

Abstract
The cerebral innate immune system is able to modulate brain functioning and cognitive processes. During activation of the cerebral innate immune system, inflammatory factors produced by microglia, such as cytokines and adenosine triphosphate (ATP), have been directly linked to modulation of glutamatergic system on one hand and learning and memory functions on the other hand. However, the cellular mechanisms by which microglial activation modulates cognitive processes are still unclear. Here, we used taste memory tasks, highly dependent on glutamatergic transmission in the insular cortex, to investigate the behavioral and cellular impacts of an inflammation restricted to this cortical area in rats. We first show that intrainsular infusion of the endotoxin lipopolysaccharide induces a local inflammation and increases glutamatergic AMPA, but not NMDA, receptor expression at the synaptic level. This cortical inflammation also enhances associative, but not incidental, taste memory through increase of glutamatergic AMPA receptor trafficking. Moreover, we demonstrate that ATP, but not proinflammatory cytokines, is responsible for inflammation-induced enhancement of both associative taste memory and AMPA receptor expression in insular cortex. In conclusion, we propose that inflammation restricted to the insular cortex enhances associative taste memory through a purinergic-dependent increase of glutamatergic AMPA receptor expression at the synapse.
Keywords: AMPA, ATP, cytokines, lipopolysaccharide, neuroinflammation
Introduction
Under pathological conditions, activation of microglia is a common early feature of most brain diseases, which is the primary stage of neuroinflammation, followed by synaptic alterations and subsequent cognitive impairments (Dantzer et al., 2008; Yirmiya and Goshen, 2011). Once activated, microglia release gliotransmitters (Färber and Kettenmann, 2005), which are small molecules, such as proinflammatory cytokines or adenosine triphosphate (ATP), all known to control neuronal function and synaptic transmission (Bessis et al., 2007; Hanisch and Kettenmann, 2007; Pascual et al., 2012; Yirmiya and Goshen, 2011). Proinflammatory cytokines, such as interleukin-1 β (IL-1β) and tumor necrosis factor α (TNF-α), have been directly linked to modulation of neuronal plasticity as well as memory functions in various animals models (Yirmiya and Goshen, 2011).
In normal quiescent conditions, immune mechanisms positively regulate neuroplasticity and promote learning and memory. Conversely, in the condition of neuroinflammation, the delicate physiological balance between immune and neural processes is disrupted, resulting in neuronal hyperexcitability, leading to impairments in neuroplasticity, learning, and memory (Yirmiya and Goshen, 2011). Recent studies suggest that neuroinflammation affects neural plasticity through the modulation of the glutamatergic system, even if the role played by the different mediators in these processes remains poorly understood. Whereas proinflammatory cytokines, such as TNF-α and IL-1β, can modify expression and/or activity of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors (Beattie et al., 2002; Ogoshi et al., 2005; Lai et al., 2006), a recent study reported that the lipopolysaccharide (LPS), a bacterial endotoxin, modifies glutamatergic neurons activity through ATP release and not proinflammatory cytokines (Pascual et al., 2012). Moreover, a clear demonstration that the cognitive alterations induced by neuroinflammation also involve the glutamatergic system is still lacking.
As a result, our study aimed at investigating the glutamatergic mechanisms of neuroinflammation-induced changes in learning and memory processes. For this purpose, we focused on taste memory, which relies mainly on glutamatergic transmission into a particular brain area, the insular cortex, which contains the gustatory cortex (Bermúdez-Rattoni et al., 2004; Gal-Ben-Ari and Rosenblum, 2011; Adaikkan and Rosenblum, 2012). The aim of our study was to investigate the cellular and behavioral impact of an inflammation restricted to the insular cortex on glutamatergic receptor expression and taste memory formation. Here, we activated microglia locally with LPS, a potent inflammatory agent (Cardona et al., 2006; Haynes, 2006; Pascual et al., 2012), and monitored cellular and behavioral consequences. The combination of in vivo pharmacology and biochemistry revealed that cortical inflammation, induced by LPS infusion into the insular cortex, enhances associative taste learning through the increase of glutamatergic AMPA receptors (AMPAR) expression and trafficking at the synapses. This effect was mediated by ATP as ATP infusion mimicked the effect of LPS on aversion and purinergic receptor blockade abolished LPS-induced enhancement of both taste memory and AMPAR expression.
Materials and Methods
Animals
All animal experiments were conducted according to the INRA Quality Reference System and to relevant French (Directive 87/148, Ministère de l'Agriculture et de la Pêche) and international legislation (Directive 86/609, November 24th 1986, European Community). This study was approved by the local ethical committee (agreement 5012047-A), and protocols adhered to those approved by Région Aquitaine Veterinary Services (Direction Départementale de la Protection des Animaux, approval). Every effort was made to minimize suffering and the number of animals used.
All experiments used adult male Wistar rats (300–350 g) obtained from Janvier. They were housed individually in polypropylene cages in a temperature (23 ± 1°C) and humidity (40%) controlled animal room on a 12 h light/dark cycle. All behavioral manipulations were performed during the light phase, approximately between 11:00 A.M. and 1 P.M. Food and water were provided ad libitum until the start of the behavioral procedures.
Drugs
LPS (from Escherichia coli serotype 0127:B8, Sigma-Aldrich) was dissolved in sterile saline (0.9%) to obtain a final concentration of 100 ng/0.5 μl per insular cortex. We ensure that the injected dose was high enough to induce a local cortical inflammation but not too elevated to avoid a generalized inflammatory reaction (see Results). We infused 0.5 μl per hemisphere at the level of the insular cortex the following drugs: ATP (Sigma-Aldrich, 10 mm); TNF-α (R&D Systems Europe, 50 or 100 ng); IL-1β (R&D Systems Europe, 5, 25, 50, or 100 ng), the peptide pepR845A (844KAMKVAKNPQ853) (Anaspec 80 ng/μl or 8 μg/μl); and suramin (Sigma-Aldrich, 20 μg). The doses of LPS, ATP, cytokines, and suramin were based on the preexisting literature and pilot experiments. For all experiments, control animals received an equivolume (0.5 μl per insular cortex) of sterile saline (0.9%).
Surgery and infusion procedure
Adult rats were anesthetized with ketamine (70 mg/kg) and xylazine (6 mg/kg) administered intraperitoneally. They were then implanted with two 5 mm stainless steel guide cannulae (Plastic One) using a standard stereotaxic frame (Kopf Instruments). Guide cannulae were bilaterally implanted above the insular cortex (anteroposterior, 1.2 mm relative to bregma; lateral, ±5.5 mm from midline; ventral, −4.1 mm from bregma). The rostrocaudal position was chosen because conditioned taste aversion (CTA) learning was altered by manipulations in the central but not in the anterior or posterior insular cortex (Nerad et al., 1996). Neurons responding to gustatory stimulations are located in the central insular cortex (Sewards and Sewards, 2001). The tips of the guide cannulae were aimed 2.0 mm above the insular cortex. The guide cannulae were fixed to the skull with dental acrylic cement and anchored with two surgical screws placed in the skull. Stylets were inserted into the guide cannulae to prevent clogging. Rats were allowed to recover from surgery for 1 week.
All animals were handled individually for ∼3 min each day during the last 3 d before infusion for habituation. On the infusion day, rats were gently restrained by hand. Stylets were removed and injection needles (30-gauge) were inserted, extending 2 mm from the tip of the guide cannulae (i.e., reaching the insular cortex). The injection needles were connected via polyethylene tubing to two 10 μl Hamilton microsyringes driven by an automated microinfusion pump (Harvard Apparatus). A total volume of 0.5 μl per insular cortex was delivered over 1 min. After injections, the needles were left in position for an additional minute to allow diffusion of the solution into the tissue and to minimize dragging of the liquid along the injection track.
Proinflammatory and anti-inflammatory cytokines quantification (BioPlex analyses)
Rats were decapitated 30, 60, or 90 min after LPS or saline infusion into the insular cortex and the brain removed. Blood samples from rats were collected in 10% EDTA-coated vials and centrifuged for 15 min at 13,000 rpm at 4°C, aliquoted, and stored at −80°C until use. Brains were frozen on dry ice and stored at −80°C; 300-μm-thick sections were cut through the areas of the insular cortex in the frontal plane using a cryostat (Leica), and bilateral punches of insular cortex from 3 sections were collected in an Eppendorf tube. Insular cortex punches (pool of 3 punches per side per animal) were homogenized in lysis buffer as recommended by the manufacturer (Bio-Rad) and protein concentration was determined by BCA (Uptima, Interchim).
