deficiency results in extremely reduced levels of APP mRNA, which could result from reduced transcription and/or reduced message stability. These VRK1 effects are most likely to be indirect, and require further investigation. Since a kinase-dead VRK1 mutant could rescue the migration phenotype caused by murine Vrk1 knockdown, we speculate that VRK1 affects APP expression or other pathways at least partially independent of its kinase activity. Many catalytic kinases also have noncatalytic activities, including scaffolding of protein complexes, competition for protein interactions, allosteric effects on other enzymes, subcellular targeting, and DNA binding (Sapir et al., 2008, Rauch et al., 2011).
VRK1 has known roles in cellular proliferation, and we show that it also affects proliferation of neural progenitors. Vrk1 knockdown significantly increased the proportion of cells in phases S, G2, or M. VRK1 knockdown in human cell lines stalls cells at G1, and decreases the proportion of cells in S (Kang et al., 2008; Molitor and Traktman, 2013). In the G2 and M phases, VRK1 depletion leads to disordered mitosis: Golgi fragmentation is inhibited (López-Sánchez et al., 2009), and BAF–chromatin interaction is prolonged, leading to aberrant nuclear envelope architecture, and increased anaphase bridges and multipolar spindles (Molitor and Traktman, 2014). Together with reported observations, our results are most consistent with Vrk1 knockdown leading to a stall at the important checkpoint for arrest at G2/M. Cell cycle progression was rescued by WT-VRK1, but not by the kinase-dead mutant K179E. This is consistent with known VRK1 actions in cell cycle regulation, which are based on phosphorylating key target proteins, such as BAF, CREB, and histone H3 (for review, see Valbuena et al., 2011). Further studies are necessary to fully delineate the proliferative role of VRK1 in neurons and to determine whether impaired proliferation underlies VRK1-associated microcephaly.
This study provides the first insight into the molecular mechanism of VRK1-related neurologic disease. We found that VRK1 plays a role in neuronal proliferation, paralleling its previously known functions, but also in neuronal migration. While the role of VRK1 in neuronal proliferation depends on its kinase activity, its role in neuronal migration is at least partly noncatalytic and upstream of APP. Further study is required to delineate the role of VRK1 in regulating APP expression and in the other features of VRK1-associated disease, including SMA.
Footnotes
This study was supported by grants from the Israel Science Foundation (Grant 702/13 to P.R. and E.L.-L., and Grant 47/10 and 322/13 to O.R. and T.S.); by the Minerva Foundation with funding from the Federal German Ministry for Education and Research, the Jérôme Lejeune Foundation, the Benoziyo Center for Neurological diseases, the Kekst Family Center for Medical Genetics, and the David and Fela Shapell Family Center for Genetic Disorders Research (to O.R.); by the JAE (Programa Junta para la Amplición de Estudios) Consejo Superior de Investigaciones Científicas Fondo Social Europeo fellowship (to L.C.); by the Ministerio de Educación, Ciencia e Innovación (SAF2010-14935 and SAF2013-44810R, to P.A.L.); and by Kutxa-Fundación Inbiomed (to P.A.L.). We thank Vennasa Zamor for technical assistance.
The authors declare no competing financial interests.
References
- Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411–422. doi: 10.1016/0387-7604(93)90080-R. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Chai X, Bock HH, Haas CA, Förster E, Zhao S. Role of Reelin in the development and maintenance of cortical lamination. J Neural Transm. 2009;116:1451–1455. doi: 10.1007/s00702-009-0228-7. [DOI] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Lotze T, Jamal L, Penney S, Campbell IM, Pehlivan D, Hunter JV, Woodbury SL, Raymond G, Adesina AM, Jhangiani SN, Reid JG, Muzny DM, Boerwinkle E, Lupski JR, Gibbs RA, Wiszniewski W. Mutations in VRK1 associated with complex motor and sensory axonal neuropathy plus microcephaly. JAMA Neurol. 2013;70:1491–1498. doi: 10.1001/jamaneurol.2013.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Müller U. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23:4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Park DY, Kim W, Kim KT. VRK1 phosphorylates CREB and mediates CCND1 expression. J Cell Sci. 2008;121:3035–3041. doi: 10.1242/jcs.026757. [DOI] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- López-Sánchez I, Sanz-García M, Lazo PA. Plk3 interacts with and specifically phosphorylates VRK1 in Ser342, a downstream target in a pathway that induces Golgi fragmentation. Mol Cell Biol. 2009;29:1189–1201. doi: 10.1128/MCB.01341-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitor TP, Traktman P. Molecular genetic analysis of VRK1 in mammary epithelial cells: depletion slows proliferation in vitro and tumor growth and metastasis in vivo. Oncogenesis. 