

Abstract
In excitable cells, ion channels are frequently challenged by repetitive stimuli, and their responses shape cellular behavior by regulating the duration and termination of bursts of action potentials. We have investigated the behavior of Shaker family voltage-gated potassium (Kv) channels subjected to repetitive stimuli, with a particular focus on Kv1.2. Genetic deletion of this subunit results in complete mortality within 2 weeks of birth in mice, highlighting a critical physiological role for Kv1.2. Kv1.2 channels exhibit a unique property described previously as “prepulse potentiation,” in which activation by a depolarizing step facilitates activation in a subsequent pulse. In this study, we demonstrate that this property enables Kv1.2 channels to exhibit use-dependent activation during trains of very brief depolarizations. Also, Kv subunits usually assemble into heteromeric channels in the central nervous system, generating diversity of function and sensitivity to signaling mechanisms. We demonstrate that other Kv1 channel types do not exhibit use-dependent activation, but this property is conferred in heteromeric channel complexes containing even a single Kv1.2 subunit. This regulatory mechanism is observed in mammalian cell lines as well as primary cultures of hippocampal neurons. Our findings illustrate that use-dependent activation is a unique property of Kv1.2 that persists in heteromeric channel complexes and may influence function of hippocampal neurons.
Keywords: gating, Kv1.2, modulation, potassium channel, signaling, use-dependence
Introduction
Voltage-gated potassium (Kv) channels in the nervous system influence threshold firing properties and propagation of action potentials. In the axon initial segment, clustered Kv channels integrate dendritic electrical signals and regulate neuronal excitability (Rasband, 2010), with this region sometimes referred to as a “hotspot” for seizure generation (Wimmer et al., 2010; Robbins and Tempel, 2012). Kv channels also exhibit marked clustering in the juxtaparanodal region around nodes of Ranvier (Rasband et al., 1998). Supporting the general physiological importance of the Kv1 family, numerous reports have linked mutations in Kv1 channels to movement disorders and epilepsy (Adelman et al., 1995; Kullmann and Hanna, 2002; Xie et al., 2010). Knock-out models of certain Kv1 channels suffer neurological consequences depending on which Kv1 subunit is deleted. Defects are especially pronounced with genetic deletion of Kv1.2, which results in complete mortality within weeks of birth, and contrasts with mild consequences of knocking out most other Kv1 channels (London et al., 1998; Smart et al., 1998; Brew et al., 2007). Overall, these findings imply distinct roles for Kv1 subtypes in neuronal excitability, although it is not known what features of Kv1.2 gating and regulation underlie its stringent physiological requirement.
Most biophysical studies of Kv channels focus on homotetrameric channels. However, it is recognized that Kv channels achieve functional diversity via their ability to assemble as heteromeric complexes (Po et al., 1993; Sheng et al., 1993; Wang et al., 1993, 1994; Plane et al., 2005). Heteromerization of Kv1 channels can involve assembly with other Kv1 subtypes and auxiliary proteins, such as the Kvβ subunits (Li et al., 1992; Bixby et al., 1999). This diversity is reflected in the biophysical details (e.g., kinetics and voltage dependence) of channel gating, but perhaps more interestingly, combination of different Kv1 subtypes may harness regulatory pathways unique to each (Huang et al., 1994; Holmes et al., 1996a,b; Tsai et al., 1997; Cachero et al., 1998; Nitabach et al., 2001). Thus, each subtype in a heteromeric channel may contribute sensitivity to specific cellular signals, generating a wide range of control mechanisms. For most regulatory mechanisms identified, it is poorly understood whether and/or how their effects are translated into altered function of heteromeric channel complexes.
Previous reports have described a unique regulatory mechanism of Kv1.2 channels described as “prepulse potentiation” (Grissmer et al., 1994; Rezazadeh et al., 2007). Although the signaling molecule(s) involved are not yet identified, this mechanism has been localized to a threonine residue in the intracellular S2–S3 linker of Kv1.2 (Rezazadeh et al., 2007). In this study, we address several questions related to the phenomenon of Kv1.2 prepulse potentiation. First, we have tested whether prepulse potentiation can lead to use-dependent behavior of Kv1.2 channels during repetitive stimulus trains. Second, we asked how assembly of Kv1.2 with other Kv1 subtypes influences use-dependent gating, and what stoichiometry of channel assembly is required for this phenomenon to occur. Last, we have investigated the presence of Kv1.2-mediated currents that exhibit use-dependent activation in primary neuronal cultures. Our findings demonstrate that Kv1.2 channels are unique in their ability to generate marked use-dependent potentiation/activation and confer this property on heteromeric Kv1.2-containing channel complexes in mammalian cell lines and hippocampal neurons.
Materials and Methods
Constructs and expression.
Kv1 channel cDNAs and dimeric constructs were expressed using the pcDNA3.1(−) vector (Invitrogen). For dimer construction, the leading protomer was subcloned into the NheI and XhoI sites in pcDNA3.1(−). The trailing protomer was subcloned into the EcoRI and HindIII sites. Kv1.1 or Kv1.4 homotetramers did not express well in the mouse ltk- fibroblast (LM) cell line. Therefore, we generated chimeras with the Kv1.5 N terminus, replacing sequence up to the N-terminal boundary of the T1 domain (these chimeras maintain the native Kv1.1 or Kv1.4 sequence in the S2–S3 linker that is important for mediating use-dependent activation). These chimeric constructs expressed well and were used to test for use-dependent activation in Figure 2. For tetramer construction, the leading two protomers (Kv1.5–Kv1.5) were constructed similarly to the dimers, except an NheI site was introduced at the 5′ end of the trailing protomer. This construct was subcloned into the NheI site of a dimeric construct (assembled as above) containing the trailing two protomers (Kv1.5–Kv1.2). Constructs were all verified by diagnostic restriction digestions and Sanger sequencing (Genewiz).
Figure 2.

