

Abstract
Slack (Slo2.2) is a sodium-activated potassium channel that regulates neuronal firing activities and patterns. Previous studies identified Slack in sensory neurons, but its contribution to acute and chronic pain in vivo remains elusive. Here we generated global and sensory neuron-specific Slack mutant mice and analyzed their behavior in various animal models of pain. Global ablation of Slack led to increased hypersensitivity in models of neuropathic pain, whereas the behavior in models of inflammatory and acute nociceptive pain was normal. Neuropathic pain behaviors were also exaggerated after ablation of Slack selectively in sensory neurons. Notably, the Slack opener loxapine ameliorated persisting neuropathic pain behaviors. In conclusion, Slack selectively controls the sensory input in neuropathic pain states, suggesting that modulating its activity might represent a novel strategy for management of neuropathic pain.
Keywords: analgesia, dorsal root ganglion, neuropathic pain, Slack channel
Introduction
Painful stimuli are detected by sensory neurons whose cell somata are located in dorsal root ganglia (DRGs) and trigeminal ganglia. Upon activation, they conduct nociceptive sensory information from peripheral sites to the dorsal horn of the spinal cord, where they form synapses with spinal neurons (Basbaum et al., 2009; Schmidtko et al., 2009). Lesions to sensory neurons can result in neuropathic pain, a major public health problem that affects 7–10% of the general population (van Hecke et al., 2014). In fact, neuropathic pain often remains refractory to treatment, with only one-third to two-thirds of patients reporting adequate pain relief (O'Connor and Dworkin, 2009; Finnerup et al., 2010). The limited treatment success reflects our incomplete understanding of the molecular pathophysiology underlying neuropathic pain processing.
Neuropathic lesions are linked to enhanced excitability of sensory neurons. This excitability is driven by altered expression and activity of ion channels (Raouf et al., 2010; Waxman and Zamponi, 2014). K+ channels, the most populous and diverse class of neuronal ion channels, are governed by some 78 genes in humans (for review, see Tsantoulas and McMahon, 2014). Based on structural and physiological characteristics, K+ channels are classically organized into voltage-gated, two-pore, inwardly rectifying, and Ca2+-activated channels. Slack channels (“sequence like a Ca2+-activated K+” channel; also termed Slo2.2) belong to the Ca2+-activated K+ channel family, but it has been shown that they are activated by intracellular Na+ rather than Ca2+ (Bhattacharjee and Kaczmarek, 2005; Zhang et al., 2010; Kaczmarek, 2013). They may control bursting and adaptation of action potential (AP) firing rates (Sanchez-Vives et al., 2000; Franceschetti et al., 2003; Wallén et al., 2007), thereby reducing neuronal activity (Nuwer et al., 2010). Interestingly, Slack channels have been recently detected in sensory neurons, and electrophysiological studies point to a role of Slack channels in pain processing (Tamsett et al., 2009; Nuwer et al., 2010; Huang et al., 2013). However, the in vivo relevance of Slack channels for acute and chronic pain remained elusive, particularly due to the requirement for high concentrations of intracellular Na+ and additional factors to activate Slack channels (Tamsett et al., 2009). Using mice with an ablation of Slack globally and in sensory neurons, this study sought to elucidate the relevance of Slack channels during pain processing in vivo. Our data suggest that Slack channels play an essential role as modulators of neuropathic pain processing.
Materials and Methods
Animals.
Slack-deficient (Slack−/−) mice were generated by homologous recombination in combination with a common Cre/loxP-based strategy in murine embryonic stem (ES) cells. In brief, we used a targeting vector that included exons 5–15 of the murine Kcnt1 gene. Exon 11, which encodes for the pore region, was flanked by a single 3′-loxP site and a floxed neo/tk cassette at the 5′ region (Fig. 1A). Correctly targeted L3/+ ES cells were identified by Southern blot analyses (Fig. 1B) and were transiently transfected with a Cre recombinase expression vector permitting limited excision of the selection cassette. Recombination resulted in ganciclovir-resistant Slack L2/+ ES cells with the pore-forming exon flanked by two loxP sites (Fig. 1A). Recombined Slack L2/+ ES cells were injected into C57BL/6 blastocysts. Chimeric male mice resulting from these injections were mated to C57BL/6 females once they reached maturity. Heterozygous Slack L2/+ offspring were subsequently crossed to Cre deleter mice (Schwenk et al., 1995) to generate the Slack L1/+ mouse line. The Slack L1/+ and Slack L2/+ mouse lines were backcrossed for ≥6 generations to the C57BL/6N background before subsequent analyses. Homozygous Slack+/+ (WT) and SlackL1/L1 (Slack−/−) progenies, which derived from interbreeding of heterozygous SlackL1/+ parental mice, were used for the biochemical and behavioral characterization. Genotyping was performed routinely by PCR on genomic DNA isolated from tail-tip biopsies using specific primer sets for the WT and different mutant Kcnt1 alleles (Fig. 1C,D).
Figure 1.
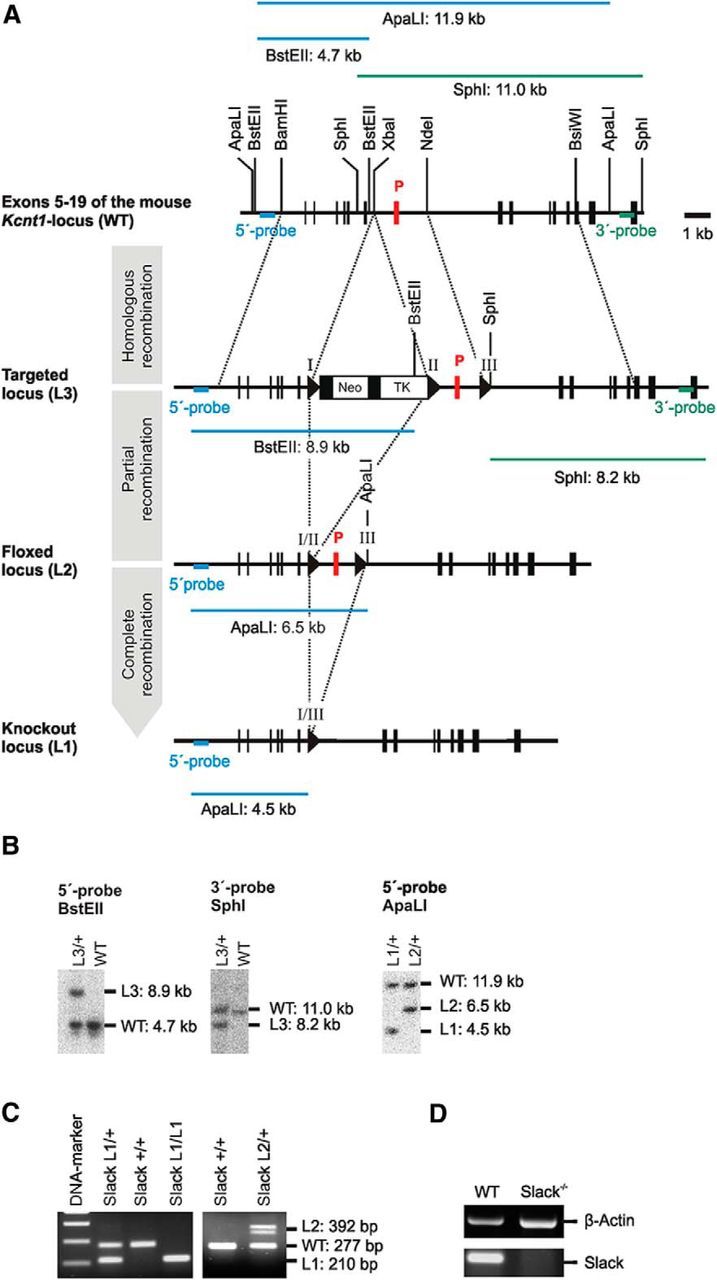
Genetic ablation of the pore-forming exon of the murine Kcnt1 gene in ES cells. A, Murine WT Kcnt1 locus with exon 11 encoding the pore (P, red box) and surrounding exons 5–19 (black boxes) including relevant restriction sites (top). In ES cells, a single loxP recombination site (triangle) and a floxed neo/tk-cassette were introduced into the 3′-intronic and 5′-intronic regions flanking exon 11 (P) by homologous recombination resulting in the Slack L3-allele. L2 ES cells with floxed pore exons were generated by Cre recombinase-mediated excision of the selection cassette. L2 ES cells were injected into C57BL/6 blastocysts to obtain the L2/+ mouse line as described in Materials and Methods. Germline excision of the pore exon (L1) was conducted in mice by crossing Slack L2/+ animals to the Cre deleter mouse strain. Selected probes, restriction sites, and fragment sizes for the identification of the correct L3, L2, and L1 alleles by Southern blot in ES cells are shown. B, Verification of correct homologous and Cre recombination in ES cells by Southern blot analyses using the different probes. C, Representative example of a genotyping PCR using genomic DNA from mouse tail-tip biopsies as templates. D, The absence of the pore-forming exon was further verified by reverse transcription PCR analyses on total RNA isolated from cerebral cortex of Slack−/− mice using a pore exon-specific primer pair.
