

Abstract
All neurodegenerative diseases are associated with oxidative stress-induced neuronal death. Forkhead box O3a (FOXO3a) is a key transcription factor involved in neuronal apoptosis. However, how FOXO3a forms complexes and functions in oxidative stress processing remains largely unknown. In the present study, we show that histone deacetylase 2 (HDAC2) forms a physical complex with FOXO3a, which plays an important role in FOXO3a-dependent gene transcription and oxidative stress-induced mouse cerebellar granule neuron (CGN) apoptosis. Interestingly, we also found that HDAC2 became selectively enriched in the promoter region of the p21 gene, but not those of other target genes, and inhibited FOXO3a-mediated p21 transcription. Furthermore, we found that oxidative stress reduced the interaction between FOXO3a and HDAC2, leading to an increased histone H4K16 acetylation level in the p21 promoter region and upregulated p21 expression in a manner independent of p53 or E2F1. Phosphorylation of HDAC2 at Ser 394 is important for the HDAC2–FOXO3a interaction, and we found that cerebral ischemia/reperfusion reduced phosphorylation of HDAC2 at Ser 394 and mitigated the HDAC2–FOXO3a interaction in mouse brain tissue. Our study reveals the novel regulation of FOXO3a-mediated selective gene transcription via epigenetic modification in the process of oxidative stress-induced cell death, which could be exploited therapeutically.
Keywords: FOXO3a, HDAC2, oxidative stress, p21, transcription
Introduction
Oxidative stress-induced neuronal death has been implicated as a crucial factor in neuronal diseases, including Alzheimer's disease (AD), Parkinson's disease, and stroke (Markesbery and Carney, 1999; Jenner, 2003; Chong et al., 2005). The primary cause of oxidative stress is an imbalance between the production of reactive oxygen species (ROS) and the elimination of ROS by antioxidants. If excessive ROS production is induced due to aging, environmental changes or ischemia/reperfusion, this balance is lost, damaging cells. Our earlier studies elucidated several pathways that respond to oxidative stress and mediate neuronal death (Lehtinen et al., 2006; Xiao et al., 2011; Liu et al., 2012; Xie et al., 2012).
FOXO3a is an important transcription factor that is involved in the responses to oxidative stress, apoptosis, metabolism, muscle atrophy, lifespan, and other cellular processes (Brunet et al., 2004; Sandri et al., 2004; Gross et al., 2008; Tao et al., 2013). The mechanism by which FOXO3a regulates its downstream genes varies depending on the cell type and the stimulus. Different conditions induce profound effects on FOXO3a activity and target selection. The classical mechanism of FOXO3a activity regulation is mediated by the PI3K/AKT pathway (Brunet et al., 1999). Acetylation and methylation of FOXO3a are also known to affect FOXO3a transcriptional activity (Wang et al., 2007; Calnan et al., 2012). Ubiquitination of FOXO by SKP2 decreases its stability (Huang et al., 2005). Recent reports have proposed that FOXO3a forms distinct complexes to differentially regulate its target genes (Li et al., 2010). The mechanism by which oxidative stress modulates the formation of FOXO3a complexes remains to be elucidated.
It is well known that epigenetic modification regulates gene transcription. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are two classes of enzymes that modulate histone acetylation. Excessive HATs increase the histone acetylation level, leading to chromatin remodeling, and therefore, enhanced transcription complex binding and gene transcription. HDACs reverse this process and silence gene transcription (Narlikar et al., 2002). HDAC inhibitors (HDACis) kill cancer cells by promoting cell-cycle repressor and apoptotic gene expression (Richon et al., 2000; Zhao et al., 2005; Khan and La Thangue, 2012). Interestingly, HDACis, such as trichostatin A and sodium butyrate, protect neurons from oxidative stress in vitro and in vivo (Langley et al., 2008). Some evidence showed that the p53/PUMA signaling pathway contributes to the neuroprotective activity of HDACis (Uo et al., 2009; Brochier et al., 2013). The endogenous HDACi d-β-hydroxybutyrate was found to protect mice from oxidative stress via FOXO3a and its downstream genes (Shimazu et al., 2013).
In this study, we reveal that HDAC1 and HDAC2 interact with FOXO3a. HDAC2 mediates oxidative stress-induced neuronal apoptosis in a FOXO3a-dependent manner. H2O2 attenuates the interaction between FOXO3a and HDAC2. The release of HDAC2 leads to increased expression of p21, and p21 is partially responsible for the beneficial effects of HDAC2 knockdown. HDAC2 phosphorylation at S394 affects its binding affinity to FOXO3a. We propose a link between FOXO3a and an epigenetic modification that mediates the cellular response to oxidative stress and neuronal apoptosis.
Materials and Methods
Drugs.
The following drugs were purchased: Ms-275 (Selleckchem), tetrabromocinnamic acid (TBCA; Millipore), and H2O2 (Acros Organics).
Animals.
p21−/− mice and p53−/−/E2F1−/− mice were maintained in our Animal Care Facility. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Institute of Biophysics at the Chinese Academy of Sciences.
Plasmids.
The use of GFP, Flag-FOXO3a, and GST-FOXO3a P1-P5 plasmids in a 3xIRS luciferase assay was described previously (Lehtinen et al., 2006). The Hdac1, Hdac2, and Sirt1 cDNAs were cloned into the pCMV10–3xFlag vector (Sigma-Aldrich). All small hairpin RNA (sh-RNA) fragments were inserted to the pLKO.1 vector between its EcoRI and AgeI sites. The targeting sequence of each construct is listed as follows:
Sh-FOXO: GAGCGTGCCCTACTTCAAGG;
Sh-HDAC1-1#: GATGTTGGAAACTACTATTAT;
Sh-HDAC1-2#: GCAGATGCAGAGATTCAATGT;
Sh-HDAC2-1#: CGAGCATCAGACAAACGGATA;
Sh-HDAC2-2#: CCCAATGAGTTGCCATATAAT;
Sh-p53: GTACATGTGTAATAGCTCCTG.
All mutations were generated using a Site-Directed Mutagenesis Kit (Stratagene). The primers used for mutagenesis were as follows:
mHDAC2-rescue-F: 5-AGAATTTCCATTCGAGCGAGCGATAAACGGATAGCTTGCGAT-3, mHDAC2-rescue-R: 5-ATCGCAAGCTATCCGTTTATCGCTCGCTCGAATGGAAATTCT-3; mHDAC2-S394A-mut-F: 5-GCTGTTCATGAAGACGCTGGAGATGAGGATGGA-3, mHDAC2-S394A-mut-R: 5-TCCATCCTCATCTCCAGCGTCTTCATGAACAGC-3;
mHDAC2-S394D-mut-F: 5-GCTGTTCATGAAGACGACGGAGATGAGGATGGA-3, mHDAC2-S394D-mut-R: 5-TCCATCCTCATCTCCGTCGTCTTCATGAACAGC-3.
Tandem affinity purification.
The tandem affinity purification (TAP) assay was described previously (Wang et al., 2011). In brief, 8 L of suspended HeLa cells stably transfected with pOZ-Flag-HA-FOXO3a were collected and washed with cold phosphate buffer solution. The cells were treated with 45 ml of hypotonic buffer (10 mm Tris-HCl, pH 7.3, 10 mm KCl, 1.5 mm MgCl2). They were homogenized, and the nuclear fraction was collected. The FOXO3a complex was immunoprecipitated from the nuclear extracts using anti-Flag (Sigma-Aldrich) and anti-HA beads (Santa Cruz Biotechnology) sequentially. Protease and phosphatase inhibitors were added to all of the solutions in this experiment. The final eluate from the anti-HA beads was digested using trypsin and subsequently analyzed via mass spectrometry.
Cell culture, transfection, and cell death assay.