Use-dependent activation is observed in Kv1.2 but not other Kv1 channel subtypes. A, LM cells expressing WT Kv1.2 or Kv1.2[T252R] channels (as indicated) were subjected to 20 Hz trains of repetitive depolarizations (60 mV for 10 ms, −80 mV for 40 ms). Exemplar currents generated from 50 pulses illustrate the slow and fast Kv1.2 phenotypes, along with exclusively fast currents generated by Kv1.2[T252R] channels. B, Similar protocols were performed with numerous Kv1 family channels. Percentage use-dependent activation was calculated (inset) as the difference in peak current between the first pulse to 60 mV and currents elicited at the end of the depolarizing train (n = 176 for WT Kv1.2, >20 for Kv1.2[T252R] and other Kv1 subtypes). Sample sweeps show pulses 1 (colored) and 50 (black) from the pulse train. Scatter/box plots illustrate the variability in percentage use-dependent activation for each channel type. * Note that Kv1.1 and Kv1.4 did not express well, and were expressed as chimeras with the distal N-terminus of Kv1.5 (see Methods) - this manipulation leaves the S2–S3 linker, previously identified as critical for use-dependent activation, unchanged. Overall, significant use-dependent activation is only observed for Kv1.2 channels. C, Structural localization of Kv1.2 residue T252 in the intracellular S2–S3 linker is highlighted on the Kv1.2 paddle chimera structure. D, Sequence alignment of the S2–S3 linker for numerous Kv1 channel subtypes, illustrating that a Thr at this position is only present in Kv1.2.
LM cells were maintained in culture in a 5% CO2 incubator at 37°C in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were split onto sterile glass coverslips and, 24 h later, were transfected with channel cDNAs using Polyplus Jetprime. Cells were cotransfected with fluorescent proteins to allow identification of cells for recording by epifluorescence. Recordings were done 1–3 d after transfection.
Electrophysiology.
Patch pipettes were manufactured from borosilicate capillary glass (World Precision Instruments) using a Sutter Instruments P-97 puller. When filled with standard recording solutions, pipettes had a tip resistance of 3–5 MΩ. This pipette size is somewhat smaller than we usually use for generation of high-fidelity recordings, but we found this to be helpful in maintaining the use-dependent activation phenotype of Kv1.2 (which is lost more rapidly in recordings that dialyze cellular contents; Rezazadeh et al., 2007). Recordings were filtered at 5 kHz, sampled at 10 kHz, with manual capacitance compensation and series resistance compensation between 60 and 80%, and stored directly on a computer hard drive using Clampex software (Molecular Devices). Bath solution had the following composition (in mm): 135 NaCl, 5 KCl, 2.8 NaCH3CO2, 1 CaCl2, 1 MgCl2, and 10 HEPES, adjusted to pH 7.4 with KOH. Pipette solution had the following composition (in mm): 135 KCl, 5 K-EGTA, and 10 HEPES, adjusted to pH 7.2 using KOH. Chemicals were purchased from Sigma-Aldrich or Thermo Fisher Scientific. Tityustoxin was purchased from Alomone Labs and was stored as a 50 μm stock (−20°C) in the standard extracellular recording solution. Toxin was added to a known solution volume in the recording chamber to achieve the desired dilutions. Throughout the text, data are presented as mean ± SEM.
Western blot analysis.
Cell lysates from transfected LM cells were harvested in RIPA buffer 3 d after transfection, separated using 7.5% SDS-PAGE gels, and transferred to nitrocellulose membranes using standard methods. Kv1.2 was detected using a monoclonal mouse Kv1.2 antibody (clone #K14/16 75-008; NeuroMab), HRP-conjugated goat anti-mouse antibody (SH023; Applied Biological Materials), and SuperSignal West Femto Max Sensitivity Substrate (Thermo Fisher Scientific). Chemiluminescence was detected using a FluorChem SP gel imager (Alpha Innotech).
MTSET [2-(trimethylammonium)ethyl methanethiosulfonate, bromide] delivery.
MTSET was dissolved in standard recording solution at a concentration of 5 mm at the beginning of each experimental day and stored on ice. A small amount of MTSET was delivered to a known volume of extracellular solution (containing no KCl) in the recording chamber, resulting in a final concentration of 100 μm. MTSET was purchased from Toronto Research Chemicals.
Hippocampal cell culture.
Hippocampi from embryonic day 18 Sprague Dawley rats of either sex were prepared as described previously (Xie et al., 2000) and plated at a density of 75 cells/mm2. Neurons were used for electrophysiology experiments at 7–16 d in vitro. Patch pipettes were manufactured from soda lime glass and pulled to a resistance of 1.5–2.5 MΩ. Tityustoxin-sensitive currents were isolated by applying tityustoxin at 100 or 300 nm and subtracting toxin-insensitive current from total current. Recordings were filtered at 5 kHz, sampled at 10 kHz, with 60–80% series resistance compensation, and stored directly on a computer hard drive using Clampex software (Molecular Devices). Bath and pipette solutions were as described above, except NaCl was replaced by 135 mm N-methyl-d-glucamine in the bath solution.
Results
Prepulse potentiation of Kv1.2 channels
Previous reports have described a gating “mode shift” in homomeric Kv1.2 channels expressed in mammalian cell lines (Rezazadeh et al., 2007). This manifests as a significant acceleration of activation kinetics and a leftward shift in the V1/2 of activation after prepulses that activate channels. These features are summarized in Figure 1 using a double-pulse protocol (Fig. 1A) in which cells were pulsed to either a low voltage (−60 mV, very few channels open) or a high voltage (100 mV, maximal channel opening), followed by a test pulse to 20 mV. Cells exhibit a wide range of responses to this protocol. On one extreme, “fast” cells (Fig. 1A, bottom) are typified by rapid activation at 20 mV, regardless of whether channels were opened during the P1 pulse. In contrast, “slow” cells (Fig. 1A, top) exhibit variable kinetics of channel opening at 20 mV, depending on whether the prepulse was sufficient to activate channels. In slow cells, if the P1 prepulse activates large numbers of channels, the P2 pulse exhibits potentiation (faster activation kinetics and larger currents). The marked variability between cells suggests that this reflects a regulatory process rather than an intrinsic property of the channels. However, the mechanisms underlying this variability are not yet known. Using a Kv1.2-selective channel toxin, tityustoxin (Werkman et al., 1993), we demonstrated that currents in cells exhibiting either the fast or slow gating phenotypes could both be inhibited (data not shown), confirming that both behaviors originate from the same channel type.
Figure 1.