Tissue-specific Slack mutants (SNS-Slack−/−) were generated by crossing double transgenic mice that carried a heterozygous ablation of the Slack pore-forming region (L1/+) together with a sensory neuron-specific (SNS) Cre recombinase under control of the Nav1.8 promoter (Agarwal et al., 2004) to SlackL2/L2 animals. In addition to the SNS conditional mutants (SNS-Slack−/−, genotype: SNS-Cretg/+; SlackL1/L1 in SNS due to the Cre-mediated recombination and L2/L1 in all other cells) these matings produced SNS-Ctrl mice (genotype: SNS-Cretg/+; SlackL2/+). Specific primer sets for the different Kcnt1 alleles and the Cre transgene were used on tail-tip DNA for genotyping.
Experiments were performed in 6–12-week-old mice of either sex on a C57BL/6 background. Animals were housed on a 12 h light/dark cycle with access to food and water ad libitum. All experiments adhered to the guidelines of the International Association for the Study of Pain and to the ARRIVE (Animal Research: Reporting on In Vivo Experiments) guidelines, and were approved by our local Ethics Committee for Animal Research (Regierungspräsidium Darmstadt, Regierungspräsidium Tübingen, and Landesamt für Natur, Umwelt und Verbraucherschutz NRW, Germany).
Behavioral testing.
Littermate mice were used in all behavioral studies. Animals were habituated to the experimental room and were investigated by an observer blinded for the genotype and treatment of the animals.
Rotarod test.
Motor coordination was assessed with a rotarod treadmill for mice (Ugo Basile) at a constant rotating speed of 13 rpm. All mice had ≥2 training sessions before the day of the experiment. The fall-off latency was averaged from two tests and the cutoff time was 120 s.
Dynamic plantar test.
Paw-withdrawal latency after light mechanical stimulation was measured using a dynamic plantar aesthesiometer (Ugo Basile). This device pushes a thin probe (0.5 mm diameter) with increasing force through a wire-gated floor against the plantar surface of the paw from beneath. When the animal withdraws the paw, the device automatically stops and records the latency time, which is the time from when the probe touched the paw to when the animal withdrew the paw. The force increased from 0 to 5 g within 10 s (0.5 g/s ramp) and was then held at 5 g for an additional 10 s (Schmidtko et al., 2008; Lu et al., 2014). The latency was calculated as the average of 4–6 consecutive exposures with ≥20 s in between.
Randall-Selitto test.
Nociceptive thresholds to noxious mechanical stimuli were determined using a Randall-Selitto digital pressure instrument (IITC Life Science). Mice were placed in a sling, and the probe was applied in the proximal part of the tail. The tail was gradually compressed until an escape response was elicited (Anseloni et al., 2003). The terminal pressure at the moment of escape response was recorded. Only one test per animal was performed.
Tail-flick test.
Mice were placed in a plantar test instrument (Hargreaves method, IITC Life Science) and the radiant heat stimulus (intensity, 13%; cutoff time, 20 s) was aimed at the tail (∼2 cm from the tip). The tail-flick latency was calculated as the mean of three measurements with ≥5 min in between.
Hot plate and cold plate test.
Mice were placed on a heated or cooled metal surface (Hot/Cold Plate, Ugo Basile). In the hot plate test, the time between placement and a nociceptive behavior (shaking or licking of a hindpaw, jumping) was recorded. Temperatures of 50, 52, and 54°C were applied with cutoff times of 40, 30, and 20 s. In the cold plate test (0°C), the time between placement and a nociceptive behavior was recorded, and the number of times the hindpaw lifted in 5 min was counted. Only one test per animal was performed.
Spared nerve injury-induced neuropathic pain.
Under isoflurane anesthesia, the tibial and common peroneal branches of the sciatic nerve were ligated and sectioned distally, while the sural nerve was left intact (Decosterd and Woolf, 2000; Bourquin et al., 2006). Mechanical hypersensitivity at the lateral surface of the hindpaw (sural nerve skin area) was determined using the dynamic plantar test.
Paclitaxel-induced neuropathic pain.
Paclitaxel (Sigma-Aldrich) was dissolved at a concentration of 6 mg/ml in a vehicle composed of 50% Cremophor EL (Fluka Chemie) and 50% absolute ethanol. The final concentration of 1 μg in 1 μl was prepared in sterile saline at the time of injection and injected intraperitoneally at 2 mg/kg once per day for 5 consecutive days (Nieto et al., 2008). The mechanical hypersensitivity was measured using the dynamic plantar test.
Formalin test.
Formalin (15 μl of a 0.5% formaldehyde solution) was injected subcutaneously into the dorsal surface of one hindpaw (Hunskaar et al., 1985; McNamara et al., 2007). The time spent licking the formalin-injected paw was recorded in 5 min intervals up to 45 min after formalin injection.
Zymosan-induced inflammatory pain.
A zymosan A suspension (20 μl, 5 mg/ml in 0.1 m PBS, pH 7.4; Sigma-Aldrich) was injected into the plantar subcutaneous space of a hindpaw (Meller and Gebhart, 1997) and the mechanical hypersensitivity was measured using the dynamic plantar test.
Drug administration.
β-Nicotinamide adenine dinucleotide (NAD+), loxapine, olanzapine, pregabalin, and niclosamide (all were dissolved in 0.9% saline; Sigma-Aldrich) were administered 14 d after spared nerve injury (SNI) or 3 h after zymosan injection. Intrathecal delivery of NAD+ was performed by direct lumbar puncture in awake, conscious mice as described in detail previously (Lu and Schmidtko, 2013).
Immunohistochemistry.
Mice were killed by CO2 and immediately perfused intracardially with 0.9% saline, followed by 1% paraformaldehyde in PBS, pH 7.4. The lumbar spinal cord (L4-L5) and DRGs (L4-L5) were dissected and cryoprotected in 20% sucrose overnight. Tissues were frozen in tissue freezing medium (Leica) on dry ice, cryostat sectioned at a thickness of 16 μm, and stored at −80°C. For immunofluorescence, sections were permeabilized for 5 min in PBST (0.1% Triton X-100 in PBS), blocked for 1 h using 10% normal goat serum and 3% bovine serum albumin (BSA) in PBS, and incubated with primary antibodies diluted in 3% BSA in PBS overnight at 4°C or for 2 h at room temperature. The following antibodies were used: mouse anti-slo2.2 (1:400; NeuroMab), rabbit anti-calcitonin gene-related peptide (anti-CGRP; 1:800; Calbiochem), mouse anti-NF200 (clone N52; 1:1000; Sigma-Aldrich), and rabbit anti-Nav1.8 (1:150; Alomone Labs). Sections were then washed in PBS and stained with secondary antibodies conjugated with Alexa Fluor 488 or 555 (Invitrogen). For staining with Griffonia simplicifolia isolectin B4 (IB4), sections were incubated with Alexa Fluor 488-conjugated IB4 (10 μg/ml in PBS buffer containing 1 mm CaCl2 · 2H2O, 1 mm MgCl2, 1 mm MnCl2, and 0.2% Triton X-100, pH 7.4; Invitrogen) for 2 h at room temperature. After immunostaining, slides were immersed for 5 min in 0.06% Sudan black B (in 70% ethanol) to reduce lipofuscin-like autofluorescence (Schmidtko et al., 2008), washed in PBS, and coverslipped. In double-labeling experiments, primary antibodies were consecutively incubated. Images were taken using an Axio Observer.Z1 microscope (Carl Zeiss) or an Eclipse Ni-U (Nikon) microscope equipped with a monochrome CCD camera, and were pseudocolored and superimposed. Adjustment of brightness and contrast was done using Adobe Photoshop CS software (Adobe Systems). Controls were performed by omitting the first and/or the second primary antibodies and by incubating tissues of Slack−/− mice.