The 293T and HT-22 cell lines were cultured in high-glucose DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 100 U/ml penicillin/streptomycin. The cells were cultured in an incubator at 37°C supplied with 5% CO2. The 293T and HT-22 cells were transfected using Lipofectamine 2000 reagent (Invitrogen). The isolation and culture of cerebellar granule neurons was previously described (Konishi et al., 2002). In brief, CGNs were obtained from 7-d-old C57BL/6 mice unless stated otherwise. After 3 d in vitro (DIV), the CGNs were transfected as described previously (Konishi et al., 2002). Under these conditions, the plasmids were effectively cotransfected into the neurons. Transfected 6 DIV CGNs were treated with H2O2 (60–100 μm) diluted in DMEM immediately before use. After H2O2 treatment for 24 h, the neuronal apoptosis assay was performed using a Zeiss Imager D1 microscope as described previously (Xie et al., 2012). The neurons were classified as live or dead neurons according to the appearance of Hoechst 33258 dye (Sigma-Aldrich) staining. GFP-positive neurons were counted in a blinded manner. In total, 150–200 cells per slide were counted for each experiment.
Lentivirus packaging and production of stable sh-RNA-expressing cell clones.
A pLKO.1 vector containing an sh-RNA sequence was cotransfected with VSV-G and pCMV-dR8.12 plasmids into 293T cells. Viruses were collected from the supernatant at 36 and 72 h after transfection. Ultracentrifugation was performed to enrich the virus concentration. Stably knocked down HT-22 cell clones were established by infecting the cells with the corresponding lentivirus. A virus produced from the pLKO.1 empty vector was used to generate control cells. The cells were selected in complete medium containing 2 μg/ml puromycin 24 h after infection.
RNA extraction, reverse transcription, and quantitative real-time PCR.
Total RNA was isolated from cells using TRIzol reagent (Invitrogen), and reverse transcription was performed using the one step first strand cDNA synthesis kit (Transgen). Quantitative real-time PCR (qRT-PCR) was performed using 2× SYBR Green PCR master mix (Transgen) in an Agilent Mx3005P qRT-PCR system. The melting temperature profiles of the final products were used to ensure amplicon specificity. The relative fold-change in the expression of each mRNA was calculated using the ddCt method relative to the expression of Gapdh. The qRT-PCR primers used are listed as follows:
mBcl6-realtime-F: 5-CCGGCACGCTAGTGATGTT-3;
mBcl6-realtime-R: 5-TGTCTTATGGGCTCTAAACTGCT-3;
mBim-realtime-F: 5-GAAGACCACCCTCAAATGGTTAT-3,
mBim-realtime-R: 5-TTCTCCATACCAGACGGAAGAT-3;
mbNIP3-realtime-F: 5-CTGGGTAGAACTGCACTTCAG-3,
mbNIP3-realtime-R: 5-GGAGCTACTTCGTCCAGATTCAT-3;
mBtg1-realtime-F: 5-CCACCATGATAGGCGAGATCG-3,
mBtg1-realtime-R: 5-TGCGAATACAACGGTAACCTG-3;
mCcnd1-realtime-F: 5-GCGTACCCTGACACCAATCTC-3,
mCcnd1-realtime-R: 5-CTCCTCTTCGCACTTCTGCTC-3;
mCcnd2-realtime-F: 5-GAGTGGGAACTGGTAGTGTTG-3,
mCcnd2-realtime-R: 5-CGCACAGAGCGATGAAGGT-3;
mFasL-realtime-F: 5-TCCGTGAGTTCACCAACCAAA-3,
mFasL-realtime-R: 5-GGGGGTTCCCTGTTAAATGGG-3;
mGadd45-realtime-F: 5-CCGAAAGGATGGACACGGTG-3,
mGadd45-realtime-R: 5-TTATCGGGGTCTACGTTGAGC-3;
mGapdh-realtime-F: 5-AGGTCGGTGTGAACGGATTTG-3,
mGapdh-realtime-R: 5-GGGGTCGTTGATGGCAACA-3;
mHdac1-realtime-F: 5-TGAAGCCTCACCGAATCCG-3,
mHdac1-realtime-R: 5-GGGCGAATAGAACGCAGGA-3;
mHdac2-realtime-F: 5-ATGGCGTACAGTCAAGGAGG-3,
mHdac2-realtime-R: 5-ATGAGGCTTCATGGGATGACC-3;
mMnsod-realtime-F: 5-CAGACCTGCCTTACGACTATGG-3,
mMnsod-realtime-R: 5-CTCGGTGGCGTTGAGATTGTT-3;
mp21-realtime-F: 5-CCTGGTGATGTCCGACCTG-3,
mp21-realtime-R: 5-CCATGAGCGCATCGCAATC-3;
mp27-realtime-F: 5-TCTCTTCGGCCCGGTCAAT-3,
mp27-realtime-R: 5-AAATTCCACTTGCGCTGACTC-3.
DNA pull-down assay.
A quantity of 1 pm biotin-labeled DNA was incubated with streptavidin-coated beads (Millipore) at room temperature for 15 min with shaking in a solution consisting of 5 mm Tris-HCl, pH 7.0, 0.5 mm EDTA and 1 m NaCl. Then, the beads were incubated in 100 μl of the cell lysates (20 mm Tris-HCl, pH 7.4, 20 mm NaF, 150 mm NaCl, 10% glycerol, 0.5% NP40) combined with 400 μl of incubation buffer (50 mm Tris-HCl, pH 7.0, 1 mm EDTA, 100 mm KCl, 5% glycerol, 0.1% Triton X-100) for 3 h at 4°C. Protease inhibitors were added to the buffers before use. Then, 6× SDS loading buffer for Western blotting was added, and the samples were boiled for electrophoresis. The primers used for the pull-down assay are listed as follows:
mp21-FHRE-biotin-F: 5′-TTCAAGCTGTTTTCTCCCAAAGTAAACAGACAGACAATGTCACTTCTATC-3′;
mp21-FHRE-biotin-R: 5′-GATAGAAGTGACATTGTCTGTCTGTTTACTTTGGGAGAAAACAGCTTGAA-3′.
Coimmunoprecipitation, GST pull-down assay, and Western blot.
The coimmunoprecipitation (co-IP), and GST pull-down protocols were previously described (Bi et al., 2010). The following antibodies were used: antibodies specific to Flag-M2 (Sigma-Aldrich), GFP (Invitrogen), FOXO3a for Western blot, p53, HA, normal mouse and rabbit IgG (Santa Cruz Biotechnology), HDAC2 (Abcam), HDAC1 (Beyotime), GAPDH (CW Biotech), pan-Acetyl (Millipore Bioscience Research Reagents), p21 (BD Biosciences), HDAC2-P394 (Bioworld), and FOXO3a for immunoprecipitation (monoclonal antibody produced by Abmart).
Luciferase reporter assay.
The luciferase reporter assay was performed using a dual luciferase reporter assay kit according to the manufacturer's instructions (Promega). The 3xIRS luciferase sequence was previously described (Tang et al., 1999).
Chromatin immunoprecipitation assay.
The chromatin immunoprecipitation (ChIP) assay was performed using a Millipore ChIP assay kit according to the manufacturer's protocol. The antibodies used were as follows: anti-HDAC2 (Abcam), anti-H4K16ac (Millipore), and normal rabbit IgG (Santa Cruz Biotechnology); 2 μg of the specified antibody was used for each ChIP assay. qRT-PCR was performed to analyze the enrichment of sequences in DNA subjected to ChIP. The primers used for ChIP are as follows:
mBcl6-ChIP-F: 5-CTAGAGTATTAGGCACCACG-3,
mBcl6-ChIP-R: 5-CGCCCCAACATAATTGTCCC-3;
mBim-ChIP-F: 5-GGGCGGGTACATTCTGAGT-3,
mBim-ChIP-R: 5-CAGCACAGGATGTCAAAACT-3;
mp21-ChIP-F: 5-GTAAAGGCACAGGAGGTCAC-3,
mp21-ChIP-R: 5-CAGCACAGGATGTCAAAACT-3;
mp27-ChIP-F: 5-ACGCATCGCTGCTACTACAG-3,
mp27-ChIP-R: 5-CTTAGCCGACCTCACTACGG-3.
In vitro acetylation and deacetylation assays.
A total of 5 μg of the protein substrate was incubated in a solution containing 50 mm Tris-HCl, pH 8.0, 10% glycerol, 0.1 mm EDTA, and 1.0 mm DTT and 1 μg of GST-P300/CBP-associated factor (PCAF) protein at 30°C for 1 h with or without 50 nm acetyl coenzyme A (Ac-CoA).