For both blood and tissue samples, the proinflammatory cytokines IL-1β, TNF-α, IL-6, and the anti-inflammatory cytokine IL-10 were measured using a BioPlex cytokine assay kit (Bio-Rad) following the manufacturer's instructions. The limit of detection was of 10 pg/ml for IL-6, 5 pg/ml for IL-10, 2 pg/ml for IL-1β, 3 pg/ml for TNF-α, and 1 pg/ml for IL-4. Briefly, samples diluted 1/4 were added to a 96-well microtiter plate (50 μl/well) coated with beads (Bio-Rad), covered with aluminum foil, and incubated for 30 min on a shaker at room temperature. After removal of the sample using a vacuum manifold, beads were incubated with detection antibodies for 30 min while shaking, followed by streptavidin-PE for 10 min. The beads were then resuspended in assay buffer and analyzed using the BioPlex 200 system (Bio-Rad). The reader was set to read a minimum of 100 beads with an identical fluorescence expressed as the median fluorescence intensity. Median fluorescence intensity readings were converted to pictograms per milliliter using calibration curves prepared with cytokine standards included in the kit. Results are expressed as relative fold change of protein expression compared with the “saline 30 min” group.
ATP assay
Rats were killed by decapitation 30 min after taste consumption, and the brains removed. For each rat, insular cortices from both sides were dissected out from fresh brains. The crossing of the rhinal fissure with the medial cerebral artery was used as an anatomical landmark, and cortical tissue 1.0 mm rostral, 0.5 mm caudal, and 1.5 mm dorsal to it was excised. The tissues were then homogenized in 200 μl TCA 10% (Sigma-Aldrich) on ice; 20 μl of the solution was diluted in 980 μl D'PBS (Dulbecco's Phosphate Buffered Saline). ATP assay was conducted on 100 μl of the mix (triplicates) according to the manufacturer's protocol (PerkinElmer). Protein contents were then determined in parallel by Bio-Rad protein assay according to the manufacturer's protocol (Bio-Rad) to check for the homogeneity of brain samples size. The data from ATPlite assays are presented as an average of more than three independent experiments. Results are expressed as the average of triplicates in μm.
Total corticosterone measurement
On the day of death, blood samples were collected in tubes containing 10% EDTA. After 15 min of centrifugation at 13,000 rpm, plasma samples were obtained and kept at −80°C. Total corticosterone was measured by an in-house RIA (for details, see Richard et al., 2010). Briefly, after steroid extraction of plasma samples with absolute ethanol, total corticosterone was measured by competition between cold corticosterone (B) and 3H-B by a specific antibody anti-corticosterone provided by Dr. H. Vaudry (University of Rouen, Rouen, France).
Western blot analyses and synaptoneurosomes preparation
Protein extraction.
Rats were killed by decapitation and the brain removed. Brains were frozen on dry ice and stored at −80°C until use. As described above, 300-μm-thick sections were cut through the areas of the insular cortex in the frontal plane with a cryostat (Leica), and bilateral punches of insular cortex from three sections were collected in an Eppendorf tube. The punches collected were then homogenized in 60 μl of buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCL, 1 mm ditriothreitol, 1 mm Na orthovanadate, protease inhibitors mixture; 10 mm Na fluoride). Homogenates were centrifuged 5 min at 3000 rpm to remove nuclei. Supernatants were stored at −80°C until use. Protein contents were determined by Bio-Rad protein assay according to the manufacturer's protocol (Bio-Rad) and heated to 100°C for 5 min in Laemmli sample buffer (2% SDS and 5% dithiothreitol) to entirely denature the proteins.
Synaptoneurosome preparation.
Rats were killed by decapitation 30 min after taste consumption and the brain removed. For each rat, insular cortices from both sides were dissected out from fresh brains as described above. Two animals of the same experimental group (4 insular cortices in total) were pooled for n = 1. The tissue was then homogenized in a glass-Teflon homogenizer in 500 μl of buffer containing 0.3 m sucrose, 20 mm HEPES, 5 mm EDTA, 1× protease inhibitor mixture, 1 mm Na orthovanadate, and 1 mm Na fluoride. The samples were centrifuged for 7 min at 4000 rpm at 4°C to remove nuclei. The supernatants were collected and centrifuged for 15 min at 10,000 × g. The pellets collected were centrifuged again for 15 min at 10,000 × g. Supernatant was then lysed with a glass-Teflon poter in 500 μl of buffer containing protease inhibitor mixture 1×, 0.1 mm Na orthovanadate, 0.1 mm Na fluoride in cold sterile water. Samples were kept for 30 min at 4°C and then centrifuged at 26,000 rpm for 20 min at 4°C. Pellets were resuspended in 100 μl of a buffer containing 0.25 m sucrose, 4 mm HEPES, 5 mm EDTA, 1× protease inhibitor mixture, 0.1 mm Na orthovanadate, 0.1 mm Na fluoride, and centrifuged on a sucrose gradient containing 0.8, 1, and 1.2 m of sucrose for 2 h at 31,000 rpm at 4°C. The 1–1.2 m of sucrose interface was removed and centrifuged for 30 min at 62,000 rpm at 4°C. The final pellets were resuspended in 70 μl of the original buffer. Protein contents were determined by Bio-Rad protein assay according to the manufacturer's protocol (Bio-Rad) and heated at 100°C for 5 min in Laemmli sample buffer (2% SDS and 5% dithiothreitol) to entirely denature the proteins.
Western blotting.
Equal quantities of proteins (10 μg/lane) were electrophoresed onto an 8% polyacrylamide gel with a 5% stacking gel. Proteins were blotted on PVDF membranes (Immobilon Millipore). Membranes were saturated by incubation with 5% milk in Tris-buffered saline-Tween 0.1% (TBS-Tween; Tris-HCl pH 7.5; 100 mm NaCl, 0.1% Tween 20) for 1 h and incubated overnight with the primary antibody. After 3 washes in TBS-Tween, membranes were incubated with the secondary antibody coupled to HRP (Southern Biotechnology Associates) diluted in TBS-Tween supplemented with 3% milk, for 2 h at room temperature. Membranes were then washed in TBS-Tween twice for 5 min, and finally in TBS alone. The complex was detected with an ECL kit (ElectroChemoLuminescence; GE Healthcare). Optical density capture of the obtained signal was performed with the Syngene Chemigenius2 apparatus (Synoptics), and intensity of the signal was quantified with the GeneTools software (Synoptics).
Behavioral procedures
The effect of LPS infusion in the insular cortex was investigated on incidental and associative taste memory.
Incidental taste memory was evaluated by presenting an unfamiliar sweet taste and then repeatedly presenting the same taste. Over time, the consumption increases serving as a measure of taste familiarity (Miranda et al., 2008). Rats were deprived of water for 24 h and then habituated to drink water from a single graded bottle (with 0.5 ml accuracy) for 15 min per day for 5 d (between 11:00 A.M. and 1 P.M.) in the home cage. On the first day of taste presentation (i.e., acquisition day), rats received a novel sweet taste composed of 0.1% saccharin solution (Sigma-Aldrich) for 15 min. For the next 2 d, rats had access to water for 15 min. After these days, taste consumption was assessed by providing saccharin solution for 15 min during 3 d. The percentage of saccharin consumption with respect to consumption during water baseline (the last 3 d before acquisition) was presented for each saccharin presentation.
Associative taste memory was evaluated using CTA. CTA consists of the association of new taste consumption with a gastric malaise. The procedure was identical to the one described above with the exception of 30 min after the first presentation of saccharin 0.1%, rats received an intraperitoneal injection of a moderate or a low dose of the visceral malaise-inducing drug lithium chloride (LiCl, Sigma-Aldrich, 0.15 m or 0.075 m, 1% of body weight). For the next 2 d, rats had access to water for 15 min to reestablish water baseline intake. After these days, taste avoidance was assessed by providing saccharin solution for 15 min, immediately followed by 15 min of water (to verify treatment did not alter total liquid ingestion). Rats underwent two other days of tests similar to the first test to evaluate extinction of the aversion. The percentage of saccharin consumption during acquisition and testing with respect to consumption during water baseline (the last 3 d before acquisition) was used as a measure of neophobia (for acquisition) and aversion strength (for testing).
Histology.
After completion of behavioral experiments, animals were deeply anesthetized with an injection of 1 ml of sodium pentobarbital (100 mg/kg i.p.), and transcardially perfused with a saline solution (0.9%). The brains were removed and stored at 4°C in a 4% PFA solution for 24 h. They were then soaked in 30% sucrose solution; 40-μm-thick sections were cut through the areas of interest in the frontal plane with a cryostat (Leica) and kept at −20°C in a cryoprotective solution. Sections were stained with cresyl violet and then examined under a microscope to verify the correct placement of the cannulae. Data from 21 rats (in addition to the 246 operated rats included in the different groups) were discarded from the final statistical analyses because of misplacement of one or both cannulae.
Statistical analyses
All data are represented as mean ± SEM. Cytokines expression and Western blot raw data, were analyzed by a two-way ANOVA (time × treatment) followed by post hoc Fisher LSD test when appropriate. For the phosphorylation level of GluN2B, a one-sample t test with comparison to 1 was used because data were expressed relative to the control group. For the synaptoneurosomal experiments and ATP assay, differences between groups were analyzed by unpaired Student's t test. Concerning the behavioral experiments, differences of saccharin consumption between groups for the acquisition day were analyzed by unpaired Student's t test, one- or two-way ANOVA and during the 3 days of tests by two- or three-way ANOVA on repeated measures (time × treatment or time × treatment × pretreatment). Differences of saccharin consumption between acquisition and test were analyzed using a paired Student's t test. Statistical significance was defined for p values <0.05.