Cell counting.
Serial sections of entire L4-L5 DRGs (three mice per genotype) were cut (16 μm), and ≥3 sections per DRG per animal were counted. Only cells showing staining clearly above background were included. Specificity of Slack immunoreactivity was confirmed by simultaneous staining of coembedded tissues of WT and Slack−/− mice. The percentage of Slack-positive cells that expressed marker was calculated by dividing the number of Slack-positive cells colocalized with marker by the total number of Slack-positive cells. The percentage of marker-positive DRG neurons in WT and Slack−/− mice is expressed as a proportion of marker-positive cells per total number of DRG neurons. Cross-sectional areas were measured using National Institutes of Health ImageJ software.
Real-time reverse transcription PCR.
Lumbar DRGs (L4-L5) were rapidly dissected and snap frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted under RNase-free conditions using a RNA isolation Kit (RNAqueous Micro Kit, Ambion) according to the manufacturer's instructions, DNase treated for 15 min to minimize genomic DNA contamination, and quantified with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). cDNA was synthesized from 200 ng of RNA, random hexamer primers, RT-Enhancer, and the Verso enzyme of the Verso Kit (Thermo Fisher Scientific). Real-time reverse transcription PCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) using Taqman gene expression assays for Kcnma1 (catalog #Mm01119505_m1), Kcnt2 (catalog #Mm01284549_m1), and GAPDH (catalog #Mm99999915_g1), purchased from Applied Biosystems. Reactions (total volume, 10 μl) were performed in duplicate or triplicate by incubating at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Water controls were included to ensure specificity. Relative expression of target gene levels was determined using the comparative 2−ΔΔCt method, with Ct indicating the cycle number at which the signal of the PCR product crosses an arbitrary threshold set within the exponential phase of the PCR. The amount of sample RNA was normalized to GAPDH.
Western blots.
Lumbar DRGs (L4-L5), lumbar spinal cord (L4-L5), and brain were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C until use. Samples were homogenized in buffer containing 100 mm Tris-HCl and 1 mm MgCl2, pH 8.0, combined with a protease inhibitor mixture (Mini Tablets, Thermo Scientific). Following freezing at −80°C for 30 min, samples were homogenized again. After adding a sixfold volume of sucrose buffer (250 mm sucrose, 10 mm Tris-HCl, pH 7.4, combined with protease inhibitor mixture), the samples were mixed and centrifuged at 1000 × g for 20 min. The supernatant was collected and centrifuged at 20,000 × g for 20 min, and the resulting pellets were resuspended in sucrose buffer. Extracted proteins (30 μg/lane) were separated by SDS-PAGE and blotted onto a nitrocellulose membrane. After blocking of nonspecific binding sites with blocking buffer (PBS with 3% low-fat milk), membranes were incubated with mouse anti-slo2.2 (1:500; NeuroMab), or mouse anti-GAPDH (1:2000; Ambion) dissolved in blocking buffer containing 0.1% Tween 20 overnight at 4°C. After incubation with secondary antibodies for 1 h at room temperature, proteins were detected using an Odyssey Infrared Imaging System or a C-DiGit blot scanner (LI-COR Bioscience).
Patch-clamp recordings.
Adult WT and Slack−/− mice (n = 4 per genotype) were killed by CO2 inhalation, and DRGs from all levels were excised and transferred to DMEM containing 50 μg/ml gentamicin (Sigma-Aldrich). Following treatment with 1 mg/ml collagenase and 0.1 mg/ml protease for 30 min (both from Sigma-Aldrich), ganglia were dissociated using a fire-polished, silicone-coated Pasteur pipette. Isolated cells were transferred onto poly-d-lysine-coated (200 μg/ml, Sigma-Aldrich) coverslips and cultured in TNB 100 medium supplemented with TNB 100 lipid protein complex, 100 μg/ml streptomycin, and penicillin (all from Biochrom) at 37°C and 5% CO2. Cells were used for experiments within 24 h after plating. Whole-cell voltage and current-clamp recordings were conducted with a HEKA Electronics USB 10 amplifier combined with Patchmaster software (HEKA Electronics). Currents were filtered at 5 kHz and sampled at 20 kHz. Offline analyses were performed using the Fitmaster software (HEKA Electronics) and Origin software. For both voltage-clamp and current recordings the pipette solution contained 140 mm KCl, 2 mm MgCl2, 5 mm EGTA, and 10 mm HEPES, pH 7.4, and was adjusted with KOH. The external solution contained 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2 and 10 mm HEPES. For Na+-free extracellular solution, NaCl was replaced with 140 mm choline chloride. The osmolarity of all solutions was adjusted with glucose to 290–300 mOsmol/L. Patch pipettes were fabricated with borosilicate glass (Science Products) using a conventional puller (DMZ-Universal Puller, Zeitz Instrumente) and heat-polished to give a pipette resistance of 2–3 MΩ. Only DRG neurons with a resting membrane potential of <−40 mV and generating reproducible APs upon 20-ms-long current injections at 2–3 times the determined threshold were used. The resting membrane potential did not differ between the two genotypes (data not shown). Shortly before a coverslip was mounted for recordings, it was dipped in extracellular solution containing 10 μg/ml FITC-conjugated IB4 (Sigma-Aldrich) for 5–10 min, and only IB4-binding DRG neurons were analyzed.
Statistical analysis.
Statistical analysis was performed with SPSS and GraphPad Prism software using the two-tailed Student's t test for paired comparisons and one-way or two-way repeated-measures ANOVA for multiple comparisons. When ANOVA showed significant difference, pairwise comparisons between means were analyzed with the Bonferroni's or Dunnett's post hoc test. Data from patch-clamp recordings were analyzed using χ2 test and unpaired t test. Rotarod fall-off latencies were analyzed with Mann–Whitney U test and are expressed as median and interquartile range. All other data are presented as the mean ± SEM. For all tests, a probability value p < 0.05 was considered as statistically significant.
Results
Slack channels are predominantly expressed in nonpeptidergic sensory neurons
We generated Slack−/− mice by homologous recombination in murine embryonic stem cells in combination with a common Cre/loxP-based strategy. The pore-forming exon 11 of the murine Kcnt1 gene was flanked by two loxP sites (Fig. 1A), and the resulting mice bearing the floxed Kcnt1 gene were crossed to Cre deleter mice (Schwenk et al., 1995) to obtain global Slack−/− mice. Correct homologous and Cre-mediated recombination was verified by Southern blot, PCR, and mRNA expression analyses (Fig. 1B–D). Slack−/− mice were fertile and showed no gross anatomical or behavioral defects. The mRNA expression levels of large conductance calcium-activated potassium (BKCa) channels and Slick potassium channels, which have been reported to interact with Slack channels (Joiner et al., 1998; Chen et al., 2009), were similar in Slack−/− and WT mice, as were the overall frequencies of DRG neuron populations positive for markers of sensory neurons (n = 3/genotype; data not shown).