After incubation, 1 μg of the acetylated protein substrate was incubated in a solution containing 25 mm Tris-HCl, pH 8.0, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2, and 1 μg purified HDAC1/2 or SIRT1 at 30°C for 1 h. An additional 100 μm NAD+ was added to the purified SIRT1. Then, 6× SDS loading buffer was added to stop the reaction. The samples were boiled and subjected to Western blot analysis.
Transient middle cerebral artery occlusion.
Adult (8- to 12-week-old) male C57BL/6 mice weighing 20–25 g were used to establish the transient middle cerebral artery occlusion (tMCAO) model. Before and after surgery, the animals were provided with ad libitum access to food and water. The animals were anesthetized via intraperitoneal injection of avertin (250 mg/kg). The body temperatures were maintained at 37 ± 0.5°C using a feedback-regulated heating pad. Cerebral blood flow was measured via laser Doppler flowmetry. The surgery method was described previously (Zhao et al., 2013). Cerebral blood flow dropped to <20% of the baseline level after placement of the filament (0.22 mm tip) and recovered to >70% after removal of the filament (1 h after ischemia). Twenty-four hours after tMCAO surgery, the mice were killed. The brains were sliced into 7 coronal sections at 1.0 mm intervals using a Brain Slicer Matrix. The third section was used for analysis.
Statistical analysis.
The intensities of the Western blot bands were determined using ImageJ software. Statistical analysis of the data were performed via one-way ANOVA followed by a Bonferroni multiple-comparisons test or via a Student's t test using GraphPad Prism 5 software. The data are presented as the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, or NS (no significant difference) denote the significance thresholds.
Results
FOXO3a interacts with HDAC1 and HDAC2
To identify the proteins that interact with FOXO3a in response to oxidative stress, we performed a TAP assay. We found that some proteins associated with histone modification, including HDAC1 and HDAC2, interact with FOXO3a (Fig. 1A). Then, we performed a co-IP assay to confirm the interactions between HDAC1/2 and FOXO3a. Under ectopic expression conditions in 293T cells, both HDAC1 and HDAC2 formed a complex with FOXO3a (Fig. 1B). To determine the region of FOXO3a that is responsible for its association with HDAC1/2, the FOXO3a sequence was divided into five parts without overlapping as described previously (Lehtinen et al., 2006), and these fragments were subjected to a GST pull-down assay (Fig. 1C). We found that GST-FOXO3a P5 was the primary region responsible for the association between FOXO3a and HDAC1/2.
Figure 1.

FOXO3a interacts with HDAC1 and HDAC2. A, Suspended HeLa cells stably transfected with the pOZ-FOXO3a expression plasmid. A final concentration of 0 or 300 μm H2O2 was added to the medium for 12 h. The final HA-eluates from the nuclear fractions were resolved on a SDS-PAGE gel and silver-stained (left). Some of the identified FOXO3a complex components based on mass-spectral analysis are listed on the right. B, 293T cells were transiently transfected with the GFP-FOXO3a expression plasmid alone or together with the Flag-HDAC1 or Flag-HDAC2 expression plasmid, as indicated. Anti-Flag beads were used to immunoprecipitate the Flag-tagged proteins; anti-GFP and anti-Flag antibodies were used for immunoblotting. C, 293T cells were transiently transfected with the Flag-HDAC1 or Flag-HDAC2 expression plasmid. Cell lysates were incubated with the GST protein or a GST-tagged FOXO3a P1–P5 protein fragment. The anti-Flag antibody was used for immunoblotting. D, 293T cells were transfected with the Flag-HDAC1 or Flag-HDAC2 expression plasmid. Twenty-four hours after transfection, 300 μm H2O2 was added for 12 h, as indicated. The anti-Flag antibody was added to immunoprecipitate the HDAC proteins; the anti-Flag and anti-FOXO3a antibodies were used for immunoblotting. E, CGNs (7 DIV) were treated with 0 or 100 μm H2O2 for 1 h. Cell lysates were immunoprecipitated using an anti-FOXO3a antibody or IgG, and an immunoblot was performed using the indicated antibodies.
Based on the TAP assay, we found that the number of peptide hits of HDAC1 and HDAC2 was dramatically decreased after H2O2 treatment (Fig. 1A). To confirm this result, we conducted co-IP experiments. The binding between HDAC1/2 and FOXO3a was also reduced in 293T cells following exposure to H2O2 (Fig. 1D), indicating that oxidative stress disrupts the interaction between the FOXO3a and HDAC1/2 proteins in cells. We also detected an interaction between endogenous HDAC1/2 and FOXO3a in CGN cells (Fig. 1E). Together, these data suggest that FOXO3a interacts with HDAC1 and HDAC2 and that these interactions are affected by oxidative stress.
HDAC2 knockdown inhibits H2O2-induced neuronal death
FOXO3a is known to mediate neuronal death (Lehtinen et al., 2006), and FOXO3a binding partners may participate in neuronal apoptosis. To determine whether HDAC1 and HDAC2 are involved in regulating H2O2-induced neuronal death, we performed an apoptosis assay using CGNs. Although HDAC1 and HDAC2 are conserved proteins that display high sequence and functional similarities, we found that HDAC1 knockdown did not affect neuronal apoptosis, whereas HDAC2 knockdown significantly protected neurons from H2O2-induced apoptosis (Fig. 2A,B). These results indicated that HDAC2 plays an important role in H2O2-induced neuronal apoptosis. Furthermore, we found that the protective effect of HDAC2 knockdown in H2O2-induced CGN apoptosis was completely reversed by the overexpression of the rescue form of HDAC2, which is completely resistant to HDAC2 RNAi (Fig. 2C,D). To further evaluate whether HDAC1 and HDAC2 are functionally replaceable, we ectopically expressed HDAC1 in CGNs with HDAC2 knocked down (Fig. 2E) and found that the expression of HDAC1 did not reverse the protective effect of HDAC2 knockdown. These results indicate that HDAC1 and HDAC2 are not redundant with respect to the regulation of oxidative stress-induced apoptosis.
Figure 2.
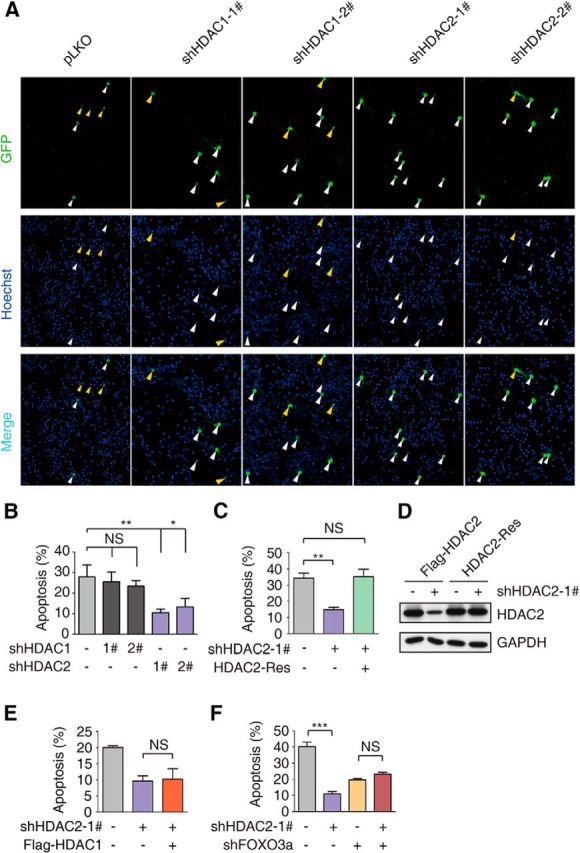
HDAC2 knockdown inhibits H2O2-induced neuronal death. A, CGNs were cotransfected with pEGFP-N1 and pLKO-HDAC1-1#/2#, pLKO-HDAC2-1#/2# or the empty vector pLKO at DIV 3. The neuronal apoptosis assay was performed 24 h after H2O2 (60–100 μm) treatment at DIV 6. Hoechst 33258 was used for nuclear staining, as indicated. The anti-GFP antibody was used for signal enhancement. Apoptotic cells are denoted by yellow arrows, and surviving cells are denoted by white arrows. B, Statistical analysis of A. HDAC2 knockdown protected neurons from oxidative stress-induced apoptosis. (ANOVA, n = 4 for the control group; n = 3 for the HDAC1 and HDAC2 knockdown groups; *p < 0.05, **p < 0.01). C, CGNs were transiently transfected with pEGFP-N1 together with the empty vector pLKO or pLKO-HDAC2–1# or the rescue vector HDAC2-Res as indicated. The neuronal apoptosis assay was performed as in A. HDAC2 knockdown protected neurons from apoptosis (ANOVA, n = 4 for the control and HDAC2 knockdown groups, **p < 0.01), but it did not protect the cells transfected with the rescue vector (ANOVA, n = 4 for the rescue-treated group, p > 0.1). A representative result of three independent experiments is shown. D, Lysates of 293T cells transfected with an expression vector encoding FLAG-HDAC2 or HDAC2-Res together with the pLKO-HDAC2–1# or control pLKO plasmid were immunoblotted with the FLAG and GAPDH antibodies. E, CGNs were transiently transfected with pEGFP-N1 together with the empty vector pLKO, pLKO-HDAC2–1#, or Flag-HDAC1 as indicated. The neuronal apoptosis assay was performed as in A. Overexpression of HDAC1 did not reverse the protective effect of HDAC2 knockdown (ANOVA, n = 3 for each group; p > 0.1). F, CGNs were transiently transfected with pEGFP-N1 together with the empty vector pLKO, pLKO-HDAC2–1#, or pLKO-FOXO3a. The neuronal apoptosis assay was performed as in A. In CGNs with FOXO3a knocked down, HDAC2 knockdown did not protect CGNs from H2O2-induced apoptosis (ANOVA, n = 3 for each group; p > 0.1). A representative result of three independent experiments is shown.