Results
Intrainsular LPS injection induces a local synthesis of proinflammatory cytokines and ATP
We first characterized the nature and the extent of inflammatory processes induced by LPS infusion in the insular cortex. Local and circulating levels of proinflammatory cytokines were evaluated 30 min, 1 h or 1h30 after bilateral infusion of either LPS (100 ng/0.5 μl/side) or saline (0.5 μl/side) in the structure (Fig. 1A). The protein concentration of the proinflammatory cytokines IL-1β and TNF-α was significantly increased after LPS infusion in a time-dependent manner (two-way ANOVA, LPS effect: F(1,29) = 16.08 and 23.1, respectively, p < 0.001; time effect: F(2,29) = 25.18 and 30.05, respectively, p < 0.001; interaction: F(2,29) = 6.48 and 5.58, respectively, p < 0.01). Post hoc analysis revealed that this effect was significant at 1h30 after LPS infusion for both IL-1β and TNF-α (p < 0.001; Fig. 1B,C). IL-6 also increased after LPS infusion (LPS effect: F(1,29) = 3.86, p = 0.05; time effect: F(2,29) = 6.08, p < 0.01; LPS × time interaction: F(2,29) = 2.26, p = 0.1228; post hoc analysis: p < 0.05 at 1h30 after LPS infusion) (data not shown). Moreover, we could not find any significant differences in the protein expression of the anti-inflammatory cytokines IL-10 and IL-4 (IL-10: LPS effect: F(1,29) = 2.23, p = 0.15; time effect: F(2,29) = 0.13, p = 0.88; interaction F(2,29) = 0.42, p = 0.66; Figure 1D; IL-4: LPS effect: F(1,29) = 1.33, p = 0.26, time effect: F(2,34) = 1.44, p = 0.26, interaction: F(2,34) = 0.35, p = 0.71, data not shown). Because a recent study reported that LPS increases brain ATP release in vitro (Pascual et al., 2012), we also quantified the levels of ATP 1 h after LPS infusion into the insular cortex. We found a significant increase of ATP concentration (unpaired t test, t(9) = 2.88, p = 0.018).
Figure 1.

LPS induces a localized inflammatory reaction. A, Schematic representation of the experimental design. Proinflammatory and anti-inflammatory cytokine expression was measured by Bioplex in insular cortices or blood samples, 30 min (saline: n = 5; LPS: n = 7), 1 h (saline: n = 5; LPS: n = 6), or 1h30 (saline: n = 5; LPS: n = 7) after saline or LPS infusion. B–D, Tissue concentrations (relative fold change) of the proinflammatory cytokines IL-1β (B), TNF-α (C), and IL-10 (D). E, Tissue concentrations of ATP (in μm). F–H, Blood concentrations (relative fold change) of the proinflammatory cytokines IL-1β (F), TNF-α (G), and IL-10 (H). I, Effect of intrainsular infusion of either vehicle (n = 8) or LPS (n = 8) on body weight 2 d before and 2 d after conditioned taste aversion acquisition. J, Effect of intrainsular infusion of either vehicle (n = 12) or LPS (n = 13) on corticosterone concentration in plasma 2h15 after LPS infusion. *p < 0.05. ***p < 0.001.
To evaluate the extent of the inflammatory reaction induced by brain infusion of LPS, cytokine expression was also measured in blood samples of the same animals. Local LPS infusion in the insular cortex did not induce any significant modification of circulating levels of proand anti-inflammatory cytokines (IL-1β, TNF-α, IL-10: Fig. 1F–H; IL-4 and IL-6: data not shown) (IL-1β: two-way ANOVA, LPS effect: F(1,27) = 0.30, p = 0.58; time effect: F(2,27) = 0.29, p = 0.75; interaction: F(2,27) = 1.2, p = 0.31; TNF-α: two-way ANOVA, LPS effect: F(1,26) = 0.34, p = 0.56; time effect: F(2,26) = 0.13, p = 0.87; interaction: F(2,26) = 0.84, p = 0.44; IL-10: two-way ANOVA, LPS effect: F(1,27) = 3.01, p = 0.09; time effect: F(2,27) = 0.49, p = 0.62; interaction: F(2,27) = 2.21 p = 0.13; IL-4: two-way ANOVA, LPS effect: F(1,28) = 0.10, p = 0.74; time effect: F(2,28) = 1.01, p = 0.38; interaction: F(2,28) = 1.36, p = 0.27; IL-6: two-way ANOVA, LPS effect: F(1,14) = 0.22, p = 0.78; time effect: F(2,14) = 0.26, p = 0.78; interaction: F(2,14) = 1.04, p = 0.38).
It has been extensively described that activation of the peripheral innate immune system also leads to an increase of glucocorticoid hormones release, the final product of the stress-related hypothalamic-pituitary-adrenal (HPA) system, as well as to a decrease of food intake and body weight (Dantzer et al., 2008). Thus, to confirm that intrainsular LPS infusion was a model of localized inflammatory reaction, we measured corticosterone levels in blood samples as well as the weight of the animals and did not find any difference between groups (corticosterone levels: unpaired t test, t(23) < 1, p = 0.9; Fig. 1I; body weight: t(17) = 1.15, p = 0.3; Fig. 1J). Overall, our data show that the infusion of LPS into the insular cortex induced a local inflammation with no activation of the peripheral immune system.
Local inflammatory reaction increases glutamatergic AMPAR expression in the insular cortex
Glutamatergic activation in the insular cortex plays an important role in taste memory formation (for review, see Bermúdez-Rattoni et al., 2004; Gal-Ben-Ari and Rosenblum, 2011; Adaikkan and Rosenblum, 2012). Consumption of a novel taste leads to hyperphosphorylation of the GluN2B subunits of NMDA receptors (NMDAR) in insular neurons (Barki-Harrington et al., 2009), and blockade of either NMDA or AMPAR in the insular cortex before novel taste exposure greatly impairs taste memory (Berman et al., 2000; Ferreira et al., 2002; Barki-Harrington et al., 2009). Moreover, some inflammatory factors are able to modulate learning and memory processes as well as glutamatergic receptor expression and activity (Yirmiya and Goshen, 2011). Consequently, we evaluated whether LPS infusion in the insular cortex modulated locally the expression and activity of the glutamatergic ionotropic NMDA and AMPAR and whether it was influenced by taste novelty.
For this purpose, animals were infused in the insular cortex with either LPS or saline 1h30 before presentation of a novel taste (saccharin) or a familiar taste (water) for 15 min. Consumption of saccharin or water did not differ between LPS- and saline-treated animals, ruling out an effect of local inflammation on drinking behavior (two-way ANOVA, no effect of LPS (F(1,17) = 1.3, p = 0.3), saccharin (F(1,17) = 3.2, p = 0.09), and interaction (F(1,17) < 1, p = 0.7); saline group: water: 20 ± 1.3 ml, saccharin: 17 ± 1.3 ml; LPS group: water: 18 ± 1.2 ml, saccharin 16 ± 1 ml). We applied Western blotting to total protein samples extracted from the insular cortex 30 min after liquid consumption (i.e., 2h15 after infusion) (Fig. 2A). First, we measured glutamatergic NMDA subunit expression. We did not find any difference in the GluN1, GluN2A, or GluN2B subunit expression in the insular cortex between the four groups (no effect of LPS, saccharin, and interaction: F(1,17) < 1; Fig. 2B, for GluN2B), indicating that consumption of a novel taste or LPS infusion did not affect the total number of glutamatergic NMDAR in the insular cortex. Exposure to a novel taste was reported to enhance phosphorylation of the GluN2B subunits in insular neurons. Therefore, we measured the GluN2B phosphorylation level to evaluate whether LPS could influence this effect. As expected, we found an increase of GluN2B phosphorylation in the insular cortex exposed to the novel taste saccharin in saline-treated rats (one-sample t test, comparison to 1, t(3) = 3.1, p < 0.05; Fig. 2C). Interestingly, saccharin intake also increased GluN2B phosphorylation in LPS-treated group (t(4) = 5.4, p < 0.01). However, there was no significant effect of LPS injection alone on GluN2B phosphorylation for rats exposed to the familiar taste (t(5) = 2.1, p = 0.09; Fig. 2C).
Figure 2.