Prepulse potentiation of Kv1.2 channels. A, Whole-cell patch-clamp recordings of Kv1.2 channels expressed in LM cells. Recordings are representative of currents elicited from cells exhibiting a fast gating phenotype with no prepulse potentiation (bottom) and a slow gating phenotype (top) in which activation of channels during a prepulse causes accelerated activation in a second pulse. B, Conductance–voltage relationships elicited before and after a 500 ms potentiating prepulse to 60 mV (protocol depicted in sample currents in C) in cells that exhibit either slow mode or fast mode gating. Cells with prominent slow mode gating (red symbols) exhibit a marked leftward shift of the V1/2 of activation after potentiation. C, Activation curves of nonpotentiated and potentiated channels were measured by pulsing cells in 10 mV steps between −70 and 70 mV in two families of sweeps (P1, P2), separated by a pulse to 60 mV. Currents from a representative cell exhibiting the slow gating phenotype are highlighted in red, and a cell exhibiting the fast gating phenotype is in blue (sweeps with test pulses to −30 mV are bolded). Activation kinetics are plotted against the shift in activation V1/2 measured between the P1 and P2 families of sweeps.
As reported previously, the prepulse-dependent shift in activation kinetics is accompanied by a shift in the V1/2 of activation (Fig. 1B). In the absence of a prepulse (open symbols), slow cells (red) exhibit a V1/2 that is displaced by 32 ± 3 mV to the right compared with fast cells (blue). This shift is normalized when the activation curves are collected after a prepulse. That is, when channels are potentiated by a prepulse, they exhibit a similar left-shifted V1/2 (filled symbols), indicating that the slow mode channels have been shifted to a fast gating mode. Figure 1C (right) highlights this behavior using a protocol to collect activation curves before (P1) and after (P2) a potentiation pulse to 60 mV. Nonpotentiated channels require strong depolarizations to open, but their V1/2 of activation shifts to more negative voltages after a potentiation pulse. In the red exemplar traces (from a cell with marked prepulse potentiation), we have bolded voltage steps to −30 mV before and after a depolarization to 60 mV. It is apparent that the −30 mV pulse elicits very little current in the “nonpotentiated” first pulse but large rapidly activating currents in the “potentiated” second pulse. The shift in V1/2 attributable to prepulse potentiation is plotted in Figure 1C, along with the activation kinetics measured at 10 mV (in the P1 pulse). Cells that exhibit slow activation in P1 (highlighted as red symbols and the red sample sweeps) are prone to strong leftward shifts in V1/2 after a potentiation pulse, often larger than 30 mV. In contrast, cells that exhibit fast gating of Kv1.2 (highlighted in blue) do not exhibit prepulse potentiation. It is noteworthy that we have observed Kv1.2 channel activation with a wide range of V1/2 (from −20 mV to nearly 40 mV). Within this palette of variable behavior, cells with slow activation kinetics (red symbols, traces) exhibit the most prominent prepulse-dependent potentiation (shifts of V1/2).
Prepulse potentiation generates use-dependent activation
Previous work has not addressed whether prepulse potentiation can affect channel function over short timescales that reflect bursting action potentials in the CNS or whether long pulses (hundreds of milliseconds, as in Fig. 1) are required to elicit the potentiated state. We hypothesized that the prepulse potentiation phenomenon would generate use-dependent increases in current during trains of repetitive brief depolarizations attributable to progressive accumulation of channels in the potentiated state. Indeed, a unique behavior of channels in the slow mode emerges in response to repetitive brief depolarizations (10 ms, 20 Hz), highlighted in Figure 2A (top trace). In this protocol, each depolarizing stimulus is too brief to activate a large number of slow mode channels (Fig. 2A). However, each depolarization activates a small fraction of channels and causes them to switch to a potentiated gating mode. With a long sequence of pulses, this pattern results in gradual recruitment of channels to the potentiated mode and a dramatic increase in the K+ current that can be activated. Hereafter, we refer to this process as “use-dependent activation”.
We quantified the “percentage use-dependent activation” as the percentage of peak current that develops between the first depolarizing pulse and the fully potentiated steady state (Fig. 2B, left). Thus, slow cells with prominent increases of peak current exhibit a large percentage use dependence, whereas fast cells with little change in current magnitude between the first and last pulses (Fig. 2A, middle trace) exhibit small percentage use dependence. Between these two extremes, there is considerable variability in cells transfected with wild-type (WT) Kv1.2 (Fig. 2B, each data point represents a measurement from a different cell). Previous work illustrated the importance of Kv1.2 residue T252 for prepulse potentiation and demonstrated that transplantation of the Kv1.2 S2 segment and S2–S3 linker into Kv1.5 could introduce prepulse potentiation (Rezazadeh et al., 2007). As expected, the T252R mutation also abolishes Kv1.2 use-dependent activation, confirming that use-dependent activation and prepulse potentiation reflect the same regulatory mechanism (Fig. 2A, bottom trace, B).
We surveyed numerous other Kv1 channel types using identical protocols to detect use-dependent activation. Because of the large cell-to-cell variability of Kv1.2, we tested at least 20 cells per channel type. Although a large fraction of Kv1.2 recordings exhibited prominent use-dependent activation, no other Kv1 channels exhibited this property (Fig. 2B). It should be noted that Kv1.1 or Kv1.4 (with a short 19 aa N-terminal deletion to remove N-type inactivation; Hoshi et al., 1990) did not express functional currents in the cell line we used for this experiment. However, when fused to the N terminus of Kv1.5 (replacing sequence preceding the conserved T1 domain), these channels exhibited robust currents. Importantly, these experiments left the S2–S3 linker (that contains Kv1.2 residue T252; Fig. 2C) unchanged. Further illustrating the importance of Kv1.2 residue T252, no other Kv1 channel exhibited use-dependent activation (all have either a Lys or Arg at the equivalent position, highlighted in Fig. 2D).
Use-dependent activation is transferred to heteromeric channels
We aimed to determine whether use-dependent activation gating of Kv1.2 could be observed in heteromeric Kv1 channel complexes. In native cells, it is common for Kv1 channel subtypes to assemble as heteromeric complexes, yet few studies have explored how various regulatory/signaling mechanisms can affect function of channels with mixed composition (Po et al., 1993; Sheng et al., 1993; Nitabach et al., 2001; Plane et al., 2005). To ensure assembly of heteromeric channels, we generated tandem dimer constructs of Kv1.2 with several other Kv1 channel subtypes and used repetitive trains of depolarizations to measure use-dependent activation (Fig. 3A–C). Currents could be detected from dimeric combinations of Kv1.2 with Kv1.1, Kv1.4, and Kv1.5 but not Kv1.3. Notably, each of the mixed dimer constructs exhibited pronounced (but variable) use-dependent activation, similar to Kv1.2 (Fig. 3A–C). These findings suggest that use-dependent activation conferred by Kv1.2 can persist in heteromeric complexes with other Kv1 subunits.