Consistent with our previous findings (Xie et al., 2012), FOXO3a knockdown significantly attenuated H2O2-induced neuronal apoptosis. Interestingly, knockdown of both FOXO3a and HDAC2 did not result in an additional protective benefit against H2O2 compared with knockdown of FOXO3a alone (Fig. 2F). In contrast, FOXO3a knockdown partially ameliorated the protection provided by HDAC2 knockdown, suggesting that FOXO3a is important for the HDAC2-mediated regulation of CGN apoptosis. Together, these results suggest that HDAC2, but not HDAC1, regulates H2O2-induced CGN apoptosis via the FOXO3a protein.
The FOXO3a acetylation and artificial transcriptional activity levels are not affected by HDAC1 or HDAC2 directly
The finding that the interaction between HDAC2 and FOXO3a regulates CGN apoptosis led us to study the molecular mechanism underlying this HDAC1/2–FOXO3a interaction. Previous reports have shown that FOXO3a can be acetylated by HATs, such as p300, CBP, and PCAF, which affects FOXO3a transcriptional activity (Brunet et al., 2004; van der Heide and Smidt, 2005). Class I HDACs deacetylate both histones and nonhistone proteins, such as p53 (Brandl et al., 2012). To determine whether HDAC1 and HDAC2 deacetylate FOXO3a directly, we performed an in vitro deacetylation assay. The acetylation sites of FOXO3a are conserved and predominantly located in P2 and P3, including the sites K241 and K244 (Fig. 3A; Greer and Brunet, 2005). The recombinant P2 and P3 fragments of FOXO3a were incubated in PCAF and Ac-CoA to add the acetyl group to the FOXO3a proteins. Then, purified HDAC1 or HDAC2 was added to the reaction mixture. SIRT1, a Class III HDAC, served as a positive control (Brunet et al., 2004). We found that HDAC1 and HDAC2 failed to deacetylate the FOXO3a proteins but that SIRT1 removed the acetyl group from FOXO3a P2 (Fig. 3B,C). We also measured the acetylation level of FOXO3a in cells. In agreement with the in vitro results, the FOXO3a acetylation level was not decreased due to coexpression with HDAC2. There is a modest increase of FOXO3a acetylation might be due to unknown indirect effect of HDAC1 overexpression. However, the data clearly showed that not like SIRT1, HDAC1, and HDAC2 could not deacetylate FOXO3a protein in cells. Together, these data suggest that neither HDAC1 nor HDAC2 deacetylates the FOXO3a protein.
Figure 3.
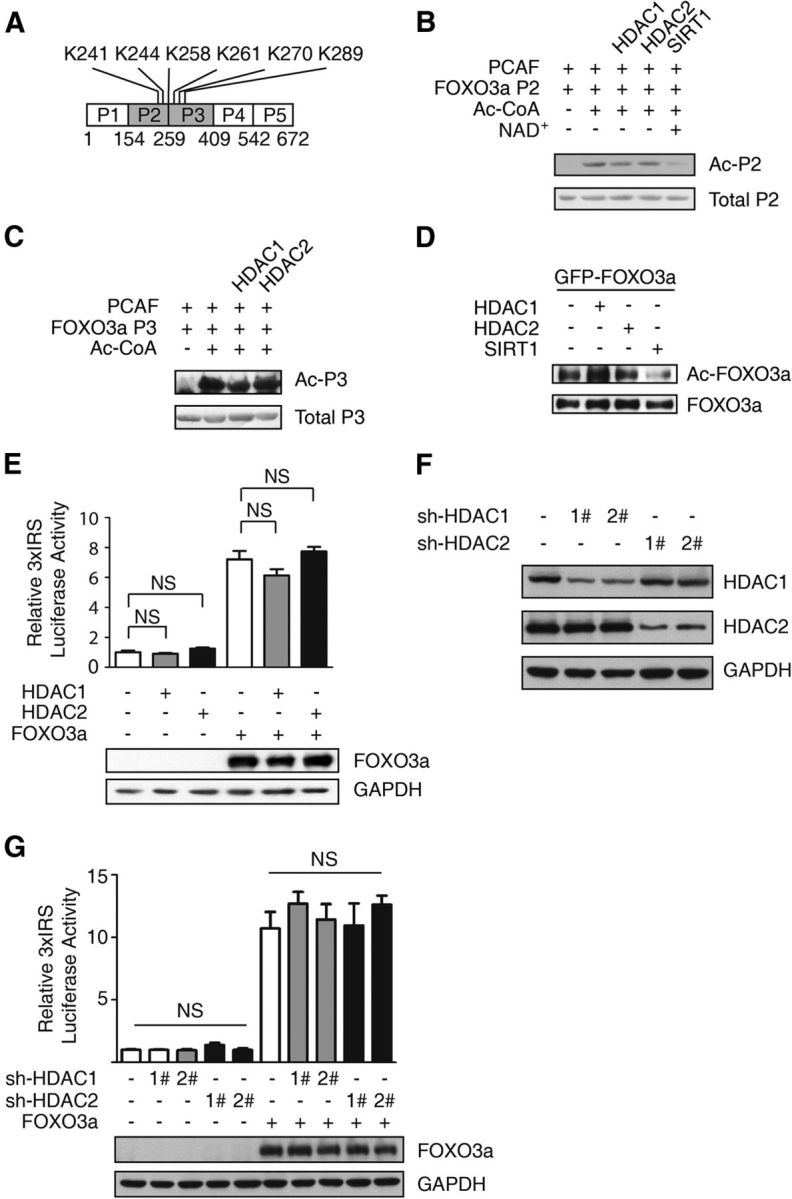
The FOXO3a acetylation and transcriptional activity levels are not directly affected by HDAC1 or HDAC2. A, The lysine sites of FOXO3a might be affected by HDACs. B, C, GST-FOXO3a P2 (B) and P3 fragments (C) were acetylated by PCAF in the presence or absence of Ac-CoA. The Flag-HDAC1, Flag-HDAC2, and Flag-SIRT1 proteins were purified from 293T cells transiently expressing the respective proteins using an anti-Flag antibody. Acetylated FOXO3a was incubated in purified HDAC1, HDAC2, or SIRT1 (supplemented with NAD+). The anti-acetyl antibody was used for immunoblotting. D, 293T cells were transfected with the GFP-FOXO3a expression plasmid alone or together with the Flag-HDAC1, Flag-HDAC2, or Flag-SIRT1 expression plasmid. The anti-GFP antibody was used to enrich the FOXO3a protein. The expression levels of the anti-acetylated FOXO3a and total FOXO3a proteins are shown. E, HT-22 cells were transiently transfected with 3xIRS, PR-TK together with the Flag-HDAC1, Flag-HDAC2, or Flag-FOXO3a expression plasmid as indicated. The relative 3xIRS luciferase activity was analyzed using a dual luciferase reporter assay system. The data are presented as the mean ± SEM. Firefly/Renilla luciferase activity was normalized to that of the control vector-transfected cells. No significant differences were detected between the control and HDAC1/2 groups with or without ectopic FOXO3a expression (ANOVA, n = 3; p > 0.05). The ectopic expressed FOXO3a protein level was blotted by anti-Flag antibody. F, Whole-cell extracts from HDAC1 or HDAC2 knockdown stable cells were resolved via SDS-PAGE; anti-HDAC1, anti-HDAC2, and anti-GAPDH antibodies were used for immunoblotting. G, HT-22 cells with HDAC1 or HDAC2 stably knocked down were transiently transfected with 3xIRS, PR-TK, and the Flag-FOXO3a expression plasmid as indicated. The relative 3xIRS luciferase activity was analyzed using the dual luciferase reporter assay system. No significant difference was detected between the knockdown and control groups. The data are presented as the mean ± SEM, and the firefly/Renilla luciferase activity level was normalized to that of the control cells (ANOVA, n = 3; p > 0.05). The ectopic expressed FOXO3a protein level was blotted by anti-Flag antibody.
