

Abstract
Axon degeneration is a programed process that takes place during development, in response to neuronal injury, and as a component of neurodegenerative disease pathology, yet the molecular mechanisms that drive this process remain poorly defined. In this study, we have developed a semi-automated, 384-well format axon degeneration assay in rat dorsal root ganglion (DRG) neurons using a trophic factor withdrawal paradigm. Using this setup, we have screened a library of known drugs and bioactives to identify several previously unappreciated regulators of axon degeneration, including lipoxygenases. Multiple structurally distinct lipoxygenase inhibitors as well as mouse DRG neurons lacking expression of 12/15-lipoxygenase display protection of axons in this context. Retinal ganglion cell axons from 12/15-lipoxygenase-null mice were similarly protected from degeneration following nerve crush injury. Through additional mechanistic studies, we demonstrate that lipoxygenases act cell autonomously within neurons to regulate degeneration, and are required for mitochondrial permeabilization and caspase activation in the axon. These findings suggest that these enzymes may represent an attractive target for treatment of neuropathies and provide a potential mechanism for the neuroprotection observed in various settings following lipoxygenase inhibitor treatment.
Keywords: axon degeneration, DRG, lipoxygenase, NGF
Introduction
The degeneration of axons is thought to underlie the functional deficits present in peripheral neuropathies, stroke, and traumatic brain injury (Werring et al., 2000; Coleman, 2005; Johnson et al., 2013) and is a feature of multiple chronic neurodegenerative disorders (Raff et al., 2002; De Vos et al., 2008; Burke and O'Malley, 2013), suggesting that inhibition of this process has significant therapeutic potential. Axon degeneration is not unique to these pathological states, however, and also occurs during development to refine neuronal connections (Luo and O'Leary, 2005). For example, in the mammalian retinotectal system, RGC axons initially overshoot their targets and are subsequently pruned via local axon degeneration (McLaughlin et al., 2003). Embryonic DRG neurons have proven to be a useful system for the investigation of axon degeneration induced through nerve growth factor (NGF) withdrawal or axonal lesion. Studies in DRG have led to the discovery that axon degeneration requires the coordinated activation of multiple signaling pathways, including DLK/JNK, GSK3β, and calpains. (Miller et al., 2009; Gerdts et al., 2011; Ghosh et al., 2011; Bhattacharya et al., 2012; Chen et al., 2012; Simon et al., 2012; Maor-Nof et al., 2013; J. Yang et al., 2013). Despite the improved understanding gained through this work, significant gaps remain in our understanding of the mechanisms underlying axon degeneration and how these pathways interact to drive axonal destruction.
Lipoxygenases are a family of enzymes that oxidize polyunsaturated fatty acids such as arachidonic acid, and were first appreciated for their pro-inflammatory function (Brash, 1999; Conrad, 1999). Pharmacological inhibition or genetic deletion of lipoxygenases is beneficial in in vivo neuronal ischemia models, suggesting a role for these enzymes in neuronal degeneration (Khanna et al., 2005; van Leyen et al., 2006; Pallast et al., 2010). Although the mechanism of lipoxygenase inhibitor-mediated neuroprotection is unclear, these enzymes have been shown to contribute to oxidative stress-induced cell death of cultured neurons (Li et al., 1997; Pallast et al., 2009). Lipoxygenases may also contribute indirectly to neurodegeneration in vivo through induction of an inflammatory response in non-neuronal cells (Xu et al., 2013), similar to its role in the periphery. No function for lipoxygenases within axons has been described, and whether lipoxygenase activity contributes to breakdown of axons is not known.
In this study, we have developed a 384-well format, high-content DRG neuron/astrocyte coculture assay and used it to identify a role for 12/15-lipoxygenases in axonal degeneration. This setup exhibits the sensitivity and reproducibility to allow rapid screening of large compound libraries and has the ability to generate 10-point dose–response curves and additional signaling-based endpoints to enable a significantly improved understanding of the potency, toxicity, and mechanism of individual compounds. A screen of known bioactives in this system led to the discovery of multiple new regulators of axon degeneration including lipoxygenases, as well as validation of several hits previously identified using both developmental and Wallerian degeneration paradigms. We then extend these findings to demonstrate that lipoxygenases may contribute more broadly to axon degeneration.
Materials and Methods
Mouse models
Alox5, Alox12/15, and caspase-3-null mice were purchased from The Jackson Laboratory.
DRG neuron and astrocyte cultures
All animals were killed according to procedures regulated by the local authorities. To generate the DRG/astrocyte cocultures, astrocyte cultures were generated as follows: cortices of E18.5 Wistar rat embryos of either sex were dissected and cell suspensions were cultured in DMEM containing 1× GlutaMAX (Invitrogen), 1× Pen/Strep (Invitrogen), and 10% FBS. Microglia were mechanically separated and astrocyte-enriched cultures were harvested after 15 d in culture and plated at a density of 5000 cells per well into collagen-coated 384-well plates (Greiner) 2 d before addition of DRG neurons.
For cocultures, DRG were dissected from Wistar rat embryos (E14.5) and treated with 0.25% Trypsin-EDTA at 37°C for 30 min while shaking at 800 rpm. After enzyme removal, DRG were triturated using a glass Pasteur pipette in DMEM/F12 media containing 10% FBS, 1× Pen/Strep, and 1% GlutaMAX. The resulting cell suspension was filtered through a 50 μm sieve (Partec) to remove remaining tissue pieces, centrifuged 5 min at 1000 rpm, and resuspended in DRG culture media (DMEM/F12 containing 1× N3 supplement, 0.18% glucose, and 25 ng/ml NGF). Astrocyte culture media was removed before plating DRG neurons at a density of 1200–2000 cells per well on top of the astrocyte monolayer. To inhibit cell proliferation, media was supplied with 200 μm uridine and 100 μm 5-fluorodeoxyuridine the next day. Rat DRG were cultured for 4 d before NGF withdrawal. Mouse DRG neurons for explant and Campenot chamber experiments were dissected from E13.5 embryos and cultured essentially as described previously (Nikolaev et al., 2009; Ghosh et al., 2011). For Western blotting experiments and MTT assay, DRG were dissociated as above and plated on PDL and laminin-coated glass chamber slides (BD Biosciences). Mouse DRG cultures were maintained for 2–5 d post plating before NGF withdrawal. The time course of degeneration following withdrawal of NGF differs somewhat due to assay setup and species of DRG neurons used.
Small molecule handling and NGF withdrawal
The small molecule compound collection screen consisted of Evotec's CSB ACL collection of 292 manually selected bioactives and known drugs as well as an additional set of 579 selected known drugs (a total of 871 compounds). Compounds were dissolved in DMSO (Sigma) and prediluted in DRG culture medium without NGF with a final compound dilution of 1:500 (0.2% final DMSO). To fully remove residual NGF, fresh DRG culture media containing 25 μg/ml anti-NGF antibody (Genentech) was added at the time of compound addition. NGF-positive controls were wells receiving DMSO alone and media containing NGF. In the coculture assay, neurons were fixed 27 h (axon quantification endpoint) or 2 h (p-c-Jun quantification endpoint) after assay start, respectively. Anti-NGF addition for mouse DRG was performed as indicated above and cultures were fixed at various times following NGF withdrawal as indicated in the main text.
ELISA assays
MTT assay was conducted as per manufacturer's protocol with the Vybrant MTT Cell Proliferation Assay Kit (Life Technologies). NGF-containing wells were used as positive controls. For the HETE ELISA, levels of 12(S)-hydroxyeicosatetraenoic acid [12(S)-HETE] released by astrocyte DRG neuron cocultures were determined 6 and 16 h after NGF withdrawal using the 12(S)-HETE ELISA kit (Enzo Life Sciences). Astrocytes were plated into collagen-coated 48-well plates to form a monolayer, and DRG neurons were plated at eightfold higher plating density than in the 384-well screening assay. Production of 12(S)-HETE was boosted by adding 4 μm arachidonic acid (AA) as substrate for the 12-LOX-catalyzed formation of 12(S)-HETE, and 12(S)-HETE was extracted from the conditioned media by C18 reverse phase extraction (Sep-Pak Cartridges; Waters) following the procedures described in the ELISA kit.
Immunofluorescence staining
DRG/astrocyte cocultures were fixed with 4% PFA (Electron Microscopy Science) and 4% sucrose (Merck) for 30 min at room temperature, followed by 20 min blocking and permeabilization in PBS containing 3% normal goat serum, 2% albumin, and 0.1% saponin at room temperature for axon staining or PBS containing 5% normal goat serum and 0.3% Triton X-100 for p-c-Jun. Cultures were then stained with either neuronal class III β-tubulin antibody (TuJ1; R&D) or p-c-Jun (rabbit anti-p-c-Jun S63 II; Cell Signaling Technology) and HuD (Life Technologies), followed by incubation with fluorescently labeled secondary antibodies (Alexa488 or 555; Life Technologies). DRAQ5 (BioStatus) was included to label nuclei. Explants and dissociated DRG from the mouse were fixed with 4% PFA and 15% sucrose for 30 min at room temperature. Cells and explants were blocked and permeabilized with 5% BSA and 0.2% Triton X-100 for 1 h and put in primary overnight at 4°C followed by incubation with secondary antibodies. Antibodies used were neuronal class III β-tubulin (TuJ1; Covance), cleaved caspase-3 (Cell Signaling Technology), TOM20, HSP60 (Santa Cruz Biotechnology), Bax 6A7 (Abcam), and cytochrome c (BD Biosciences).
