

Abstract
Nerve injury is associated with microvascular disturbance; however, the role of the vascular system has not been well characterized in the context of neuropathic pain. Furthermore, ischemia is thought to play a role in a number of neuropathic pain conditions, and yet the role of hypoxia has also not been characterized in neuropathic pain conditions. In this study, we observed the presence of persistent endoneurial hypoxia in a mouse model of traumatic peripheral nerve injury, causing painful mononeuropathy. We attribute the ongoing hypoxia to microvascular dysfunction, endoneurial fibrosis, and increased metabolic requirements within the injured nerve. Increased lactate levels were observed in injured nerves, as well as increased oxygen consumption and extracellular acidification rates, suggesting that anaerobic glycolysis is required to maintain cellular ATP levels. Hypoxia causes a reduction in levels of the Na+/K+ ATPase ion transporter in both cultured primary dorsal root ganglion neurons and injured peripheral nerve. A reduction of Na+/K+ ATPase ion transporter levels likely contributes to the hyperexcitability of injured nerves. Physiological antagonism of hypoxia with hyperbaric oxygen alleviated mechanical allodynia in nerve-injured animals. These results suggest that hypoxia and the Na+/K+ ATPase ion transporter may be a novel mechanistic target for the treatment of neuropathic pain. In addition, the findings support the possibility of using hypoxia activated pro-drugs to localize treatments for neuropathic pain and nerve injury to injured nerves.
Keywords: drug development, hypoxia, Na+/K+ ATPase ion transporter, nerve injury, neuropathic pain, vascular dysfunction
Introduction
Experiments in animal models have greatly enriched our understanding of the molecular and cellular mechanisms underlying the pathogenesis of nerve injury-induced neuropathic pain. Undoubtedly, structural and functional alterations on neuronal pathways play determinant roles. In addition, findings from the past 20 years have demonstrated the importance of the immune system in modulating neuronal activities (Scholz and Woolf, 2007). Although the involvement of both neuronal and immune systems has been characterized in the context of neuropathic pain, very little is known about the role that the vascular system plays in the development and maintenance of debilitating chronic pain conditions.
Although the direct contribution of vascular dysfunction to neuropathic pain has not been explored fully, evidence of microvascular disturbances has been reported in both humans having neuropathic pain and in neuropathic pain animal models. For example, in diabetes, pathological changes result in basement membrane thickening and reduced luminal sizes in endoneurial capillaries (Yasuda and Dyck, 1987). Such alterations were also observed in patients with nerve compression (Mackinnon et al., 1986). Similar microvascular disturbances have been observed in traumatic models of painful neuropathy (Sommer et al., 1993; Sommer and Myers, 1996). Additionally, vascular dysfunction itself has been proposed to be a potential mechanism in the production of some types of chronic pain. Restriction of blood flow and tissue ischemia results in complex regional pain syndrome-like symptoms in rats (Coderre et al., 2004). In rodent models of nerve ischemia, photochemical damage to nerve blood vessels generates neuropathic pain behavior (Kupers et al., 1998; Hao et al., 2000). In humans, those with sickle cell anemia experience extreme pain during vaso-occlusive episodes whereby sickle-shaped red blood cells obstruct blood vessels, resulting in tissue ischemia (Ballas et al., 2012). Chronic ischemic pain is also reported by patients with peripheral arterial diseases (Rüger et al., 2008). Clearly, the vascular system can influence pain, and its role in pain pathogenesis needs to be investigated further.
Whether hypoxia plays a contributing factor to the genesis of pain is not known. However, studies in animal models (Thompson et al., 2010) and patients (Kiralp et al., 2004; Gu et al., 2012) have demonstrated that hyperbaric oxygen therapy is of therapeutic benefit to neuropathic pain conditions, suggesting a role of hypoxia in pain pathogenesis. In this study, our first goal was to investigate whether traumatic nerve injury leads to persistent endoneurial hypoxia, and thus we characterized the spatiotemporal pattern of hypoxia after nerve injury. The mechanisms involved in producing the persistent hypoxic state of the nerve were examined, which consist of microvascular dysfunction, endoneurial fibrosis, and increased metabolic loads. We also explored potential mechanisms by which hypoxia could contribute to neuropathic pain, uncovering a novel role of the Na+/K+ ATPase ion transporter in neuropathic pain pathogenesis. We demonstrate that abrogating hypoxia by hyperbaric oxygen therapy leads to an alleviation of neuropathic pain behaviors. Our results suggest that hypoxia has functional effects on both the immune and peripheral nervous systems and that these effects in turn contribute to the pathobiology of neuropathic pain.
Materials and Methods
Animals.
Experiments were performed in adult C57BL/6 mice (males, weighing 20–25 g) purchased from Charles River Laboratories. Mice were housed four to five per cage in a temperature- and humidity-controlled vivarium on a 12 h light/dark cycle beginning at 7:00 A.M., with access to rodent chow and water ad libitum. Behavioral experiments were conducted between 8:00 A.M. and 4:00 P.M. All protocols were conducted according to the guidelines of the Canadian Council on Animal Care and the International Association for the Study of Pain, as administrated by the McGill University animal care committee.
Nerve injury model.
Partial sciatic nerve ligation (PSNL) was performed according to the method of Seltzer et al. (1990), adapted to mice (Malmberg and Basbaum, 1998). Briefly, mice were anesthetized with isoflurane (3% for induction and maintenance; Baxter), and, under aseptic conditions, the left sciatic nerve was exposed at high-thigh level. The dorsum of the nerve was freed carefully from the surrounding connective tissues at a site near the trochanter just distal to the point at which the posterior biceps semitendinosus nerve branches off the common sciatic nerve. An 8-0 silk suture was inserted into the nerve with a ⅜ curved, reversed-cutting mini-needle and tightly ligated so that the dorsal one-third to one-half of the nerve thickness was trapped in the ligature. The wound was then closed with two to three skin sutures (4-0).
Drugs and materials.
Hypoxyprobe-1 was obtained from Hypoxyprobe. Fluorescein isothiocyanate (FITC)-labeled dextran was obtained from Sigma-Aldrich (average molecular weight, 2 m).
Behavioral monitoring.
Mice were habituated to the testing environment daily for at least 2 d before baseline testing. For measurements of mechanical sensitivity, paw-withdrawal thresholds were measured with calibrated von Frey fibers (Stoelting) using the up–down method (Chaplan et al., 1994). Mice were placed on a metal mesh floor with small Plexiglas cubicles (9 × 5 × 5 cm high) and kept for at least 1 h for habituation in their individual Plexiglas observation chamber before testing. A set of eight calibrated von Frey fibers (ranging from 0.008 to 1.40 g of force) were applied to the plantar surface of the hindpaw until they bent. The threshold force required to elicit withdrawal of the paw (median 50% paw withdrawal) was determined as the average of two tests separated by at least 1 h.
Hyperbaric oxygen.
Mice were placed in a custom-made 10 L hyperbaric chamber. A 25 cm2 polystyrene cell culture flask with a vented 0.2 μm cap (Becton Dickinson Labware) containing 10 g of calcium oxide (Acros Organics) was placed inside the hyperbaric chamber to sequester CO2. The hyperbaric chamber was filled with 100% O2 (Megs), and chamber pressure was slowly increased to 2.5 atmospheric (atm) total pressure over 5 min. Flow rate of O2 was maintained at 10 L/min, and animals were treated with hyperbaric O2 for 120 min. The hyperbaric chamber was then depressurized over 5 min to atmospheric pressure. In sham-treated animals, animals were treated with 100% O2 at normal atmospheric pressure.
Tissue preparation and histology.
For hypoxyprobe-1 experiments, mice were injected with 60 mg/kg hypoxyprobe-1 intraperitoneally. One hour later, mice were then deeply anesthetized with a ketamine/xylazine mixture and then perfused transcardially with 0.9% NaCl (Sigma-Aldrich), followed by 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) in 0.1 m sodium phosphate buffer (Sigma-Aldrich) at pH 7.4. Sciatic nerves were removed and placed in 4% PFA overnight and then transferred to 30% sucrose (Sigma-Aldrich) for cryoprotection. For histological experiments of hypoxia inducible factor-1α (HIF-1α), animals were perfused transcardially with 0.9% NaCl, and sciatic nerves were extracted, postfixed in 4% PFA for 5 min, and transferred to 30% sucrose for cryoprotection. For experiments with FITC–dextran angiography, 4 mg/kg FITC–dextran (average molecular weight, 2 m) was injected intravenously through the tail vein. Animals were killed 1 min after injection, and nerves were immediately extracted and postfixed overnight in 4% PFA before being transferred to 30% sucrose for cryoprotection. Tissue was embedded in VWR Clear Frozen Section Compound (VWR International) and either cut longitudinally into 14 μm sections or at 5 μm cross-sections using a Leica CM3050 S cryostat). Sections were mounted on SuperFrost+ slides (VWR International) and stored at −20°C until use.
For electron microscopy and toluidine blue histology, mice were perfused with 0.5% PFA and 2.5% glutaraldehyde in 0.1 m phosphate buffer. Sciatic nerves were extracted and maintained in perfusion buffer overnight. For postfixation, nerves were transferred to 2% osmium tetroxide (Canemco) for 2 h. Tissue was then dehydrated by serial incubations in solutions of increasing alcohol concentration, with a final incubation in 100% propylene oxide (Canemco). Tissues were embedded in Epon. Cross-sections of the nerve were cut at 0.5 μm thickness with glass knives and stained with 0.5% toluidine blue O (Thermo Fisher Scientific) in 1% sodium borate to assess tissue with light microscopy. Ultrathin sections were cut and stained with lead citrate and examined with a Philips transmission electron microscope.
Immunohistochemistry.