Furthermore, an artificial FOXO luciferase reporter assay revealed that ectopically expressed HDAC1/2 did not affect FOXO-mediated luciferase reporter activity in HT-22 cells, which are derived from mouse hippocampal cells (Fig. 3E). Similarly, knockdown of HDAC1 or HDAC2 did not alter FOXO3a overexpression-induced luciferase activity in HT-22 cells (Fig. 3F,G). Together, these results indicate that HDAC1/2 does not directly deacetylate FOXO3a or alter its transcriptional activity.
HDAC2 is recruited to the p21 promoter by FOXO3a and regulates p21 expression
To elucidate the mechanism of HDAC2 knockdown-mediated neuroprotection, we measured the mRNA levels of several putative FOXO3a target genes in HT-22 cells in which HDAC1 or HDAC2 was stably knocked down (Greer and Brunet, 2005). We found that the mRNA levels of several genes associated with apoptosis and the cell cycle were significantly increased (Fig. 4A). Because we found that HDAC1/2 did not directly affect the transactivation of FOXO3a, we hypothesized that FOXO3a might act as a recruiter of HDAC2 to the specific gene promoter region, inducing a change in histone acetylation (Karadedou et al., 2012; Keniry et al., 2013). First, we performed a ChIP assay using an anti-HDAC2 antibody to examine the abundance of HDAC2 in the promoter region of p21, an important target of FOXO3a (Seoane et al., 2004). When FOXO3a is knocked down, the enrichment of HDAC2 is decreased on p21 Forkhead response element (FHRE) (Fig. 4B). Histone 4 lysine 16 (H4K16) is a site that is regulated by HDAC2 (Miller et al., 2010). The H4K16 acetylation level of the p21 FHRE was increased due to HDAC2 or FOXO3a knockdown (Fig. 4C). In agreement with our finding that oxidative stress inhibits the interaction between FOXO3a and HDAC1/2 (Fig. 1D), we also detected a decrease in HDAC2 binding to the p21 promoter following H2O2 treatment (Fig. 4D). Interestingly, the interactions between HDAC2 and the FHREs of Bcl6, Bim, and p27 were not affected by H2O2 treatment. In agreement with this result, oxidative stress enhanced the p21 FHRE acetylation level but not that of other FHREs (Fig. 4E). Among the putative FOXO3a targets, only HDAC2-mediated p21 expression was affected by H2O2 treatment and the regulation is dependent on FOXO3a, arguing that FOXO3a dynamically recruits HDAC2 to p21 promoter and regulates its expression in response to oxidative stress. The other targets including Bcl6, Bim, and p27 were increased upon HDAC2 knockdown, indicating there might be other regulatory pathway that is HDAC2 dependent, but FOXO3a independent in the transcriptional regulation of these genes. Next we performed an in vitro DNA pull-down assay (Fig. 4F,G). The interaction between HDAC2 and the p21 FHRE was promoted by FOXO3a, whereas H2O2 treatment attenuated this binding (Fig. 4G). To determine whether p21 contributes to the protective effect of HDAC2 deficiency, we knocked down HDAC2 in p21−/− CGNs (Fig. 4H). The protective effect of HDAC2 knockdown was partially attenuated in p21−/− CGNs compared with the wild-type control. These results suggest that p21 plays a role in HDAC2 knockdown-induced neuroprotection, and they demonstrate that HDAC2 and p21 play an important role in the FOXO3a-dependent regulation of H2O2-induced neuronal death.
Figure 4.
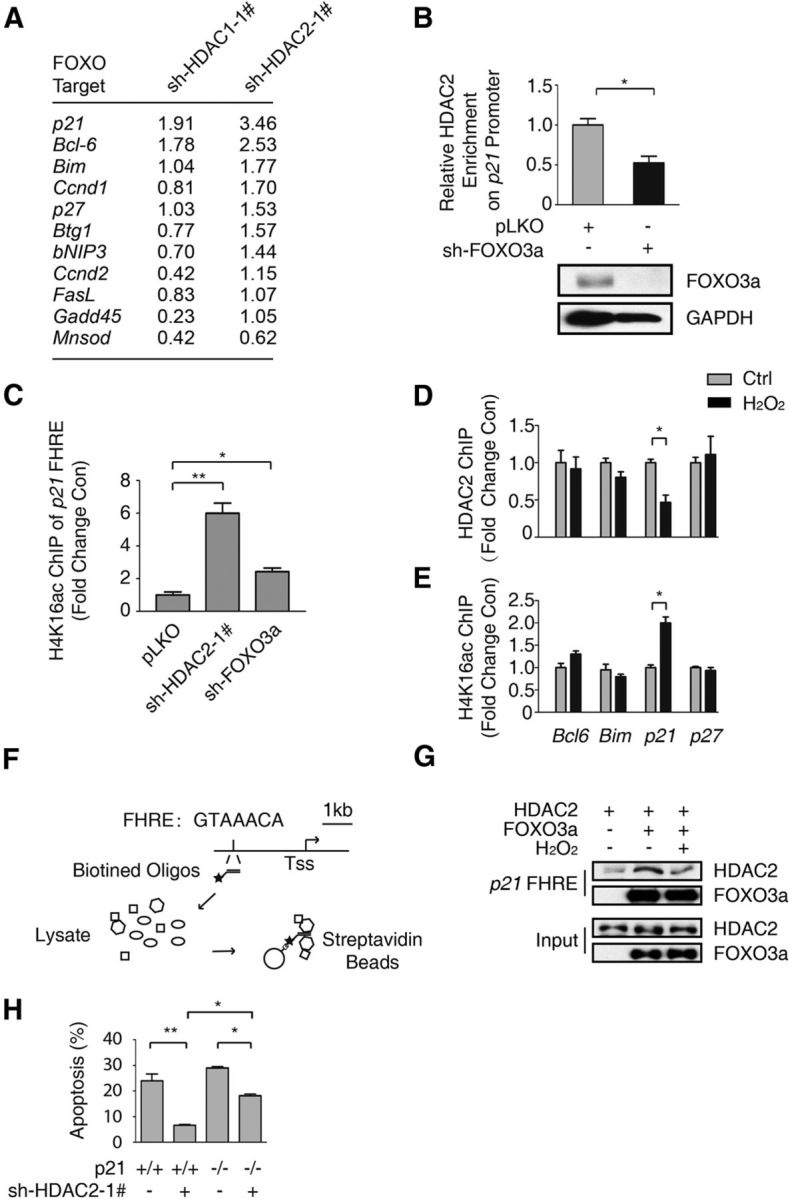
HDAC2 is recruited to the p21 promoter by FOXO3a and regulates p21 expression. A, Total RNA was extracted from HT-22 cells with HDAC1 or HDAC2 stably knocked down. The relative mRNA level was detected via qRT-PCR. The fold-change in the RNA level compared with pLKO empty cells was determined. B, HT-22 cells with FOXO3a stably knocked down and control HT-22 cells were subjected to ChIP using an anti-HDAC2 antibody. qRT-PCR was performed to detect the abundance of HDAC2 at the p21 FHRE (top). The HDAC2 abundance was decreased due to FOXO3a knockdown (Student's t test, n = 3; *p < 0.05). Lysates from FOXO3a knockdown and pLKO-transfected control HT-22 cells were immunoblotted using anti-FOXO3a and anti-GAPDH antibodies (bottom). C, Lysates from HT-22 cells with HDAC2 or FOXO3a stably knocked down and pLKO control HT-22 cells were subjected to ChIP using an anti-H4K16ac antibody. The p21 promoter acetylation level was analyzed via qRT-PCR using primers specific to the p21 FHRE. HDAC2 knockdown (Student's t-test, n = 3, **p < 0.01) and FOXO3a knockdown (Student's t-test, n = 3, *p < 