We examined the Slack distribution in DRGs using an anti-Slack antibody whose immunoreactivity was absent in Slack−/− mice (Fig. 2A,B), and observed that 31.6% of DRG neurons in WT mice expressed Slack. The detailed Slack distribution in DRGs was investigated by double-labeling experiments with established markers. We found that 86.5% of Slack-positive cells bound the lectin IB4, a marker of the nonpeptidergic population of unmyelinated nociceptors, whereas 93.2% of IB4-positive cells expressed Slack (Fig. 2C,E). A population (12.6%) of Slack-positive cells was immunoreactive for CGRP, a marker of peptidergic unmyelinated nociceptors (Fig. 2D,E). In accordance to these double-labeling experiments, cell size analyses revealed that Slack is mostly expressed in nociceptive small-diameter to medium-diameter DRG neurons (Fig. 2F). In the spinal cord, Slack immunoreactivity was concentrated in the superficial dorsal horn (Fig. 3A) and the specificity of anti-Slack antibody was further confirmed by Western blot analyses with spinal cord protein extracts (Fig. 3B). Double-labeling revealed a high degree of Slack colocalization with central terminals of IB4-positive sensory neurons in lamina II (Fig. 3C). Hence, our results indicate that Slack channels are predominately expressed in IB4-positive nonpeptidergic nociceptors and their central terminals in the dorsal horn of the spinal cord, pointing to a contribution of Slack channels to the processing of pain.
Figure 2.
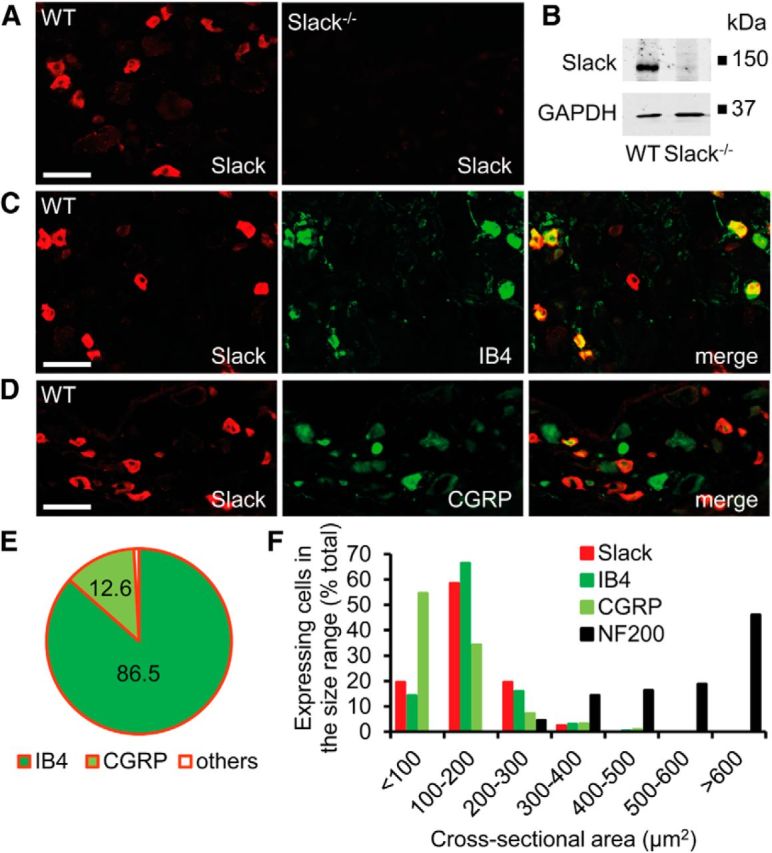
Expression of Slack channels in DRG neurons. A, Immunofluorescence of Slack in lumbar DRGs of WT and Slack−/− mice revealed specific Slack expression in 31.6 ± 1.7% (2571 cells counted, n = 3 mice) of DRG neurons. B, Western blot analysis of Slack (140 kDa) in DRG homogenates of WT and Slack−/− mice confirms the specificity of the anti-Slack antibody. GAPDH (36 kDa) was used as loading control. C, D, Typical examples of Slack immunoreactivity in nociceptor subpopulations positive for IB4 and CGRP. E, Quantitative summary of DRG neuron populations expressing Slack. Most Slack-positive cells bind IB4 (86.5 ± 1.6%, 2108 cells counted, n = 3 mice) and are therefore nonpeptidergic, whereas a few Slack-positive cells colocalize with CGRP in peptidergic DRG neurons (12.6 ± 0.33%, 1114 cells counted, n = 3 mice). F, Size distribution of Slack-positive DRG neurons compared with those expressing IB4, CGRP, and neurofilament 200 (NF200), a marker of large myelinated DRG neurons. The data demonstrate that Slack channels are nearly exclusively expressed in nociceptive DRG neurons of small and medium diameter. Scale bars, 50 μm.
Figure 3.

Expression of Slack channels in the spinal cord. A, Immunofluorescence of Slack in the lumbar spinal cord of WT and Slack−/− mice reveals specific Slack expression in the superficial dorsal horn. B, Western blot analysis of Slack (140 kDa) in spinal cord homogenates of WT and Slack−/− mice confirms the specificity of the anti-Slack antibody. GAPDH (36 kDa) was used as loading control. C, Colocalization of Slack with IB4 shows that Slack channels are present in central terminals of sensory neurons entering the superficial dorsal horn. Scale bars, 100 μm.
Slack channels generate sodium-activated potassium currents and regulate AP firing in sensory neurons
Previous reports suggest that Slack is important for generating sodium-activated potassium (KNa) currents and thus for the excitability of sensory neurons (Nuwer et al., 2010). To examine the functional role of Slack for the generation of KNa in sensory neurons, we performed whole-cell voltage-clamp recordings on IB4-positive DRG neurons of adult Slack−/− and WT mice. At a holding potential of −70 mV, series of 1000-ms-long test pulses ranging from −80 to +110 mV in intervals of 10 mV were applied in presence of 140 mm extracellular NaCl and after replacement of NaCl with 140 mm choline chloride (i.e., Na+-free; Fig. 4A). This removal of extracellular Na+ resulted in a prominent reduction of the peak amplitude of total outward K+ currents (IK) in 8 of 17 (47%) recorded WT DRG neurons, but only in 2 of 16 (13%) DRG neurons from Slack−/− mice (p = 0.031, χ2 test; Fig. 4D). In these DRG neurons, digital subtraction of the total currents recorded in 140 mm choline chloride from the currents obtained in 140 mm NaCl revealed a large Na+-activated K+ current (WT, 7 ± 1 nA, n = 8; Slack−/−, 2 ± 1 nA, n = 2; p = 0.029, unpaired t test; Fig. 4E). In DRG neurons in which removal of extracellular Na+ did not result in a marked reduction of the total K+ current (WT, 53%; Slack−/−, 87%), the same digital subtraction more or less resulted in the isolation of voltage-gated Na+ currents without any prominent outward currents (Fig. 4B).
Figure 4.