Western blotting
DRG cultures were lysed in 100 μl of Triton lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 0.1% Triton X-100, and protease and phosphatase inhibitors) for 30 min at 4°C and quantified using BCA assay. Similar amounts of protein were then loaded on 4–12% Bis-Tris gels and subjected to standard immunoblotting procedures: caspase-3, caspase-6, cleaved caspase-9, Bcl-2, Bcl-xl, p-JNK, p-c-Jun, GAPDH, and actin (Cell Signaling Technology). Blots were developed using SuperSignal West Dura Chemiluminescent substrate (Thermo Scientific) images were taken using the ChemiDoc system from Bio-Rad.
Optic nerve crush
All experiments with mice were performed under animal protocols approved by the Animal Care and Use Committee at Genentech. Age-matched, 3-month-old male C57BL6 and Alox12/15 KO mice were deeply anesthetized using intraperitoneal injection of 0.5 mg/g Avertin. The left eye of each animal was subjected to optic nerve crush injury via an intra-orbital approach. An incision was made on the superior conjunctiva of the surgery eye and the optic nerve was exposed. The crush injury was inflicted for 1 s using a 45° angled fine-tip forceps 1–2 mm from the eyeball and unoperated right eyes were used as controls. Three weeks post crush the proximal optic nerve was harvested and fixed in half Karnovsky's Fixative: 2% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 m cacodylate buffer for 24 h. Nerves were washed in 0.1 m Sorenson's buffer, postfixed in 1.0% osmium tetroxide in 0.1 m Sorenson's buffer for 24 h and washed in 0.1 m Sorenson's buffer. Optic nerves were dehydrated with agitation at 70, 95, and 100% in 200 proof ethanol ×2 for 10 min and agitated in propylene oxide ×2 for 10 min. The nerves were then infiltrated with Eponate 12 (PELCO, no. 18012) with agitation as follows: 1:1 Eponate/propylene oxide for 12 h, followed by 2:1 Eponate/propylene oxide for 4 h, followed by pure Eponate for 4 h. The nerves were embedded in Eponate 12 in flat silicone embedding molds and polymerized in Eponate at 70°C overnight. Blocks were sectioned on an automated ultramicrotome (Leica model EM UC7) set to cut 1 μm at 6 degrees knife angle at 1.60 mm/s. Slides were stained with 2% p-phenylenediamine (Sigma-Aldrich) in 50% ethanol for 15 min, rinsed in water for 10 min, air dried, and coverslipped with Cytoseal 60 (Thermo Scientific).
Image acquisition and data analysis
For high rat DRG assay plates, images were acquired with an Opera High Content Screening System using 10× lens and laser lines 488, 532, and 635 nm. Six images were taken per well and evaluated using Acapella script-based algorithms to detect nuclei and axon area. Detected nuclei were used to identify and count cells in all assays, and a minimum nuclear area threshold criterion was applied to select for live cells. The nuclear stencil was also used to quantify the mean intensities of p-c-Jun and HuD, respectively. Intensity threshold criteria were set for p-c-Jun and HuD to flag positive cells and determine the proportion of neurons (HuD threshold-positive cells) labeled with p-c-Jun (p-c-Jun threshold-positive cells).
For stained optic nerve sections, digital whole-slide images of paraphenylenediamine (PPD)-stained optic nerve sections were acquired using the NanoZoomer (Olympus) system at ×400 magnification with a resolution of 0.23 μm/pixel. ROIs outlining the cross-sectional area of the nerve were manually drawn for five to six sections per animal on each slide. Image analysis was performed using MATLAB 8.1 (The MathWorks). Positive pixel classifiers for myelin staining were generated in hue, saturation, and intensity space by sampling 8–10 positive subregions per section from different animals and applied to all samples. Segmentation of total myelin area was further processed using morphological operations. The percentage of stained area was calculated by normalizing the total myelin area to the cross-sectional area of the nerve, and this value was designated as intact axons, with the remaining area representing degenerated axons. All image analyses and selection of ROIs were performed blind to the experimental groupings.
Hit definition and hit profiling
Primary screening of compounds was conducted at 20 μm in plate duplicate. The mean axon area of NGF control wells was defined as 100% protection and the mean axon area of anti-NGF controls was defined as 0% protection. To monitor cell health the nuclei count of NGF controls was defined as 100% and a tenth of this was defined as 0%. A treatment resulting in reduction of nuclei count to <70% (i.e., 30% hit threshold) was defined as cytotoxic. Neurite area data points were excluded from the fit if the cytotoxicity criteria was met. Neuroprotection hits were defined as compounds showing larger than 25% preservation of axon area under NGF withdrawal in at least one of two replicate measurements. Pharmacological inhibition of p-c-Jun was determined in an equivalent manner with minimal p-c-Jun levels in NGF controls (full inhibition, 0%) and maximal in anti-NGF controls (no inhibition, 100%). Concentration–response curves were analyzed with Excel Fit software using the 4 Parameter Logistic Model or Sigmoidal Dose–Response Model. On some occasions neuroprotection data fits were constrained to the top value to improve the fit. To quantify data from mouse DRG explants and Campenot chamber cultures, degeneration was assessed blindly on a scale from 1 to 5. Values from greater than three independent experiments were averaged to generate the values presented.
Biostatistical methods
Data preparation.
For the comparison three datasets were selected: (1) 221,055 unique molecules with defined structure and molecular weight <1500 were retrieved from the MDDR (version 01.2013), and this set had been selected as a reference catalog defining the known bioactive chemical space; (2) 871 molecules from the Evotec library tested in the primary screen; and (3) 19 structurally dissimilar compounds targeting the same target as the original primary screening hits. The sets were combined forming a 221,944 compound set. All compounds were processed using the MOE wash routine with default parameters (version 2012.10; Molecular Operating EnVironment, Version 2012.10; Chemical Computing Group) MOE physicochemical 2D descriptors were calculated for all compounds and further processed using an unsupervised forward selection method (Whitley et al., 2000).
Algorithm.
Self-organizing maps (SOMs) were used for the chemical space analysis. The SOM technique is a nonlinear clustering technique, allowing visualization of a d-dimensional dataset as a 2D (or more) map containing n clusters (or neurons). Employing Kohonen's algorithm, a topology-preserving projection of the d-dimensional space is obtained (Saarinen and Kohonen, 1985). Each cluster is represented by a neuron ci with d weights. In a distance and time-dependent iterative optimization procedure neuron weights are adopted that best represent the dataset. In the present study, toroidal (borderless) SOMs were calculated using the ESOM algorithm with 500 × 820 neurons and 10 training cycles. All other parameters were left at default.
Results
Development of a screen to measure axon degeneration in 384-well format
Cultured embryonic DRG neurons have commonly been used to study axonal degeneration because of their pseudo-unipolar morphology consisting of a single two- branched axon with no dendrites and the reproducibility of degeneration observed in these axons following NGF withdrawal or axonal lesion. To date, studies in DRG have largely been performed manually, limiting the ability to conduct larger screening efforts. To develop a semi-automated high-content assay for axon degeneration in DRG capable of screening large compound or siRNA libraries, we first aimed to adapt established NGF withdrawal paradigms (Chen et al., 2012) to dissociated rat DRG cultures in a multiwell format. Multiple plate-coating protocols on glass and plastic bottom-fitted 96- and 384-well plates were tested, but no plate/coating combination was found to be compatible with automated screening as the neuronal meshwork either grew unevenly or detached from wells during standard manual or automated washing procedures. We rationalized that a DRG neuron/astrocyte coculture system may circumvent this issue. Indeed, plating of DRG neurons on a monolayer of primary astrocytes generated cultures with excellent spatial distribution of neuron bodies, axon outgrowth pattern, and compatibility with fixation and staining procedures (Fig. 1A). Using this setup, DRG neuron cell density could be titrated to as little as 1200 DRG neurons per well, which were able to produce viable and healthy DRG/astrocyte cocultures in a 384-well format. We next tested the ability of NGF withdrawal through the addition of an anti-NGF antibody to induce axonal degeneration in cocultures. The effects of NGF-withdrawal in the coculture system were visualized by combining a DRAQ5 nuclear stain with immunofluorescence staining for Tuj1 to label axons and GFAP to label astrocytes (Fig. 1B). Axonal degeneration in DRG/astrocyte cocultures progressed at a timescale similar to that observed with isolated DRG. Based on analysis of axonal integrity as judged by Tuj1 staining at various time points, we selected an endpoint 27 h following anti-NGF treatment because the degeneration of DRG axons was largely complete by this time with only a few intact axons remaining. We next developed an Acapella script-based detection of Tuj1-positive axons and DRAQ5-positive nuclei generating simple but robust measures of axon degeneration (axon area) and cytotoxicity (nuclei count), respectively (Fig. 1C,D). NGF withdrawal resulted in a significant and highly reproducible reduction in axon area of >80% without affecting nuclei count, as the majority of nuclei visualized in this setup are present in the astrocyte monolayer. In contrast, the addition of toxic compounds to the coculture system resulted in a reduction of both axon area and nuclei count as astrocyte viability was compromised (Fig. 1C,D).
Figure 1.