Slide-mounted sections were incubated in blocking buffer consisting of 3% goat serum (Vector Laboratories), 1% bovine serum albumin (Sigma-Aldrich), and 0.25% Triton X-100 (Sigma-Aldrich, St. Louis, MO) in Tris-buffered saline (TBS) for 1 h at room temperature. Slides were then incubated overnight at 4°C with rabbit anti-hypoxyprobe-1 (1:500; Hypoxyprobe), rabbit anti-glucose transporter 1 (GLUT1; 1:6000; Millipore), rabbit anti-collagen IV (1:400; Abcam), rabbit anti-HIF-1α (1:100; Santa Cruz Biotechnology), FITC-conjugated mouse anti-neurofilament 160/200 (1:1000), rat anti-F4/80 (1:1000; AbD Serotec), or 4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene (BODIPY 493/503) (1 μg/ml; Life Technologies). FITC-conjugated mouse anti-neurofilament 160/200 was prepared using an FITC conjugation kit (Abcam) with mouse anti-neurofilament 160/200 (Sigma-Aldrich). Sections were then incubated with Alexa Fluor 594 or 488-conjugated goat anti-rabbit secondary antibodies (1:500; Invitrogen) for 1 h at room temperature. Selected sections were also counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear labeling. Slides were coverslipped with Vectashield mounting medium (Vector Laboratories).
Dorsal root ganglion primary culture.
Male adult mice (20–25 g) were overdosed with isoflurane and decapitated. The spinal cord was cut out above the coxa and muscle tissue and costal bones were trimmed away. Spinal cords were washed twice in sterile cold Dulbecco's PBS (Corning Life Sciences). The spinal cord was cut along the sagittal plane, and spinal cord tissue was displaced to reveal dorsal root ganglions (DRGs). DRGs were isolated on ice and transferred to cold sterile HBSS without magnesium or calcium (Life Technologies). DRGs were digested with 40 U/ml papain (Worthington Biochemical) and then with 1 kU/ml collagenase-II (Worthington Biochemical). After digestion, ganglia were triturated, and the suspension was passed through a 100 μm nylon cell strainer (Corning). The DRG neurons were plated on glass chamber slides (Nunc) for viability tests or on 25 cm2 polysterene vented cell culture flasks (BD Labware) for Na+/K+ ATPase quantification by Western blot. DRGs from two mice were plated on two chamber slides or on two 25 cm2 flasks. Both chamber slides and cell culture flasks were coated with 50 mg/ml poly-d-lysine (BD Biosciences) and 5 mg/ml laminin (Sigma-Aldrich) overnight at 4°C before use. Neurons were plated in Ham's F-12 nutrient mixture (HyClone Laboratories) with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1× penicillin/streptomycin (Life Technologies). After 24 h of plating, media were exchanged with 10% FBS F-12 media containing 20 μm uridine, 20 μm 5-fluoro-2′-deoxyuridine, and 0.5 μm cytosine β-d-arabinofuranoside (all purchased from Sigma-Aldrich) to remove non-neuronal cells (Liu et al., 2013). Neurons were maintained under normoxic conditions in a Heracell 150 incubator (Thermo Fisher Scientific) at 37°C, 5% CO2 and ambient oxygen for 6 d. For hypoxic conditions, cells were transferred to a separate Heracell 150 tri-gas incubator set at 1% O2, 94% N2, 5% CO2 and cultured for 48 h. A control flask was maintained under normoxic conditions for the same amount of time.
Neuronal viability assay.
DRG neurons were cultured in chamber slides as described above. F-12 media was removed from chamber slides and replaced with HBSS with 3 μm calcien AM (eBioscience) at 37°C for 30 min. The following procedures were then performed at room temperature and in the dark: cells were fixed by incubation with 2% PFA for 10 min. PFA was removed and washed with PBS. Blocking buffer consisting of 3% goat serum, 1% BSA, and 0.25% Triton X-100 in PBS was applied for 30 min. A 1:500 dilution of mouse monoclonal anti-neurofilament 160/200 antibody (Sigma-Aldrich) in blocking buffer was incubated on slides for 1 h in the dark. After washing with PBS, 1:500 goat anti-mouse Alexa Fluor 594-conjugated secondary antibody in blocking buffer was applied to the cells for 30 min. Slides were washed with PBS, dehydrated under a vacuum, and coverslipped with Vectashield mounting medium. Cell death control slides were prepared by applying 0.25% Triton X-100 in HBSS for 10 min at 37°C before application of calcien AM. Neurons were imaged using an Olympus BX51 microscope equipped with a color digital camera (Olympus DP71), with attention to constant exposure time and ISO sensitivity settings. Images were color segmented with Image-Pro Plus 6 (Media Cybernetics) software. Green thresholds were set to determine live cells that sequestered calcien AM. Red thresholds were set to identify neuronal cells from non-neuronal cells. Cell viability was defined as the number of live neuronal cells divided by the total number of neuronal cells observed.
Western blot.
To prepare nerve lysates from mice, animals were overdosed with a ketamine/xylazine mixture and perfused transcardially with 0.9% NaCl. Sciatic nerves 1 cm in length (excised at 3 mm proximal, 7 mm distal of suture) were extracted and homogenized in a buffer containing 2% SDS (BioShop Canada), 95 mm NaCl, 10 mm ethylenediaminetetraacetic acid (Sigma-Aldrich), and 1 Mini cOmplete protease inhibitor tablet (Roche Diagnostics) per 10 ml. Homogenization was performed with 1.4 mm zirconium oxide beads (Cayman Chemical) and a Precellys 24 tissue homogenizer (Bertin Technologies) using two 20 s pulses at 5600 rpm. Protein determination of homogenates was performed by bicinchoninic acid assay (Thermo Fisher Scientific). Equal amounts of homogenate (30 μg) were loaded into 8% polyacrylamide mini-protean (Bio-Rad) hand-cast gels.
For DRG neuronal lysates, DRG neurons were cultured as described above in 25 cm2 flasks. Media was removed, and the flask was washed several times with PBS. RIPA buffer (Sigma-Aldrich) containing protease inhibitors (Mini cOmplete; Roche Diagnostics) was added to the flask, and a cell scraper was used to dislodge neurons. Lysate was transferred to a microcentrifuge tube, and it was agitated for 30 min at 4°C. Protein determination of lysate was performed by bicinchoninic acid assay (Thermo Fisher Scientific). Equal amounts of lysate (5 μg) was loaded into 8% polyacrylamide mini-protein (Bio-Rad) hand-cast gels.
Polyacrylamide gels underwent electrophoresis, and proteins were transferred onto nitrocellulose membranes (Bio-Rad). Successful protein transfer was confirmed with 5% Ponceau red (Sigma-Aldrich), and membranes were blocked with 5% milk (BioShop Canada) in TBS (Sigma-Aldrich) with 0.1% Tween 20 (BioShop Canada) for 1 h at room temperature. Membranes were incubated overnight at 4°C with 1:200 rabbit pan anti-Na+/K+ ATPase α (Santa Cruz Biotechnology), 1:100 rabbit HIF-1α (Santa Cruz Biotechnology), or 1:10,000 mouse anti-β-actin (Sigma-Aldrich). Membranes were then incubated for 1 h at room temperature with 1:5000 goat anti-rabbit or 1:10,000 goat anti-mouse horseradish peroxidase-conjugated secondary antibodies (Millipore Bioscience Research Reagents). Membranes were imaged using Supersignal West Pico Chemiluminescence Substrate (Thermo Fisher Scientific) on Biomax Light film (Carestream Health), using a Kodak X-OMAT 2000A developer (Carestream Health). Films were digitized with a CanoScan LiDE 110 scanner (Canon) and quantified using NIH ImageJ (version 1.44p).
Image analysis.
Images were captured using an Olympus BX51 microscope equipped with a color digital camera (Olympus DP71). Images were digitized using constant exposure time and ISO sensitivity settings across samples. Quantification of hypoxyprobe and HIF-1α was performed by fluorescence intensity analysis in longitudinal nerve sections. An area of interest (AOI) was defined within the middle of the nerve, at areas 2 mm distal to the injury site. For GLUT1 quantification, an AOI was defined in the middle of nerve cross-sections at various distances along the nerve. The intensity of fluorescence within the AOI was measured using Image-Pro Plus 6 (Media Cybernetics). Blood vessel luminal areas were measured using NIH ImageJ (version 1.44p) using data from blood vessels throughout the entire nerve cross-section. Nerve cross-sectional areas and intercapillary distances were measured with Image-Pro Plus 6 (Media Cybernetics). The intercapillary distance was defined as the minimum distance between nonadjacent capillaries (Less et al., 1991).
3D reconstruction of sciatic nerve.
FITC–dextran angiography was performed as described in the histology section above. Serial cross-sections of a PSNL sciatic nerve were cut at 5 μm with a Leica CM3050 S cryostat. Every second section was slide mounted on SuperFrost plus slides (VWR International), so that serial cross-sections were 10 μm apart in distance. GLUT1 immunohistochemistry was performed as described in the immunohistochemistry section above. Images were digitized as described in the image analysis section above. Serial cross-sectional images were aligned manually using Amira 3D analysis software (version 5.4.1; FEI Visualization Sciences Group). Aligned cross-sectional images were extruded to a 3D scalar field according to the z distances of each cross-section. The 3D scalar field was rendered as orange to represent GLUT1 staining of the perineurium. Endoneurial blood vessels were traced through the nerve by manual tracing of FITC–dextran-positive blood vessels through the endoneurium. Traced blood vessels were extruded as a 3D mesh and rendered in green.
Lactate assay.
On day 14, mice were perfused transcardially with 0.9% saline after ketamine/xylazine overdose. Equal-length nerves of 2 cm were extracted. Nerves were homogenized with 1.4 mm zirconium oxide beads in lactate assay buffer (Sigma-Aldrich). Nerve homogenate was deproteinated by passing homogenate through a 10,000 Da molecular weight cutoff centrifugal filtration device (Vivaspin 500; Sartorius Stedim) at 15,000 × g for 40 min at 4°C, using a 5415R centrifuge (Eppendorf). Filtrate was stored at −80°C until analyzed by fluorometry. Samples were thawed on ice and analyzed using a lactate assay kit (MAK064; Sigma-Aldrich) according to the protocol of the manufacturer. Samples were diluted and incubated with lactate enzyme and lactate probe in a 96-well clear flat-bottom medium binding microplate (Greiner Bio-One) along with lactate standards. Fluorescence intensity (λex = 535 nm, λem = 587 nm) was measured using a microplate reader (Spectramax M2E; Molecular Devices).