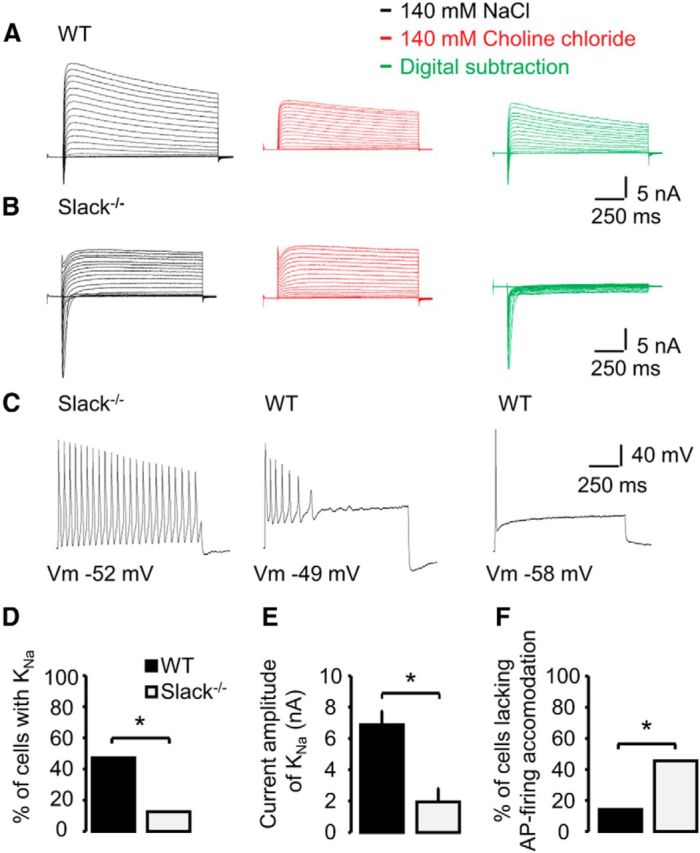
Slack generates KNa and regulates AP firing in sensory neurons. A, B, Representative current traces from whole-cell voltage-clamp recordings on IB4-positive WT (n = 17) and Slack−/− (n = 16) DRG neurons monitoring outward K+ currents in presence of 140 mm NaCl (black traces) and after replacement of NaCl with 140 mm choline chloride (red traces). Currents were elicited by protocols consisting of 1000-ms-long test pulses ranging from −80 to +110 mV in steps of 10 mV. Holding potential was −70 mV. All neurons were first recorded in presence of NaCl. To visualize the KNa current, the current traces obtained with 140 mm NaCl and 140 mm choline chloride were digitally subtracted and depicted as green traces. Please note that the absence of extracellular Na+ was verified by the lack of Na+ inward currents (red traces). C, Representative current traces from whole-cell current-clamp recordings on IB4-positive WT (n = 21) and Slack−/− (n = 22) DRG neurons. APs were elicited by 1000 ms current injections corresponding to 2–3 times the threshold of a single AP. Pictured are three observed patterns of AP firing during the current injection, i.e., no accommodation (left), accommodation (middle), and strong accommodation without repetitive firing (right). D, Percentage of DRG neurons from WT and Slack−/− mice generating outward K+ currents (KNa). E, Mean KNa current amplitudes in DRG neurons from WT (n = 8) and Slack−/− (n = 2) mice. F, Percentage of DRG neurons from WT and Slack−/− mice generating APs without accommodation, i.e., cells with repetitive firing of APs. *p < 0.05.
We next sought to determine whether the deficiency of Slack changed the excitability of DRG neurons. For that purpose, we monitored the pattern of AP firing during 1000-ms-long step-current injections corresponding to 2–3-fold of the threshold for firing a single AP. As shown in Figure 4C, we observed three different patterns of AP firing in DRG neurons from WT and Slack−/− mice. While only 14% (3 of 21) of WT DRG neurons produced APs throughout the current injection (i.e., no accommodation), 45% (10 of 22) of the investigated neurons from Slack−/− mice produced APs without a prominent accommodation (p = 0.026, χ2 test; Fig. 4F). A similar fraction of neurons from both genotypes produced APs that accommodated during the current injection, resulting in ≥2 APs (WT, 38%, 8 of 21; Slack−/−, 41%, 9 of 22; p = 0.85, χ2 test). Finally, 48% (10 of 21) of WT DRG neurons only produced one AP in the course of the current injection, i.e., they displayed a strong accommodation and were not able to produce repetitive AP firing. In contrast, only 3 of 22 (14%) examined DRG neurons from Slack−/− mice also failed to produce >1 AP (p = 0.016, χ2 test). Together, our data obtained by whole-cell voltage and current-clamp recordings imply that Slack channels generate a substantial fraction of the total KNa current in nonpeptidergic sensory neurons, and that they seem to regulate the pattern of AP firing in these neurons.
Neuropathic pain behavior is increased in Slack mutant mice
To determine the functional role of Slack channels for pain processing in vivo, we analyzed the behavior of Slack−/− and littermate WT mice in various animal models of pain. Having established that motor coordination is intact in Slack−/− mice [median fall-off latencies: Slack−/− mice, 112.2 s (interquartile range, 69.9–120.0 s); WT mice, 120.0 s (interquartile range, 106.3–120.0 s); p = 0.226; n = 14/genotype], we assessed their immediate responses to noxious stimuli. Slack−/− mice displayed similar pain thresholds compared with control littermates after applying light and intense noxious mechanical stimuli using the dynamic plantar and the Randall-Selitto tests, respectively (Fig. 5A,B). Similar responses in both genotypes also were observed after applying thermal stimuli in the tail-flick, 50–54°C hot plate, and 0°C cold plate tests (Fig. 5C–E). These data suggest that Slack channels are not involved in the processing of acute nociceptive pain.
Figure 5.

Basal sensitivity is normal in Slack−/− mice. A, B, Mechanical pain sensitivity was measured using the dynamic plantar (A; n = 15–17/genotype) and the Randall-Selitto tests (B; n = 8/genotype). C, D, Thermal heat pain sensitivity was assessed by tail-flick (C; n = 8/genotype) and hot plate tests (D; n = 14/genotype). E, Cold pain sensitivity was investigated on a 0°C cold plate (n = 14/genotype). No significant differences were observed between Slack−/− and WT mice in any of these tests.
We then examined the behavior of Slack−/− mice after SNI, a model of peripheral nerve injury-induced neuropathic pain that produces a pronounced, long-lasting mechanical hypersensitivity (Decosterd and Woolf, 2000; Bourquin et al., 2006). Interestingly, as shown in Figure 6A, the extent of SNI-induced mechanical hypersensitivity was significantly increased in Slack−/− mice compared with WT mice (p = 0.0206; Fig. 6A), suggesting that Slack modulates neuropathic pain processing in response to nerve injury. We then tested the behavior of Slack−/− mice in another model of neuropathic pain, in which mechanical hypersensitivity is induced by repeated intraperitoneal injection of the cytostatic drug paclitaxel (Nieto et al., 2008). Similar to SNI, increased hindpaw hypersensitivity in Slack−/− mice did also occur after paclitaxel administration (p = 0.0212; Fig. 6B). These data imply that Slack channels control neuropathic pain processing in vivo.
Figure 6.

Increased neuropathic pain behavior in Slack−/− mice. A, B, Paw-withdrawal latencies of Slack−/− and WT mice after mechanical stimulation in the SNI model (n = 8/genotype; A) and the paclitaxel model (2 mg/kg, i.p., for 5 consecutive days; n = 8–9/genotype; B) of neuropathic pain. Slack−/− mice demonstrated increased mechanical hypersensitivity compared with WT littermates. *p < 0.05, WT versus Slack−/−, repeated-measures ANOVA with Bonferroni's post hoc test.
Neuropathic pain behavior is increased in sensory neuron-specific Slack mutants
We next investigated the specific contribution of Slack channels expressed in sensory neurons to neuropathic pain processing. For this purpose, we crossed mice carrying the floxed exon 11 of the Kcnt1 gene with SNS-Cre mice (Agarwal et al., 2004), to obtain mice lacking Slack channels specifically in NaV1.8-positive sensory neurons. In agreement with our observation that virtually all Slack-positive DRG neurons are also positive for NaV1.8 (Fig. 7A), the Cre/loxP-mediated deletion led to a nearly complete ablation of Slack expression in DRGs of the conditional knock-outs (referred to as SNS-Slack−/− mice; controls were referred to as SNS-Ctrl mice; Fig. 7B). Slack expression was also reduced in the spinal cord of SNS-Slack−/− mice, in accordance to its localization in central terminals of sensory neurons in the dorsal horn (Fig. 7C; compare with Fig. 3). In contrast, the Slack expression in the brain was similar between SNS-Slack−/− and SNS-Ctrl mice (Fig. 7C), as expected (Agarwal et al., 2007; Y. Liu et al., 2010; Lu et al., 2014). We then analyzed the neuropathic pain behavior of SNS-Slack−/− mice after SNI. Notably, similar to the results obtained with the global Slack−/− mice, the SNI-induced mechanical hypersensitivity was significantly increased in SNS-Slack−/− mice compared with SNS-Ctrl mice (p = 0.0397; Fig. 7D). These results demonstrate that Slack channels expressed in sensory neurons control the processing of persistent neuropathic pain.
Figure 7.