High-content screening assay setup. A, Schematic DRG assay setup. Precultured astrocytes from E18 rat brain are combined with dissociated E14.5 rat DRG neurons in 384-well assay plates and cultured in the presence of NGF for 4 d before assay start. NGF is withdrawn and compounds are incubated for 27 h (Anti-NGF). Plates are fixed and stained to reveal nuclei (DRAQ5) and axons (Tuj1). An example axon area heat map of a 384-well hit profiling plate is shown on the right. The level of neuroprotection is indicated by color coding, ranging from small (white) to large (green) total axon area. B, Example NGF and anti-NGF control wells are stained to reveal nuclei (DRAQ5), axons (Tuj1), and astrocytes (GFAP). Many Tuj1-positive axons can be visualized in the presence of NGF, while only sparse axons remain after anti-NGF treatment. GFAP staining is unaffected by anti-NGF treatment. Scale bar, 50 μm. C, Automated process for quantification of axon integrity and cell viability. DRAQ5 images are processed with a nuclei detection algorithm to produce nuclei count data while Tuj1 images are processed with a texture detection algorithm to produce axon pixel area data. Bottom, Pseudocoloring demarcates detected axon area and detected nuclei. Examples of NGF, anti-NGF, and a toxic (tox) compound (20 μm PD-146176) are shown. Scale bar, 50 μm. D, Quantification of example data shown in C. Anti-NGF treatment results in a significant reduction in axon area (NGF: 127,794 ± 6208, anti-NGF: 19,342 ± 3831; ***p < 0.001) but not nuclei count (NGF: 1354 ± 72, anti-NGF: 1446 ± 55) as the majority of nuclei represent astrocytes. In contrast, the toxic compound reduced both axon area (toxic compound: 648 ± 746) and nuclei count (toxic compound: 51 ± 19, n = 7 for NGF and anti-NGF, n = 4 for PD-146176).
To validate the ability of our semi-automated DRG assay to identify compounds that protect axons from degeneration, we next tested 10-point concentration ranges of protective molecules previously identified in NGF-withdrawal or axon lesion-based assays in DRG, including small molecule inhibitors of JNK, p38MAPK, and calpains (Miller et al., 2009; Gerdts et al., 2011; Chen et al., 2012; J. Yang et al., 2013). We also included Gliotoxin, a compound that has been shown to protect axons in a previous screen but had known toxicity in astrocytes (Gerdts et al., 2011) to determine how astrocyte health would affect axonal integrity. All three of the known protective compounds improved axonal integrity in our assay, yet with varying potencies and toxicities (Fig. 2). The most potent of these compounds, the JNK inhibitor AS601245, exhibited levels of axonal protection approaching that of NGF-containing positive control cultures at 1 μm, but showed less axonal protection and reduced nuclei count at concentrations >20 μm (Fig. 2A,A′), suggesting onset of toxicity at these doses. This observation highlights the value of (1) multiplexing readouts and (2) testing several concentrations in identifying the balance between activity and toxicity with individual compounds. Both the p38MAPK and calpain inhibitors exhibited robust protection of axons with no evidence of toxicity at the concentrations tested (Fig. 2B,C,B′,C′). In contrast, addition of Gliotoxin resulted primarily in loss of astrocytes at concentrations >1 μm, thus masking any axonal protection above the 0.8 and 1.7 μm concentrations where only modest protection was observed (Fig. 2D,D′).
Figure 2.
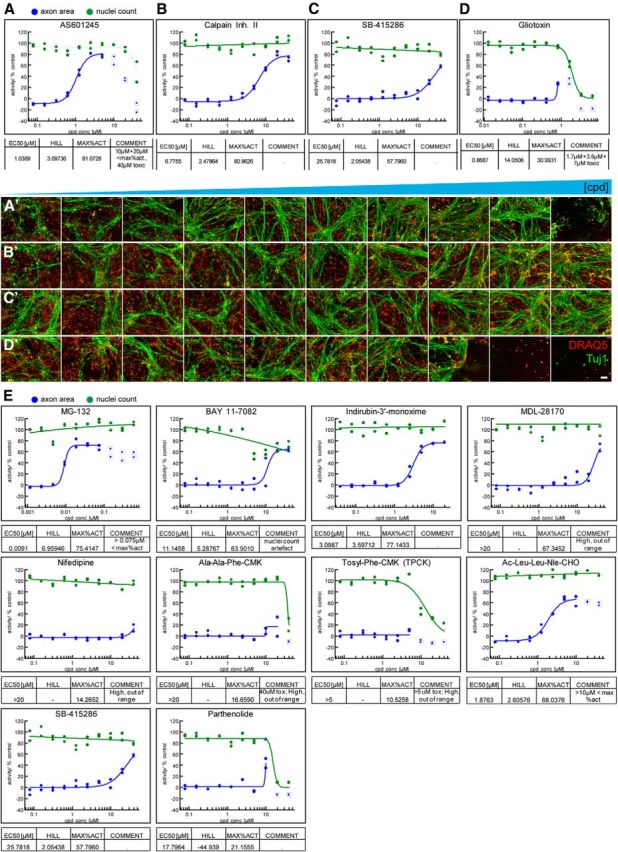
Assay validation with known neuroprotective compounds. A–D, Concentration–response curves for validation compounds. Representative curves for JNK inhibitor AS601245, calpain inhibitor-II, GSK3 inhibitor SB-415286, and the mycotoxin Gliotoxin are shown (N = 2 independent experiment with n = 2 each). Nuclei count and axon area data are expressed as percentage inhibition and percentage protection relative to controls. Maximal activation (MAX%ACT) represents the percentage of axonal protection achieved relative to NGF-positive controls. AS601245 demonstrates potent neuroprotection at ∼1 μm and toxic side effects at >5 μm. Calpain Inhibitor II displays a clean neuroprotection profile with a 7 μm EC50 and 80% maximal activation. SB-415286 displays modest neuroprotection with an EC50 of ∼25 μm. Gliotoxin is not protective and displays toxicity at >1 μm. Data points surrounded by squares were masked when generating curves. A′–D′, Single-imaging fields from the assay plate corresponding to the concentration–response curves shown in A–D compound (cpd; concentrations ascending from left to right). DRAQ5 is shown in red while Tuj1 is shown in green (N = 2). Scale bar, 100 μm. E, Protective effects of compounds previously identified in an axon lesion paradigm. MG-132, BAY 11–7082, Indirubin 3′-monoxime, MDL-28170, Ac-Leu-Leu-Nle-CHO, SB-415286, and parthenolide protected axons from degeneration while nifedipine, Ala-Ala-Phe-CMK, and Tosyl-Phe-CMK did not.
We then completed the validation of our 384-well coculture NGF withdrawal assay by conducting dose–response analysis on all 11 of the compounds previously identified as protective in a 96-well plate-based DRG neuron axotomy assay (Gerdts et al., 2011). The comparison between these two axon degeneration paradigms provided important validation for the approach: 7 of 11 hit compounds from the axotomy assay also showed concentration-dependent axonal protection in the NGF withdrawal assay (Fig. 2E). Three of the remaining four compounds were cytotoxic to our astrocyte/DRG coculture leading to loss of the astrocyte bed. The fourth compound, nifedipine, suppressed axon degeneration in the axon lesion-based study at only the early 6 h time point and not the 24 h time point, which is consistent with our results. These data demonstrate the robustness of this assay and suggest that screening for regulators of axon degeneration via NGF withdrawal is likely to be equally suitable to Wallerian degeneration-based paradigms in identifying regulators of this process.
A screen for novel regulators of axon degeneration
Having established an assay that is suitable to detect and quantify both toxic and protective effects of compounds, we next used it to search for new pathways that may be involved in axon degeneration. To do this, we screened 871 compounds consisting of known drugs as well as an Evotec collection of bioactives directed against key pathways and targets (CSB ACL; Fig. 3A). The primary screen was conducted at 10 μm in duplicate plates to minimize the occurrence of false positives and negatives. With a mean Z′ value of 0.63 for the axon area readout, the assay performance was robust and each replicate generated a similar dataset as indicated by the correlation plot (Fig. 3B,C; R2 = 0.53). Of 42 primary hit compounds, 34 were repurchased and submitted to 10 point dose–response profiling in replicate (Table 1). Database searching was conducted to collect information on compound mode of action and resulted in segregation of the confirmed hit compounds into multiple targets and pathways. The largest group was comprised of bioactives and known drugs addressing kinase signaling pathways (GW 5074, Indirubin-3′-oxime, SB 239063, a Sutent analog, and Arctigenin; Table 1). Many of these inhibitors are known to target kinases identified as regulators of axon degeneration in earlier studies (GSK3β, p38MAPK, and IKB) or have previously been shown to have neuroprotective activity through inhibition of on- or off-target kinases (Weishaupt et al., 2003; Chin et al., 2004; Gerdts et al., 2011; Chen et al., 2012; Welsbie et al., 2013). Other hits found in the screen included a number of anticancer agents, namely daunorubicin and teniposide (DNA intercalators and topoisomerase II inhibitors), the microtubule stabilizer taxol, and the protein synthesis inhibitor lycorine. Well known neurobiological targets were also identified, with the cannabinoid receptor CB2 agonist JWH 015, the vanilloid receptor antagonist capsazepine, the adenylyl cyclase activators forskolin and NKH 477, and the dopamine uptake inhibitor GBR 12783 all appearing as confirmed hits (Table 1). While adenylyl cyclase activity has previously been implicated as a regulator of axon degeneration (Chen et al., 2012), the other drug targets have not been associated with this process in earlier studies. One additional class of enzymes, lipoxygenases, caught our attention as it was identified through the protective activity of two structurally unrelated compounds, baicalein, a 5- and 12-lipoxygenase inhibitor, and 2-TEDC, a 5-, 12-, 15-lipoxygenase inhibitor. As lipoxygenase activity has been shown to regulate neuronal cell death due to oxidative stress (Li et al., 1997; van Leyen et al., 2008), we decided to investigate lipoxygenase inhibitors to elucidate whether this family of enzymes also contributes to axon degeneration following NGF withdrawal.