Figure 3.

Use-dependent activation persists in Kv1.2-containing heteromeric channel complexes with other Kv1 channel subtypes. A–C, Dimeric channel constructs were built with Kv1.2 and various other Kv1 channel subtypes, with protomers arranged in both orientations. Use-dependent activation was measured with trains of repetitive depolarizations (10 ms, 60 mV, 20 Hz), as described in Figures 3 and 4. D–F, Cell lysates were probed with a monoclonal anti-Kv1.2 antibody to confirm and validate synthesis of full-length dimer constructs (LM cells indicate mock transfected cells).
Certain previous reports have questioned the appropriate assembly of concatenated K+ channel subunits (McCormack et al., 1992). This prompted us to rule out the possibility that monomeric Kv1.2 subunits were generating the use-dependent currents observed. For example, monomeric Kv1.2 subunits could conceivably be synthesized from spurious internal translation start sites in the dimeric constructs. It has also been suggested that inappropriate assembly of concatenated subunits can lead to unpredictable channel stoichiometry. Thus, we tested dimeric constructs with both orientations of the protomers (i.e., for the Kv1.1–Kv1.2 dimer, we used a dimer with Kv1.1 in the protomer A position and a second dimer with Kv1.2 in the protomer A position). Dimers generated in both orientations exhibited similar use-dependent activation. Importantly, we also performed Western blots of lysates from transfected cells to confirm the generation of dimeric channels and absence of Kv1.2 monomeric subunits (Fig. 3D–F). Typically, lysates from cells transfected with monomeric Kv1.2 generated two bands (Fig. 3D), reflecting mature and immature glycosylated forms (Fujita et al., 2006). Transfection with dimeric constructs generated protein with approximately double the molecular weight of these characteristic Kv1.2 bands (Fig. 3D–F), in all cases but one (the A:Kv1.4,B:Kv1.2 dimer; Fig. 3E). More importantly, virtually no monomeric Kv1.2 protein was observed in dimer lysates, negating the possibility that the use dependence of mixed dimers arises from unexpected expression of monomeric Kv1.2. However, an important exception to these findings was the A:Kv1.4,B:Kv1.2 dimer, which yielded fairly weak Kv1.2 immunoreactivity in the expected dimeric molecular weight range and some signal slightly larger than monomeric Kv1.2 (Fig. 3E). These observations suggest that use-dependent currents detected in cells transfected with this particular dimer (A:Kv1.4,B:Kv1.2) arise from spurious translation of subunits missing a large amount of Kv1.4 sequence rather than the full-length dimer construct. These findings indicate that, in most cases, full-length dimers (containing Kv1.2) are synthesized as expected, with use-dependent activation conferred to these heteromeric channels by Kv1.2. However, these observations also highlight the importance of controls to ensure appropriate translation of dimer constructs.
Functional demonstration of dominance of Kv1.2 use-dependence in dimeric channels
Uncertainty related to appropriate synthesis and assembly of transfected dimeric channel constructs (Fig. 3E) prompted additional investigation to confirm Kv1.2 contributions to heteromeric channels. We adopted a functional approach to demonstrate that Kv1.2 can confer use-dependent activation to other Kv1 subunits based on cysteine modification in the channel pore mouth. C-type inactivation of Shaker family channels is associated with a conformational change in the outer pore mouth that changes the accessibility of substituted cysteines (Liu et al., 1996). We generated dimeric channels comprising Kv1.2[V381T] (to match the Shaker T449 position), in tandem with Kv1.5[P488C] (equivalent to the Shaker P450 position), bearing a cysteine substitution that enables pronounced current inhibition after MTSET modification (Liu et al., 1996). With this design (Fig. 4A), we tested for channels that exhibited use-dependent activation (arising from the presence of Kv1.2) in combination with MTSET sensitivity (arising from the presence of Kv1.5[P488C]). Cells were first exposed to a train of depolarizations, demonstrating use-dependent activation attributable to the presence of Kv1.2 in the functional channels (Fig. 4Bi). The presence of a cysteine at position P488 does not affect the observation of use-dependent activation (Fig. 4C). Next, cells were depolarized for 400 ms at 4 s intervals, in the presence of extracellular MTSET (100 μm; Fig. 4Bii). In channels lacking a modifiable cysteine in the Kv1.5 subunit, no rundown was observed (Fig. 4C, black symbols). However, the P488C substitution in the Kv1.5 protomer rendered channels highly sensitive to MTSET and complete channel rundown (Fig. 4C, red symbols). Because cysteine modification was sufficient to abolish all use-dependent currents, we conclude that virtually all of the channels that exhibit use-dependent activation (arising from Kv1.2) also contain a modifiable cysteine (contributed by Kv1.5[P488C]). These experiments demonstrate that both subunits in the dimeric construct contribute to functional channels and that use-dependent activation features of Kv1.2 persist in heteromeric channels.
Figure 4.