0.05) promoted H4K16ac enrichment in the p21 promoter. D, HT-22 cells were treated with 0 or 300 μm H2O2 for 1 h. Then, the samples were subjected to ChIP using the anti-HDAC2 antibody, which was followed by qRT-PCR analysis of the Bcl6, Bim, p21, and p27 promoter FHRE regions. HDAC2 was specifically enriched in the p21 promoter (Student's t test, n = 3; *p < 0.05). E, HT-22 cells were treated with 0 or 300 μm H2O2 for 1 h. Then, the samples were subjected to ChIP using an anti-H4K16ac antibody, which was followed by qRT-PCR analysis of the Bcl6, Bim, p21, and p27 promoter FHRE regions. The H4K16ac level was increased in the p21 promoter (Student's t test, n = 3, *p < 0.05). F, Flow chart of the DNA pull-down assay for the p21 FHRE. G, 293T cells were transfected with the Flag-HDAC2 or Flag-FOXO3a expression plasmid. The cells were treated with 300 μm H2O2 for 1 h before they were harvested, as indicated. The cell lysates were pulled down using the biotin-labeled p21 FHRE. Western blotting was performed using an anti-HDAC2 antibody. H, CGNs prepared from WT or p21 knock-out mice were transfected with pEGFP-N1 together with the pLKO-HDAC2–1# plasmid, as indicated. The neuronal apoptosis assay was performed as in Figure 2A. The neuronal protection of HDAC2 knockdown in p21−/− CGNs was less than in WT CGNs (ANOVA, n = 4 for each group; *p < 0.05).
HDAC2 knockdown induces p21 upregulation in a p53-independent manner, and HDAC2 knockdown-mediated neuronal protection occurs independently of p53 and E2F1
p53 is a known p21 regulator that participates in oxidative stress-induced apoptosis. Therefore, we investigated whether HDAC2 knockdown-induced protection is p53-dependent. We found that the Class I HDAC inhibitor Ms-275 promoted p21 expression at both the mRNA and protein levels in HT-22 cells with p53 knocked down (Fig. 5A,B). HDAC2 knockdown significantly protected CGNs after p53 knockdown (Fig. 5C).
Figure 5.

HDAC2 knockdown can induce p21 upregulation in a p53-independent manner, and neuronal protection occurs independently of p53 and E2F1. A, HT-22 cells with p53 stably knocked down and control cells were treated with DMSO or 2 μm Ms-275 for 12 h. Total RNA was extracted, and the p21 mRNA level was detected. GAPDH served as a control. The p21 mRNA level was significantly higher when the cells in which p53 was knocked down were treated with Ms-275 (Student's t test, n = 3; ***p < 0.001). B, HT-22 cell lines were treated as in A, and cell lysates were resolved on an SDS-PAGE gel and analyzed (top). The statistical analysis of the protein level is shown in the bottom panel. In the cells in which p53 was knocked down as well as in the control cells, Ms-275 increased the p21 protein level (Student's t test, n = 3; **p < 0.01). C, CGNs were transfected with pEGFP-N1 together with pLKO-HDAC2–1#, pLKO-p53, and pLKO-FOXO3a, as indicated. A neuronal apoptosis assay was performed. Knocking down HDAC2 still protected the CGNs in the p53 knockdown groups (ANOVA, n = 4; ***p < 0.001). When FOXO3a was knocked down, HDAC2 RNAi could not protect the cells. (ANOVA, n = 3; p > 0.1). D, CGNs prepared from E2F1−/−, p53−/− mice were transfected with pEGFP-N1, together with pLKO-HDAC2–1#, and this step was followed by a neuronal apoptosis assay and analysis as in Figure 2A. HDAC2 knockdown protected CGNs from apoptosis (Student's t test, n = 3; *p < 0.05).
A previous study revealed a HDAC-E2F1-Bim axis that affects apoptosis (Zhao et al., 2005). We performed the apoptosis assay using p53/E2F1 double-knock-out CGNs and obtained similar results to those using wide-type (WT) CGNs (Fig. 5D). We found that Class I HDACs upregulate p21 expression in a p53-independent manner and that HDAC2 knockdown protects CGNs from oxidative stress-induced apoptosis via a mechanism that occurs independently of p53 and E2F1.
HDAC2 phosphorylation at S394 is important for the HDAC2–FOXO3a interaction
HDAC2 phosphorylation at S394 has been shown to affect the binding affinity of HDAC2 to the transcription factor Klf5 (Zheng et al., 2011). We investigated the hypothesis that the HDAC2–FOXO3a interaction is affected by oxidative stress-mediated dephosphorylation of HDAC2 at S394. Oxidative stress diminished the phosphorylation level of HDAC2 at S394 in HT-22 cells and in mice (Fig. 6A–C). A co-IP assay revealed that the interaction between FOXO3a and HDAC2 is weakened in the brain following ischemia/reperfusion (Fig. 6D).
Figure 6.