Figure 3.

Overview of small molecule screen for regulators of axon degeneration. A, Compounds selected for the screen. Emergent self-organizing maps (SOM) obtained for the MDDR collection and the Evotec known drugs and bioactives. Each dot corresponds to a compound. X-SOM and Y-SOM correspond to the X and Y direction of the calculated self-organizing map. The distance between dots is an expression of structural similarity between compounds. Green dots represent molecules from the MDDR classified launched drugs while gray dots represent the remainder from the MDDR collection (221,055 compounds total). Blue dots represent the 871 compounds screened that consist of known drugs as well as additional compounds with known bioactivity (CSB ACL collection). B, Scatter plot showing results from primary DRG screen. Compound plates were screened at 20 μm in duplicate and a correlation of the respective percentage neuroprotection values is shown. The R2 value of the replicate data points was 0.57. The mean Z′ value of the screening plates was 0.61. Hits (blue squares) versus nonhits (red squares) were qualified based on the hit threshold of 25% protection. Compounds reducing nuclei number <70% of NGF control were defined as cytotoxic (X shaped) and excluded from the hit set. Forty-two compounds fulfilled these hit criteria. C, Example of results from primary DRG screen. A screening plate is shown as a heat map indicating axonal protection in shades of green color (top). High (positive) controls (H; 100%) are NGF wells located in column 24, and low (negative) controls (L; 0%) are anti-NGF wells located in column 23. A neuroprotective compound served as a quality control (C). Representative primary data from the screen with Tuj1 in green and DRAQ5 in red. Some wells have a degree of Tuj1 staining indicating protection of axons while others do not. Scale bar, 50 μm.
Table 1.
Hits from small molecule screen confirmed by EC50 profiling
| Compound name | HCS |
Hit profiling |
Therapeutic class/bioactivity information | |
|---|---|---|---|---|
| % Act. ([20 μm]) | Mean % Act | Mean EC50 ([μm]) | ||
| GW 5074 | 30.65 | 86.1 | 0.31 | Potent, selective cRaf1 kinase inhibitor |
| Indirubin-3′-oxime | 94.12 | 75.9 | 2.25 | GSK-3b inhibitor; also inhibits other protein kinases |
| JWH 015 | 37.67 | 72.2 | 4.31 | Cannabinoid, selective CB2 agonist |
| Daunorubicin hydrochloride | 77.09 | 71.0 | 0.81 | Anticancer agent; increases expression of gangliosides, especially GQ1b, in differentiating neuronal cells |
| Baicalein | 28.81 | 67.6 | 1.84 | 5- and 12-Lipoxygenase inhibitor |
| Lycorine hydrochloride | 45.6 | 67.4 | 3.74 | Anticancer agent, activates p21CIP1/WAF1, which inhibits caspase-3, as well as the kinases ASK1 and JNK |
| 2-TEDC | 58.42 | 66.1 | 2.79 | 5-, 12-, 15-Lipoxygenase inhibitor |
| Demeclocycline hydrochloride | 43.56 | 64.8 | >20 | Antibacterial, calpain inhibitor |
| SB 239063 | 64.93 | 64.6 | 9.80 | Potent, selective p38MAPK inhibitor; orally active |
| Capsazepine | 73.78 | 63.0 | 6.81 | Selective vanilloid receptor antagonist |
| SU 6668-like (SU 11248) | 69.27 | 61.7 | 0.88 | Anticancer agent; inhibitor of multiple receptor transduction kinases |
| Arctigenin | 49.25 | 57.9 | 2.18 | Antioxidant, anti-inflammatory, antiproliferative, and antiviral agent; MKK1and IKBa inhibitor, neuroprotective by binding to kainate receptors |
| Luteolin | 59.58 | 56.0 | 8.34 | Flavinoid, multitarget actions including ROS-scavenging, Nrf2 induction, inhibition of caspase-3 activation |
| NKH 477 | 45.31 | 54.1 | 1.61 | Adenylyl cyclase activator |
| Teniposide | 58.91 | 48.7 | 2.70 | Anticancer agent |
| GBR 12783 dihydrochloride | 29.88 | 48.1 | 4.98 | Dopamine uptake inhibitor |
| Tolnaftate | 41.59 | 45.8 | 6.01 | Antifungal, may inhibit squalene epoxidase |
| Taxol | 56.85 | 40.9 | 1.29 | Anticancer agent; promotes assembly and inhibits disassembly of microtubules |
| Forskolin | 25.57 | 40.5 | 4.13 | Adenylyl cyclase activator |
HCS, high-content screening. Summary of top primary hit compounds confirmed by 10-point EC50 profiling. A compound was identified as a hit based on >25% maximal percentage protection (%Act) in both replicates of the primary HCS. Mean maximal percentage protection (mean %Act) and potency values (mean EC50 [μm]) for hit profiling are also shown. Compound annotations were collected based on therapeutic class and bioactivity from public databases.
Lipoxygenase activity is required for axon degeneration
To verify the involvement of lipoxygenase activity in NGF withdrawal-induced axon degeneration and to confirm that the protective effect was indeed due to on-target activity of the identified hits, we first expanded the group of lipoxygenase inhibitors to a total of eight structurally unrelated compounds (Fig. 4A). Seven of eight small molecule lipoxygenase (LOX) inhibitors demonstrated concentration-dependent protection of axons in the low micromolar range (Fig. 4B), which provided strong pharmacological evidence that lipoxygenase activity contributes to axon degeneration in our system. However, given the limited isoform selectivity of many of the lipoxygenase inhibitor compounds available (Pergola and Werz, 2010), we were not able to ascribe the observed effects to a specific lipoxygenase enzyme.
Figure 4.
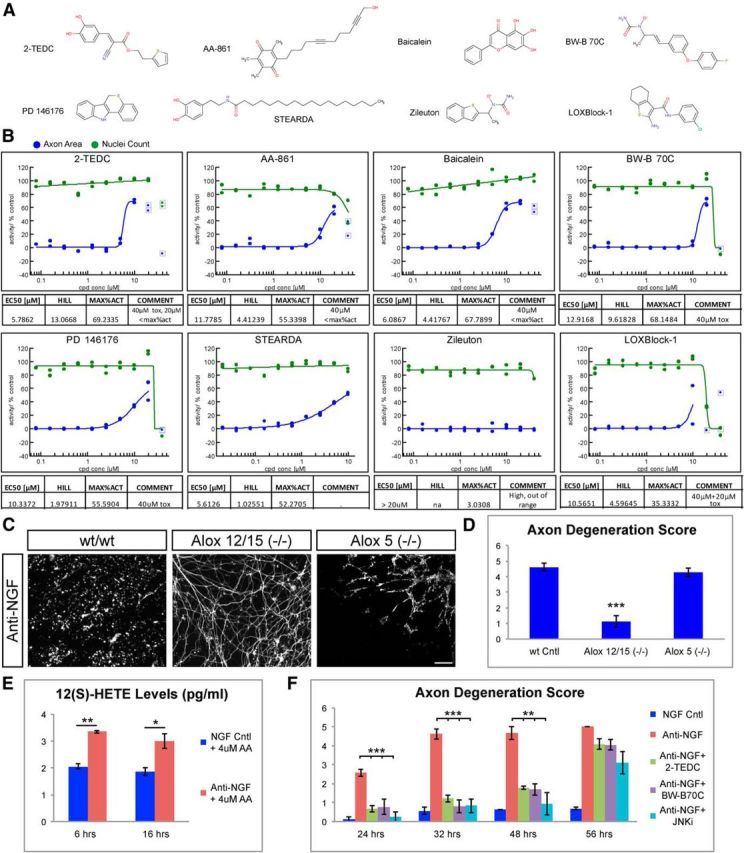
Lipoxygenases are required for axon degeneration. A, Structures of lipoxygenase inhibitors tested in the assay. 2-TEDC and baicalein were hits identified in the primary screen, while the other structurally distinct lipoxygenase inhibitors were selected for follow-up studies based on published data. B, EC50 profiles of tested lipoxygenase inhibitors are shown. Graphs are representative of two to six test replicates. Compounds were tested in 10 point 1:2 dilution series with 10 μm, 20 μm[scap], or 40 μm top concentration. All tested lipoxygenase inhibitors except Zileuton showed some degree of protection. Potencies ranged between ∼6 (2-TEDC) and ∼13 μm (BW-B 70C). C, Mouse DRG neurons cultured from Alox5(−/−) and Alox12/15(−/−) mice. NGF withdrawal resulted in robust axon degeneration and loss of axons (Tuj1 staining, white) in control and Alox5(−/−) but not in Alox12/15(−/−). Scale bar, 100 μm. D, Quantification of axonal degeneration observed in C. WT = 4.63 ± 0.26, Alox12/15(−/−) = 1.23 ± 0.37, Alox5(−/−) = 4.29 ± 0.27, N = 3 independent experiments, ***p < 0.001(Student's t test, error bars indicate SEM). E, Quantification of 12(S)-HETE levels in DRG-astrocyte cocultures either 6 h or 16 h following NGF withdrawal (Anti-NGF), and in controls grown in presence of NGF (NGF Cntl). AA (4 μm) was added to the media to have an excess of lipoxygenase substrate. NGF withdrawal resulted in increased 12(S)-HETE at both time points. 6 h: NGF = 2.05 ± 0.09, anti-NGF = 3.34 ± 0.05; 16 h: NGF = 1.864 ± 0.14, anti-NGF = 3.0 ± 0.27 pg/ml. Data are representative of two independent experiments (N = 4), **p < 0.01, *p < 0.05 (Student's t test, error bars indicate SEM). F, Quantification of NGF withdrawal-induced axon degeneration at various time points following treatment with lipoxygenase inhibitors (2-TEDC or BW-B70C) or the JNK inhibitor AS601245 (JNKi). Both lipoxygenase and JNK inhibitors protect axons against degeneration up to 48 h but not 56 h post-NGF withdrawal. Relative axon degeneration score: 24 h: NGF = 0.12 ± 0.12, anti-NGF = 2.58 ± 0.19, anti-NGF + 2-TEDC = 0.66 ± 0.16, anti-NGF + BW-B 70C = 0.77 ± 0.39, anti-NGF + JNK inhibitor = 0.25 ± 0.25; 32 h: NGF = 0.56 ± 0.18, anti-NGF = 4.61 ± 0.26, anti-NGF + 2-TEDC = 1.22 ± 0.18, anti-NGF + BW-B 70C = 0.80 ± 0.35, anti-NGF + JNK inhibitor = 0.83 ± 0.37; 48 h: NGF = 0.6 ± 0, anti-NGF = 4.66 ± 0.33, anti-NGF + 2-TEDC = 1.77 ± 0.07, anti-NGF + BW-B 70C = 1.69 ± 0.29, anti-NGF + JNK inhibitor = 0.94 ± 0.59; 56 h: NGF = 0.68 ± 0.06, anti-NGF = 5 ± 0, anti-NGF + 2-TEDC = 4.08 ± 0.27, anti-NGF + BW-B 70C = 4.05 ± 0.29, anti-NGF+JNK inhibitor = 3.11 ± 0.59; ***p < 0.001, **p < 0.01 (Student's t test, error bars indicate SEM; N = 3 independent experiments, where >3 explants were scored).