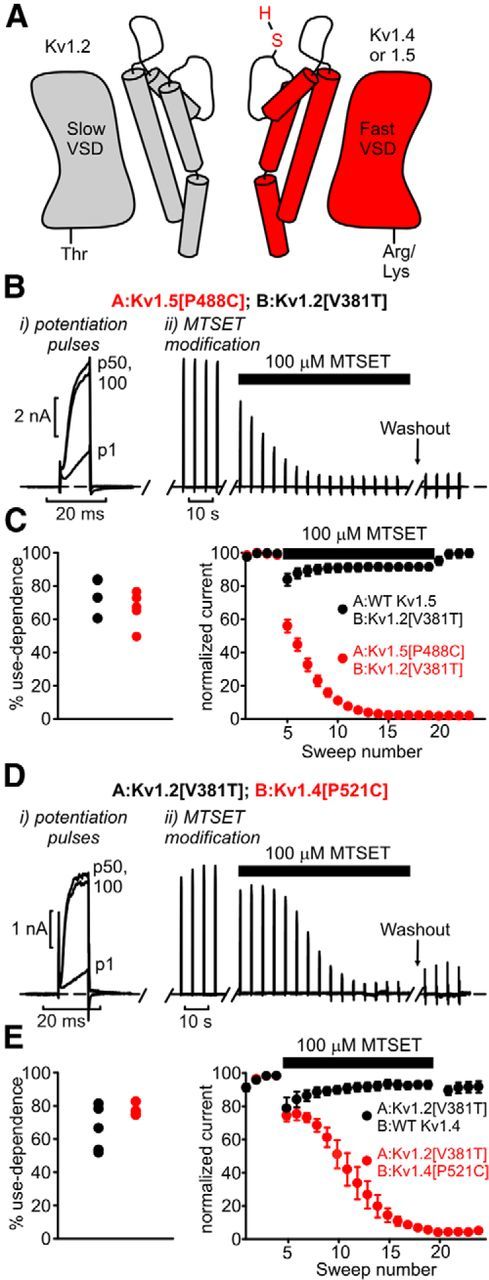
Functional contribution of Kv1.2 to heteromeric channel complexes that exhibit use-dependent activation. A, Dimeric channel constructs were generated comprising Kv1.2[V381T] and Kv1.5 (with or without a substituted cysteine at position P488) or Kv1.4 (cysteine substitution at P521). This design enables us to test whether channels that exhibit use-dependent activation (attributable to Kv1.2) also contain Kv1.5/Kv1.4 subunits in the functional channel. VSD indicates voltage-sensing domain. B, Cells transfected with A:Kv1.5[P488C]; B:Kv1.2[V381T] dimeric constructs were subjected to a series of 100 repetitive brief depolarizations (potentiation pulses of 10 ms, 60 mV, 20 Hz) to demonstrate use-dependent activation (Bi). Next, in Bii (MTSET modification), cells were exposed to 100 μm MTSET in the bath solution and pulsed to 60 mV for 400 ms, every 4 s. C, Mean data describing the experiments in B, with the distribution of percentage use-dependent activation for both constructs (left) and the MTSET sensitivity of both constructs (right). In dimers containing WT Kv1.5 (with no modifiable cysteines), no rundown is observed (black symbols). However, dimers comprising Kv1.5[P488C] exhibit virtually complete channel rundown, n = 4–5 per construct. D, E, Similar experiments as in B and C were performed using A:Kv1.2[V381T]; B:Kv1.4[P521C] dimeric channels, n = 4–5 per construct.
The dimeric constructs with trafficking and synthesis issues were the Kv1.4–Kv1.2 dimers (Fig. 3E). Therefore, we performed similar experiments using Kv1.2[V381T]–Kv1.4[P521C] tandem dimers, first confirming the presence of Kv1.2 by demonstrating use-dependent activation and next confirming the contribution of Kv1.4 to the functional dimer by triggering rundown by MTSET modification of residue P521C. We observed similar results as described for the Kv1.5[P488C]–Kv1.2[V381T] dimer (Fig. 4D,E), thereby demonstrating the presence of use-dependent activation in heteromeric channels containing Kv1.2 and Kv1.4.
Use-dependent activation in heteromeric channels containing one Kv1.2 subunit
Heteromeric channel assemblies containing two subunits of Kv1.2 are able to recapitulate strong use-dependent activation. We also investigated the stoichiometry of this effect and asked whether subunits with only one Kv1.2 subunit could also exhibit use-dependent activation. We generated a concatenated tetrameric channel containing three subunits of Kv1.5 and one Kv1.2 subunit (protomer A:Kv1.5, B:Kv1.5, C:Kv1.5, D:Kv1.2) and used repetitive depolarizations to elicit use-dependent activation (Fig. 5A). These tandem constructs generate robust ionic currents, and some cells exhibited prominent use-dependent activation. However, the majority of cells exhibited less use-dependent activation relative to parallel experiments on Kv1.2 monomeric channels (Fig. 5A). Western blots of cells transfected with the tetrameric construct exhibited two bands at approximately four times the weight of the monomeric construct (Fig. 5B). The lack of spurious bands at weights lower than expected indicates appropriate expression of the tetrameric construct. Overall, the presence of one subunit of Kv1.2 appears to be sufficient to confer use-dependent activation in a heteromeric channel. However, there may be a dosing effect in which increased numbers of Kv1.2 subunits renders channels more susceptible to use-dependent activation.
Figure 5.

Use-dependent activation persists in channels containing a single Kv1.2 subunit. A, Use-dependent activation was measured in mouse ltk- cells as described in Figure 2B, comparing monomeric Kv1.2 channels with a tandem-linked tetramer comprising three Kv1.5 subunits and one Kv1.2 subunit. Sample currents for the tandem tetramer construct illustrate the variability of use-dependent activation observed for these channels. B, Cell lysates were probed using a monoclonal anti-Kv1.2 antibody, illustrating the faithful generation of tetrameric channels from the tandem-linked construct (LM cells indicate mock transfected cells).
Use-dependent activation is evident in a primary culture of hippocampal neurons
Use-dependent activation can arise in homomeric and heteromeric channel assemblies, even those containing just one Kv1.2 subunit. Furthermore, this behavior has been observed in all mammalian cell lines tested, including LM, COS, and human embryonic kidney cells, prompting us to ask whether use-dependent activation might be observed in native cell types. To this end, we isolated currents generated from Kv1.2-containing channels in cultured primary rat hippocampal neurons (embryonic day 18) using tityustoxin (Werkman et al., 1993). We validated the specificity of this toxin by applying 100 or 300 nm to heterologously expressed Kv1.2 and found that it was fully sensitive, whereas Kv1.1, Kv1.4, and Kv1.5 were insensitive (Fig. 6A). It should be noted that Kv1.3 has been reported to be susceptible to tityustoxin, with a KD of 19.8 nm (Rodrigues et al., 2003), although the more prominent inactivation and absence of use-dependent activation of Kv1.3 can be distinguished clearly from Kv1.2. We also tested whether dimeric and tetrameric constructs containing two or one Kv1.2 subunits, respectively, were tityustoxin sensitive. The effect of tityustoxin was variable depending on subtype composition, with the Kv1.1–Kv1.2 dimer being especially sensitive. Nevertheless, each of these constructs was partially inhibited by 100 nm tityustoxin (Fig. 6A). Overall, these findings demonstrate that tityustoxin inhibits homomeric and heteromeric (partially) channels containing Kv1.2 subunits, indicating this is an effective tool to isolate currents generated by Kv1.2-containing channels.
Figure 6.

Isolation of tityustoxin-sensitive currents in dissociated hippocampal neurons. A, The tityustoxin (100 nm) sensitivity of Kv1.2 and various Kv1.2-containing heteromeric channels was measured for channels expressed in LM cells. B, K+ currents were measured from dissociated hippocampal neurons in the presence and absence of 100 or 300 nm tityustoxin to isolate tityustoxin-sensitive currents by subtraction.