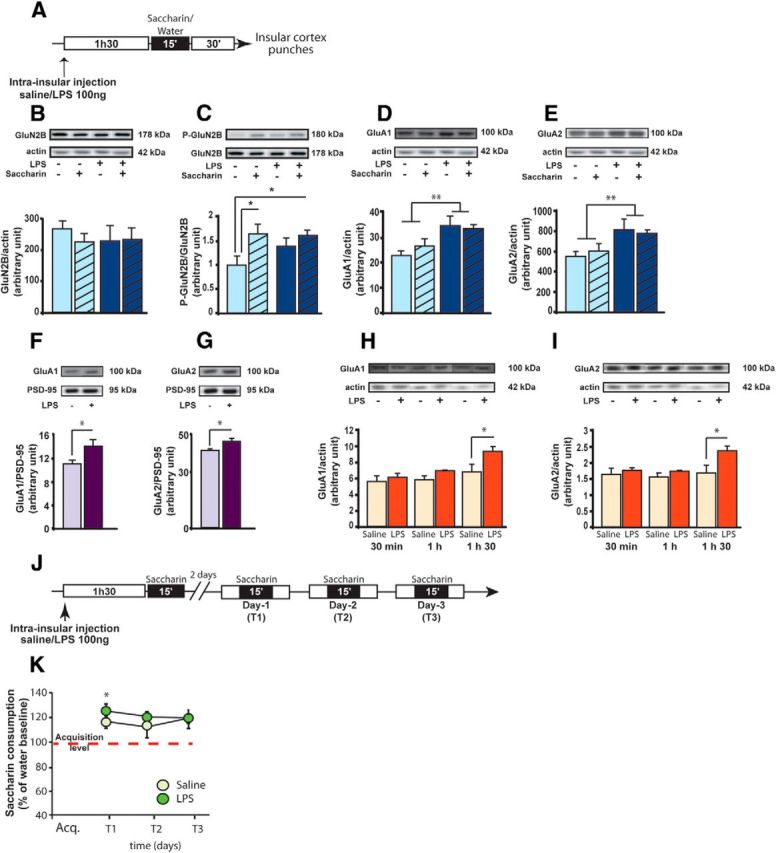
LPS modulates glutamatergic receptors expression in the insular cortex with no effect on incidental taste memory. A, Schematic representation of the experimental design. Rats were infused 1h30 before a novel (saccharin) or a familiar (water) taste consumption and killed 30 min later. After decapitation, insular cortices were punched and protein expression measured by Western blot in total or synaptoneurosomal fractions. B–E, Expression levels of GluN2B (B), phospho-GluN2B (C), GluA1 (D), and GluA2 (E) in total extracts. Familiar taste-saline: n = 5; familiar taste-LPS: n = 6; novel taste-saline: n = 5; novel taste-LPS: n = 5. F, G, Expression levels of GluA1 (F) and GluA2 (G) in synaptoneurosomal fractions (saline: n = 5; LPS: n = 4). H, I, Kinetics of expression of GluA1 and GluA2 30 min (saline: n = 5; LPS: n = 4), 1h (saline: n = 5; LPS: n = 5), and 1h30 (saline: n = 4; LPS: n = 6) after LPS infusion in the insular cortex. J, Schematic representation of the experimental design for incidental taste memory evaluation. Two groups of rats were infused either with saline (n = 7) or LPS (n = 8) in the insular cortex 1h30 before the first saccharin presentation (acquisition). K, Saccharin consumption measured daily over the 3 d of test as a percentage of water intake at baseline. Saccharin consumption is significantly increased in both saline- and LPS-treated animals at T1 compared with acquisition day. *p < 0.05. **p < 0.01.
We then measured the impact of LPS infusion on AMPAR expression in the insular cortex. We found an increase of GluA1 and GluA2 subunits expression in the insular cortex of LPS-treated animals, independently on the taste presented (LPS effect: F(1,17) = 10.8, F(1,17) = 8.4, p < 0.01 for GluA1 and GluA2, respectively; no saccharin effect and interaction: F(1,17) < 1; Fig. 2D,E). Our results show that this effect was specific to the insular cortex as we did not find any difference in NMDA or AMPAR expression at the level of the somatosensory cortex, which is in close proximity to the insular cortex (no effect of LPS, saccharin and interaction: F(1,17) < 1; data not shown). Overall, our data show that intrainsular infusion of LPS induced site-specific increase of AMPAR expression without affecting NMDAR.
The Western blot experiments presented above were performed on the total contents of insular cortex extracts. To determine whether LPS effect on AMPAR expression was found at the synapses, a new cohort of animals was infused with either LPS or saline 1h30 before saccharin consumption; and 30 min later, the insular cortex was dissected and the synaptoneurosomal fraction was extracted. On this material, we demonstrated that expression of GluA1 and GluA2 subunits was significantly increased at the synaptic level (unpaired t test, t(7) = 2.5, t(6) = 2.6, p < 0.05 for GluA1 and GluA2, respectively; Fig. 2F,G) without changing the PSD-95 expression (t(6) < 1, saline: 19,073 ± 1687; LPS: 21,018 ± 1922).
We finally determined the kinetics of the LPS-induced increase of AMPAR in the insular cortex. We measured GluA1 and GluA2 AMPAR subunits expression at 30, 60, or 90 min after LPS infusion on total insular cortex extract. For both AMPAR subunits, we found a treatment and time effect (for GluA1: LPS effect: F(1,24) = 8.9, p < 0.01, time effect: F(1,24) = 7.9, p < 0.01, no interaction: F(2,24) = 1.65, p = 0.21; for GluA2: LPS effect: F(1,23) = 6.9, p < 0.05; time effect: F(1,23) = 3.7, p < 0.05; no interaction: F(1,24) = 2.1, p = 0.14). The increase of GluA1 and GluA2 protein expression was significant at 1h30 after LPS infusion (t(8) = 2.5, p < 0.05 for both GluA1 and GluA2; Fig. 2H,I). This indicates that AMPAR expression starts to increase 1h30 min after LPS infusion and is still increased 2h15 after infusion, at the synaptic level (Fig. 2F,G). According to the important role played by AMPAR in the insular cortex for taste memory formation (Berman et al., 2000), LPS-induced increase of glutamatergic AMPAR expression at the insular synapses may affect taste learning.
Cortical inflammation enhances associative taste memory through GluA2 AMPAR trafficking
We first evaluated whether LPS infusion into the insular cortex before novel taste consumption was able to modulate incidental taste memory (i.e., increase consumption of a sweet taste after repeated exposures) (Miranda et al., 2008) (Fig. 2J). As expected, repeated presentations led to an increase in the consumption of saccharin in control animals, which was also present in LPS-infused rats (LPS effect: F(1,39) < 1; test effect F(3,39) = 8.2, p < 0.001; LPS × test interaction: F(3,39) < 1; Figure 2J). For both groups, the increased consumption of saccharin was evident between the first and the second saccharin presentations (paired t test: saline group: t(6) = 3.1, p = 0.02; LPS group: t(7) = 3.7, p = 0.008; Figure 2K), indicating that LPS infusion did not affect incidental taste memory. This result also excluded an aversive effect of intracortical LPS infusion that could induce taste aversion on subsequent taste presentation, as previously reported after peripheral LPS injection.
Associative taste learning, like the aversion induced by taste-malaise pairing, could rely more heavily on glutamatergic transmission in insular cortex than incidental taste learning as gastric malaise induces an important release of glutamate in the insular cortex, whereas novel taste alone does not (Miranda et al., 2002; Barki-Harrington et al., 2009). We thus evaluated whether intracortical LPS infusion could affect CTA. A bilateral infusion of either LPS or saline was performed within the insular cortex 90 min before CTA acquisition (Fig. 3A). Saccharin intake was not affected by intracortical LPS infusion (saline group: 12.4 ± 0.5 ml; LPS group: 12.7 ± 0.7 ml; t(17) < 1, p = 0.7). Over the 2 d following CTA acquisition, both groups showed water consumption similar to baseline conditions (mean for saline group: 13.2 ± 0.5 ml; mean for LPS group: 13.3 ± 0.8 ml; t(17) < 1, p = 0.8). On the first day of test, LPS-treated animals showed a lower consumption of saccharin than control animals, indicating a higher aversion after LPS infusion (T1; Fig. 3B, left). Figure 3B (right) shows that this increase of aversion strength in LPS-treated animals lasted over the 3 d of extinction tests (two-way ANOVA on repeated measures: LPS effect: F(1,34) = 4.7, p = 0.04; time effect: F(2,34) = 168.2, p < 0.0001; no interaction F(2,34) = 1.4, p = 0.2).
Figure 3.