To identify which lipoxygenase isoform was involved in degeneration, we next examined axon degeneration in DRG from Alox12/15(−/−) mice lacking expression of 12/15-lipoxygenase or Alox5(−/−) mice, which encode the known lipoxygenase enzymes. DRG axons from Alox12/15(−/−) but not Alox5(−/−) embryos demonstrated reduced degeneration compared with wild-type axons after NGF withdrawal (Fig. 4C,D). These findings provide strong evidence that activity of 12/15-lipoxygenase but not 5-lipoxygenase contributes to trophic factor-induced axon degeneration and are consistent with previous results from in vivo ischemia models, which demonstrated reduced infarct size in brains from Alox12/15(−/−) mice (Khanna et al., 2005; van Leyen et al., 2006) but not from Alox5(−/−) mice (Kitagawa et al., 2004). The protection in Alox12/15(−/−) mice suggests that 12/15-lipoxygenase activity is increased following NGF withdrawal. This hypothesis was tested directly through measuring levels of 12(S)-HETE, a major 12/15-lipoxygenase product, following NGF withdrawal in DRG cultures. Because 12(S)-HETE levels in the DRG cultures were below the detection limit of the ELISA assay, 4 μm arachidonic acid was also added to the culture to provide excess 12/15-lipoxygenase substrate. This concentration of arachidonic acid was selected as it was found not to be toxic to DRG cultures at either time point and resulted in detectable 12(S)-HETE (Fig. 4E; data not shown). Consistent with our observation from Alox12/15 −/−) mice, 12(S)-HETE levels were significantly increased both 6 and 16 h after NGF deprivation indicating 12/15-lipoxygenase activity was indeed increased (Fig. 4E).
To determine how robustly lipoxygenase inhibitors protect axons from degeneration, we next conducted a time course at longer periods following NGF withdrawal with two structurally distinct lipoxygenase inhibitors: 2-TEDC and BW-B 70C. The JNK inhibitor AS601245, a compound known to potently protect axons from degeneration (Fig. 2A; Chen et al., 2012), was also included as a control. All three compounds provided clear protection of axons up to 48 h after NGF withdrawal, but this protection was not sustained at 56 h (Fig. 4F). These data indicate that lipoxygenase activity is an important contributor to axon degeneration in DRG neurons.
Lipoxygenases act locally in the axon to regulate degeneration
As our screening assay used a neuron/astrocyte coculture setup, it is possible that lipoxygenase activity in either cell type could contribute to axon degeneration. To discriminate between these possibilities, we tested these compounds in mouse DRG explants, which have few astrocytes associated with axons. Treatment of DRG explants with 2-TEDC or BW-B 70C following NGF withdrawal resulted in axonal protection at concentrations similar to those used in the coculture setup (Fig. 5A), suggesting lipoxygenases act cell autonomously within neurons to direct degeneration of axons.
Figure 5.
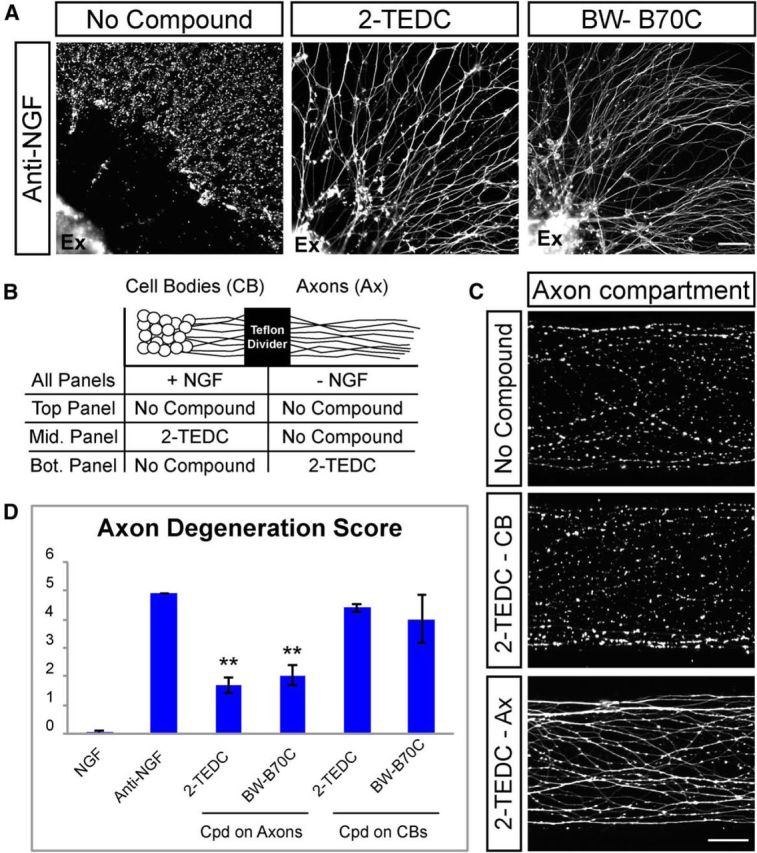
Lipoxygenases act locally within axons to regulate axon degeneration. A, DRG explants (Ex) cultured in the absence of astrocytes and stained with Tuj1 to visualize axons. Axons are protected following NGF withdrawal in the presence of lipoxygenase inhibitors (2-TEDC, 18 μm; BW-B 70C, 30 μm; n = 5). Scale bar, 100 μm. B, Schematic of experiments using compartmentalized culture chambers shown in C and D in which anti-NGF is added to distal axons only and lipoxygenase inhibitor is added to either the cell body (CB) compartment or the distal axonal (Ax) compartment. C, DRG neurons grown in a compartmentalized chamber and stained with Tuj1. Axons in the distal axonal compartment are largely degenerated 28 h after NGF withdrawal (top). Addition of 2-TEDC to the cell body compartment does not protect axons from degeneration (middle), but addition to the distal axon compartment results in protection of axons (bottom). Scale bar, 50 μm. D, Quantification of axon degeneration in compartmentalized chamber treated with lipoxygenase inhibitors. NGF = 0.05 ± 0.07, anti-NGF = 4.9 ± 0, 2-TEDC on cell bodies = 4.4 ± 0.14, 2-TEDC on axons = 1.85 ± 0.07, BW-B 70C on cell bodies = 4 ± 0.84, BW-B 70C on axons = 2.35 ± 0.07; **p = 0.01, N = 2 independent experiments with multiple cultures in each experiment (Student's t test, error bars indicate SEM). Cpd, Compound.