Seahorse XF extracellular flux analyzer.
At day 7 after PSNL injury, mice were killed by isoflurane overdose and cervical dislocation. Approximately 1.5-cm-long lengths of sciatic nerves were immediately extracted and placed in ice-cold XF base medium minimal DMEM at pH 7.4, supplemented with 5.5 mm glucose (Sigma-Aldrich), 0.5 mm sodium pyruvate (Invitrogen), and 1 mm glutaMAX (Invitrogen). Nerves were cut into three 3 mm segments using an acrylic V-shaped 1.0 mm slicer matrix (Zivic Instruments). Nerve segments were then cut in half longitudinally with a razor blade, and the two halves were transferred into a single well of a XF96 cell culture microplate (Seahorse Bioscience), with 140 μl of cold supplemented XF base medium. Each nerve was tested in triplicate. The distal portions of nerves were processed in all analysis. Basal oxygen consumption rates and extracellular acidification rates were measured with an XF 96 Analyzer (Seahorse Bioscience). A 3 min mix, 3 min measure protocol was used. Basal values were obtained by averaging the triplicate values at the third and fourth measurements.
Statistics.
All data are presented as mean ± SEM. For behavioral tests with a time course, the thresholds of the injured paw at different time points were compared by one-way ANOVA, and Bonferroni's multiple comparison test was performed post hoc to compare various time points to baseline thresholds. For experiments of DAPI nuclei counts, nerve cross-sectional area, and maximum intercapillary distance, two-way repeated-measures ANOVA was used, with matching across measures at different locations of the same nerve. A Bonferroni's post hoc test was then performed to compare ipsilateral and contralateral values. For comparisons of blood vessel area, lactate concentrations in nerve homogenate, nerve volume, Na+/K+ ATPase levels, oxygen consumption rate, or extracellular acidification rate, the unpaired t test was used. For experiments of hypoxyprobe-1 and HIF-1α fluorescence intensity in nerves, unmatched two-way ANOVA was used, followed by Bonferroni's post hoc tests. The criterion for statistical significance was p < 0.05.
Results
Nerve injury causes persistent endoneurial hypoxia
Hypoxyprobe-1 is an exogenously administered probe that is activated under conditions of hypoxia (pO2 < 10 mmHg; Gross et al., 1995), creating thiol adducts with proteins that can then be examined by immunohistochemical techniques. Figure 1A illustrates the time course of endoneurial hypoxia determined using hypoxyprobe-1. After nerve injury, an area of severe hypoxia at the injury site was observed by 2 h. This hypoxic area was present from 2 h to 3 d. Then from 3 d onward, hypoxia was detected at the injury site and distally. The presence of hypoxyprobe-1 was persistent and still present by day 28. Hypoxyprobe-1 was not detected on contralateral nerves (Fig. 1A–D). Hypoxyprobe-1 was colocalized with axons (Fig. 1E), macrophages (Fig. 1F), and Schwann cells (Fig. 1G).
Figure 1.

Traumatic nerve injury causing painful peripheral neuropathy induces endoneurial accumulation of the exogenous hypoxia probe hypoxyprobe-1. A, Under hypoxic conditions, hypoxyprobe-1 forms thiol adducts on cellular proteins. Immunohistochemistry against hypoxyprobe-1 demonstrated that hypoxia was present at the injury site as early as 2 h. Starting at ∼3 d after injury, hypoxia was also observed distal to the injury site. Hypoxia was persistent and still observable by 28 d after injury. Hypoxyprobe-1 was not observed in uninjured contralateral nerve. B–D, Contralateral nerves were stained for hypoxyprobe-1, DAPI, and neurofilament 160/200 (axons), F4/80 (macrophages), or BODIPY (myelin/Schwann cells). E–G, Ipsilateral nerves were subjected to the same staining. The area in proximity to the injury site is shown. Hypoxia, as shown by hypoxyprobe-1 staining, is observed in a region close to the injury site, in which axons, macrophages, and Schwann cells are all found to be hypoxic. This hypoxic region, in which nerve fibers were ligated, is delineated by the white dotted line. Areas outside of the dotted regions containing nonligated nerve fibers still have more hypoxyprobe deposition than contralateral nerves.
HIF-1 is a transcription factor that acts as an endogenous sensor of O2, enabling organisms and cells to adapt to conditions of hypoxia (Hellwig-Bürgel et al., 2005). HIF-1 is a heterodimeric protein made up of α and β subunits, both of which are constitutively and ubiquitously expressed at constant levels regardless of O2 levels (Ke and Costa, 2006). However, under normal O2 conditions, HIF-1α is degraded rapidly with a half-life of ∼5–10 min. In contrast, under conditions of hypoxia, HIF-1α protein levels are stabilized (Salceda and Caro, 1997). Immunohistochemistry for HIF-1α (Fig. 2A) showed results similar to those of hypoxyprobe-1. After nerve injury, there was an area at the injury site that stained positively for HIF-1α as early as 2 h after injury. By day 3 after injury, HIF-1α was also observed distal to the injury. The presence of HIF-1α was still detected in injured nerves 28 d after injury, and its presence in injured nerves was also confirmed by Western blot (Fig. 2B,C). Similar to what was observed with hypoxyprobe-1, HIF-1α staining was found colocalized in axons (Fig. 2G), macrophages (Fig. 2H), and Schwann cells (Fig. 2I) in injured nerve but not in contralateral nerve (Fig. 2A,D–F).
Figure 2.
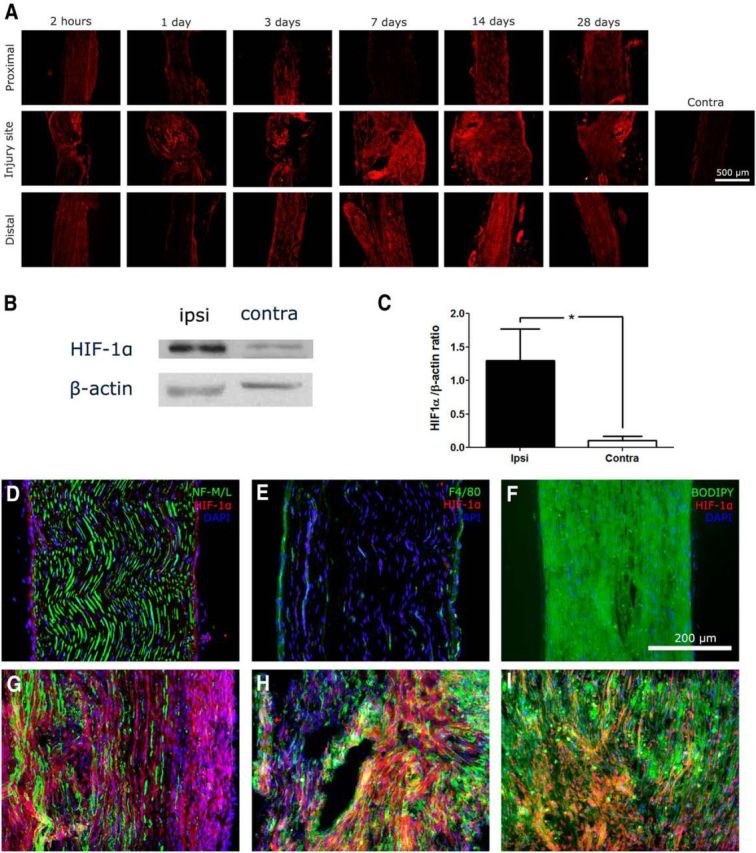
Traumatic nerve injury causing painful peripheral neuropathy induces increased levels of HIF-1α. A, HIF-1α is a constitutively expressed endogenous protein that is normally degraded under normal conditions. However, hypoxic conditions prevent the degradation of HIF-1α, permitting it to accumulate. Immunohistochemistry against HIF-1α demonstrated that hypoxia is present at the nerve injury site as early as 2 h after injury. By day 3, hypoxia could be seen distal to the injury site. HIF-1α was observed as late as 28 d after injury and was not observed in contralateral nerves. B, C, Western blot was performed on nerve lysates for HIF-1α at day 28 after injury (n = 4; *p < 0.05). D–F, Contralateral nerves were stained for HIF-1α, DAPI, and neurofilament 160/200 (axons), F4/80 (macrophages), or BODIPY (myelin/Schwann cells). G–I, Ipsilateral nerves were subjected to the same staining. HIF-1α was colocalized with axons, macrophages, and Schwann cells.
Nerve injury causes alterations in endoneurial blood vessel structure
Nerve cross-sections of sciatic nerves demonstrated alterations in endoneurial blood vessel morphology after PSNL injury. Figure 3A shows a cross-section from a normal nerve. High-magnification micrographs demonstrate that endoneurial capillaries from uninjured nerves are small and thin (Fig. 3D). In uninjured nerve, endothelial nuclei were not readily observed in cross-sections, and if they were observed, a maximum of one nuclei was observed at a time. In each case, one or two cells formed the single-layered capillary. In injured nerve proximal (2 mm) to the nerve injury, endoneurial capillaries are similar in size and morphology to those in uninjured nerve (Fig. 3B,E). Conversely, cross-sections taken ∼2 mm distal to the injury site at day 14 contain a number of abnormally large blood vessels within the endoneurial area (Fig. 3C,F). In injured nerves distal to the injury site, the majority of capillaries were formed by multiple hypertrophic endothelial cells with large nuclei, with some capillaries having multiple layers. Analysis on the distribution of blood vessel size (Fig. 3G; n = 5) showed that, after nerve injury, there is a clear shift in blood vessel size from small vessels toward larger vessels, along with an increase in variance. The average blood vessel cross-sectional area of a nerve was significantly increased distal to the injury when compared with uninjured nerve (Fig. 3H). In addition, electron microscopy revealed that the basal lamina was thicker in blood vessels distal to the injury site compared with normal nerves (Fig. 3I,J). To determine whether the large blood vessels observed were functional, fluorescent FITC–dextran was injected intravenously, and nerves were extracted for analysis. Figure 3K shows that active vessels are positive for FITC in uninjured nerve. Indeed, large blood vessels in injured nerves were positive for FITC (Fig. 3L), demonstrating that blood flows through the large vessels.