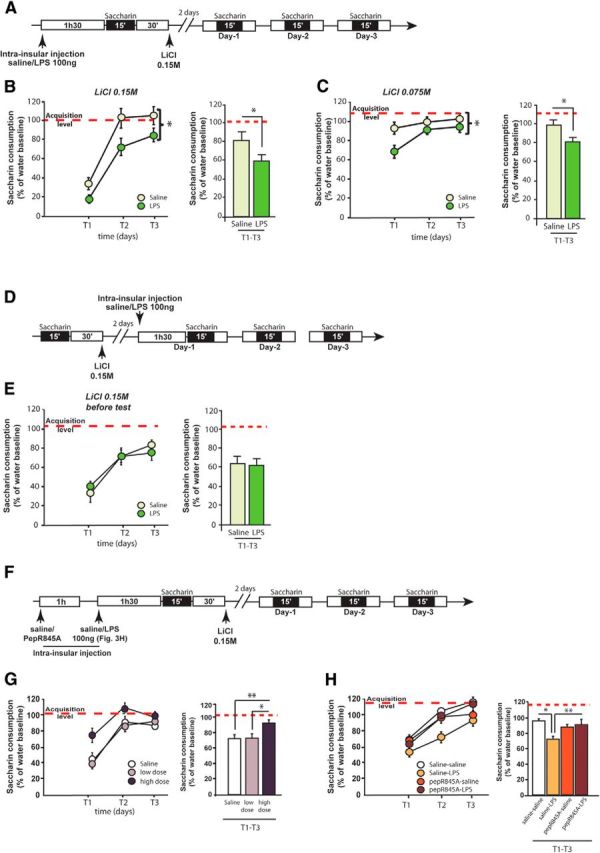
LPS enhances associative taste memory. A, Schematic representation of the experimental design. B, Effect of LPS infusion in the insular cortex on conditioned taste aversion acquisition induced by a moderate dose of LiCl (0.15 m). Left, Kinetics of saccharin consumption over the 3 d of test (T1–T3). Two groups of rats were infused with either saline (n = 9) or LPS (n = 10) in the insular cortex before conditioned taste aversion acquisition. *(apposed to bracket), LPS increased aversion strength over the 3 d of extinction tests (significant LPS effect and no interaction with time). Right, Averaged saccharin consumption of rats over the 3 d of test. Red dotted line indicates the level of saccharin consumption on the acquisition day. C, Effect of LPS infusion in the insular cortex on conditioned taste aversion acquisition induced by a low dose of LiCl (0.075 m). Left, Kinetics of saccharin consumption over the 3 d of test (T1–T3). Two groups of rats were infused either with saline (n = 9) or LPS (n = 8) in the insular cortex before conditioned taste aversion acquisition. *(apposed to bracket), LPS increased aversion strength over the 3 d of extinction tests (significant LPS effect and no interaction with time). Right, Averaged saccharin consumption of rats over the 3 d of test. Red dotted line indicates the level of saccharin consumption on the acquisition day. D, Schematic representation of the experimental design. E, Effect of intrainsular LPS infusion on retrieval of conditioned taste aversion. Three days after pairing of saccharin with a moderate dose of LiCl (0.15 m), LPS was infused 1h30 before exposure to saccharin on the first retrieval test. Left, Kinetics of saccharin consumption over the 3 d of test (T1-T3). Two groups of rats were infused with either saline (n = 9) or LPS (n = 8) in the insular cortex. F, Schematic representation of the experimental design. G, Effect of GluA2 AMPAR trafficking blocking peptide (pepR845A) on conditioned taste aversion induced by a moderate dose of LiCl (0.15 m). Left, Kinetics of saccharin consumption over the 3 d of extinction test (T1-T3). Three groups of rats were infused with saline (n = 9), low dose of peptide (80 ng/μl, n = 8), or high dose of peptide (8 μg/μl, n = 9) in the insular cortex before conditioned taste aversion acquisition. Right, Averaged saccharin consumption of rats over the 3 d of test. Red dotted line indicates the level of saccharin consumption on the acquisition day. H, Effect of pepR845A on LPS-induced conditioned taste aversion enhancement. Left, Kinetics of saccharin consumption over the 3 d of extinction test (T1-T3). Four groups of rats were infused with saline-saline (n = 5), saline-LPS (n = 5), pepR845A-saline (n = 6), or pepR845A-LPS (n = 7) in the insular cortex before conditioned taste aversion acquisition. Right, Averaged saccharin consumption of rats over the 3 d of test. Red dotted line indicates the level of saccharin consumption on the acquisition day. *p < 0.05. **p < 0.01.
To assess the robustness of this behavioral effect, we used the same paradigm as before, except that the dose of LiCl was halved to induce a lower aversion (0.075 m instead of 0.15 m). As described above, LPS infusion enhanced associative taste memory, as revealed by a significantly higher aversion on the first test (Fig. 3C, left) in LPS-infused animals compared with controls ones. Importantly, this effect was enduring because it lasted over the 3 d of extinction tests (Fig. 3C, right) (two-way ANOVA on repeated measure, treatment effect: F(1,30) = 6.4, p = 0.02; time effect: F(2,30) = 9.5, p = 0.006; no interaction between factors F(2,30) = 2.3, p = 0.11). Overall, our data show that LPS infusion into the insular cortex before the first taste presentation enhances associative, but not incidental, taste memory. This did not rely on aversive effect of LPS per se. These results also suggest that the dose of LPS we used (100 ng) was not neurotoxic as it is known that insular cortex lesion induces CTA memory impairment but not enhancement (Nerad et al., 1996; Stehberg et al., 2011).
Our results also show that the effects of LPS were specific to the acquisition phase because the infusion of LPS 1h30 before the first day of test did not impact the expression of the taste aversion (Fig. 3D; two-way ANOVA on repeated measure, treatment effect: F(1,17) = 0.02, p = 0.90; time effect: F(2,34) = 49.58, p < 0.0001; no interaction between factors F(2,34) = 0.76, p = 0.47). Thus, LPS exerts its effects specifically on the acquisition of CTA.
Because LPS infusion in the insular cortex enhanced both associative taste memory and AMPAR expression at the synapses, we assessed whether blockade of AMPAR trafficking was able to reverse the LPS-induced CTA enhancement. We therefore used the PepR845A interfering peptide previously described as a specific blocker of the GluA2 subunit trafficking (Joels and Lamprecht, 2010; Danielson et al., 2012; Wang et al., 2013; Migues et al., 2014) and infused this peptide in the insular cortex 1 h before LPS infusion to optimally block the effects of LPS on GluA2 subunit trafficking (Fig. 3F). We first looked at the involvement of the GluA2 trafficking in the CTA acquisition and thus infused rats with a low dose (80 ng/μl) or a high dose (8 μg/μl) of the PepR845A (Joels and Lamprecht, 2010; Danielson et al., 2012; Migues et al., 2014), without LPS infusion. We infused animals 150 min before taste-malaise association (i.e., 60 min earlier than previously used for LPS injection). On the first day of test, only animals treated with the high dose of PepR845A showed a higher consumption of saccharin compared with controls, indicating a decrease of aversion (T1; Fig. 3G, left). This decrease of aversion strength in high dose-treated animals lasted over the 3 d of extinction tests (one-way ANOVA on repeated measure, treatment effect: F(2,23) = 4.98, p < 0.05; time effect: F(2,46) = 64.03, p < 0.0001; no interaction F(4,46) = 2.14, p = 0.09; Figure 3G).
Then, we determined whether the blockade of GluA2 subunit trafficking could restore normal CTA after LPS infusion. The low dose of PepR845A, which had no effect by itself on CTA, was infused in the insular cortex 1 h before LPS (or saline; Fig. 3F). We found an interaction between LPS and peptide treatment (F(1,18) = 6.1, p < 0.05) with a time effect indicating that all groups extinguished (F(2,36) = 47.2, p < 0.0001). Post hoc analysis first revealed that we were able to replicate the enhancing effects of LPS infusion on CTA memory (p < 0.05; Fig. 3H). Interestingly, pretreatment with pepR845A abolished these effects of LPS on aversion strength (p < 0.01). Together, these results indicate that the blockade of GluA2 subunit trafficking was able to inhibit the LPS-induced CTA enhancement.
Purinergic receptors activation, but not the proinflammatory cytokines IL-1β and TNF-α, mediate the effect of LPS on GluA2 AMPAR expression and CTA acquisition
The cellular localization of the LPS receptor Toll-Like Receptor (TLR4) in the brain has been reported to be confined to glial cells, ruling out a direct action of LPS on neurons (Chakravarty and Herkenham, 2005). Proinflammatory cytokines are good candidates as intermediaries of action of LPS on neuronal cells because they have been described to modulate glutamatergic receptors expression in vitro (Beattie et al., 2002; Viviani et al., 2003; Ogoshi et al., 2005; Lai et al., 2006; Yirmiya and Goshen, 2011). We thus determined whether infusion of the proinflammatory cytokines TNF-α and IL-1β was able to replicate the effect of LPS (i.e., to enhance associative taste memory). Based on the literature and pilot experiments, we injected TNF-α at a dose of 100 ng/0.5 μl/side, 90 min before CTA acquisition (Fig. 4A). Intrainsular infusion of TNF-α did not significantly modify aversion strength compared with control animals on the 3 d of extinction tests (treatment effect: F(1,34) = 2.2, p = 0.15, time effect: F(2,34) = 28.9, p < 0.0001, interaction: F(2,34) < 1 Figure 4B). The literature on intracerebral infusion of IL-1β being scattered, we injected IL-1β at different doses comprised in the full range of what is generally used in vivo (i.e., 5, 25, 50, and 100 ng/0.5 μl/side). Cortical IL-1β infusion did not affect the CTA memory, even at the highest dose, because no difference in saccharin intake was found over the 3 d of test (For 100 ng: treatment effect: F(1,38) = 1.9, p = 0.2; time effect: F(1,38) = 29.4, p < 0.0001, interaction: F(1,38) < 1; Figure 4B; data not shown for 5, 25, and 50 ng).