To better define the mechanism by which lipoxygenases contribute to axonal degeneration, we used compartmentalized culture chambers that allow isolated treatment of DRG cell bodies and axons (Fig. 5B). In this experimental paradigm, withdrawal of NGF exclusively from the axonal compartment results in extensive axonal degeneration in this compartment without affecting neuronal viability or proximal axon integrity (Campenot, 1977; Ghosh et al., 2011). This setup can be used both to (1) generate local axon degeneration that is independent from neuronal cell death and (2) spatially define where specific contributors to axon degeneration act through addition of inhibitors solely to either the cell body or axon compartment (Chen et al., 2012). Interestingly, addition of lipoxygenase inhibitors 2-TEDC or BW-B 70C to only the axonal compartment significantly reduced the amount of degeneration, while addition of these compounds only to the cell body compartment did not affect the localized degeneration of the distal axon (Fig. 5B–D). These results indicate that lipoxygenases do not drive axon degeneration simply through promoting neuronal apoptosis, and that lipoxygenase activity is required locally within axons to drive degeneration.
Lipoxygenases do not affect JNK activity but are required for caspase activation in the axon
Work from multiple groups has revealed that NGF withdrawal in DRG neurons initiates a programmed process involving the activation of multiple signaling pathways to induce the degeneration of axons. The DLK/JNK pathway has been identified as an essential early component of this response that is required for activation of several other regulators of degeneration such as caspases and calpains (Ghosh et al., 2011; J. Yang et al., 2013). To determine whether lipoxygenases influence the DLK/JNK signaling pathway, we examined the effect of lipoxygenase inhibition on phosphorylation of c-Jun (p-c-Jun), a specific marker of stress-induced DLK/JNK activity (Ghosh et al., 2011) at 2 h after NGF withdrawal, before the onset of degeneration. Through adapting our coculture system to allow p-c-Jun staining along with HuD to label neuronal nuclei, compounds could be profiled in a 10-point dose–response as was conducted when measuring axonal protection (Fig. 6A,B). While NGF withdrawal resulted in a significant increase in the number of p-c-Jun-positive neuronal nuclei that was prevented by addition of the JNK inhibitor SP600125 in a concentration-dependent fashion, neither of the two lipoxygenase inhibitors tested (2-TEDC and BW-B 70C) reduced the number of p-c-Jun-positive cells at nontoxic concentrations (Fig. 6D). These observations were confirmed by measuring p-JNK and p-c-Jun levels in DRG from Alox12/15(−/−) embryos 3 h after NGF withdrawal by Western blot, which revealed no difference in JNK pathway activation between genotypes (Fig. 6C). These data indicate that lipoxygenase activity is either downstream or independent of DLK/JNK activity.
Figure 6.

Lipoxygenases do not protect axons via alteration of JNK signaling. A, B, Measurement of p-c-Jun induction 2 h after NGF withdrawal in a 384-well setup. A, NGF and anti-NGF control wells are stained to reveal nuclei (DRAQ5), p-c-Jun, axons (Tuj1), and neuronal nuclei (HuD). Two hours after NGF withdrawal there is robust phosphorylation of c-Jun in neuronal nuclei without signs of axon degeneration. Scale bar, 50 μm. B, Quantification of example data shown in A. Anti-NGF treatment results in a significant increase in p-c-Jun-positive neurons (percentage p-cJun-positive neurons; NGF: 12.4 ± 3.9 and anti-NGF: 83.5 ± 4.0; ***p < 0001, Student's t test, error bars indicate SD) but does not alter nuclei count (NGF: 1511 ± 28, anti-NGF: 1495 ± 42) nor neuron content (percentage neurons; NGF: 16.1 ± 0.6%, anti-NGF: 14.9 ± 1.5%), while the toxic compound (tox cpd; 40 μm PD-146176) abolished all readouts (data from one experiment is shown, n = 7 for NGF and anti-NGF, n = 4 for PD-146176). C, Western blots measuring phosphorylation of JNK and c-Jun in Alox12/15(−/−) and wild-type DRG neurons in the presence of NGF (+ NGF) or 3 h after NGF withdrawal (− NGF). A similar induction of JNK pathway activity occurs in both genotypes. D, Dose–response curves showing effect of treatment with JNK inhibitor SP600125, 2-TEDC, or BW-B 70C on p-c-Jun-positive cell number. SP600125 resulted in p-c-Jun reduction with an IC50 of 0.31 μm while LOX inhibitors 2-TEDC and BW-B 70C had no effect on p-c-Jun.
Although evidence of JNK activation is apparent early after NGF withdrawal, some downstream components such as caspases act relatively late in the axon degeneration process (∼6–8 h; Schoenmann et al., 2010; Ghosh et al., 2011). To narrow down the pathways that may be affected by lipoxygenase activity, we next conducted a time course where lipoxygenase inhibitors were added at various intervals (0–8 h) following NGF withdrawal. Degeneration of mouse DRG explants was then assayed at 24 h after NGF withdrawal in all cases. The JNK inhibitor SP600125 was also tested in the same time course as a control. Consistent with the timing of JNK pathway activation observed in earlier studies (Ghosh et al., 2011), we found that JNKs must be inhibited at the time of NGF withdrawal to protect axons from degeneration (Fig. 7A). In contrast, the lipoxygenase inhibitors 2-TEDC and BW-B 70C could be added as late as 3 h after NGF withdrawal and still elicit significant protection, while mild protection of axons was observed even when inhibitors were added 6 h following anti-NGF treatment (Fig. 7A). These data suggest that lipoxygenase activity occurs between 3 and 6 h following NGF withdrawal, consistent with the timing of local caspase activation within axons. To evaluate if lipoxygenases indeed contribute to caspase activation, levels of caspase-3 activity were examined in axons from Alox12/15(−/−) and C57BL6 wild-type control DRG explants 8 h after NGF withdrawal via immunofluorescence. Interestingly, while C57BL6 DRG showed a significant increase in active caspase-3 staining within axons 8 h after NGF withdrawal, this staining was largely absent in Alox12/15(−/−) DRG axons (Fig. 7B), indicating that lipoxygenase activity is required for activation of caspase-3 in the axon.
Figure 7.
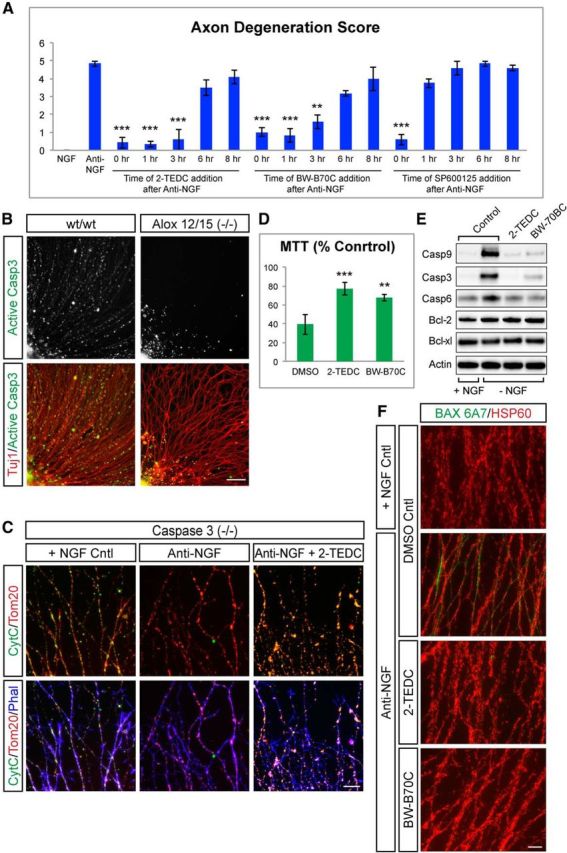
Lipoxygenases are required for release of cytochrome c from mitochondria and activation of caspase-3 in axons. A, Quantification of axon protection achieved through addition of lipoxygenase inhibitor added at the times indicated following NGF withdrawal. Lipoxygenase inhibitors can be added as late as 6 h after NGF withdrawal and still protect axons from degeneration, while the JNK inhibitor (SP600125) must be added at the time of NGF withdrawal. (Anti-NGF = 4.83 ± 0.14; 2-TEDC: 0 h = 0.41 ± 0.28, 1 h = 0.33 ± 0.14, 3 h = 0.58 ± 0.57, 6 h = 3.5 ± 0.43, 8 h = 4.08 ± 0.38; BW-B 70C: 0 h = 1 ± 0.25, 1 h = 0.83 ± 0.38, 3 h = 1.58 ± 0.38, 6 h = 3.16 ± 0.14, 8 h = 4 ± 0.66; JNK inhibitor: 0 h = 0.58 ± 0.28, 1 h = 3.75 ± 0.25, 3 h = 4.58 ± 0.38, 6 h = 4.83 ± 0.14, 8 h = 4.58 ± 0.14; ***p < 0.001, **p < 0.01, *p < 0.05 (Student's t test, error bars indicate SEM; N = 3 independent experiments). B, DRG explants from wt and Alox12/15(−/−) stained for activated caspase-3 (green) and Tuj1 (red) 8 h after NGF withdrawal. Caspase-3 is activated in many wt axons, but staining is greatly reduced in Alox12/15(−/−) axons. Data are representative of four independent experiments. Scale bar, 100 μm. C, Caspase-3(−/−) DRG explants stained with cytochrome c (green), Tom20 (red), and phalloidin (blue). In the presence of NGF (left), Tom20 and cytochrome c colocalize within axonal mitochondria. Eight hours following NGF withdrawal in untreated cultures (middle), cytochrome c staining is lost, but this does not occur when cultures are treated with a lipoxygenase inhibitor (right). Data are representative of four independent experiments. Scale bar, 50 μm. D, Quantification of mitochondrial activity via an MTT assay 16 h after NGF withdrawal with or without lipoxygenase inhibitors. Mitochondrial activity is shown as percentage NGF control. MTT signal is reduced after NGF withdrawal while the addition of lipoxygenase inhibitors significantly improved mitochondrial activity. Anti-NGF = 39.45 ± 6.48%, anti-NGF + 2-TEDC = 77.15 ± 6.84%, anti-NGF + BW-B 70C = 67.62 ± 3.71%; ***p < 0.001 (Student's t test, error bars indicate SEM; N > 3 independent experiments). E, Western blots for components of the intrinsic apoptosis pathway in DRG 8 h after NGF withdrawal (− NGF) with or without lipoxygenase inhibitors compared with NGF controls (+ NGF). Levels of cleaved caspase-3, -6, and -9 increase after NGF withdrawal but are reduced in the presence of 2-TEDC or BW-B 70C. No change is observed in Bcl-xl and Bcl-2 levels in any condition. F, Immunostaining with a Bax antibody specific for the active conformation in wt DRG explants 6 h following NGF withdrawal with or without lipoxygenase inhibitors. Bax 6A7 (green), HSP60 (red). NGF withdrawal results in increased axonal 6A7 staining that does not occur when cultures are treated with 2-TEDC or BW-B 70C. Data are representative of four independent experiments. Scale bar, 50 μm.