Figure 3.
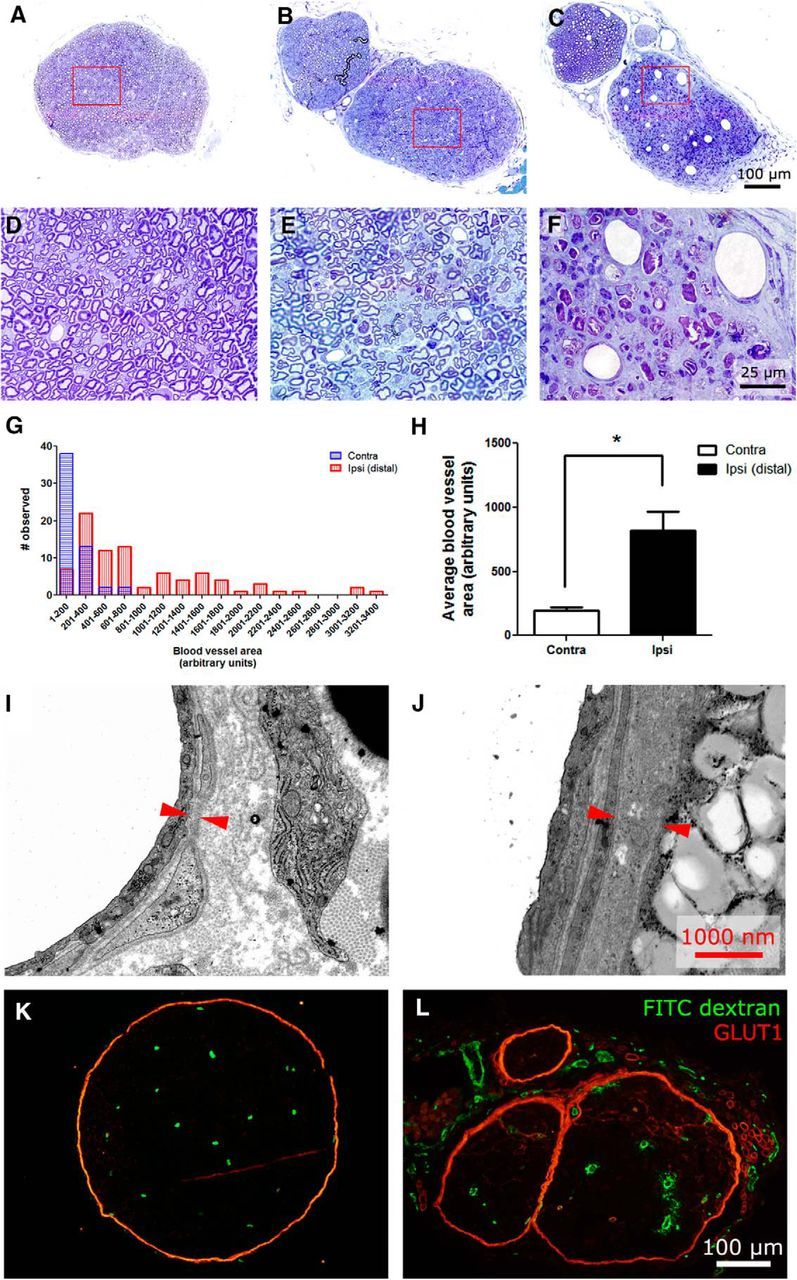
Endoneurial blood vessel structure is altered by traumatic nerve injury causing painful peripheral neuropathy. A, Toluidine blue staining of uninjured contralateral nerve shows that endoneurial blood vessels are small and capillary like. B, After PSNL, blood vessels proximal to the nerve injury site look similar to those in uninjured nerves. C, Distal to the nerve injury site, abnormally sized blood vessels with unusually large lumens are observed. High-magnification photographs of blood vessels in uninjured contralateral nerve (D) and proximal to the nerve injury site (E) demonstrate that endoneurial blood vessels are thin and small. F, High-magnification photographs of endoneurial blood vessels distal to the injury site show enlarged blood vessels, some of which have multiple endothelial cell bodies observable within cross-sections. The cell bodies of the endothelial cells were hypertrophic. G, Lumen sizes of blood vessels were measured in uninjured nerve and distal injured nerve. A histogram of data collected from five contralateral and five distal ipsilateral nerves is shown and clearly demonstrates an altered distribution in blood vessel luminal areas after nerve injury. H, Average luminal area of ipsilateral nerves was significantly increased when compared with contralateral nerve (n = 5 per group; *p < 0.05). I, Electron microscopy of endoneurial capillaries in contralateral uninjured nerve demonstrates that microvessels have a thin basal lamina. The interfaces of the basal lamina are denoted with red arrows. J, Electron microscopy of endoneurial capillaries distal to the injury site demonstrate that blood vessels have a thickened basal lamina, as denoted by the red arrows. K, Intravenously injected FITC–dextran can be used to examine the functionality of blood vessels within the endoneurium. L, The large vascular structures are filled with FITC–dextran, demonstrating that they are functional blood vessels.
Nerve injury causes endoneurial fibrosis and compartmentalization
GLUT1 is expressed normally on perineural cells and endoneurial microvessels (Fig. 4A; Gerhart and Drewes, 1990). By day 14 after injury, fibrosis of GLUT1-positive cells can be observed within the endoneurium in proximity to the injury site (Fig. 4B). The high-magnification inset demonstrates that some fibrotic cells envelop blood vessels. By day 28, fibrosis is extensive, and compartmentalization of the endoneurium into many small fascicles occurs (Fig. 4C). A quantification of fibrosis was performed on nerve cross-sections at day 28 at various lengths along the injured nerve (Fig. 4D). Fibrosis was restricted essentially to 1 mm proximal and distal to the injury site. Similar results were observed using immunohistochemical staining of collagen IV, a type of collagen found on basal lamina, such as the perineurium (Lowry et al., 1997) and blood vessels (Ugrenović et al., 2011). Indeed, collagen IV staining performed in uninjured nerve labels the perineurium (Fig. 4E) and blood vessels (inset). At day 14 after injury, great amounts of collagen IV is detected within the endoneurium (Fig. 4F) and also seen around distended blood vessels (Fig. 4F, inset). A number of DAPI-positive cells can be observed within the blood vessels. By day 28, collagen IV surrounds peripheral nerve fibers, compartmentalizing the nerve into mini-fascicles (Fig. 4G).
Figure 4.

Nerve fibrosis and compartmentalization is induced by traumatic nerve injury causing painful peripheral neuropathy. A, In healthy nerve, GLUT1 is a marker for the perineurium. B, Fourteen days after ligation, fibrotic growth of GLUT1-positive cells were found throughout the endoneurium. Perineural thickening is also observed. C, By day 28, fibrotic cells encapsulate regions of the endoneurium into mini-fascicles. D, GLUT1 intensity at day 28 was measured within an AOI placed within the endoneurium at different distances from the nerve injury site. On average, fibrosis was restricted mainly to a distance around 1000 μm of the injury site (n = 3). E, Collagen IV is a type of collagen found normally on basal lamina and is found on the perineurium in healthy nerve. F, Nerve injury results in increased collagen IV synthesis within the endoneurium at day 14 after injury. Increased collagen IV amounts are also seen around blood vessels. G, By day 28, collagen IV encapsulates regions of the endoneurium into mini-fascicles.
Nerve injury increases metabolic demands of the nerve
DAPI, a nuclear stain, was used to count the number of DAPI-positive cells within 5-μm-thick cross-sections of the endoneurium (Fig. 5A). There were vastly increased numbers of cells within injured nerves distal to the injury (Fig. 5B). The increase was approximately fourfold higher by day 14 after injury (Fig. 5C). In addition, lactate, the end product of anaerobic glycolysis, was assayed in nerve homogenates at day 14 after injury. Injured nerves had significantly increased lactate levels when compared with uninjured nerves (Fig. 5D). Nerves were tested ex vivo for oxygen consumption and extracellular acidification rate using the Seahorse extracellular flux analyzer. Injured nerves (day 7 after injury) consumed greater amounts of oxygen than uninjured nerves (Fig. 5E) and also produced more lactic acid (Fig. 5F).
Figure 5.
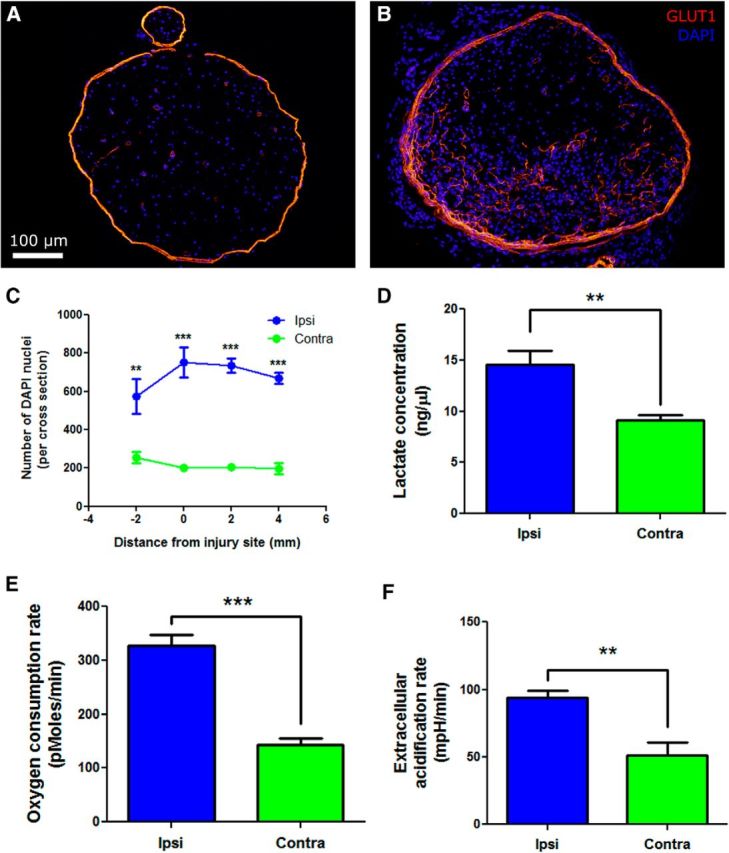
Increased metabolic load after traumatic nerve injury causing painful peripheral neuropathy. A, DAPI was used to stain nuclei in uninjured nerve as a proxy measure of the number of cells within the endoneurium. Only cells within the GLUT1-stained perineurium were counted. B, In injured nerve, the number of cells within the endoneurium increase markedly. C, The number of DAPI-stained nuclei within the nerve was quantified on day 14 after injury, demonstrating a fourfold increase in the number of cells in ipsilateral nerves (n = 6 per group; **p < 0.01, ***p < 0.001). D, Lactate, an indicator of anaerobic glycolysis, was quantified in nerve homogenates at day 14 after injury. Injured nerves had significantly greater lactate levels than in uninjured nerves. E, Basal oxygen consumption rates were measured ex vitro in sciatic nerves at day 7 after PSNL by extracellular flux. At basal conditions, injured nerves consumed more oxygen than contralateral nerves (n = 4 per group; ***p < 0.001). F, Extracellular acidification rates were also measured ex vitro in injured nerves at day 7 after PSNL by extracellular flux. At basal conditions, injured nerves produced more lactic acid than uninjured nerves (n = 4 per group; **p < 0.01).