To test for the presence of use-dependent activation in dissociated hippocampal neurons, cells were stimulated with short (10 ms, 10 Hz) repetitive depolarizing voltage steps before and after addition of tityustoxin (Fig. 6B). Typically, the tityustoxin-sensitive current made up a significant proportion of the total voltage-gated K+ current, although this was quite variable from cell to cell and ranged from 10 to 80%. Much like in mammalian cell lines, a variety of current phenotypes were observed (Fig. 7A,B). Interestingly, we frequently observed use-dependent activation in the tityustoxin-sensitive current, highlighted in the top traces in Figure 7B. We also often observed currents with a prominent inactivating component (Fig. 7B, bottom traces), which might emerge from coassembly of Kv1.2 with Kv1.4 or a Kvβ subunit. The percentage use-dependence was calculated as described in previous figures and is summarized in Figure 7A, illustrating the wide range of biophysical properties emerging from Kv1.2-containing channels in these cells. Please note that only cells that exhibited use-dependent activation in either the 10 or 2 Hz stimulation protocols (discussed below) are included in Figure 7A (some degree of use-dependent activation was observed in 45% of the neurons tested). Overall, cells that exhibit use-dependent activation in neurons mimic closely the effect seen in mammalian cells and so it seems reasonable to speculate that the underlying mechanism (although unknown at present) is likely the same.
Figure 7.

Variability of gating properties of tityustoxin-sensitive channels in dissociated hippocampal neurons. A, Summary of use-dependent properties of neurons subjected to repetitive depolarizations at the indicated frequencies. Cells exhibit a range of use-dependent activation and/or inactivation. B, Sample traces of 10 Hz recordings of tityustoxin-sensitive currents from hippocampal neurons, illustrating extreme cases of either use-dependent activation (top) or inactivation (bottom). C, Sample traces from longer pulse durations, illustrating the cell-to-cell variability of inactivation, and the emergence of use-dependent potentiation in the sustained current component of some cells with pronounced inactivation (middle and right).
We also used longer (100 ms) repetitive depolarizations to reveal other features of elicited K+ currents (Fig. 7A,C), and this was helpful in revealing use-dependent activation in cells with a prominent inactivating component in the tityustoxin-sensitive current. We have presented several sample traces to contrast the variable phenotypes that were observed (Fig. 7C). In cells with a very small A-type component, use-dependent activation was observed frequently in trains of both short (10 ms) and/or long (100 ms) depolarizations. However, in some cells, particularly those with a prominent A-type component, use-dependent activation was not observed with repetitive 10 ms pulses but emerged with 100 ms pulses as an increase in the sustained current (Fig. 7C, middle and right traces). Other phenotypes resulting from a mixture of A-type current and use-dependent activation were observed between these extremes. Together, these data illustrate that Kv1.2 can assemble with other Kv1 subunits to generate marked cell-to-cell diversity in current properties (combining inactivation and use-dependent activation). As with mammalian cell lines expressing Kv1.2, use-dependent activation is one of the predominant phenotypes.
Discussion
Expression of Kv1.2 in mammalian cell lines generates remarkable plasticity in the kinetic and thermodynamic properties of channel activation. From cell to cell, varying degrees of prepulse potentiation are observed, but a consistent feature is that channels can be shifted from a slow gating mode to a fast gating mode by delivering a depolarizing stimulus (Rezazadeh et al., 2007). In this study, we have demonstrated that this prepulse potentiation translates into a mechanism of use-dependent activation in trains of repetitive short pulses designed to more closely reflect the brief stimulations that might be encountered by Kv1.2-containing channels in the CNS. We extend previous findings in several important ways, by demonstrating that Kv1.2 channel potentiation can occur over very brief timescales, can persist in heteromeric Kv1 channels with even a single Kv1.2 subunit, and can be observed in cultured hippocampal neurons. Use-dependent activation is clearly unique to Kv1.2 subunits and relies on the presence of a threonine in the intracellular S2–S3 linker (it is a Lys or Arg in all other Kv1 channel types and most other Kv channels). Moreover, this property can be transferred to the fast gating Kv1.5 subtype by substituting the S2 segment and S2–S3 linker of Kv1.2. The regulatory mechanism involved has not been identified, although this property appears to be attributable to a diffusible cellular signal rather than an intrinsic property of the channel (Rezazadeh et al., 2007).
Kv1.2 regulation of excitability in the hippocampus
Kv1.2 plays a key role in regulating excitability in the hippocampus. At the neuronal level, its presence provides a strong dampening force to regulate action potential firing (Palani et al., 2010). Pathologies associated with abnormal Kv1.2 function and expression also highlight its importance. In a seizure-prone gerbil model, Kv1.2 and other Kv1 channels are downregulated, whereas neuronal Kv2, Kv3, and Kv4 channels are unaffected (Lee et al., 2009). As mentioned previously, complete knock-out of Kv1.2 in mice leads to death by generalized seizure within 2 weeks of birth (Brew et al., 2007). Also, limbic encephalitis and Morvan syndrome, both of which lead to increased seizure susceptibility, are associated with autoantibodies to Kv1, particularly anti-Kv1.1 and anti-Kv1.2 antibodies (Kleopa et al., 2006). At the other end of the spectrum, hypothyroidism in rats leads to increased Kv1.2 (and Kv4) expression, with associated action potential shortening (Buckley et al., 2001). These gross changes in regulation and expression of Kv1.2 are associated with severe diseases of excitability, hinting that more subtle and controlled changes in gating and/or expression could provide a powerful mechanism for tuning cellular excitability.
Ion channel responses to repetitive stimuli
Effects of repetitive or prolonged stimuli of Kv channels have been considered normally in the context of inactivation mechanisms (Kurata and Fedida, 2006). Central to this is the concept of “cumulative inactivation,” in which a series of repetitive stimuli causes the accumulation of channels in an inactivated state (Aldrich, 1981; Klemic et al., 1998; Kurata et al., 2001). This phenomenon arises because the time between stimuli is insufficient to allow complete recovery of channels that have inactivated in a preceding depolarization. Accumulation of inactivation is especially important in the context of Kv channel contributions to cellular excitability, because it will determine the number of channels “available” to activate during an action potential or at subthreshold voltages and thereby influence the strength of the repolarizing influence that can develop from these channel types.