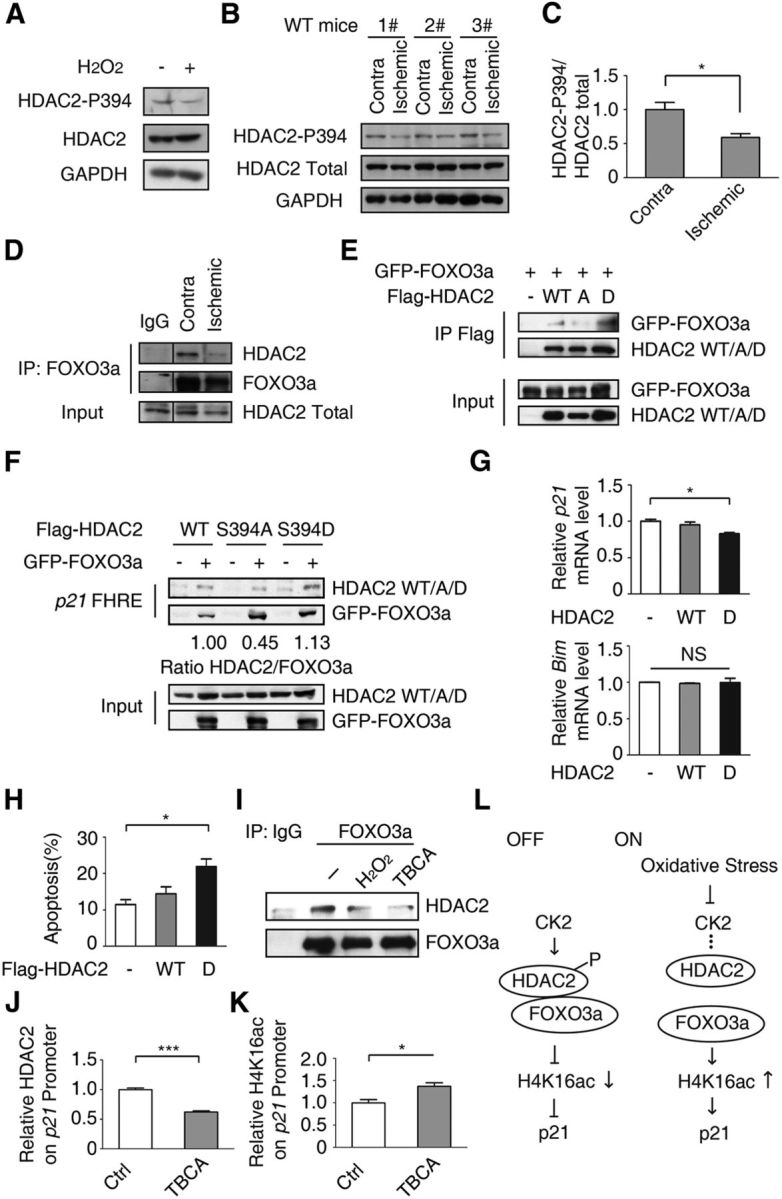
HDAC2 phosphorylation at S394 is important for the HDAC2-FOXO3a interaction. A, HT-22 cells were treated with 300 μm H2O2 for 1 h. The cell lysates were immunoblotted using an anti-HDAC2–p394, anti-HDAC2, or anti-GAPDH antibody. B, Brain lysates of the contralateral and ipsilateral hemispheres from MCAO model mice were resolved on a SDS-PAGE gel and immunoblotted using anti-HDAC2–p394, anti-HDAC2, and anti-GAPDH antibodies. Representative results of six independent mice are shown. C, Statistical analysis of B (Student's t test, n = 6, *p < 0.05). D, Brain lysates as in B were immunoprecipitated using an anti-FOXO3a antibody or normal mouse IgG. Anti-HDAC2 and anti-FOXO3a antibodies were used for immunoblotting. Representative results of three mice are shown. E, The Flag-HDAC2 WT, or S394A or S394D expression plasmids together with the GFP-FOXO3a plasmid were transiently transfected into 293T cells. Twenty-four hours after transfection, the cell lysates were immunoprecipitated using an anti-Flag antibody. Anti-GFP and anti-Flag antibodies were used for immunoblotting. F, 293T cells were transfected with the Flag-HDAC2 WT or S394A or S394D expression plasmid together with the GFP-FOXO3a plasmid. Then, the DNA pull-down assay was performed using biotin-labeled p21 FHRE DNA. The ratio of HDAC2/FOXO3a abundance was calculated. G, HT-22 cells were transfected with Flag-HDAC2 WT or S394D expression plasmid. Thirty-six hours after transfection, cells were harvested. The mRNA level of p21 and Bim were analyzed. HDAC2 S394D repressed p21 expression (Student's t test, n = 3; *p < 0.05), whereas Bim level remains no change (Student's t test, n = 3; p > 0.1). H, CGNs were transfected with pEGFP-N1 together with Flag-HDAC2 WT, S394D mutation or control vector as indicated. A neuronal apoptosis assay was performed. Ectopic HDAC2 S394D promotes oxidative stress-induced neuron apoptosis. (ANOVA, n = 3; *p < 0.05). I, HT-22 cells were treated with 300 μm H2O2 or 20 μm TBCA for 6 h. The cell lysates were immunoprecipitated using an anti-FOXO3a antibody or IgG, followed by an immunoblot using the indicated antibodies. J, K, HT-22 cells were treated with 20 μm TBCA for 6 h. Fixed cell lysates were immunoprecipitated using an anti-HDAC2 (J) or anti-H4K16ac antibody (K). qRT-PCR was performed. The relative abundance of HDAC2 (J; Student's t test, n = 3; ***p < 0.001) and H4K16ac (K; Student's t test, n = 3; *p < 0.05) was calculated. L, Model of the mechanism by which HDAC2 phosphorylation at S394 mediates the suppression of p21 expression via FOXO3a.
Next, we examined whether the HDAC2–FOXO3a interaction is associated with the phosphorylation of HDAC2 at S394. HDAC2 S394A mutation (mimicking the nonphosphorylated state) abolished the HDAC2–FOXO3a interaction, whereas HDAC2 S394D mutation (mimicking the consistent phosphorylation) increased the interaction (Fig. 6E). In accordance with the co-IP result, the p21 FHRE displayed a tendency to bind to WT HDAC2 or HDAC2 S394D mutation, whereas its binding affinity to the S394A mutant was reduced (Fig. 6F). Because HDAC2 S394D showed a stronger interaction with FOXO3a, we next tested whether expression of p21 is repressed by overexpression of HDAC2 S394D (Fig. 6G). The mRNA level of p21 was decreased when HDAC2 S394D was overexpressed. Interestingly, the Bim mRNA level is not altered, indicating the HDAC2-mediated inhibition is specific to p21. In agreement with these results, the CGNs ectopically expressed HDAC2 S394D were sensitive to oxidative stress (Fig. 6H).
S394 of HDAC2 has been reported to be primarily phosphorylated by casein kinase 2 (CK2; Adenuga and Rahman, 2010; Eom et al., 2011). In HT-22 cells, treatment with the CK2 inhibitor TBCA significantly disrupted the HDAC2–FOXO3a interaction (Fig. 6I). This result indicated that HDAC2–FOXO3a depends on CK2-mediated phosphorylation. In accordance with this result, HDAC2 dissociated from the p21 FHRE following TBCA treatment (Fig. 6J), and the H4K16ac level of the p21 FHRE was increased (Fig. 6K). Therefore, the HDAC2–FOXO3a interaction is dependent on CK2-mediated phosphorylation of HDAC2 at S394, which is affected by oxidative stress (Fig. 6L).
Discussion
In this study, we demonstrated that the HDAC2-FOXO3a complex responds to oxidative stress and mediates neuronal apoptosis via epigenetic modification. FOXO3a recruits HDAC2 to the p21 promoter and deacetylates neighboring histones, reducing p21 expression (Fig. 7, top). Oxidative stress abolishes the interaction between HDAC2 and FOXO3a, thus increasing the histone acetylation level of the p21 promoter and upregulating p21 mRNA expression, but not the other putative FOXO3a targets. Similarly, HDAC inhibition led to histone hyperacetylation at the p21 promoter and increased p21 expression (Fig. 7, bottom). Furthermore, we elucidated that the S394 phosphorylation of HDAC2 is important for the FOXO3a–HDAC2 interaction and regulation of p21 expression (Fig. 6L). Together, we identified HDAC2-p21 pathway that is important for oxidative stress-induced neuronal death.
Figure 7.

Working model by which oxidative stress regulates p21 expression via the HDAC2-FOXO3a complex. In “OFF” state, HDAC2 is recruited by FOXO3a and selectively enriched in p21 promoter region, which causes the hypoacetylation levels of histone and inhibits p21 transcription. In “ON” stage, the reduced interaction of HDAC2-FOXO3a by oxidative stress in p21 promoter region or HDAC inhibition increases histone acetylation and promotes p21 expression, which protects neuron from oxidative stress-induced neuronal cell death. (⊣ stands for inhibit and → stands for promote).
The mechanism by which p21 protects cells from apoptosis has been elucidated. For example, p21 binds to apoptosis signal-regulating kinase-1 and caspase-3 in the cytoplasm, thereby protecting neurons from oxidative stress-induced apoptosis (Langley et al., 2008). Alternatively, p21 acts as a cell cycle repressor, potentially preventing cell-cycle re-entry and protecting neurons from apoptosis (Klein and Ackerman, 2003; Herrup et al., 2004; Kruman et al., 2004).
Recent reports suggest that HDACs are potential therapeutic targets for neurological diseases, including AD and stroke. For example, in ischemic stroke, HDAC inhibition decreased the infarct volume and improved neurologic function in rodents (Langley et al., 2009; Kilgore et al., 2010). We propose an important mechanism underlying the neuroprotection provided by HDAC inhibition. (Biswas et al., 2005).
In the present study, we also found that HDAC1 and HDAC2 exhibit distinct effects on neuronal death. Several reports have shown that these two similar enzymes function differentially in neurological diseases. In AD patients, HDAC2, but not HDAC1 or HDAC3, is upregulated in the brain, thus mediating cognitive dysfunction (Gräff et al., 2012). HDAC2, but not HDAC1, has been demonstrated to negatively regulate synaptic plasticity and memory formation (Guan et al., 2009). In addition, HDAC1 knockdown causes DNA damage-related neuronal death, whereas HDAC2 knockdown does not induce neuronal toxicity (Kim et al., 2008). These results, in combination with our findings, suggest that HDAC2, but perhaps not other HDACs, plays an important role in oxidative stress-induced neuronal apoptosis and may be exploited as a therapeutic target for neurological diseases.