Nerve injury results in edema and increased intercapillary distances proximal to the injury site
3D reconstruction of PSNL-injured sciatic nerve shows that the region proximal to the injury is swollen when compared with the distal side (Fig. 6A). Blood vessels are shown in green, and the perineurium is shown in orange. GLUT1 was used to delineate the endoneurial area of nerve cross-sections in uninjured (Fig. 6B) and injured (Fig. 6C) nerves at day 14. Spatial analysis of sciatic nerve cross-sectional area demonstrated that the nerve is larger at the injury site and proximally when compared with uninjured nerve (Fig. 6D). Distal to the nerve injury site, cross-sectional area was not significantly altered by PSNL injury when compared with uninjured nerves (Fig. 6D). Volume was calculated by integrating the area under the curve of cross-sectional areas, from 2 mm proximal to the injury to 4 mm distal to the injury. Indeed, injured nerves had significantly larger volumes than injured nerves (Fig. 6E). The number of FITC–dextran-positive blood vessels was quantified in injured and uninjured nerves at various distances along the nerve (Fig. 6F). No significant change in the number of blood vessels was observed. The maximum intercapillary distance between blood vessels within the endoneurium was measured at various distances from the PSNL injury site. Intercapillary distances were only increased proximal to the injury site (Fig. 6G). Average intercapillary distances were not affected by PSNL injury (data not shown).
Figure 6.

Edema is not the main contributing factor to endoneurial hypoxia after traumatic nerve injury. A, 3D reconstruction after PSNL shows that the nerve injury site and proximal side is larger when compared with the distal side. Blood vessels are shown in green, and the perineurium as determined by GLUT1 staining is shown in orange. B, GLUT1, a marker of the perineurium, was used to determine the cross-sectional area of the endoneurium in an uninjured nerve. C, At the nerve injury site, the endoneurium is larger in size. D, Nerve cross-sectional area was measured at various locations. Nerve cross-sectional area is significantly increased proximal to the injury and at the injury site (n = 6 per group; **p < 0.01, ***p < 0.001). E, The volume of uninjured and injured nerves was evaluated by integration of cross-sectional area over a distance of 6 mm, from 2 mm proximal to the injury to 4 mm distal to the injury. The injured nerve was significantly larger in volume (n = 4–6 per group; ***p < 0.001). F, The number of FITC-positive blood vessels was quantified across various locations along the sciatic nerve. No significant differences were observed in the number of blood vessels per nerve cross-section (n = 6 per group). G, Non-adjacent intercapillary distances were measured in nerve cross-sections. The maximum distances were compared and were significantly increased proximal to the site of nerve injury (n = 6 per group; ***p < 0.001).
Na+/K+ ATPase levels are reduced in sciatic nerve after PSNL injury and in DRG neurons after exposure to hypoxia
Na+/K+ ATPase exists as a diprotomeric protein consisting of two α subunits and two β subunits (Linnertz et al., 1998). Levels of the α subunits (pan) were quantitated by Western blot in sciatic nerve lysates on day 14 after PSNL. PSNL injury significantly reduced the amount of Na+/K+ ATPase detected in nerve lysates at day 14 after injury (Fig. 7A,B).
Figure 7.
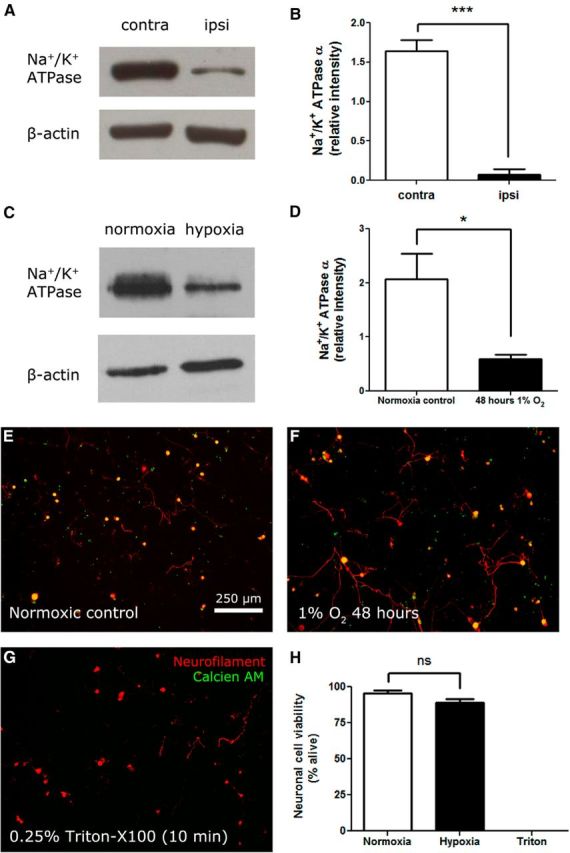
Levels of the Na+/K+ ATPase ion transporter are reduced after traumatic nerve injury causing painful neuropathy or after exposure to hypoxia. A, Sciatic nerve lysates at day 14 after injury were prepared. Example immunoblots of pan Na+/K+ ATPase α and β-actin are shown. B, Quantification of Western blot bands demonstrates a reduction in Na+/K+ ATPase α in injured nerve (n = 3 per group; ***p < 0.001). C, DRG neurons were cultured and exposed to 48 h of hypoxia (1% O2) or to ambient oxygen (21% O2). Example immunoblots of pan Na+/K+ ATPase α and β-actin from cultured neurons are shown. D, Quantification of Western blot bands demonstrates that Na+/K+ ATPase α levels in cultured neurons were significantly reduced by hypoxia (n = 3 per group; *p < 0.05). E, Calcien AM is a viability dye that is cleaved by intracellular esterases to become cell impermeable. Cells with intact membranes sequester calcien AM. Neurons cultured in normoxic conditions express neurofilament 160/200 and are positive for calcien AM. F, Neurons cultured in hypoxic conditions (48 h, 1% O2) also express neurofilament 160/200 and are positive for calcien AM. G, Neurons killed by exposure to 0.25% Triton X-100 for 10 min do not sequester calcien AM. H, Quantification of viable neurons by calcien AM demonstrates that 48 h 1% O2 exposure does not affect neuronal viability (n = 6 per group).
To determine whether the reduction of Na+/K+ ATPase levels after nerve injury was a result of endoneurial hypoxia, primary DRG neurons were cultured and exposed to hypoxic conditions (1% O2) for 48 h. DRG neurons cultured under these conditions expressed reduced levels of Na+/K+ ATPase compared with control neurons cultured in normoxic (ambient atmosphere, 21%) oxygen levels (Fig. 7C,D). Hypoxia at these conditions did not affect neuronal cell viability, as assessed by calcien AM sequestration (Fig. 7E–H). This is in agreement with past experiments that have shown that neurons and neuronal stem cells can tolerate 1% O2 without loss of viability (Felfly et al., 2011).
Physiological antagonism of endoneurial hypoxia with hyperbaric oxygen alleviates neuropathic pain after PSNL
To examine whether hyperbaric or normobaric oxygen had any effect on mechanical thresholds or motor responses, naive animals received 5 d daily treatment with 2 h of room air, normobaric oxygen, or hyperbaric oxygen, and neither changes in their responses to von Frey hair stimulation (Fig. 8A) nor alterations in the rotarod test (Fig. 8B) were observed (n = 4 per group).
Figure 8.