Our study highlights an entirely different response of a Kv channel to repetitive depolarizations, in which channels accumulate in a potentiated state. Rather than progressive loss of Kv channel availability, use-dependent activation enhances Kv channel activity later in trains of depolarizations. Potential physiological consequences of this behavior might be to suppress cellular excitability in a use-dependent manner during bursts of action potentials in certain cell types. One report has described use-dependent activation of a Kv channel (KVS-1) cloned from Caenorhabditis elegans (Rojas et al., 2008), but this does not appear to share similar structural determinants or cellular mediators (for example, use-dependent activation mechanisms of KVS-1 persists in Xenopus laevis oocytes but is absent for Kv1.2 in this expression system (data not shown)). Use-dependent activation of Kv1.2 also appears to be highly contextual, because it varies dramatically from cell to cell in the same preparation and thus may depend on cell-cycle stage or other unidentified regulatory processes. Several noteworthy studies have proposed signaling pathways that control gating of Kv1.2 channels, including regulation by the sigma receptor cortactin and the M1 ACh receptor (Tsai et al., 1997; Williams et al., 2007; Kourrich et al., 2013). However, channel properties influenced by these signaling mechanisms do not appear similar to the use-dependent activation features that we observe. Most recently, phosphatidic acid was shown to induce a marked rightward shift in the activation curve of the Kv1.2/2.1 paddle chimera, although the role of phosphatidic acid in use-dependent activation of Kv1.2 has not been tested explicitly (Hite et al., 2014). Notable similarities are also apparent between the prepulse potentiation observed for Kv1.2 and prepulse-dependent relief of Gβγ inhibition of N- and P/Q-type Cav channels (Zamponi and Snutch, 1998). However, preliminary experiments in our laboratory suggest that these candidate regulatory mechanisms do not affect use-dependent activation of Kv1.2.
Integration of signaling mechanisms in heteromeric Kv channels
Although it is recognized widely that different Kv1 channel subtypes assemble into heteromeric channels in numerous tissues (Vacher et al., 2008), it is not commonplace to study the behavior of heteromeric channels or to examine the mechanisms by which regulation of one Kv1 channel subtype influences other subunits in a functional channel. In the case of use-dependent activation of Kv1.2, this gating behavior clearly persists in channels of mixed subtype composition and can appear with even a single Kv1.2 subunit in the tetrameric channel (Figs. 4, 5). Moreover, in primary hippocampal neurons, use-dependent activation was observed frequently in combination with fast inactivation in a given cell, demonstrating the “comingling” of these regulatory mechanisms. A previous study used a variety of recording modes to infer that use-dependent activation relied on interaction of the channel with a diffusible cellular component. For example, excision of membrane patches or prolonged dialysis of intracellular contents led to a loss of this effect (Rezazadeh et al., 2007). Our findings suggest that interaction of this hypothetical regulator with a single-channel subunit is enough to decelerate activation kinetics and shift the V1/2 of channel activation. Thus, an interesting possibility is that Kv1.2 may act to recruit this regulatory mechanism to heteromeric channels, enabling a mechanism for use-dependent modulation of neuronal excitability. Whether this unique gating mechanism plays a role in the stringent requirement of Kv1.2 in animal models is not established but could be tested in knock-in models using a T252 mutation to ablate use-dependent activation.
Conclusion
We report that Kv1.2 channels exhibit a unique mechanism of use-dependent activation. This mechanism acts in a “dominant” manner, in that it confers use-dependent activation to heteromeric channels comprising Kv1.2 and other (non-use-dependent) Kv1 subtypes, with as few as one Kv1.2 subunit. This regulatory mechanism persists across mammalian cell lines and primary cultures of hippocampal neurons.
Footnotes
This work was supported by Canadian Institutes of Health Research Operating Grants MOP-97988 (H.T.K.) and MOP-81158 (S.X.B.). V.A.B. was supported by a Natural Sciences and Engineering Research Council of Canada Undergraduate Research Award and the University of British Columbia MD/PhD program. B.R.M. was supported by the University of British Columbia Faculty of Medicine Summer Student Research Program. R.Y.K. was supported by the University of British Columbia Work Study program, and a Canada Graduate Scholarships–Master's award. H.T.K. received salary support from the Canadian Institutes of Health Research, Heart and Stroke Foundation of Canada, and the Michael Smith Foundation for Health Research. We thank Arohumam Kan for assistance with validating tetrameric Kv channel constructs.
The authors declare no competing financial interests.
References
- Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Aldrich RW. Inactivation of voltage-gated delayed potassium current in molluscan neurons. A kinetic model. Biophys J. 1981;36:519–532. doi: 10.1016/S0006-3495(81)84750-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bixby KA, Nanao MH, Shen NV, Kreusch A, Bellamy H, Pfaffinger PJ, Choe S. Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat Struct Biol. 1999;6:38–43. doi: 10.1038/4911. [DOI] [PubMed] [Google Scholar]
- Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, Robbins CA, McKee-Johnson J, Chiu SY, Messing A, Tempel BL. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J Neurophysiol. 2007;98:1501–1525. doi: 10.1152/jn.00640.2006. [DOI] [PubMed] [Google Scholar]
- Buckley C, Oger J, Clover L, Tüzün E, Carpenter K, Jackson M, Vincent A. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001;50:73–78. doi: 10.1002/ana.1097. [DOI] [PubMed] [Google Scholar]
- Cachero TG, Morielli AD, Peralta EG. The small GTP-binding protein RhoA regulates a delayed rectifier potassium channel. Cell. 1998;93:1077–1085. doi: 10.1016/S0092-8674(00)81212-X. [DOI] [PubMed] [Google Scholar]