Figure 4.
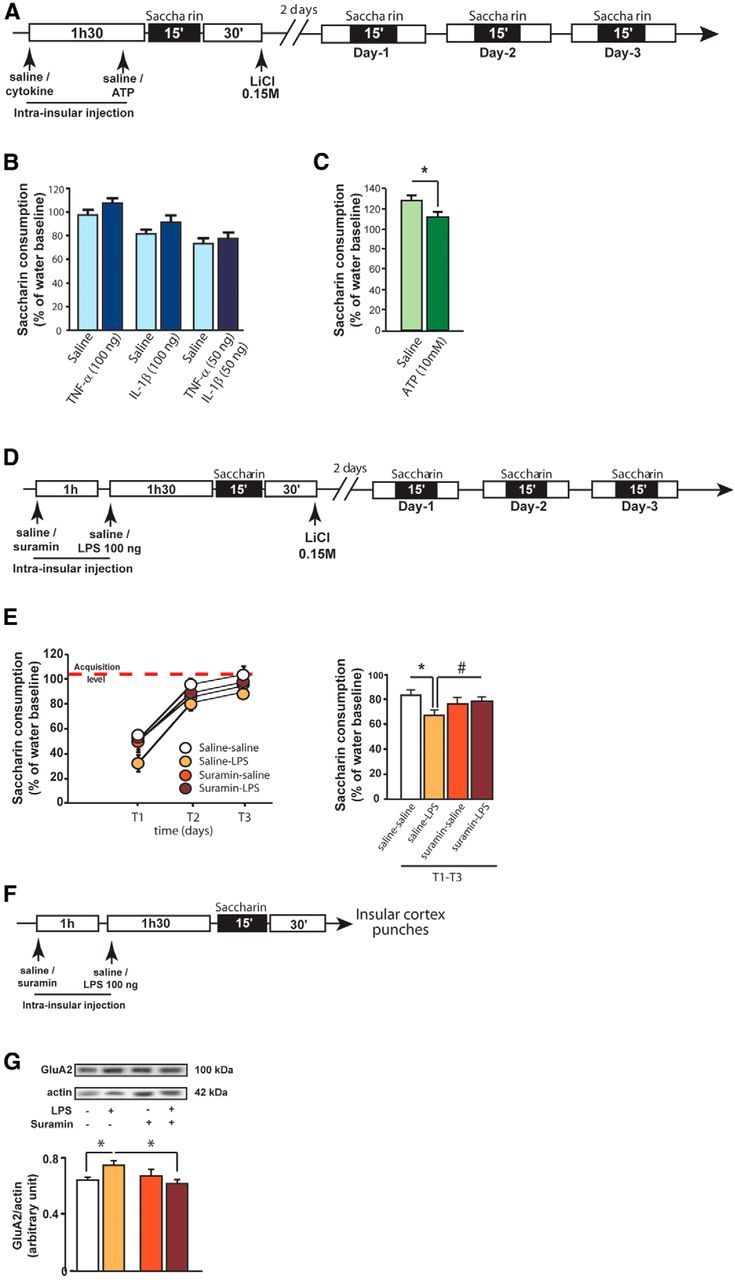
ATP, but not proinflammatory cytokines, is the main inflammatory mediator of LPS-induced associative taste memory enhancement. A, Schematic representation of the experimental design. B, Averaged saccharin consumption of rats over the 3 d of test after infusion of TNF-α (100 ng), IL-1β (100 ng), or a mix of TNF-α and IL-1β (50 ng each) in the insular cortex. C, Averaged saccharin consumption of rats over the 3 d of test after infusion of ATP (10 mm) in the insular cortex. D, Schematic representation of the experimental design. Conditioned taste aversion acquisition in rats infused either with saline or suramin (20 μg/0.5 μl/side) in the insular cortex as a pretreatment, followed by infusion of either saline or LPS (100 ng/0.5 μl/side) as a treatment (saline-saline: n = 7; saline-LPS: n = 7; suramin-saline: n = 6; suramin-LPS: n = 7). E, Left, Kinetics of saccharin consumption over the 3 d of extinction test (T1-T3). Right, Averaged saccharin consumption of rats over the 3 d of test. Red dotted line indicates the level of saccharin consumption on the acquisition day. F, Schematic representation of the experimental design. GluA2 AMPAR expression levels in punched insular cortices of rats infused either with saline or suramin (20 μg/0.5 μl/side) in the insular cortex as a pretreatment, followed by infusion of either saline or LPS (100 ng/0.5 μl/side) as a treatment (saline-saline: n = 4; saline-LPS: n = 5; suramin-saline: n = 4; suramin-LPS: n = 6). G, Expression levels of GluA2 in total fractions. *p < 0.05. #p = 0.06.
Some works have reported that the action of proinflammatory cytokines is potentiated when they are infused in combination (Brebner et al., 2000). We thus infused a mix of TNF-α and IL-1β (50 ng each, as described previously, Brebner et al., 2000) 90 min before taste-malaise pairing, but it had no effect on CTA (treatment effect: F(1,40) < 1; time effect: F(1,40) = 66.9, p < 0.0001, interaction: F(2,40) < 1; Figure 4B). Together, our data showed that TNF-α and IL-1β, alone or in combination, were unable to modulate associative taste memory, suggesting that LPS acts via different actors than proinflammatory cytokines to exert its effect.
ATP is an important and ubiquitous inflammatory factor involved in neuroinflammatory processes (Perregaux and Gabel, 1994; Ferrari et al., 1997; Mehta et al., 2001). Interestingly, a recent study reported that LPS modulates activity in glutamatergic neurons via ATP release by microglia and not proinflammatory cytokines (Pascual et al., 2012). The present study shows that the concentration of ATP was significantly increased 1 h after LPS infusion (Fig. 1E). We thus investigated whether ATP was necessary for the LPS effect on associative taste memory. Because ATP is a very labile molecule that acts rapidly on cells, ATP was infused 15 min before exposure to saccharin (i.e., 60 min before malaise induction) at a concentration of 10 mm based on the literature (Davalos et al., 2005) and pilot experiments. We found that saccharin consumption was significantly decreased over the 3 d of test when ATP was infused before CTA acquisition (unpaired t test, t(15) = 2.2, p = 0.043; Fig. 4C).
Based on these data, we next investigated whether ATP signaling was necessary for the LPS effect on associative taste memory. We blocked all purinergic receptors (P2Y and P2X) activation by using the nonspecific purinergic blocker suramin. We postulated that blockade of purinergic receptors activation would prevent LPS-induced CTA exaggeration. Thus, on the day of CTA acquisition, animals received a bilateral infusion of either suramin, a nonspecific P2Y and P2X receptors blocker, or sterile saline 60 min before infusion of either LPS or saline (Fig. 4D). During the 3 d of tests, an interaction between LPS and suramin treatment was found on saccharin consumption (interaction between suramin and LPS F(1,25) = 4.7 p < 0.05; time effect: F(2,50) = 103.4, p < 0.001; Figure 4E). Post hoc analysis revealed here again that LPS infusion enhanced taste aversion compared with vehicle infusion (p < 0.01). Importantly, suramin infusion by itself did not affect CTA (p = 0.27) but tended to reverse the enhancement of aversion induced by LPS (p = 0.06; Fig. 4E).
Finally, we quantified the levels of GluA2 AMPAR subunit expression in the insular cortex in the same paradigm as before to assess the role of ATP in LPS-enhanced AMPAR expression (Fig. 4F). A two-way ANOVA analysis revealed an interaction between suramin and LPS treatment (F(1,15) = 5.4, p < 0.05). Post hoc analysis confirmed that LPS enhanced GluA2 AMPAR expression (p < 0.05; Fig. 4F), whereas preinfusion with suramin prevented it (p < 0.05; Fig. 4G). These results indicate that the activation of purinergic receptors induced by LPS infusion was responsible for GluA2 AMPAR overexpression in neurons of the insular cortex.
Discussion
Together, our results showed that an inflammation restricted to the insular cortex enhances associative taste memory through purinergic modulation of GluA2 AMPAR expression and trafficking. This represents the first in vivo study demonstrating a causal link between neuroinflammation-induced glutamatergic transmission and cortical memory modulation.