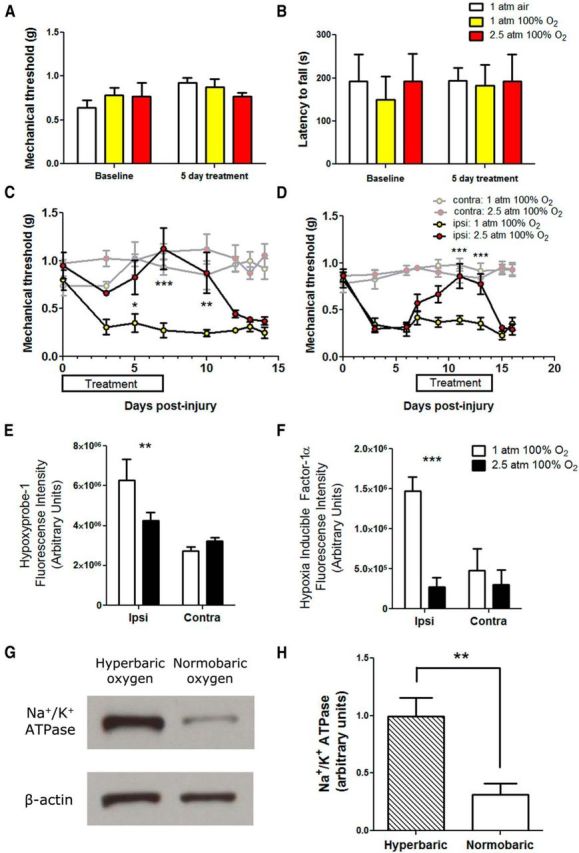
Hyperbaric oxygen treatment alleviates painful peripheral neuropathy induced by traumatic injury. A, Naive animals were exposed to air, normobaric oxygen, or hyperbaric oxygen, and mechanical thresholds were measured. There was no significant differences of either treatment condition on mechanical thresholds. B, Motor responses were also measured by the rotarod test. Normobaric oxygen or hyperbaric oxygen had on effects of motor responses on naive animals. C, After PSNL, mice received hyperbaric oxygen (100% O2 at 2.5 atm) or sham treatment (100% O2 at 1 atm) for 2 h daily, from day 0 to day 7 after injury. Hyperbaric oxygen significantly alleviated mechanical allodynia (n = 5 per group; *p < 0.05, **p < 0.01, ***p < 0.001). Contralateral mechanical thresholds were not altered by hyperbaric or normobaric oxygen treatment. D, Hyperbaric oxygen administered from day 7 to day 14 after injury was also able to significantly alleviate mechanical allodynia (n = 5 per group; ***p < 0.001). Hyperbaric or normobaric oxygen treatment did not alter contralateral mechanical thresholds. E, Hyperbaric oxygen treatment from day 0 to day 7 after injury significantly reduced the amount of hypoxyprobe-1 adducts observed in injured nerve at day 7 (n = 3 per group; **p < 0.01). F, Hyperbaric oxygen treatment from day 0 to day 7 after injury significantly reduced the amount of immunoreactivity in HIF-1α-stained nerve sections (n = 3 per group; ***p < 0.001). G, Example immunoblots of pan Na+/K+ ATPase and β-actin in sciatic nerve lysates from ipsilateral nerves from animals that received hyperbaric oxygen or sham treatment (normobaric oxygen). H, Hyperbaric oxygen treatment from day 0 to day 7 after injury significantly increased the amount of Na+/K+ ATPase α protein levels in injured nerves as determined by Western blot (n = 6–8 per group; **p < 0.01).
Mice having PSNL were administered normobaric or hyperbaric oxygen for 2 h daily at 2.5 atm, from day 0 to day 7 after injury. Hyperbaric oxygen, but not normobaric oxygen, was able to alleviate mechanical allodynia in PSNL nerve-injured mice. The alleviation of mechanical hypersensitivity persisted a few days after hyperbaric oxygen treatment was stopped (Fig. 8C). Hyperbaric oxygen treatment did not affect contralateral mechanical thresholds. Additionally, in PSNL nerve-injured mice treated at day 7 to day 14, hyperbaric oxygen was able to reverse existing mechanical allodynia for the duration of treatment, without affecting contralateral mechanical thresholds (Fig. 8D). The amount of hypoxyprobe-1 deposition in the nerve was quantified at day 7 in PSNL-injured mice treated with hyperbaric oxygen from day 0 to day 7. Hyperbaric oxygen significantly reduced the amount of hypoxyprobe-1 deposition in the nerve (Fig. 8E). Similarly, hyperbaric oxygen significantly reduced the amount of HIF-1α in the endoneurial space as well (Fig. 8F). The expression of the Na+/K+ ATPase was examined by Western blot after hyperbaric oxygen treatment, and hyperbaric oxygen significantly rescued the amount of Na+/K+ ATPase detected in nerve lysates at day 14 after PSNL injury (Fig. 8G,H).
Discussion
Peripheral nerve injury-inducing painful mononeuropathy is associated with endoneurial hypoxia
In the PSNL model of neuropathic pain, part of the sciatic nerve is tightly ligated. Constricting blood vessels within the ligated segment of the nerve is likely to immediately produce endoneurial ischemia. In agreement with this, increased labeling with hypoxyprobe-1 and expression of HIF-1α were observed at the injury site as early as 2 h after injury.
By day 3, a second phase of hypoxia developed. Hypoxia is now observed distal to the injury site and persisted until at least day 28 after injury. The mechanisms leading to the development of this second phase of hypoxia were characterized and discussed below. The presence of persistent endoneurial hypoxia suggests that hypoxia activated analgesic pro-drugs (similar to the hypoxia pro-drugs developed for cancer) may be developed for the treatment of neuropathic pain.
Vascular dysfunction contributes to endoneurial hypoxia
To facilitate gas, nutrient, and waste exchange, endothelial cells are thin structures. After nerve injury, the increase in endothelial cell basal lamina thickness likely reduces nutrient and gas transport. Additionally, the small lumen of healthy capillaries causes red blood cells to deform from their resting biconcave disk shape into a parabaloid shape resembling a bell. This deformation facilitates rapid gas exchange by simultaneously increasing the surface area available for gas exchange and reducing the distance of hemoglobin to the tissues (Guest et al., 1963). After nerve injury, the increase in capillary lumen sizes will prevent this process from occurring. Also, it is likely that nerve injury causes a reduction in the flow rate within endoneurial capillaries, which also reduces the effectiveness of oxygen, nutrient, and waste exchange.
Nerve fibrosis contributes to endoneurial hypoxia
The growth of fibrotic tissue within the endoneurial space after nerve injury further reduces the ability of oxygen and nutrients to diffuse through the endoneurium, contributing to persistent hypoxia. Fibrotic cells were GLUT1 positive (Hirose et al., 2003) and collagen IV positive (Van Agtmael and Bruckner-Tuderman, 2010), suggesting that they are perineurial cells. Indeed, both GLUT1 (Chen et al., 2001) and collagen deposition (Gilkes et al., 2013) are regulated downstream of HIF-1α.
Increased metabolic needs contributes to endoneurial hypoxia
Nerve injury results in opening of the blood–nerve barrier (BNB), which permits the entry of blood-borne immune cells into the endoneurial space. This mechanism is mediated through vascular endothelial growth factor (VEGF) expressed in resident macrophages (Lim et al., 2014). Hypoxia likely drives the expression of VEGF, downstream of HIF-1 (Burke et al., 2002), in macrophages, and penultimately, the opening of the BNB. The massive infiltration of immune cells, together with the proliferation of Schwann cells (Thomas, 1948) and resident macrophages (Mueller et al., 2001), increase the metabolic demands of the nerve, resulting in a higher oxygen consumption of injured nerves when compared with uninjured nerves. Increased lactate levels in injured nerves suggest that anaerobic glycolysis is required to maintain cellular ATP levels. In agreement, injured nerves had higher extracellular acidification rates than uninjured nerves. Lactate acidosis may produce excitatory effects on TRPV1 and acid-sensing ion channels. Lactate can also affect inflammatory responses, upregulating Toll-like receptor 4 and nuclear factor-κB transcriptional activity in macrophages (Samuvel et al., 2009).
Macrophages respond to hypoxia with altered gene expression mediated through HIFs (Murdoch et al., 2005). Many of the genes regulated by HIFs in hypoxic conditions are known to be expressed in the nerve after nerve injury and have been implicated in pain. Interleukin-1β (Scannell, 1996), interleukin-6 (Albina et al., 1995), tumor necrosis factor-α (Cramer et al., 2003), and CC chemokine ligand 3 (Bosco et al., 2004) are upregulated in macrophages by hypoxia. These same molecules are upregulated in macrophages in models of neuropathic pain (Shi et al., 2011; Lee and Zhang, 2012; Lim et al., 2014). It is possible that targeting hypoxia may abrogate many unwanted effects of macrophages, without affecting their beneficial functions.
Effects of endoneurial hypoxia on the Na+/K+ ATPase ion transporter
In the CNS, ischemia leads to anoxic depolarization, whereby neurons depolarize because of a deficit in the ATP required to run the Na+/K+ ATPase ion transporter. Membrane potentials can no longer be maintained and neurons reach action potential firing thresholds, resulting in depolarization (Balestrino, 1995). After traumatic nerve injury, it is likely that a similar phenomenon occurs at the nerve injury site, producing depolarization in injured axons. The fact that ischemic peripheral nerve in healthy subjects generates ectopic activity and spontaneous discharge during reperfusion (Ochoa and Torebjörk, 1980) supports this hypothesis.
In response to hypoxia, levels of the Na+/K+ ATPase ion transporter is reduced as part of ATP conservation mechanisms (Hochachka and Lutz, 2001). This has been shown to occur in alveolar epithelial cells (Planès et al., 1996; Dada et al., 2003) via an HIF-1α independent mechanism (Zhou et al., 2008), and is thought to contribute to pulmonary edema under hypoxic conditions. Our experiments have demonstrated that hypoxia mediated regulation of Na+/K+ ATPase also occurs in DRG neurons also. We hypothesize that prolonged reduction in Na+/K+ ATPase activity may result in spontaneous depolarization of the axons as a result of failed maintenance of the resting membrane potential. This may lead to the generation of ectopic activity characteristic of painful peripheral neuropathy (Wall and Gutnick, 1974). Indeed, there are chronic pain phenotypes that arise out of defective Na+/K+ ATPase, such as familial hemiplegic migraine (Koenderink et al., 2005). Polymorphisms in humans that confer reduced Na+/K+ ATPase activity are more frequently associated with diabetic neuropathy (Vague et al., 1997). Reductions in Na+/K+ ATPase activities also underlie increased strain-specific sensitivity to the Formalin test in mice (LaCroix-Fralish et al., 2009). Reversing the reduction in or stimulating the Na+/K+ ATPase in peripheral sensory neurons may benefit neuropathic pain conditions.
Effects of hypoxia on ion channels
Hypoxia may also contribute to neuropathic pain through various ion channels. TRPA1 is activated by hypoxia, through prolyl hydroxylase inhibition (Takahashi et al., 2011). TRPV1 activity is also potentiated by hypoxia through HIF-1α (Ristoiu et al., 2011). Both TRPA1 and TRPV1 are present in human neuromas (Biggs et al., 2007; Morgan et al., 2009) and may contribute to hyperexcitability and ectopic activity after nerve injury through hypoxic sensitization. Voltage-gated potassium channels may also be involved, because hypoxia blocks a dentrotoxin-sensitive potassium current in DRG neurons, increasing excitability and sometimes producing spontaneous activity (Gruss et al., 2006).