Increased neuropathic pain behavior after conditional deletion of Slack channels in sensory neurons. A, Double-labeling immunostaining revealed that virtually all Slack-positive DRG neurons are also positive for NaV1.8. B, Slack immunofluorescence was not detectable in DRGs of SNS-Slack−/− mice, in which the Cre/loxP-mediated recombination of the Slack encoding gene Kcnt1 occurred in NaV1.8-positive sensory neurons. C, Western blot analysis demonstrated that Slack expression was considerably reduced in the spinal cord but normal in the brain of SNS-Slack−/− mice. GAPDH was used as loading control. D, Paw-withdrawal latencies of SNS-Slack−/− and SNS-Ctrl mice after mechanical stimulation in the SNI model of neuropathic pain (n = 8/genotype). SNS-Slack−/− mice demonstrated increased mechanical hypersensitivity compared with SNS-Ctrl mice. *p < 0.05, WT versus Slack−/−, repeated-measures ANOVA with Bonferroni's post hoc test. Scale bars, 50 μm.
Slack channel activation ameliorates neuropathic pain behavior
A previous study revealed that the sensitivity of Slack channels for the activation by Na+ can be modulated by NAD+ through a NAD+ binding site (Tamsett et al., 2009). To assess whether Slack channel modulation by NAD+ affects neuropathic pain processing in vivo, we intrathecally injected NAD+ (50 μg; Pittelli et al., 2011) in WT and Slack−/− mice 14 d after SNI and analyzed the neuropathic pain behavior over 45 min. Before NAD+ injection, Slack−/− mice demonstrated an increased SNI-induced mechanical hypersensitivity compared with WT mice, as expected (Fig. 8; compare with Fig. 6A). Notably, the NAD+ administration significantly ameliorated the hypersensitivity in WT mice (p = 0.004) but did not affect the hypersensitivity in Slack−/− mice (Fig. 8). These data point to NAD+ as an endogenous modulator of Slack channels during neuropathic pain processing.
Figure 8.

Intrathecal (i.t.) NAD+ ameliorates neuropathic pain in a Slack-dependent manner. Animals were subjected to SNI to induce neuropathic pain, and NAD+ was intrathecally administered 14 d thereafter. Time courses of paw-withdrawal latencies after mechanical stimulation (n = 7–8/genotype). *p < 0.05, before NAD+ injection (“after SNI”) versus after NAD+ injection in WT mice, repeated-measures ANOVA with Dunnett's post hoc test.
We next analyzed the effects of the atypical antipsychotic drug loxapine on persisting neuropathic pain behavior. A recent study demonstrated that loxapine activates Slack channels (Biton et al., 2012), in addition to its blocking activities at dopamine, serotonin, histamine, muscarinic, and adrenergic receptors (Chakrabarti et al., 2007). Interestingly, systemic administration of a low dose of loxapine (0.175 mg/kg, i.p.), which did not impair the motor coordination in rotarod tests (data not shown), significantly alleviated the SNI-induced mechanical hypersensitivity in WT mice (p < 0.0001; Fig. 9A). In contrast, the paw hypersensitivity was not affected in Slack−/− mice (Fig. 9A), indicating that the loxapine-induced analgesia depends on Slack. Administration of higher doses of loxapine (>0.35 mg/kg, i.p.) increased the paw-withdrawal latency in both genotypes, suggesting that additional mechanisms independent from Slack activation may affect the paw-withdrawal latency if higher loxapine doses were used. In contrast to loxapine, the neuropathic pain behavior of WT mice was not ameliorated by olanzapine (0.175 mg/kg, i.p.; Fig. 9B), which has a similar pharmacological profile compared with loxapine but lacks Slack-activating properties (Biton et al., 2012). The neuropathic pain behavior was, however, inhibited in both genotypes by pregabalin (60 mg/kg, i.p.; p < 0.0001; Fig. 9C), which was used as a positive control. Moreover, the neuropathic pain behavior in WT mice was inhibited after oral administration of the anthelmintic drug niclosamide (200 mg/kg; data not shown), another Slack activator (Biton et al., 2012). Together, these data implicate that Slack channel activators can ameliorate neuropathic pain once it has been already established.
Figure 9.
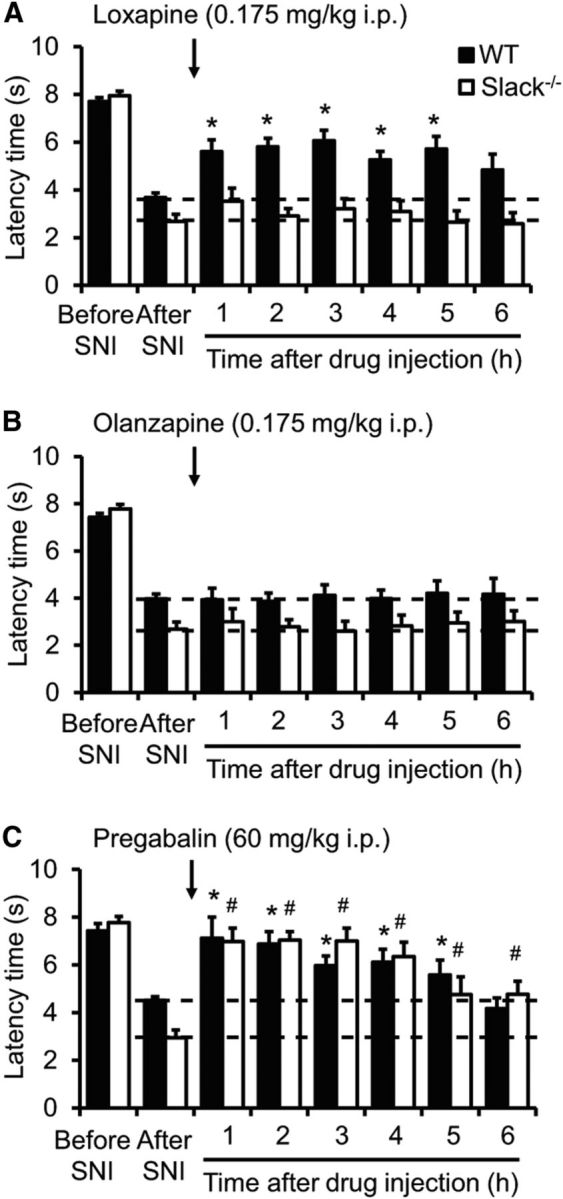
Loxapine ameliorates neuropathic pain in a Slack-dependent manner. A–C, Neuropathic pain was induced in WT and Slack−/− mice by SNI surgery. Fourteen days thereafter, animals were intraperitoneally injected with loxapine (A; n = 12–13/genotype), olanzapine (B; n = 7–8/genotype), or pregabalin (C; n = 7/genotype), and the mechanical paw-withdrawal latencies were assessed over 6 h. Note that loxapine inhibited the neuropathic pain behavior in WT but not in Slack−/− mice, whereas olanzapine did not affect the pain behavior in both genotypes. Pregabalin was used as a positive control. *#p < 0.05, before drug injection (“after SNI”) versus after drug injection in WT and Slack−/− mice, respectively; repeated-measures ANOVA with Bonferroni's post hoc test.
Inflammatory pain behavior is normal in Slack mutant mice
We next investigated the inflammatory pain behavior in Slack−/− mice. In the 0.5% formalin test, we found no significant differences in formalin-induced paw-licking behavior between Slack−/− mice and WT littermates (Fig. 10A,B). Similarly, the extent of mechanical hypersensitivity evoked by injection of zymosan into a hindpaw was indistinguishable between both genotypes (Fig. 10C). Moreover, the zymosan-induced hypersensitivity, unlike the SNI-induced hypersensitivity, was not affected by intraperitoneal administration of 0.175 mg/kg loxapine (Fig. 10D). These data suggest that Slack channels, despite their distinctive localization in IB4-positive sensory neurons, are not critically involved in the processing of inflammatory pain in vivo.
Figure 10.