Recent studies have demonstrated that local initiation of the intrinsic apoptosis pathway within the axon triggers caspase-3 activation and is an essential step in trophic factor withdrawal-induced axonal degeneration (Nikolaev et al., 2009; Schoenmann et al., 2010; Ghosh et al., 2011; Simon et al., 2012). It has also been suggested that lipoxygenase activity can damage mitochondria following oxidative stress (Pallast et al., 2009), so we next examined whether lipoxygenases contributed to mitochondrial permeabilization in the axon. To visualize this process in intact axons at later time points, we used DRG derived from caspase-3(−/−) animals as these axons display strong protection from trophic factor withdrawal-induced degeneration yet should not show disruption of upstream signaling events (Schoenmann et al., 2010; Simon et al., 2012). Caspase-3(−/−) neurons were stained for the mitochondrial membrane protein Tom20 and cytochrome c, a mitochondrial luminal protein, whose colocalization serves as an indicator of mitochondrial integrity in the axon (Cusack et al., 2013). Consistent with previous studies, Tom20 and cytochrome c were colocalized in healthy axons cultured in the presence of NGF, but cytochrome c staining was lost from Tom20-labeled mitochondria 16 h after NGF withdrawal (Fig. 7C). Interestingly, treatment of these cultures with the lipoxygenase inhibitors 2-TEDC or BW-B 70C resulted in cytochrome c staining retained within mitochondria, suggesting that intact mitochondria remained at this time point (Fig. 7C; data not shown).
To further confirm that lipoxygenase inhibitor treatment results in the maintenance of intact mitochondria following NGF withdrawal, mitochondrial function was measured in these cultures using an MTT assay. Treatment of cultures with either 2-TEDC or BW-B 70C significantly increased mitochondrial function, supporting the hypothesis that lipoxygenases act upstream of mitochondrial permeabilization (Fig. 7D). To better define the mechanism by which this occurs, the components of this pathway were examined by Western blot. Consistent with our observations by staining (Fig. 7B), activation of the caspases in this pathway (caspase-9, -3, and -6) were attenuated by lipoxygenase inhibitor treatment (Fig. 7E). Levels of the anti-apoptotic Bcl family members Bcl-2 and Bcl-xl were unchanged under the same conditions, making it unlikely that these factors contributed to this effect (Fig. 7E). As conformational changes in Bax are thought to be required for disruption of mitochondrial integrity (Hsu and Youle, 1998), we next stained DRG neurons with an active conformation-specific 6A7 Bax antibody to examine whether lipoxygenase activity influences this process in combination with HSP60 staining to label mitochondria. Six hours after NGF withdrawal, an increase in 6A7 staining could be readily observed in a subset of DRG axons, which was not present when DRG were treated with 2-TEDC or BW-B 70C, suggesting lipoxygenases act upstream of Bax activation (Fig. 7F).These data support a model in which lipoxygenase activity either directly or indirectly modulates Bax-dependent mitochondrial permeabilization and subsequent activation of caspases within the axon to drive degeneration.
Axons of lipoxygenase 12/15-null animals display reduced degeneration following optic nerve crush
The identification of lipoxygenase inhibitors as regulators of axon degeneration in our NGF withdrawal-based assay suggests that these enzymes are important contributors to developmental degeneration in DRG neurons. To determine whether 12/15-lipoxygenases are also involved in axon degeneration following neuronal injury in the adult, we examined nerves of Alox12/15(−/−) following optic nerve crush. Proximal optic nerves were stained with PPD 3 weeks after nerve crush when significant degeneration has taken place (Watkins et al., 2013; Fig. 8A) to measure the number of intact axon bundles compared with uncrushed controls. Both genotypes displayed degeneration of axons at this time point, but Alox12/15(−/−) displayed reduced degeneration and an increase in intact axon bundles compared with wild-type mice (Fig. 8A,B), indicating that 12/15-lipoxygenase activity is important for degeneration of retinal ganglion cell axons following axon injury in vivo.
Figure 8.

Alox12/15(−/−) displays reduced axon degeneration after optic nerve crush. A, Representative images of PPD-stained proximal optic nerve sections from wt and Alox12/15(−/−) animals 3 weeks after nerve crush. Loss of intact axons can be readily observed 3 weeks after nerve crush in both genotypes compared with uncrushed contralateral nerves, though the amount of axonal degeneration in Alox12/15(−/−) mice was reduced compared with and without controls. Arrow indicates areas devoid of axons in crushed nerves. Low-magnification (Low Mag) scale bar, 50 μm; high-magnification (High Mag) scale bar, 20 μm. B, Quantification of images shown in A. wt = 32.19 ± 2.29% and Alox12/15(−/−) = 24.04 ± 4.97%; *p < 0.05 (Student's t test, error bars indicate SEM, N = 5 animals/genotype, with 10 sections quantified from each nerve).
Discussion
In this study, we sought to identify new regulators of axon degeneration using an NGF withdrawal paradigm and therefore developed a 384-well, high-content imaging assay using DRG neurons in coculture with astrocytes that is amenable for compound library screening. Although the assay is designed to mimic the competition for NGF that DRG neurons encounter during development, the demonstrated applicability of pathways identified using this paradigm to other developmental and disease contexts suggests that mechanisms identified in these neurons may have broad applicability and are not limited to this peripherally projecting neuronal population (D. Yang et al., 1997; Li et al., 2000; Libby et al., 2005; Schoenmann et al., 2010; Simon et al., 2012; Pozniak et al., 2013; Watkins et al., 2013; J. Yang et al., 2013). In addition, the observation that robust and reproducible degeneration of axons is elicited simply by depletion of NGF argues that this is a highly programmed process, perhaps more so than those based on the addition of toxins at levels higher than neurons likely encounter in vivo. Consistent with this notion, the results obtained in DRG-based assay paradigms with regard to the signaling pathways underlying the degeneration process have proven to be largely reproducible between groups (Schoenmann et al., 2010; Gerdts et al., 2011; Ghosh et al., 2011; Chen et al., 2012; Simon et al., 2012). In addition, the overlap between the pathways identified here and those that contribute to Wallerian degeneration suggest that although key differences in the pathways exist, NGF depletion represents an attractive approach to study the molecular mechanisms that underlie axon degeneration.
Conversion of the NGF withdrawal degeneration assay into a semi-automated, 384-well platform has facilitated screening of compound libraries and allowed a more comprehensive characterization of compound activity than previously achievable. Although the throughput for screening purposes is undoubtedly valuable and provides capacity to conduct large-scale phenotypic screens, the ability to conduct follow-up studies for specific compounds should not be discounted. For example, our results demonstrate that the protective activity of certain compounds is highly dose dependent, and testing protective compounds at too high a concentration can result in significant cytotoxicity, which may occlude protective effects (Figs. 2A, 4B). These observations highlight the value of conducting multipoint dose ranging with individual compounds. Similarly, the ability to conduct multipoint dose–response profiling on multiple related compounds and investigate activation of specific pathways in DRG neurons can quickly provide a much more comprehensive idea of a specific compound's mechanism of action (Figs. 4A,B, 6A–C). Indeed, the use of this setup validated many of the hits identified in previous screening efforts but also identified a number of compounds whose ability to protect axons was not previously appreciated, including lipoxygenase inhibitors, cannabinoid receptor agonists, and TRP channel antagonists. As small molecule inhibitors often have off-target activities that may complicate interpretation of phenotypic results, the ability to conduct a more thorough evaluation before follow-up studies as was done here provides a more appealing starting point for future study.