Hyperbaric oxygen alleviates endoneurial hypoxia and neuropathic pain
Under physiological conditions, the solubility of oxygen in plasma is 0.3%, making up ∼2% of the total oxygen content in blood (Pittman, 2011). At a pressure of 3 atm, the solubility of oxygen increases toward 6%, which is sufficient to supply resting tissues without a contribution from hemoglobin (Leach et al., 1998). Thus, hyperbaric oxygen physiologically antagonizes endoneurial hypoxia, resulting in an alleviation of neuropathic pain. If hyperbaric oxygen is administered early after traumatic injury, alleviation of mechanical hypersensitivity persists for a few days after hyperbaric oxygen is removed. It is likely that early hyperbaric oxygen treatment prevented the generation of persistent endoneurial hypoxia. In agreement, the same intervention attenuated the reduction of endoneurial Na+/K+ ATPase triggered by nerve injury. When hyperbaric oxygen is administered after neuropathic pain was established, hyperbaric oxygen could still alleviate mechanical allodynia. However, in this paradigm, once hyperbaric oxygen treatment was interrupted, no persistent alleviation of mechanical hypersensitivity was observed. Our results were consistent with previous reports (Gu et al., 2012; Gibbons et al., 2013), demonstrating the effectiveness of hyperbaric oxygen in animal models of neuropathic pain. Although we have focused on the effects of hyperbaric oxygen on hypoxia, it is possible the observed alleviation of mechanical allodynia may be mediated in part through central effects of hyperbaric oxygen (Zelinski et al., 2009; Chung et al., 2010).
Edema does not necessarily contribute to endoneurial hypoxia
Past experiments have attributed endoneurial edema as a critical factor in the genesis of endoneurial ischemia (Sommer et al., 1993). Accumulation of fluid in the endoneurial space increases endoneurial fluid pressure, which is thought to cause the deformation and collapse of blood vessels passing through the perineurium to enter the endoneurium (Myers et al., 1986). Nerve edema may also produce ischemic areas within the endoneurium by increasing the distance between capillaries within the nerve, thereby creating zones that are not adequately served by blood vessels (Low et al., 1985). In the PSNL model of painful peripheral neuropathy, both edema and increased intercapillary distances were observed. However, these changes were localized proximally and thus cannot explain the observation of hypoxia distal to the injury site. Endoneurial fluid moves in a proximodistal direction along peripheral nerves (Weiss et al., 1945). After traumatic injury, endoneurial fluid flow likely becomes interrupted, leading to a buildup of fluid at the proximal site of injury. However, the resulting edema does not appear to be sufficient to produce hypoxia, at least as detected with our methods. We contend that nerve edema is not the main contributing factor to persistent endoneurial hypoxia after traumatic injury.
Significance
Traumatic nerve injury resulting in painful peripheral neuropathy produces persistent endoneurial hypoxia. After nerve injury, endoneurial capillaries enlarge and thicken, and perineurial cells undergo aberrant fibrosis. Together, this results in a reduced gas, nutrient, and waste transport through the nerve. Nerve injury also increases the metabolic needs of the nerve, leading to increased oxygen consumption and acidosis. Persistent endoneurial hypoxia reduces Na+/K+ ATPase levels, which likely contributes to axonal excitability. Physiological antagonism of endoneurial hypoxia with hyperbaric oxygen was able to reduce mechanical hypersensitivity. Our results suggest that hypoxia is a viable target for the treatment of neuropathic pain. Therapeutics that alleviate endoneurial hypoxia likely will have a wide therapeutic window, because oxygen is an endogenous molecule required for proper functioning of tissues.
Footnotes
T.K.Y.L. is supported by the Louise and Alan Edwards Foundation. J.J. is supported by the Canadian Institutes for Health Research Neuroinflammation Training Program. This work was supported by funding from the Canadian Institutes for Health Research MOP-111129 (J.Z.). We thank Bruno Mietto, Scott Cameron, Margaret Attiwell, Emerson Krock, and Seunghwan Lee for technical assistance. We thank Walter Kucharski for designing and manufacturing the hyperbaric oxygen chamber used in this study. Seahorse XF experiments were recorded at the Rosalind and Morris Goodman Cancer Research Centre Metabolomics Core Facility supported by the Canada Foundation for Innovation, the Dr. John R. and Clara M. Fraser Memorial Trust, Terry Fox Foundation Oncometabolism Team Grant 116128, and McGill University.
The authors declare no competing financial interests.
References
- Albina JE, Henry WL, Jr, Mastrofrancesco B, Martin BA, Reichner JS. Macrophage activation by culture in an anoxic environment. J Immunol. 1995;155:4391–4396. [PubMed] [Google Scholar]
- Balestrino M. Pathophysiology of anoxic depolarization: new findings and a working hypothesis. J Neurosci Methods. 1995;59:99–103. doi: 10.1016/0165-0270(94)00199-Q. [DOI] [PubMed] [Google Scholar]
- Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120:3647–3656. doi: 10.1182/blood-2012-04-383430. [DOI] [PubMed] [Google Scholar]
- Biggs JE, Yates JM, Loescher AR, Clayton NM, Boissonade FM, Robinson PP. Vanilloid receptor 1 (TRPV1) expression in lingual nerve neuromas from patients with or without symptoms of burning pain. Brain Res. 2007;1127:59–65. doi: 10.1016/j.brainres.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Bosco MC, Puppo M, Pastorino S, Mi Z, Melillo G, Massazza S, Rapisarda A, Varesio L. Hypoxia selectively inhibits monocyte chemoattractant protein-1 production by macrophages. J Immunol. 2004;172:1681–1690. doi: 10.4049/jimmunol.172.3.1681. [DOI] [PubMed] [Google Scholar]
- Burke B, Tang N, Corke KP, Tazzyman D, Ameri K, Wells M, Lewis CE. Expression of HIF-1alpha by human macrophages: implications for the use of macrophages in hypoxia-regulated cancer gene therapy. J Pathol. 2002;196:204–212. doi: 10.1002/path.1029. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–9525. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- Chung E, Zelinski LM, Ohgami Y, Shirachi DY, Quock RM. Hyperbaric oxygen treatment induces a 2-phase antinociceptive response of unusually long duration in mice. J Pain. 2010;11:847–853. doi: 10.1016/j.jpain.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Xanthos DN, Francis L, Bennett GJ. Chronic post-ischemia pain (CPIP): a novel animal model of complex regional pain syndrome-type I (CRPS-I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain. 2004;112:94–105. doi: 10.1016/j.pain.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/S0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. Hypoxia-induced endocytosis of Na, K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest. 2003;111:1057–1064. doi: 10.1172/JCI200316826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felfly H, Zambon AC, Xue J, Muotri A, Zhou D, Snyder EY, Haddad GG. Severe hypoxia: consequences to neural stem cells and neurons. J Neurol Res. 2011;1(5) doi: 10.4021/jnr70w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart DZ, Drewes LR. Glucose transporters at the blood-nerve barrier are associated with perineurial cells and endoneurial microvessels. Brain Res. 1990;508:46–50. doi: 10.1016/0006-8993(90)91115-W. [DOI] [PubMed] [Google Scholar]
- Gibbons CR, Liu S, Zhang Y, Sayre CL, Levitch BR, Moehlmann SB, Shirachi DY, Quock RM. Involvement of brain opioid receptors in the anti-allodynic effect of hyperbaric oxygen in rats with sciatic nerve crush-induced neuropathic pain. Brain Res. 2013;1537:111–116. doi: 10.1016/j.brainres.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013;288:10819–10829. doi: 10.1074/jbc.M112.442939. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer. 1995;61:567–573. doi: 10.1002/ijc.2910610422. [DOI] [PubMed] [Google Scholar]
- Gruss M, Ettorre G, Stehr AJ, Henrich M, Hempelmann G, Scholz A. Moderate hypoxia influences excitability and blocks dendrotoxin sensitive K+ currents in rat primary sensory neurones. Mol Pain. 2006;2:12. doi: 10.1186/1744-8069-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Niu JY, Liu WT, Sun YY, Liu S, Lv Y, Dong HL, Song XJ, Xiong LZ. Hyperbaric oxygen therapy attenuates neuropathic hyperalgesia in rats and idiopathic trigeminal neuralgia in patients. Eur J Pain. 2012;16:1094–1105. doi: 10.1002/j.1532-2149.2012.00113.x. [DOI] [PubMed] [Google Scholar]
- Guest MM, Bond TP, Cooper RG, Derrick JR. Red blood cells: change in shape in capillaries. Science. 1963;142:1319–1321. doi: 10.1126/science.142.3597.1319. [DOI] [PubMed] [Google Scholar]
- Hao JX, Blakeman KH, Yu W, Hultenby K, Xu XJ, Wiesenfeld-Hallin Z. Development of a mouse model of neuropathic pain following photochemically induced ischemia in the sciatic nerve. Exp Neurol. 2000;163:231–238. doi: 10.1006/exnr.2000.7373. [DOI] [PubMed] [Google Scholar]
- Hellwig-Bürgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W. Review: hypoxia-inducible factor-1 (HIF-1): a novel transcription factor in immune reactions. J Interferon Cytokine Res. 2005;25:297–310. doi: 10.1089/jir.2005.25.297. [DOI] [PubMed] [Google Scholar]