2013;2:e48. doi: 10.1038/oncsis.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitor TP, Traktman P. Depletion of the protein kinase VRK1 disrupts nuclear envelope morphology and leads to BAF retention on mitotic chromosomes. Mol Biol Cell. 2014;25:891–903. doi: 10.1091/mbc.E13-10-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, Zecha A, Mohseni M, Püttmann L, Vahid LN, Jensen C, Moheb LA, Bienek M, Larti F, Mueller I, Weissmann R, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- Nezu J, Oku A, Jones MH, Shimane M. Identification of two novel human putative serine/threonine kinases, VRK1 and VRK2, with structural similarity to vaccinia virus B1R kinase. Genomics. 1997;45:327–331. doi: 10.1006/geno.1997.4938. [DOI] [PubMed] [Google Scholar]
- Ramaker JM, Swanson TL, Copenhaver PF. Amyloid precursor proteins interact with the heterotrimeric G protein Go in the control of neuronal migration. J Neurosci. 2013;33:10165–10181. doi: 10.1523/JNEUROSCI.1146-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J, Volinsky N, Romano D, Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun Signal. 2011;9:23. doi: 10.1186/1478-811X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renbaum P, Kellerman E, Jaron R, Geiger D, Segel R, Lee M, King MC, Levy-Lahad E. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet. 2009;85:281–289. doi: 10.1016/j.ajhg.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Rice HC, Townsend M, Bai J, Suth S, Cavanaugh W, Selkoe DJ, Young-Pearse TL. Pancortins interact with amyloid precursor protein and modulate cortical cell migration. Development. 2012;139:3986–3996. doi: 10.1242/dev.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S, Sztriha L, Aithala GR, Houge G, Laegreid LM, Seeger J, Huppke M, Wirth B, Zerres K. Extended phenotype of pontocerebellar hypoplasia with infantile spinal muscular atrophy. Am J Med Genet A. 2003;117a:10–17. doi: 10.1002/ajmg.a.10863. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Santos CR, Rodríguez-Pinilla M, Vega FM, Rodríguez-Peralto JL, Blanco S, Sevilla A, Valbuena A, Hernández T, van Wijnen AJ, Li F, de Alava E, Sánchez-Céspedes M, Lazo PA. VRK1 signaling pathway in the context of the proliferation phenotype in head and neck squamous cell carcinoma. Mol Cancer Res: MCR. 2006;4:177–185. doi: 10.1158/1541-7786.MCR-05-0212. [DOI] [PubMed] [Google Scholar]
- Sanz-García M, Vázquez-Cedeira M, Kellerman E, Renbaum P, Levy-Lahad E, Lazo PA. Substrate profiling of human vaccinia-related kinases identifies coilin, a Cajal body nuclear protein, as a phosphorylation target with neurological implications. J Proteomics. 2011;75:548–560. doi: 10.1016/j.jprot.2011.08.019. [DOI] [PubMed] [Google Scholar]
- Sapir T, Sapoznik S, Levy T, Finkelshtein D, Shmueli A, Timm T, Mandelkow EM, Reiner O. Accurate balance of the polarity kinase MARK2/Par-1 is required for proper cortical neuronal migration. J Neurosci. 2008;28:5710–5720. doi: 10.1523/JNEUROSCI.0911-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valbuena A, López-Sánchez I, Vega FM, Sevilla A, Sanz-García M, Blanco S, Lazo PA. Identification of a dominant epitope in human vaccinia-related kinase 1 (VRK1) and detection of different intracellular subpopulations. Arch Biochem Biophys. 2007;465:219–226. doi: 10.1016/j.abb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Valbuena A, Sanz-García M, López-Sánchez I, Vega FM, Lazo PA. Roles of VRK1 as a new player in the control of biological processes required for cell division. Cell Signal. 2011;23:1267–1272. doi: 10.1016/j.cellsig.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Wee CD, Kong L, Sumner CJ. The genetics of spinal muscular atrophies. Curr Opin Neurol. 2010;23:450–458. doi: 10.1097/WCO.0b013e32833e1765. [DOI] [PubMed] [Google Scholar]
- Yip YP, Mehta N, Magdaleno S, Curran T, Yip JW. Ectopic expression of reelin alters migration of sympathetic preganglionic neurons in the spinal cord. J Comp Neurol. 2009;515:260–268. doi: 10.1002/cne.22044. [DOI] [PubMed] [Google Scholar]
- Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ. A critical function for β-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. 2007;27:14459–14469. doi: 10.1523/JNEUROSCI.4701-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]