The culmination of a large body of recent work has identified a number of pathways that contribute to axon degeneration, though many gaps in our understanding remain (Coleman, 2005). However, it is clear that the later stages of NGF withdrawal-induced axon degeneration are characterized by Bax-dependent mitochondrial permeabilization and activation of apoptotic caspases such as caspase-3 and -9 (Nikolaev et al., 2009; Schoenmann et al., 2010; Simon et al., 2012). Our results indicate that lipoxygenases also contribute to loss of mitochondrial integrity, and suggest that lipoxygenase activity leads either directly or indirectly to Bax activation through promoting the conformational changes and exposure of hydrophobic domains thought to be important for function (Cartron et al., 2005). Indeed, the observation that this conformational switch can be induced through detergent treatment is consistent with a role for lipids in this process (Hsu and Youle, 1998). Previous studies have also shown that lipoxygenases can bind directly to membranes of mitochondria and other cellular organelles and result in oxidation of membrane lipids (van Leyen et al., 1998), which could facilitate Bax insertion into the mitochondrial membrane. Alternatively, lipoxygenases may contribute to conformational changes in Bax before its interaction with mitochondria and distinguishing between these and other possibilities will require further study.
Lipoxygenase function has been studied in a range of neurodegenerative disorders ranging from stroke to Alzheimer's disease (Praticò et al., 2004; van Leyen et al., 2006; Pallast et al., 2010; Chu and Praticò, 2011), yet the mechanisms by which they exert their function in these paradigms is not well characterized. Our observations provide important additions to the understanding of lipoxygenase function in neurons. First, we demonstrate that the functional contribution of lipoxygenases to neuronal degeneration is not limited to the oxidative stress-based paradigms evaluated to date (Li et al., 1997; Zhang et al., 2004; Pallast et al., 2009) and suggests that lipoxygenases may function more broadly to regulate neurodegeneration in various contexts. Second, we show that lipoxygenases have an essential axon intrinsic function, and can be activated locally to elicit axonal degeneration without resulting in neuronal apoptosis (Fig. 5B). Last, we propose a mechanism that incorporates lipoxygenase activity into a known pathway leading to axon degeneration, thus providing a better understanding of how lipoxygenases may contribute to neuronal degeneration in other contexts. These results support future studies to better understand the contribution of lipoxygenases to axonal degeneration in chronic neurodegenerative disease and suggest that treatment with lipoxygenase inhibitors may represent a viable therapeutic strategy for prevention of axonal degeneration in these indications.
Footnotes
We thank Mark Brewer and Mark Whittaker for assembling the CSB ACL collection and to Evotec compound management. We also thank Ina Sternberger, David Hackos, and Baris Bingol for their comments on this manuscript.
The authors declare no competing financial interests.
References
- Bhattacharya MR, Gerdts J, Naylor SA, Royse EX, Ebstein SY, Sasaki Y, Milbrandt J, DiAntonio A. A model of toxic neuropathy in Drosophila reveals a role for MORN4 in promoting axonal degeneration. J Neurosci. 2012;32:5054–5061. doi: 10.1523/JNEUROSCI.4951-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- Burke RE, O'Malley K. Axon degeneration in Parkinson's disease. Exp Neurol. 2013;246:72–83. doi: 10.1016/j.expneurol.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci U S A. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartron PF, Arokium H, Oliver L, Meflah K, Manon S, Vallette FM. Distinct domains control the addressing and the insertion of Bax into mitochondria. J Biol Chem. 2005;280:10587–10598. doi: 10.1074/jbc.M409714200. [DOI] [PubMed] [Google Scholar]
- Chen M, Maloney JA, Kallop DY, Atwal JK, Tam SJ, Baer K, Kissel H, Kaminker JS, Lewcock JW, Weimer RM, Watts RJ. Spatially coordinated kinase signaling regulates local axon degeneration. J Neurosci. 2012;32:13439–13453. doi: 10.1523/JNEUROSCI.2039-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin PC, Liu L, Morrison BE, Siddiq A, Ratan RR, Bottiglieri T, D'Mello SR. The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J Neurochem. 2004;90:595–608. doi: 10.1111/j.1471-4159.2004.02530.x. [DOI] [PubMed] [Google Scholar]
- Chu J, Praticò D. 5-lipoxygenase as an endogenous modulator of amyloid beta formation in vivo. Ann Neurol. 2011;69:34–46. doi: 10.1002/ana.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Conrad DJ. The arachidonate 12/15 lipoxygenases. A review of tissue expression and biologic function. Clin Rev Allergy Immunol. 1999;17:71–89. doi: 10.1007/BF02737598. [DOI] [PubMed] [Google Scholar]
- Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun. 2013;4:1876. doi: 10.1038/ncomms2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Gerdts J, Sasaki Y, Vohra B, Marasa J, Milbrandt J. Image-based screening identifies novel roles for IkappaB kinase and glycogen synthase kinase 3 in axonal degeneration. J Biol Chem. 2011;286:28011–28018. doi: 10.1074/jbc.M111.250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Slivka A, Craft TK, Chaki S, Rink C, Notestine MA, DeVries AC, Parinandi NL, Sen CK. Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke. 2005;36:2258–2264. doi: 10.1161/01.STR.0000181082.70763.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Hori M. Cerebral ischemia in 5-lipoxygenase knockout mice. Brain Res. 2004;1004:198–202. doi: 10.1016/j.brainres.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/S0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71:209–213. doi: 10.1006/exer.2000.0873. [DOI] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Maor-Nof M, Homma N, Raanan C, Nof A, Hirokawa N, Yaron A. Axonal pruning is actively regulated by the microtubule-destabilizing protein kinesin superfamily protein 2A. Cell Rep. 2013;3:971–977. doi: 10.1016/j.celrep.2013.03.005. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, Torborg CL, Feller MB, O'Leary DD. Retinotopic map refinement requires spontaneous retinal waves during a brief critical period of development. Neuron. 2003;40:1147–1160. doi: 10.1016/S0896-6273(03)00790-6. [DOI] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pallast S, Arai K, Wang X, Lo EH, van Leyen K. 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J Neurochem. 2009;111:882–889. doi: 10.1111/j.1471-4159.2009.06379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallast S, Arai K, Pekcec A, Yigitkanli K, Yu Z, Wang X, Lo EH, van Leyen K. Increased nuclear apoptosis-inducing factor after transient focal ischemia: a 12/15-lipoxygenase-dependent organelle damage pathway. J Cereb Blood Flow Metab. 2010;30:1157–1167. doi: 10.1038/jcbfm.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergola C, Werz O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert Opin Ther Pat. 2010;20:355–375. doi: 10.1517/13543771003602012. [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Sengupta Ghosh A, Gogineni A, Hanson JE, Lee SH, Larson JL, Solanoy H, Bustos D, Li H, Ngu H, Jubb AM, Ayalon G, Wu J, Scearce-Levie K, Zhou Q, Weimer RM, Kirkpatrick DS, Lewcock JW. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J Exp Med. 2013;210:2553–2567. doi: 10.1084/jem.20122832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praticò D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VM. 12/15-lipoxygenase is increased in Alzheimer's disease: possible involvement in brain oxidative stress. Am J Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Saarinen J, Kohonen T. Self-organized formation of colour maps in a model cortex. Perception. 1985;14:711–719. doi: 10.1068/p140711. [DOI] [PubMed] [Google Scholar]
- Schoenmann Z, Assa-Kunik E, Tiomny S, Minis A, Haklai-Topper L, Arama E, Yaron A. Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J Neurosci. 2010;30:6375–6386. doi: 10.1523/JNEUROSCI.0922-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Weimer RM, McLaughlin T, Kallop D, Stanger K, Yang J, O'Leary DD, Hannoush RN, Tessier-Lavigne M. A caspase cascade regulating developmental axon degeneration. J Neurosci. 2012;32:17540–17553. doi: 10.1523/JNEUROSCI.3012-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leyen K, Duvoisin RM, Engelhardt H, Wiedmann M. A function for lipoxygenase in programmed organelle degradation. Nature. 1998;395:392–395. doi: 10.1038/26500. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Lo EH. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg PA, Holman TR, Lo EH. Novel lipoxygenase inhibitors as neuroprotective reagents. J Neurosci Res. 2008;86:904–909. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weishaupt JH, Neusch C, Bähr M. Cyclin-dependent kinase 5 (CDK5) and neuronal cell death. Cell Tissue Res. 2003;312:1–8. doi: 10.1007/s00441-003-0703-7. [DOI] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L, Jr, Fuller J, Fu J, Cao LH, Han B, Auld D, Xue T, Hirai S, Germain L, Simard-Bisson C, Blouin R, Nguyen JV, Davis CH, et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A. 2013;110:4045–4050. doi: 10.1073/pnas.1211284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werring DJ, Toosy AT, Clark CA, Parker GJ, Barker GJ, Miller DH, Thompson AJ. Diffusion tensor imaging can detect and quantify corticospinal tract degeneration after stroke. J Neurol Neurosurg Psychiatry. 2000;69:269–272. doi: 10.1136/jnnp.69.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley DC, Ford MG, Livingstone DJ. Unsupervised forward selection: a method for eliminating redundant variables. J Chem Inf Comput Sci. 2000;40:1160–1168. doi: 10.1021/ci000384c. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhang Y, Xiao Y, Ma S, Liu Q, Dang S, Jin M, Shi Y, Wan B, Zhang Y. Inhibition of 12/15-lipoxygenase by baicalein induces microglia PPARbeta/delta: a potential therapeutic role for CNS autoimmune disease. Cell Death Dis. 2013;4:e569. doi: 10.1038/cddis.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincón M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yang J, Weimer RM, Kallop D, Olsen O, Wu Z, Renier N, Uryu K, Tessier-Lavigne M. Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron. 2013;80:1175–1189. doi: 10.1016/j.neuron.2013.08.034. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]