- Fujita T, Utsunomiya I, Ren J, Matsushita Y, Kawai M, Sasaki S, Hoshi K, Miyatake T, Taguchi K. Glycosylation and cell surface expression of Kv1.2 potassium channel are regulated by determinants in the pore region. Neurochem Res. 2006;31:589–596. doi: 10.1007/s11064-006-9056-4. [DOI] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Hite RK, Butterwick JA, MacKinnon R. Phosphatidic acid modulation of Kv channel voltage sensor function. Elife. 2014;3 doi: 10.7554/eLife.04366. 04366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes TC, Fadool DA, Levitan IB. Tyrosine phosphorylation of the Kv1.3 potassium channel. J Neurosci. 1996a;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes TC, Fadool DA, Ren R, Levitan IB. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science. 1996b;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Huang XY, Morielli AD, Peralta EG. Molecular basis of cardiac potassium channel stimulation by protein kinase A. Proc Natl Acad Sci U S A. 1994;91:624–628. doi: 10.1073/pnas.91.2.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic KG, Shieh CC, Kirsch GE, Jones SW. Inactivation of Kv2.1 potassium channels. Biophys J. 1998;74:1779–1789. doi: 10.1016/S0006-3495(98)77888-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleopa KA, Elman LB, Lang B, Vincent A, Scherer SS. Neuromyotonia and limbic encephalitis sera target mature Shaker-type K+ channels: subunit specificity correlates with clinical manifestations. Brain. 2006;129:1570–1584. doi: 10.1093/brain/awl084. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su TP, Bonci A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell. 2013;152:236–247. doi: 10.1016/j.cell.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Hanna MG. Neurological disorders caused by inherited ion-channel mutations. Lancet Neurol. 2002;1:157–166. doi: 10.1016/S1474-4422(02)00071-6. [DOI] [PubMed] [Google Scholar]
- Kurata HT, Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog Biophys Mol Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Kurata HT, Soon GS, Fedida D. Altered state dependence of c-type inactivation in the long and short forms of human Kv1.5. J Gen Physiol. 2001;118:315–332. doi: 10.1085/jgp.118.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Kim JE, Sohn JH, Choi HC, Lee JS, Kim SH, Kim MJ, Choi IG, Kang TC. Down-regulation of delayed rectifier K+ channels in the hippocampus of seizure sensitive gerbils. Brain Res Bull. 2009;80:433–442. doi: 10.1016/j.brainresbull.2009.07.016. [DOI] [PubMed] [Google Scholar]
- Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1230. doi: 10.1126/science.1519059. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/S0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- London B, Wang DW, Hill JA, Bennett PB. The transient outward current in mice lacking the potassium channel gene Kv1.4. J Physiol. 1998;509:171–182. doi: 10.1111/j.1469-7793.1998.171bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K, Lin L, Iverson LE, Tanouye MA, Sigworth FJ. Tandem linkage of Shaker K+ channel subunits does not ensure the stoichiometry of expressed channels. Biophys J. 1992;63:1406–1411. doi: 10.1016/S0006-3495(92)81703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitabach MN, Llamas DA, Araneda RC, Intile JL, Thompson IJ, Zhou YI, Holmes TC. A mechanism for combinatorial regulation of electrical activity: Potassium channel subunits capable of functioning as Src homology 3-dependent adaptors. Proc Natl Acad Sci U S A. 2001;98:705–710. doi: 10.1073/pnas.98.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palani D, Baginskas A, Raastad M. Bursts and hyperexcitability in non-myelinated axons of the rat hippocampus. Neuroscience. 2010;167:1004–1013. doi: 10.1016/j.neuroscience.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Plane F, Johnson R, Kerr P, Wiehler W, Thorneloe K, Ishii K, Chen T, Cole W. Heteromultimeric Kv1 channels contribute to myogenic control of arterial diameter. Circ Res. 2005;96:216–224. doi: 10.1161/01.RES.0000154070.06421.25. [DOI] [PubMed] [Google Scholar]
- Po S, Roberds S, Snyders DJ, Tamkun MM, Bennett PB. Heteromultimeric assembly of human potassium channels. Molecular basis of a transient outward current? Circ Res. 1993;72:1326–1336. doi: 10.1161/01.RES.72.6.1326. [DOI] [PubMed] [Google Scholar]
- Rasband MN. Clustered K+ channel complexes in axons. Neurosci Lett. 2010;486:101–106. doi: 10.1016/j.neulet.2010.08.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, Shrager P. Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci. 1998;18:36–47. doi: 10.1523/JNEUROSCI.18-01-00036.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezazadeh S, Kurata HT, Claydon TW, Kehl SJ, Fedida D. An activation gating switch in Kv1.2 is localized to a threonine residue in the S2–S3 linker. Biophys J. 2007;93:4173–4186. doi: 10.1529/biophysj.107.116160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CA, Tempel BL. Kv1.1 and Kv1.2: similar channels, different seizure models. Epilepsia. 2012;53(Suppl 1):134–141. doi: 10.1111/j.1528-1167.2012.03484.x. [DOI] [PubMed] [Google Scholar]
- Rodrigues AR, Arantes EC, Monje F, Stühmer W, Varanda WA. Tityustoxin-K(alpha) blockade of the voltage-gated potassium channel Kv1.3. Br J Pharmacol. 2003;139:1180–1186. doi: 10.1038/sj.bjp.0705343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas P, Garst-Orozco J, Baban B, de Santiago-Castillo JA, Covarrubias M, Salkoff L. Cumulative activation of voltage-dependent KVS-1 potassium channels. J Neurosci. 2008;28:757–765. doi: 10.1523/JNEUROSCI.3825-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Liao YJ, Jan YN, Jan LY. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Nature. 1993;365:72–75. doi: 10.1038/365072a0. [DOI] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/S0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- Tsai W, Morielli AD, Peralta EG. The m1 muscarinic acetylcholine receptor transactivates the EGF receptor to modulate ion channel activity. EMBO J. 1997;16:4597–4605. doi: 10.1093/emboj/16.15.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88:1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 1993;365:75–79. doi: 10.1038/365075a0. [DOI] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. 1994;14:4588–4599. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werkman TR, Gustafson TA, Rogowski RS, Blaustein MP, Rogawski MA. Tityustoxin-K alpha, a structurally novel and highly potent K+ channel peptide toxin, interacts with the alpha-dendrotoxin binding site on the cloned Kv1.2 K+ channel. Mol Pharmacol. 1993;44:430–436. [PubMed] [Google Scholar]
- Williams MR, Markey JC, Doczi MA, Morielli AD. An essential role for cortactin in the modulation of the potassium channel Kv1.2. Proc Natl Acad Sci U S A. 2007;104:17412–17417. doi: 10.1073/pnas.0703865104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer VC, Reid CA, So EY, Berkovic SF, Petrou S. Axon initial segment dysfunction in epilepsy. J Physiol. 2010;588:1829–1840. doi: 10.1113/jphysiol.2010.188417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Markesbery WR, Lovell MA. Survival of hippocampal and cortical neurons in a mixture of MEM+ and B27-supplemented neurobasal medium. Free Radic Biol Med. 2000;28:665–672. doi: 10.1016/S0891-5849(99)00268-3. [DOI] [PubMed] [Google Scholar]
- Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. 2010;285:32160–32173. doi: 10.1074/jbc.M110.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998;8:351–356. doi: 10.1016/S0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]