The general idea that inflammatory mediators can markedly affect learning and memory arose from a limited number of studies focusing on hippocampal-dependent memories (Pugh et al., 1999; Barrientos et al., 2002, 2004; Matsumoto et al., 2002; Gonzalez et al., 2009; for review, Yirmiya and Goshen, 2011). In the same vein, most of the biochemical evidence on excitatory neurotransmission was obtained in the hippocampus. Here, we provide new insights on the response of cortical neurons to inflammation and we show an enhancement of memory performances in a paradigm of associative taste learning. Interestingly, our data are the first to suggest that cytokines do not play the same role in the cortex compared with the hippocampus. Second, even though cytokines are not the main inflammatory mediators, we confirm that glutamatergic neurotransmission is altered by neuroinflamamtion and this sustains the behavioral effects. Finally, we show enhancement of cortical-dependent memory rather than impairment. This result demonstrates that the neuronal response to neuroinflammation is not unspecific but rather depends on the information learned (taste cue vs context) and consequently on the structure involved and may lead to a plethora of behavioral outcomes.
Previous studies demonstrating that immune activation (induced by LPS or proinflammatory cytokines) alters learning and memory processes have often failed to account for features of sickness-related behavior that induce potential confounding factors (for review, see Cunningham and Sanderson, 2008). To cautiously interpret our results, we thus evaluated the extent of the inflammatory reaction in animals infused with LPS locally in the insular cortex as stimulation of the peripheral immune system, and the subsequent release of proinflammatory cytokines in the bloodstream, is known to produce gastric malaise (Cross-Mellor et al., 2004), changes in taste palatability (Aubert and Dantzer, 2005) as well as lethargy in animals (Franklin et al., 2003, 2007). To rule out nonspecific behavioral effects, we first measured systemic cytokines expression and could not find any effect of LPS treatment on blood levels of proinflammatory and anti-inflammatory factors. We also measured the activity of the HPA axis as another marker of systemic immune reaction and because glucocorticoid receptor activation in the insular cortex is known to modulate CTA memory formation (Miranda et al., 2008). Corticosterone concentration was similar between LPS-treated animals and controls, indicating that the HPA axis was not abnormally activated in these animals. Moreover, our local LPS infusion was not associated with decrease in body weight and food intake indicating an absence of an anorexic effect. Finally, we showed that LPS treatment did not affect neophobic responses as both saline and LPS-treated animals displayed a similar liquid consumption during the first presentation of the sweet taste. Conversely, in the absence of gastric malaise, both groups similarly increased their saccharin consumption between the first and the second presentation. These results indicate that LPS did not alter incidental taste learning (i.e., taste familiarity). Together, our results suggest that the enhancement of taste aversion learning we observed was linked to a localized cortical immune-stimulation and was independent from a more generalized inflammatory reaction.
The development of local inflammatory processes in the insular cortex also affected glutamatergic transmission, in the form of higher expression of AMPA receptor subunits in neurons. It is important to note that NMDAR expression and phosphorylation were not affected by cortical LPS infusion. The effects of AMPAR expression were specific to the insular cortex as we did not find any modification of these receptors expression in adjacent cortical areas. It is well known that the synaptic, but not extrasynaptic, localization of AMPA receptors controls the general activity of neurons (Malinow and Malenka, 2002). More specifically, elevation of AMPA receptors expression at the synaptic level leads to an increase of glutamatergic-dependent synaptic strength (Kauer and Malenka, 2006). By applying Western blotting on synaptoneurosomes and total extracts, we showed that LPS infusion in the insular cortex enhanced total AMPA receptors expression as it has already been demonstrated in various inflammatory context (Beattie et al., 2002; Ogoshi et al., 2005; Lai et al., 2006). Moreover, synaptoneurosome experiments demonstrate that a significant portion of these AMPAR is translocated at the synaptic level, suggesting that LPS increased AMPA receptors trafficking to the synapses. As a confirmation, by pharmacologically interfering with AMPA receptors trafficking, we were able to block LPS-induced exaggeration of associative taste memory. This indicates that increased AMPA receptors levels at the synapse is responsible for inflammation-induced enhancement of associative taste memory. Because the gastric malaise induces glutamatergic release in the insular cortex (Miranda et al., 2002), it is likely that LPS potentiates the glutamatergic-dependent processing of gastric malaise, thus leading to enhanced aversion.
Previous works have shown that inflammatory mediators modulate glutamatergic receptor expression and activity. Although we found a significant increase of IL-1β and TNF-α proteins expression in the insular cortex 60–90 min after LPS infusion, a role of IL-1β in our model was unexpected. IL-1β is known to phosphorylate GluN2B subunits in the hippocampus (Viviani et al., 2003; Yang et al., 2005) and to reduce GluA1 subunits in vitro in hippocampal neurons cultures (Lai et al., 2006), whereas we found no effect on GluN2B subunits and an enhancement of GluA1 and GluA2 subunits. Indeed, we were unable to modulate associative taste memory by locally injecting IL-1β in the insular cortex, whatever the dose used. Conversely, a previous study showed that TNF-α increases surface expression of AMPA receptors when applied on hippocampal neurons cultures, leading to increased synaptic strength (Beattie et al., 2002). However, here again, TNF-α infusion in the insular cortex did not reproduce the effects of LPS.
Finally, we focused on ATP and their purinergic P2 receptors. Purinergic P2 receptors are widely expressed throughout the CNS, including neurons of the cerebral cortex (Pankratov et al., 2002, 2007), and ATP regulates many key cellular functions, including rapid AMPA receptor function and trafficking at synapses (Nishimune et al., 1998; Song et al., 1998; Lüscher et al., 1999; Lüthi et al., 1999; Noel et al., 1999; Hanley et al., 2002; Lee et al., 2002; Lim and Isaac, 2005). Interestingly, whereas ATP induces a decrease of AMPA currents on hippocampal slices in noninflammatory condition (Pougnet et al., 2014), Pascual et al. (2012) were the first to show that LPS-induced ATP increased EPSC frequency. This suggested that microglial ATP is the primary messenger of the LPS-mediated modulation of glutamatergic neurotransmission. Here, we showed that LPS induced an increase of ATP concentration locally and that intrainsular ATP infusion enhanced aversion strength, mimicking LPS effects. We also examined the involvement of purines in local inflammation-induced exaggeration of associative taste memory and glutamatergic neurotransmission by pretreating animals with suramin, a nonselective antagonist of P2Y and P2X receptors (Windscheif, 1996), before LPS infusion. Suramin abolished both the biochemical and behavioral effects of LPS, indicating that purinergic receptors are necessary for the modulation of associative taste memory.
In conclusion, our data represent the first in vivo study demonstrating a causal link between neuroinflammation-induced modulation of glutamatergic transmission and memory involving ATP as a major mediator of these neuronal and behavioral alterations.
Footnotes
This work was supported by the Grant Emergence de Jeune Equipe INRA 2010–2012 to G.F., the Foundation for Medical Research, INRA, and the University of Bordeaux. The Biochemistry and Biophysics Facility of the Bordeaux Neurocampus is supported by the LABEX BRAIN (ANR-10-LABX-43). J.-C.D. was the recipient of a fellowship from the French Ministry of Research and Higher Education and from the Région Aquitaine. We thank Philippe Birac and Mathieu Cadet for technical assistance and for taking care of the animals; and Dr. V. Migues for helping with pepR845A experiments.
The authors declare no competing financial interests.