- Hirose T, Tani T, Shimada T, Ishizawa K, Shimada S, Sano T. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod Pathol. 2003;16:293–298. doi: 10.1097/01.MP.0000062654.83617.B7. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Lutz PL. Mechanism, origin, and evolution of anoxia tolerance in animals. Comp Biochem Physiol B Biochem Mol Biol. 2001;130:435–459. doi: 10.1016/S1096-4959(01)00408-0. [DOI] [PubMed] [Google Scholar]
- Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol Pharmacol. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- Kiralp MZ, Yildiz S, Vural D, Keskin I, Ay H, Dursun H. Effectiveness of hyperbaric oxygen therapy in the treatment of complex regional pain syndrome. J Int Med Res. 2004;32:258–262. doi: 10.1177/147323000403200304. [DOI] [PubMed] [Google Scholar]
- Koenderink JB, Zifarelli G, Qiu LY, Schwarz W, De Pont JJ, Bamberg E, Friedrich T. Na, K-ATPase mutations in familial hemiplegic migraine lead to functional inactivation. Biochim Biophys Acta. 2005;1669:61–68. doi: 10.1016/j.bbamem.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Kupers R, Yu W, Persson JK, Xu XJ, Wiesenfeld-Hallin Z. Photochemically-induced ischemia of the rat sciatic nerve produces a dose-dependent and highly reproducible mechanical, heat and cold allodynia, and signs of spontaneous pain. Pain. 1998;76:45–59. doi: 10.1016/S0304-3959(98)00022-0. [DOI] [PubMed] [Google Scholar]
- LaCroix-Fralish ML, Mo G, Smith SB, Sotocinal SG, Ritchie J, Austin JS, Melmed K, Schorscher-Petcu A, Laferriere AC, Lee TH, Romanovsky D, Liao G, Behlke MA, Clark DJ, Peltz G, Séguéla P, Dobretsov M, Mogil JS. The beta3 subunit of the Na+, K+-ATPase mediates variable nociceptive sensitivity in the formalin test. Pain. 2009;144:294–302. doi: 10.1016/j.pain.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach RM, Rees PJ, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317:1140–1143. doi: 10.1136/bmj.317.7166.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Zhang J. Heterogeneity of macrophages in injured trigeminal nerves: cytokine/chemokine expressing vs. phagocytic macrophages. Brain Behav Immun. 2012;26:891–903. doi: 10.1016/j.bbi.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Less JR, Skalak TC, Sevick EM, Jain RK. Microvascular architecture in a mammary carcinoma: branching patterns and vessel dimensions. Cancer Res. 1991;51:265–273. [PubMed] [Google Scholar]
- Lim TK, Shi XQ, Martin HC, Huang H, Luheshi G, Rivest S, Zhang J. Blood-nerve barrier dysfunction contributes to the generation of neuropathic pain and allows targeting of injured nerves for pain relief. Pain. 2014;155:954–967. doi: 10.1016/j.pain.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Linnertz H, Urbanova P, Obsil T, Herman P, Amler E, Schoner W. Molecular distance measurements reveal an (alpha beta)2 dimeric structure of Na+/K+-ATPase. High affinity ATP binding site and K+-activated phosphatase reside on different alpha-subunits. J Biol Chem. 1998;273:28813–28821. doi: 10.1074/jbc.273.44.28813. [DOI] [PubMed] [Google Scholar]
- Liu R, Lin G, Xu H. An efficient method for dorsal root ganglia neurons purification with a one-time anti-mitotic reagent treatment. PLoS One. 2013;8:e60558. doi: 10.1371/journal.pone.0060558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low PA, Nukada H, Schmelzer JD, Tuck RR, Dyck PJ. Endoneurial oxygen tension and radial topography in nerve edema. Brain Res. 1985;341:147–154. doi: 10.1016/0006-8993(85)91482-9. [DOI] [PubMed] [Google Scholar]
- Lowry A, Wilcox D, Masson EA, Williams PE. Immunohistochemical methods for semiquantitative analysis of collagen content in human peripheral nerve. J Anat. 1997;191:367–374. doi: 10.1046/j.1469-7580.1997.19130367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon SE, Dellon AL, Hudson AR, Hunter DA. Chronic human nerve compression—a histological assessment. Neuropathol Appl Neurobiol. 1986;12:547–565. doi: 10.1111/j.1365-2990.1986.tb00159.x. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Basbaum AI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain. 1998;76:215–222. doi: 10.1016/S0304-3959(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Morgan CR, Bird EV, Robinson PP, Boissonade FM. TRPA1 expression in human lingual nerve neuromas in patients with and without symptoms of dysaesthesia. Neurosci Lett. 2009;465:189–193. doi: 10.1016/j.neulet.2009.08.055. [DOI] [PubMed] [Google Scholar]
- Mueller M, Wacker K, Ringelstein EB, Hickey WF, Imai Y, Kiefer R. Rapid response of identified resident endoneurial macrophages to nerve injury. Am J Pathol. 2001;159:2187–2197. doi: 10.1016/S0002-9440(10)63070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175:6257–6263. doi: 10.4049/jimmunol.175.10.6257. [DOI] [PubMed] [Google Scholar]
- Myers RR, Murakami H, Powell HC. Reduced nerve blood flow in edematous neuropathies: a biomechanical mechanism. Microvasc Res. 1986;32:145–151. doi: 10.1016/0026-2862(86)90050-6. [DOI] [PubMed] [Google Scholar]
- Ochoa JL, Torebjörk HE. Paraesthesiae from ectopic impulse generation in human sensory nerves. Brain. 1980;103:835–853. doi: 10.1093/brain/103.4.835. [DOI] [PubMed] [Google Scholar]
- Pittman RN. Regulation of tissue oxygenation. San Rafael, CA: Morgan and Claypool Life Sciences; 2011. Oxygen transport. [PubMed] [Google Scholar]
- Planès C, Friedlander G, Loiseau A, Amiel C, Clerici C. Inhibition of Na-K-ATPase activity after prolonged hypoxia in an alveolar epithelial cell line. Am J Physiol. 1996;271:L70–L78. doi: 10.1152/ajplung.1996.271.1.L70. [DOI] [PubMed] [Google Scholar]
- Ristoiu V, Shibasaki K, Uchida K, Zhou Y, Ton BH, Flonta ML, Tominaga M. Hypoxia-induced sensitization of transient receptor potential vanilloid 1 involves activation of hypoxia-inducible factor-1 alpha and PKC. Pain. 2011;152:936–945. doi: 10.1016/j.pain.2011.02.024. [DOI] [PubMed] [Google Scholar]
- Rüger LJ, Irnich D, Abahji TN, Crispin A, Hoffmann U, Lang PM. Characteristics of chronic ischemic pain in patients with peripheral arterial disease. Pain. 2008;139:201–208. doi: 10.1016/j.pain.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol. 2009;182:2476–2484. doi: 10.4049/jimmunol.0802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell G. Leukocyte responses to hypoxic/ischemic conditions. New Horiz. 1996;4:179–183. [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Shi XQ, Lim TK, Lee S, Zhao YQ, Zhang J. Statins alleviate experimental nerve injury-induced neuropathic pain. Pain. 2011;152:1033–1043. doi: 10.1016/j.pain.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Sommer C, Myers RR. Vascular pathology in CCI neuropathy: a quantitative temporal study. Exp Neurol. 1996;141:113–119. doi: 10.1006/S0014-4886(96)92000-2. [DOI] [PubMed] [Google Scholar]
- Sommer C, Galbraith JA, Heckman HM, Myers RR. Pathology of experimental compression neuropathy producing hyperesthesia. J Neuropathol Exp Neurol. 1993;52:223–233. doi: 10.1097/00005072-199305000-00006. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kuwaki T, Kiyonaka S, Numata T, Kozai D, Mizuno Y, Yamamoto S, Naito S, Knevels E, Carmeliet P, Oga T, Kaneko S, Suga S, Nokami T, Yoshida J, Mori Y. TRPA1 underlies a sensing mechanism for O2. Nat Chem Biol. 2011;7:701–711. doi: 10.1038/nchembio.640. [DOI] [PubMed] [Google Scholar]
- Thomas GA. Quantitative histology of Wallerian degeneration: II. Nuclear population in two nerves of different fibre spectrum. J Anat. 1948;82:135–145. [PubMed] [Google Scholar]
- Thompson CD, Uhelski ML, Wilson JR, Fuchs PN. Hyperbaric oxygen treatment decreases pain in two nerve injury models. Neurosci Res. 2010;66:279–283. doi: 10.1016/j.neures.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Ugrenović S, Jovanović I, Vasović L. Morphometric analysis of human sciatic nerve perineurial collagen type IV content. Microsc Res Tech. 2011;74:1127–1133. doi: 10.1002/jemt.21004. [DOI] [PubMed] [Google Scholar]
- Vague P, Dufayet D, Coste T, Moriscot C, Jannot MF, Raccah D. Association of diabetic neuropathy with Na/K ATPase gene polymorphism. Diabetologia. 1997;40:506–511. doi: 10.1007/s001250050708. [DOI] [PubMed] [Google Scholar]
- Van Agtmael T, Bruckner-Tuderman L. Basement membranes and human disease. Cell Tissue Res. 2010;339:167–188. doi: 10.1007/s00441-009-0866-y. [DOI] [PubMed] [Google Scholar]
- Wall PD, Gutnick M. Properties of afferent nerve impulses originating from a neuroma. Nature. 1974;248:740–743. doi: 10.1038/248740a0. [DOI] [PubMed] [Google Scholar]
- Weiss P, Wang H, Taylor AC, Edds MV. Proximo-distal fluid convection in the endoneurial spaces of peripheral nerves, demonstrated by colored and radioactive (isotope) tracers. Am J Physiol. 1945;143:521–540. [Google Scholar]
- Yasuda H, Dyck PJ. Abnormalities of endoneurial microvessels and sural nerve pathology in diabetic neuropathy. Neurology. 1987;37:20–28. doi: 10.1212/WNL.37.1.20. [DOI] [PubMed] [Google Scholar]
- Zelinski LM, Ohgami Y, Chung E, Shirachi DY, Quock RM. A prolonged nitric oxide-dependent, opioid-mediated antinociceptive effect of hyperbaric oxygen in mice. J Pain. 2009;10:167–172. doi: 10.1016/j.jpain.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Dada LA, Chandel NS, Iwai K, Lecuona E, Ciechanover A, Sznajder JI. Hypoxia-mediated Na-K-ATPase degradation requires von Hippel Lindau protein. FASEB J. 2008;22:1335–1342. doi: 10.1096/fj.07-8369com. [DOI] [PubMed] [Google Scholar]