Inflammatory pain behavior is normal in Slack−/− mice. A, B, Formalin test. A, The time course of paw licking induced by 0.5% formalin injection was similar between Slack−/− and WT mice. B, Statistical analyses revealed no significant differences in the first (1–10 min) and the second (11–45 min) phase of paw licking (n = 6/genotype). C, Zymosan model. Paw-withdrawal latencies after mechanical stimulation after injection of zymosan into a hindpaw did not differ between genotypes (n = 7–8/genotype). D, The zymosan-induced mechanical hypersensitivity in WT mice was not affected by intraperitoneal administration of the Slack channel opener loxapine (0.175 mg/kg) 3 h after zymosan injection (n = 8/genotype).
Discussion
We here demonstrate that Slack channels are distinctly expressed in a population of nociceptive sensory neurons where they are important for the generation of KNa currents and for firing of APs. Behavioral analysis of Slack−/− mice points to an important function of these channels in the control of neuropathic pain, but to a limited role in the processing of acute nociceptive or inflammatory pain.
KNa currents likely to be generated by Slack channels have previously been identified in mouse DRG neurons (Tamsett et al., 2009; Nuwer et al., 2010). Our electrophysiological data from DRG neurons of WT and Slack−/− mice reveal Slack as an important molecule for the generation of KNa in IB4-positive DRG neurons, and that Slack is at least a contributing factor for the strong accommodation of AP firing, which has been previously observed in this neuronal subpopulation (Choi et al., 2007). Despite electrophysiological evidence for an important function of Slack in sensory neurons, the relevance of Slack for pain processing in vivo remained elusive, in particular due to their requirement for high concentrations of intracellular Na+ to activate. In fact, the normal resting level of intracellular Na+ in neurons lies between 4 and 15 mm (Rose, 2002), whereas the half-maximal effective concentration (EC50) for Na+ activation of Slack channels in excised patch recordings ranges between 40 and 75 mm (Bhattacharjee et al., 2003; Yuan et al., 2003; Huang et al., 2013). It is therefore plausible that additional factors are required to allow Slack channels to operate in their native environment in response to physiological relevant levels of intracellular Na+.
Our behavioral analysis demonstrated that Slack contributes to the processing of neuropathic pain after SNI and paclitaxel injection. The SNI model is produced by transection of two branches of the sciatic nerve, resulting in prolonged mechanical hypersensitivity in the skin area of the intact third branch (the sural nerve in our experiments). SNI induces many changes in the peripheral nerve system and at the level of the spinal cord dorsal horn (Basbaum et al., 2009). So far, identified peripheral mechanisms include transganglionic degenerative atrophy of the nerve terminals of primary afferents (Shields et al., 2003), dysmyelination of peripheral nerves (Kallenborn-Gerhardt et al., 2012), and recruitment of immune cells to the lesion site and to DRGs (Scholz and Woolf, 2007; Ellis and Bennett, 2013). Similarly, systemic delivery of paclitaxel causes changes in peripheral nerves and in the spinal cord. These various changes contribute to the induced mechanical hypersensitivity (for review, see Sisignano et al., 2014). Peripheral mechanisms include, for example, loss of peripheral nerve fibers in epidermis (Siau et al., 2006; C. C. Liu et al., 2010), impairment of axonal transport and mitochondria function in primary afferent neurons (Flatters and Bennett, 2006), and infiltration of macrophages into peripheral nerves and DRGs (Peters et al., 2007). As Slack channels are selectively expressed in the IB4-binding population of primary afferents, our data implicate a contribution of this cell population to neuropathic pain induced by peripheral nerve injury and paclitaxel injection. This finding is supported by an earlier observation that mechanical hypersensitivity after peripheral nerve injury is reduced when the IB4-binding population is deleted by injection of IB4 conjugated to the toxin saporin (Tarpley et al., 2004).
We here provide several lines of evidence that manipulation of Slack, either by knock-out or by pharmacological agents, selectively influences the animal behaviors in response to a neuropathic pain stimulus. First, the pain behavior in Slack−/− mice was increased after SNI or paclitaxel injection, but not in models of inflammatory pain (formalin and zymosan) or acute nociceptive pain (dynamic plantar, Randall-Selitto, tail flick, hot plate, and cold plate). Second, increased neuropathic pain behavior was also observed after conditional knock-out of Slack channels in sensory neurons. As significant differences between groups occurred at later stages in the sensory neuron-specific knock-outs compared with the global knock-outs (21 vs 11 d after SNI), it seems possible that Slack channels expressed in the CNS might also contribute to neuropathic pain processing. Third, the Slack channel opener loxapine (0.175 mg/kg, i.p.) ameliorated neuropathic but not inflammatory pain behavior. The fact that such a low dose of loxapine was effective further points to additional factors that sensitize Slack channels in neuropathic pain conditions, given that the EC50 of loxapine to open recombinant Slack channels in cell culture is relatively high (3.5 μm; Biton et al., 2012).
One putative factor that might sensitize Slack channels during neuropathic pain processing is the metabolic coenzyme NAD+. Slack channels contain a putative NAD+ binding site, and modulation by NAD+ in inside-out excised-patch recordings decreased the Na+ EC50 of Slack channels toward the physiological neuronal range (Tamsett et al., 2009). It has been reported that neurons upregulate NAD+ biosynthetic enzymes after injury (Sasaki et al., 2006; Ying, 2007), and we here demonstrate that neuropathic pain behavior is attenuated in a Slack channel-dependent manner after intrathecal delivery of NAD+. Hence, intracellular NAD+ might contribute to the selective activation of Slack channels in neuropathic pain conditions. Another recently identified modulator that may enhance the Na+ sensitivity of Slack channels is TMEM16C (also referred to as ANO3; Huang et al., 2013). However, TMEM16C knock-out rats exhibited increased thermal and mechanical sensitivity under basal conditions, in contrast to the Slack−/− mice, and neuropathic pain behaviors were not investigated in TMEM16C knock-outs. It remains, therefore, to be determined whether TMEM16C contributes to Slack channel activation during neuropathic pain.
Altogether, targeting Slack channels could be a new strategy for the treatment of neuropathic pain.
Footnotes
This work was supported by grants from the Doktor Robert Pfleger-Stiftung (to A.S.), the Witten/Herdecke University (to A.S.), the Deutsche Forschungsgemeinschaft (SFB 815-A14; to G.G. and A.S.), and the Landesapothekerkammer Baden-Württemberg (to A.B. and P.R.). We thank Prof. Rohini Kuner (Institute of Pharmacology, Heidelberg) for providing SNS-Cre mice and Natalie Wolfsdorff, Christine Manderscheid, Karin Schilling, and Clement Kabagema-Bilan for excellent technical assistance.
The authors declare no competing financial interests.