References
- Adaikkan C, Rosenblum K. The role of protein phosphorylation in the gustatory cortex and amygdala during taste learning. Exp Neurobiol. 2012;21:37–51. doi: 10.5607/en.2012.21.2.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert A, Dantzer R. The taste of sickness: lipopolysaccharide-induced finickiness in rats. Physiol Behav. 2005;84:437–444. doi: 10.1016/j.physbeh.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Barki-Harrington L, Elkobi A, Tzabary T, Rosenblum K. Tyrosine phosphorylation of the 2B subunit of the NMDA receptor is necessary for taste memory formation. J Neurosci. 2009;29:9219–9226. doi: 10.1523/JNEUROSCI.5667-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res. 2002;134:291–298. doi: 10.1016/S0166-4328(02)00043-8. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Sprunger DB, Campeau S, Watkins LR, Rudy JW, Maier SF. BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1beta administration. J Neuroimmunol. 2004;155:119–126. doi: 10.1016/j.jneuroim.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Berman DE, Hazvi S, Neduva V, Dudai Y. The role of identified neurotransmitter systems in the response of insular cortex to unfamiliar taste: activation of ERK1–2 and formation of a memory trace. J Neurosci. 2000;20:7017–7023. doi: 10.1523/JNEUROSCI.20-18-07017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermúdez-Rattoni F, Ramírez-Lugo L, Gutiérrez R, Miranda MI. Molecular signals into the insular cortex and amygdala during aversive gustatory memory formation. Cell Mol Neurobiol. 2004;24:25–36. doi: 10.1023/B:CEMN.0000012722.45805.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- Brebner K, Hayley S, Zacharko R, Merali Z, Anisman H. Synergistic effects of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha: central monoamine, corticosterone, and behavioral variations. Neuropsychopharmacology. 2000;22:566–580. doi: 10.1016/S0893-133X(99)00166-9. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Mellor SK, Kavaliers M, Ossenkopp KP. Comparing immune activation (lipopolysaccharide) and toxin (lithium chloride)-induced gustatory conditioning: lipopolysaccharide produces conditioned taste avoidance but not aversion. Behav Brain Res. 2004;148:11–19. doi: 10.1016/S0166-4328(03)00181-5. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Sanderson DJ. Malaise in the water maze: untangling the effects of LPS and IL-1beta on learning and memory. Brain Behav Immun. 2008;22:1117–1127. doi: 10.1016/j.bbi.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson E, Zhang N, Metallo J, Kaleka K, Shin SM, Gerges N, Lee SH. S-SCAM/MAGI-2 is an essential synaptic scaffolding molecule for the GluA2-containing maintenance pool of AMPA receptors. J Neurosci. 2012;32:6967–6980. doi: 10.1523/JNEUROSCI.0025-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Färber K, Kettenmann H. Physiology of microglial cells. Brain Res Brain Res Rev. 2005;48:133–143. doi: 10.1016/j.brainresrev.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol. 1997;159:1451–1458. [PubMed] [Google Scholar]
- Ferreira G, Gutiérrez R, De La Cruz V, Bermúdez-Rattoni F. Differential involvement of cortical muscarinic and NMDA receptors in short- and long-term taste aversion memory. Eur J Neurosci. 2002;16:1139–1145. doi: 10.1046/j.1460-9568.2002.02174.x. [DOI] [PubMed] [Google Scholar]
- Franklin AE, Engeland CG, Kavaliers M, Ossenkopp KP. Lipopolysaccharide-induced hypoactivity and behavioral tolerance development are modulated by the light-dark cycle in male and female rats. Psychopharmacology (Berl) 2003;170:399–408. doi: 10.1007/s00213-003-1554-3. [DOI] [PubMed] [Google Scholar]
- Franklin AE, Engeland CG, Kavaliers M, Ossenkopp KP. The rate of behavioral tolerance development to repeated lipopolysaccharide treatments depends upon the time of injection during the light-dark cycle: a multivariable examination of locomotor activity. Behav Brain Res. 2007;180:161–173. doi: 10.1016/j.bbr.2007.03.021. [DOI] [PubMed] [Google Scholar]
- Gal-Ben-Ari S, Rosenblum K. Molecular mechanisms underlying memory consolidation of taste information in the cortex. Front Behav Neurosci. 2011;5:87. doi: 10.3389/fnbeh.2011.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez PV, Schiöth HB, Lasaga M, Scimonelli TN. Memory impairment induced by IL-1beta is reversed by alpha-MSH through central melanocortin-4 receptors. Brain Behav Immun. 2009;23:817–822. doi: 10.1016/j.bbi.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Khatri L, Hanson PI, Ziff EB. NSF ATPase and alpha-/beta-SNAPs disassemble the AMPA receptor-PICK1 complex. Neuron. 2002;34:53–67. doi: 10.1016/S0896-6273(02)00638-4. [DOI] [PubMed] [Google Scholar]
- Haynes DR. Emerging and future therapies for the treatment of bone loss associated with chronic inflammation. Inflammopharmacology. 2006;14:193–197. doi: 10.1007/s10787-006-0006-1. [DOI] [PubMed] [Google Scholar]
- Joels G, Lamprecht R. Interaction between N-ethylmaleimide-sensitive factor and GluR2 is essential for fear memory formation in lateral amygdala. J Neurosci. 2010;30:15981–15986. doi: 10.1523/JNEUROSCI.1872-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. LTP: AMPA receptors trading places. Nat Neurosci. 2006;9:593–594. doi: 10.1038/nn0506-593. [DOI] [PubMed] [Google Scholar]
- Lai AY, Swayze RD, El-Husseini A, Song C. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol. 2006;175:97–106. doi: 10.1016/j.jneuroim.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/S0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Lim W, Isaac JT. ATP hydrolysis is required for the rapid regulation of AMPA receptors during basal synaptic transmission and long-term synaptic plasticity. Neuropharmacology. 2005;48:949–955. doi: 10.1016/j.neuropharm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/S0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Lüthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, Kidd FL, Henley JM, Isaac JT, Collingridge GL. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF-GluR2 interaction. Neuron. 1999;24:389–399. doi: 10.1016/S0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Watanabe S, Suh YH, Yamamoto T. Effects of intrahippocampal CT105, a carboxyl terminal fragment of beta-amyloid precursor protein, alone/with inflammatory cytokines on working memory in rats. J Neurochem. 2002;82:234–239. doi: 10.1046/j.1471-4159.2002.00944.x. [DOI] [PubMed] [Google Scholar]
- Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem. 2001;276:3820–3826. doi: 10.1074/jbc.M006814200. [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Finnie P, Wang YW, Nader K. The maintenance of long-term memory in the hippocampus depends on the interaction between N-ethylmaleimide-sensitive factor and GluA2. Hippocampus. 2014;24:1112–1119. doi: 10.1002/hipo.22295. [DOI] [PubMed] [Google Scholar]
- Miranda MI, Ferreira G, Ramirez-Lugo L, Bermúdez-Rattoni F. Glutamatergic activity in the amygdala signals visceral input during taste memory formation. Proc Natl Acad Sci U S A. 2002;99:11417–11422. doi: 10.1073/pnas.182200499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda MI, Quirarte GL, Rodriguez-Garcia G, McGaugh JL, Roozendaal B. Glucocorticoids enhance taste aversion memory via actions in the insular cortex and basolateral amygdala. Learn Mem. 2008;15:468–476. doi: 10.1101/lm.964708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerad L, Ramírez-Amaya V, Ormsby CE, Bermúdez-Rattoni F. Differential effects of anterior and posterior insular cortex lesions on the acquisition of conditioned taste aversion and spatial learning. Neurobiol Learn Mem. 1996;66:44–50. doi: 10.1006/nlme.1996.0042. [DOI] [PubMed] [Google Scholar]
- Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/S0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- Noel J, Ralph GS, Pickard L, Williams J, Molnar E, Uney JB, Collingridge GL, Henley JM. Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron. 1999;23:365–376. doi: 10.1016/S0896-6273(00)80786-2. [DOI] [PubMed] [Google Scholar]
- Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH. Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol. 2005;193:384–393. doi: 10.1016/j.expneurol.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Krishtal O, Verkhratsky A. Ionotropic P2X purinoreceptors mediate synaptic transmission in rat pyramidal neurones of layer II/III of somato-sensory cortex. J Physiol. 2002;542:529–536. doi: 10.1113/jphysiol.2002.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Verkhratsky A, North RA. Quantal release of ATP in mouse cortex. J Gen Physiol. 2007;129:257–265. doi: 10.1085/jgp.200609693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2012;109:E197–E205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin: evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. 1994;269:15195–15203. [PubMed] [Google Scholar]
- Pougnet JT, Toulme E, Martinez A, Choquet D, Hosy E, Boué-Grabot E. ATP P2X receptors downregulate AMPA receptor trafficking and postsynaptic efficacy in hippocampal neurons. Neuron. 2014;83:417–430. doi: 10.1016/j.neuron.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Pugh CR, Nguyen KT, Gonyea JL, Fleshner M, Wakins LR, Maier SF, Rudy JW. Role of interleukin-1 beta in impairment of contextual fear conditioning caused by social isolation. Behav Brain Res. 1999;106:109–118. doi: 10.1016/S0166-4328(99)00098-4. [DOI] [PubMed] [Google Scholar]
- Richard EM, Helbling JC, Tridon C, Desmedt A, Minni AM, Cador M, Pourtau L, Konsman JP, Mormède P, Moisan MP. Plasma transcortin influences endocrine and behavioral stress responses in mice. Endocrinology. 2010;151:649–659. doi: 10.1210/en.2009-0862. [DOI] [PubMed] [Google Scholar]
- Sewards TV, Sewards MA. Cortical association areas in the gustatory system. Neurosci Biobehav Rev. 2001;25:395–407. doi: 10.1016/S0149-7634(01)00021-5. [DOI] [PubMed] [Google Scholar]
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/S0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- Stehberg J, Moraga-Amaro R, Simon F. The role of the insular cortex in taste function. Neurobiol Learn Mem. 2011;96:130–135. doi: 10.1016/j.nlm.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dong Q, Xu XF, Feng X, Xin J, Wang DD, Yu H, Tian T, Chen ZY. Phosphorylation of cofilin regulates extinction of conditioned aversive memory via AMPAR trafficking. J Neurosci. 2013;33:6423–6433. doi: 10.1523/JNEUROSCI.5107-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windscheif U. Purinoceptors: from history to recent progress. A review. J Pharm Pharmacol. 1996;48:993–1011. doi: 10.1111/j.2042-7158.1996.tb05891.x. [DOI] [PubMed] [Google Scholar]
- Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. 2005;1034:172–179. doi: 10.1016/j.brainres.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]