Interestingly, a recent study demonstrated that DAF-16, the FOXO3a homolog in Caenorhabditis elegans, employs the SWI/SNF complex to manipulate chromatin remodeling, thereby regulating the expression of its target genes (Riedel et al., 2013). Based on our TAP results regarding FOXO3a, we identified a set of proteins that participates in histone modification, including EP300, a histone acetyltransferase (Ogryzko et al., 1996). These proteins may cooperatively regulate the acetylation balance of histones and epigenetically control gene expression by interacting with FOXO3a. We also found that phosphorylation of HDAC2 dynamically regulates the HDAC2–FOXO3a interaction. CK2, a kinase responsible for HDAC2 phosphorylation, is considered as an important regulator of the HDAC2 and FOXO3a interaction. Interestingly, it has been reported that CK2 is modulated by oxidative stress and its subunit is decreased in the ischemic brain (Kim et al., 2012). Our findings suggest that CK2-mediated HDAC2 phosphorylation affects the HDAC2–FOXO3 interaction, and CK2 inhibitors could potentially be used for neuroprotection.
In summary, our study elucidates the mechanism by which HDAC2 physically interacts with FOXO3a and differentially regulates the expression of FOXO3a target genes under oxidative stress conditions, thus mediating oxidative stress-induced apoptosis (Fig. 7). Therefore, HDAC2 may be a potential therapeutic target for neuronal apoptosis-related diseases, including stroke and neurodegenerative diseases.
Footnotes
This work was supported by the National Science Foundation of China (Grant Nos. 81125010 and 81030025), the National Basic Research Program of China (973-2012CB910701 and 2013DFA31990), the Beijing Natural Science Foundation (Grant No. 7132147), and Cross-disciplinary Collaborative Teams Program for Science, Technology and Innovation (2014–2016) from Chinese Academy of Sciences. We thank the members of the Yuan laboratory for critical reading of the paper and helpful discussion.
The authors declare no competing financial interests.
References
- Adenuga D, Rahman I. Protein kinase CK2-mediated phosphorylation of HDAC2 regulates co-repressor formation, deacetylase activity and acetylation of HDAC2 by cigarette smoke and aldehydes. Arch Biochem Biophys. 2010;498:62–73. doi: 10.1016/j.abb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Xiao L, Jia Y, Wu J, Xie Q, Ren J, Ji G, Yuan Z. c-Jun N-terminal kinase enhances MST1-mediated pro-apoptotic signaling through phosphorylation at serine 82. J Biol Chem. 2010;285:6259–6264. doi: 10.1074/jbc.M109.038570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SC, Liu DX, Greene LA. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl A, Wagner T, Uhlig KM, Knauer SK, Stauber RH, Melchior F, Schneider G, Heinzel T, Krämer OH. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. J Mol Cell Biol. 2012;4:284–293. doi: 10.1093/jmcb/mjs013. [DOI] [PubMed] [Google Scholar]
- Brochier C, Dennis G, Rivieccio MA, McLaughlin K, Coppola G, Ratan RR, Langley B. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J Neurosci. 2013;33:8621–8632. doi: 10.1523/JNEUROSCI.5214-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Calnan DR, Webb AE, White JL, Stowe TR, Goswami T, Shi X, Espejo A, Bedford MT, Gozani O, Gygi SP, Brunet A. Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging (Albany NY) 2012;4:462–479. doi: 10.18632/aging.100471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Eom GH, Cho YK, Ko JH, Shin S, Choe N, Kim Y, Joung H, Kim HS, Nam KI, Kee HJ, Kook H. Casein kinase-2alpha1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation. 2011;123:2392–2403. doi: 10.1161/CIRCULATIONAHA.110.003665. [DOI] [PubMed] [Google Scholar]
- Gräff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang XY, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, Tindall DJ. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005;102:1649–1654. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003;53:S26–S36; discussion S36–S38. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- Karadedou CT, Gomes AR, Chen J, Petkovic M, Ho KK, Zwolinska AK, Feltes A, Wong SY, Chan KY, Cheung YN, Tsang JW, Brosens JJ, Khoo US, Lam EW. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene. 2012;31:1845–1858. doi: 10.1038/onc.2011.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keniry M, Pires MM, Mense S, Lefebvre C, Gan B, Justiano K, Lau YK, Hopkins B, Hodakoski C, Koujak S, Toole J, Fenton F, Calahan A, Califano A, DePinho RA, Maurer M, Parsons R. Survival factor NFIL3 restricts FOXO-induced gene expression in cancer. Genes Dev. 2013;27:916–927. doi: 10.1101/gad.214049.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, Broodie N, Mazitschek R, Delalle I, Haggarty SJ, Neve RL, Lu Y, Tsai LH. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GS, Jung JE, Narasimhan P, Sakata H, Yoshioka H, Song YS, Okami N, Chan PH. Release of mitochondrial apoptogenic factors and cell death are mediated by CK2 and NADPH oxidase. J Cereb Blood Flow Metab. 2012;32:720–730. doi: 10.1038/jcbfm.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Ackerman SL. Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest. 2003;111:785–793. doi: 10.1172/JCI200318182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/S1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R, Jr, Gorospe M, Mattson MP. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–561. doi: 10.1016/S0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 2009;40:2899–2905. doi: 10.1161/STROKEAHA.108.540229. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villén J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Li J, Du W, Maynard S, Andreassen PR, Pang Q. Oxidative stress-specific interaction between FANCD2 and FOXO3a. Blood. 2010;115:1545–1548. doi: 10.1182/blood-2009-07-234385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Wu J, Xiao L, Bai Y, Qu A, Zheng Z, Yuan Z. Regulation of neuronal cell death by c-Abl-Hippo/MST2 signaling pathway. PLoS One. 2012;7:e36562. doi: 10.1371/journal.pone.0036562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery WR, Carney JM. Oxidative alterations in Alzheimer's disease. Brain Pathol. 1999;9:133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/S0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/S0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA, Bowman SK, Kingston RE, Dillin A, Asara JM, Ruvkun G. DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol. 2013;15:491–501. doi: 10.1038/ncb2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/S0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/S0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV, Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ED, Nuñez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- Tao L, Xie Q, Ding YH, Li ST, Peng S, Zhang YP, Tan D, Yuan Z, Dong MQ. CAMKII and calcineurin regulate the lifespan of Caenorhabditis elegans through the FOXO transcription factor DAF-16. Elife. 2013;2:e00518. doi: 10.7554/eLife.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Veenstra TD, Morrison RS. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. 2009;29:2824–2832. doi: 10.1523/JNEUROSCI.6186-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci. 2005;30:81–86. doi: 10.1016/j.tibs.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- Wang P, Bai Y, Song B, Wang Y, Liu D, Lai Y, Bi X, Yuan Z. PP1A-mediated dephosphorylation positively regulates YAP2 activity. PLoS One. 2011;6:e24288. doi: 10.1371/journal.pone.0024288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Chen D, Hu P, Wu J, Liu W, Zhao Y, Cao M, Fang Y, Bi W, Zheng Z, Ren J, Ji G, Wang Y, Yuan Z. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. J Neurosci. 2011;31:9611–9619. doi: 10.1523/JNEUROSCI.0035-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Hao Y, Tao L, Peng S, Rao C, Chen H, You H, Dong MQ, Yuan Z. Lysine methylation of FOXO3 regulates oxidative stress-induced neuronal cell death. EMBO Rep. 2012;13:371–377. doi: 10.1038/embor.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Wang J, Gao L, Wang R, Liu X, Gao Z, Tao Z, Xu C, Song J, Ji X, Luo Y. MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke. 2013;44:1706–1713. doi: 10.1161/STROKEAHA.111.000504. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci U S A. 2005;102:16090–16095. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Han M, Shu YN, Li YJ, Miao SB, Zhang XH, Shi HJ, Zhang T, Wen JK. HDAC2 phosphorylation-dependent Klf5 deacetylation and RARalpha acetylation induced by RAR agonist switch the transcription regulatory programs of p21 in VSMCs. Cell Res. 2011;21:1487–1508. doi: 10.1038/cr.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]