References
- Agarwal N, Offermanns S, Kuner R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis. 2004;38:122–129. doi: 10.1002/gene.20010. [DOI] [PubMed] [Google Scholar]
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anseloni VC, Ennis M, Lidow MS. Optimization of the mechanical nociceptive threshold testing with the Randall-Selitto assay. J Neurosci Methods. 2003;131:93–97. doi: 10.1016/S0165-0270(03)00241-3. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Kaczmarek LK. For K+ channels, Na+ is the new Ca2+ Trends Neurosci. 2005;28:422–428. doi: 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci. 2003;23:11681–11691. doi: 10.1523/JNEUROSCI.23-37-11681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton B, Sethuramanujam S, Picchione KE, Bhattacharjee A, Khessibi N, Chesney F, Lanneau C, Curet O, Avenet P. The antipsychotic drug loxapine is an opener of the sodium-activated potassium channel slack (Slo2.2) J Pharmacol Exp Ther. 2012;340:706–715. doi: 10.1124/jpet.111.184622. [DOI] [PubMed] [Google Scholar]
- Bourquin AF, Süveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain. 2006;122:14.e11–14. doi: 10.1016/j.pain.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A, Bagnall A, Chue P, Fenton M, Palaniswamy V, Wong W, Xia J. Loxapine for schizophrenia. Cochrane Database Syst Rev. 2007:CD001943. doi: 10.1002/14651858.CD001943.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Kronengold J, Yan Y, Gazula VR, Brown MR, Ma L, Ferreira G, Yang Y, Bhattacharjee A, Sigworth FJ, Salkoff L, Kaczmarek LK. The N-terminal domain of Slack determines the formation and trafficking of Slick/Slack heteromeric sodium-activated potassium channels. J Neurosci. 2009;29:5654–5665. doi: 10.1523/JNEUROSCI.5978-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Dib-Hajj SD, Waxman SG. Differential slow inactivation and use-dependent inhibition of Nav1.8 channels contribute to distinct firing properties in IB4+ and IB4− DRG neurons. J Neurophysiol. 2007;97:1258–1265. doi: 10.1152/jn.01033.2006. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Ellis A, Bennett DL. Neuroinflammation and the generation of neuropathic pain. Br J Anaesth. 2013;111:26–37. doi: 10.1093/bja/aet128. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150:573–581. doi: 10.1016/j.pain.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122:245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschetti S, Lavazza T, Curia G, Aracri P, Panzica F, Sancini G, Avanzini G, Magistretti J. Na+-activated K+ current contributes to postexcitatory hyperpolarization in neocortical intrinsically bursting neurons. J Neurophysiol. 2003;89:2101–2111. doi: 10.1152/jn.00695.2002. [DOI] [PubMed] [Google Scholar]
- Huang F, Wang X, Ostertag EM, Nuwal T, Huang B, Jan YN, Basbaum AI, Jan LY. TMEM16C facilitates Na(+)-activated K+ currents in rat sensory neurons and regulates pain processing. Nat Neurosci. 2013;16:1284–1290. doi: 10.1038/nn.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunskaar S, Fasmer OB, Hole K. Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods. 1985;14:69–76. doi: 10.1016/0165-0270(85)90116-5. [DOI] [PubMed] [Google Scholar]
- Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, Gribkoff VK, Kaczmarek LK. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat Neurosci. 1998;1:462–469. doi: 10.1038/2176. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK. Slack, Slick and sodium-activated potassium channels. ISRN Neurosci. 2013;pii:354262. doi: 10.1155/2013/354262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallenborn-Gerhardt W, Schröder K, Del Turco D, Lu R, Kynast K, Kosowski J, Niederberger E, Shah AM, Brandes RP, Geisslinger G, Schmidtko A. NADPH oxidase-4 maintains neuropathic pain after peripheral nerve injury. J Neurosci. 2012;32:10136–10145. doi: 10.1523/JNEUROSCI.6227-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Lu N, Cui Y, Yang T, Zhao ZQ, Xin WJ, Liu XG. Prevention of paclitaxel-induced allodynia by minocycline: effect on loss of peripheral nerve fibers and infiltration of macrophages in rats. Mol Pain. 2010;6:76. doi: 10.1186/1744-8069-6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Abdel Samad O, Zhang L, Duan B, Tong Q, Lopes C, Ji RR, Lowell BB, Ma Q. VGLUT2-dependent glutamate release from nociceptors is required to sense pain and suppress itch. Neuron. 2010;68:543–556. doi: 10.1016/j.neuron.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Schmidtko A. Direct intrathecal drug delivery in mice for detecting in vivo effects of cGMP on pain processing. Methods Mol Biol. 2013;1020:215–221. doi: 10.1007/978-1-62703-459-3_14. [DOI] [PubMed] [Google Scholar]
- Lu R, Lukowski R, Sausbier M, Zhang DD, Sisignano M, Schuh CD, Kuner R, Ruth P, Geisslinger G, Schmidtko A. BKCa channels expressed in sensory neurons modulate inflammatory pain in mice. Pain. 2014;155:556–565. doi: 10.1016/j.pain.2013.12.005. [DOI] [PubMed] [Google Scholar]
- McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain. 1997;1:43–52. doi: 10.1016/S1090-3801(97)90052-5. [DOI] [PubMed] [Google Scholar]
- Nieto FR, Entrena JM, Cendán CM, Pozo ED, Vela JM, Baeyens JM. Tetrodotoxin inhibits the development and expression of neuropathic pain induced by paclitaxel in mice. Pain. 2008;137:520–531. doi: 10.1016/j.pain.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Nuwer MO, Picchione KE, Bhattacharjee A. PKA-induced internalization of slack KNa channels produces dorsal root ganglion neuron hyperexcitability. J Neurosci. 2010;30:14165–14172. doi: 10.1523/JNEUROSCI.3150-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122:S22–S32. doi: 10.1016/j.amjmed.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Peters CM, Jimenez-Andrade JM, Kuskowski MA, Ghilardi JR, Mantyh PW. An evolving cellular pathology occurs in dorsal root ganglia, peripheral nerve and spinal cord following intravenous administration of paclitaxel in the rat. Brain Res. 2007;1168:46–59. doi: 10.1016/j.brainres.2007.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, Chiarugi A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol. 2011;80:1136–1146. doi: 10.1124/mol.111.073916. [DOI] [PubMed] [Google Scholar]
- Raouf R, Quick K, Wood JN. Pain as a channelopathy. J Clin Invest. 2010;120:3745–3752. doi: 10.1172/JCI43158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR. Na+ signals at central synapses. Neuroscientist. 2002;8:532–539. doi: 10.1177/1073858402238512. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Nowak LG, McCormick DA. Cellular mechanisms of long-lasting adaptation in visual cortical neurons in vitro. J Neurosci. 2000;20:4286–4299. doi: 10.1523/JNEUROSCI.20-11-04286.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, Sausbier M, Rauhmeier I, Sausbier U, Niederberger E, Scholich K, Huber A, Neuhuber W, Allescher HD, Hofmann F, Tegeder I, Ruth P, Geisslinger G. Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J Neurosci. 2008;28:1320–1330. doi: 10.1523/JNEUROSCI.5037-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Tegeder I, Geisslinger G. No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci. 2009;32:339–346. doi: 10.1016/j.tins.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, 3rd, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain. 2003;4:465–470. doi: 10.1067/S1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Siau C, Xiao W, Bennett GJ. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: loss of epidermal innervation and activation of Langerhans cells. Exp Neurol. 2006;201:507–514. doi: 10.1016/j.expneurol.2006.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisignano M, Baron R, Scholich K, Geisslinger G. Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat Rev Neurol. 2014;10:694–707. doi: 10.1038/nrneurol.2014.211. [DOI] [PubMed] [Google Scholar]
- Tamsett TJ, Picchione KE, Bhattacharjee A. NAD+ activates KNa channels in dorsal root ganglion neurons. J Neurosci. 2009;29:5127–5134. doi: 10.1523/JNEUROSCI.0859-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpley JW, Kohler MG, Martin WJ. The behavioral and neuroanatomical effects of IB4-saporin treatment in rat models of nociceptive and neuropathic pain. Brain Res. 2004;1029:65–76. doi: 10.1016/j.brainres.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Tsantoulas C, McMahon SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 2014;37:146–158. doi: 10.1016/j.tins.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain. 2014;155:654–662. doi: 10.1016/j.pain.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Wallén P, Robertson B, Cangiano L, Löw P, Bhattacharjee A, Kaczmarek LK, Grillner S. Sodium-dependent potassium channels of a Slack-like subtype contribute to the slow afterhyperpolarization in lamprey spinal neurons. J Physiol. 2007;585:75–90. doi: 10.1113/jphysiol.2007.138156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Zamponi GW. Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci. 2014;17:153–163. doi: 10.1038/nn.3602. [DOI] [PubMed] [Google Scholar]
- Ying W. NAD+ and NADH in neuronal death. J Neuroimmune Pharmacol. 2007;2:270–275. doi: 10.1007/s11481-007-9063-5. [DOI] [PubMed] [Google Scholar]
- Yuan A, Santi CM, Wei A, Wang ZW, Pollak K, Nonet M, Kaczmarek L, Crowder CM, Salkoff L. The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron. 2003;37:765–773. doi: 10.1016/S0896-6273(03)00096-5. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Rosenhouse-Dantsker A, Tang QY, Noskov S, Logothetis DE. The RCK2 domain uses a coordination site present in Kir channels to confer sodium sensitivity to Slo2.2 channels. J Neurosci. 2010;30:7554–7562. doi: 10.1523/JNEUROSCI.0525-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]