

Abstract
Although the migration of hepatic stellate cells (HSCs) is important for hepatic fibrosis, the regulation of this migration is poorly understood. Notably, transforming growth factor (TGF)-β1 induces monocyte migration to sites of injury or inflammation during the early phase, but inhibits cell migration during the late phase. In the present study, the role of transforming protein RhoA signaling in TGF-β1-induced HSC migration was investigated. TGF-β1 was found to increase the protein and mRNA levels of smooth muscle actin and collagen type I in HSC-T6 cells. The level of RhoA-GTP in TGF-β1-stimulated cells was significantly higher than that in control cells. Furthermore, the phosphorylation of cofilin and formation of filamentous actin (F-actin) were more marked in TGF-β1-stimulated cells than in control cells. Additionally, TGF-β1 induced the activation of nuclear factor-κB, and the expression of extracellular matrix proteins and several cytokines in HSC-T6 cells. The active form of Rap1 (Rap1 V12) suppressed RhoA-GTP levels, whereas the dominant-negative form of Rap1 (Rap1 N17) augmented RhoA-GTP levels. Therefore, the data confirmed that Rap1 regulated the activation of RhoA in TGF-β1-stimulated HSC-T6 cells. These findings suggest that TGF-β1 regulates Rap1, resulting in the suppression of RhoA, activation of and formation of F-actin during the migration of HSCs.
Keywords: cell migration, hepatic stellate cell, transforming growth factor-β1, Rap1, transforming protein RhoA, nuclear factor-κB
Introduction
Hepatic fibrosis is characterized by the excessive deposition of extracellular matrix (ECM) mediated by activated hepatic stellate cells (HSCs) (1). Activated HSCs are considered to serve a major role via proliferation and the production of ECM, and they may migrate to fibrotic regions by transforming into myofibroblasts (2). Although the majority of studies investigating hepatic fibrosis have focused on the proliferation and production of ECM of activated HSCs, studies have demonstrated that the migration of HSCs serves an important role in hepatic fibrosis, including ECM and growth factor production (3,4). Cell migration is essential in a variety of biological events. Cell migration is a dynamic and cyclic process that is regulated by small GTPase proteins (5). Cdc42 regulates the direction of movement, and Rac induces membrane protrusion at the leading edge of the cell through actin polymerization and the stimulation of integrin adhesion complexes (6). Rho regulates actin-myosin contraction and retraction in the cell body and lagging edge (7,8). Among the cytoskeletal components, actin filaments serve a major role in the formation of stress fibers during cell adhesion and migration. Rho GTPase is known to regulate the actin cytoskeleton, cell polarity, gene expression, microtubule dynamics and vesicular trafficking (9).
Transforming growth factor (TGF)-β1 is known to be a multifunctional cytokine that regulates cell proliferation, differentiation, migration, survival, carcinogenesis, wound healing, immune reactions and fibrosis (10). In particular, TGF-β1 is the most important profibrotic cytokine in hepatic fibrosis. Of note, TGF-β1 activates Rho subfamily GTPases, including transforming protein RhoA, Ras-related C3 botulinum toxin substrate 1 (Rac1) and Cdc42, in various cell lines, including the HSC-T6 cell line (11-14). Furthermore, a previous study demonstrated that the TGF-β1-induced activation of RhoA in activated HSCs mediates the migration of HSCs via the Smad/JNK/p38 signaling pathway (15).
In addition to the Smad-dependent TGF-β1 signaling pathway, TGF-β1 can induce activation of the nuclear factor-κB (NF-κB) signaling pathway, leading to the migration of cells (11). NF-κB is a well-known transcription factor that regulates the expression of cytokines, chemokines, cell adhesion molecules and inducible proteins to control inflammation, apoptosis and malignancy (16). Kim et al demonstrated that inhibitor of NF-κB (IκB) kinase (IKK)-γ stimulates the activation of RhoA, which leads to the direct phosphorylation of IKKβ and subsequent activation of NF-κB, and induces chemokine expression and cell migration in response to TGF-β1 (17). As a member of the Ras superfamily of small GTPases, Rap1 is regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). Rap1 is associated with the regulation of both inte-grin- and cadherin-mediated cell adhesion, and the recycling, avidity and affinity of integrins via the inside-out activation process (18). Rap1 signaling can positively and negatively modulate the activity of Rho family proteins, including Cdc42, Rac1 and RhoA. Moon et al demonstrated that Rap1 inhibits cell migration by regulating the activity of RhoA in response to TGF-β1 (19). Rap1 is an important modulator of the NF-κB signaling pathway (20,21). Although the influence of Rho GTPase signaling on HSC migration during hepatic fibrosis has been reported (15), the role of NF-κB signaling in response to TGF-β1 via RhoA GTPase activation has not been investigated in activated HSCs. Therefore, the present study investigated the mechanism by which Rap1 regulates the activity of RhoA by NF-κB signaling during TGF-β1-induced HSC migration.
Materials and methods
Materials
Bovine serum albumin (BSA; cat. no. A2058), Y27632 (cat. no. Y0503), BAY11-7085 (cat. no. 196309-76-9) and the recombinant TGF-β1 protein (cat. no. T7039) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Details of antibodies are shown in Table I.
Table I.
Antibodies and dilutions used for western blotting.
| Primary antibody (Company, cat. no.) | Dilution/incubation | Secondary antibody (horseradish peroxidase-conjugated) | Dilution/incubation | Blocking buffer and antibody diluent (in 1X PBS with 0.1% Tween-20) |
|---|---|---|---|---|
| anti-Rap1 (Santa Cruz, sc-65) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-RhoA (Santa Cruz, sc-418) | 1:1,000 4°C/overnight | Anti-mouse (Enzo Life Sciences, ADI-SAB-100-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-GAPDH (Santa Cruz, sc-32233) | 1:3,000 4°C/overnight | Anti-mouse (Enzo Life Sciences, ADI-SAB-100-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-cofilin (Santa Cruz, sc-33779) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-IKKα/β (Santa Cruz, sc-7607) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000RT/1 h | 5% SkM |
| anti-SMA (CST, #19245S) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-IL-1β (Abcam, ab2105) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-fibronectin (Abcam, ab2413) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-collagen type1 (CST, 84336S) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% SkM |
| anti-p-p65 (CST, 3033S) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% BSA |
| anti-p65 (CST, 8242S) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% BSA |
| anti-p-IκBα (CST, 9246S) | 1:1,000 4°C/overnight | Anti-mouse (Enzo Life Sciences, ADI-SAB-100-J) | 1:5,000 RT/1 h | 5% BSA |
| anti-IκBα (CST, 4812S) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% BSA |
| anti-p-IKKα/β (CST, 2697S) | 1:1,000 4°C/overnight | Anti-rabbit (Enzo Life Sciences, ADI-SAB-300-J) | 1:5,000 RT/1 h | 5% BSA |
BSA, bovine serum albumin; RT, room temperature; SkM, skimmed milk; CST, Cell Signaling Technology, Inc. (Danvers, MA, USA); Santa Cruz, Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture and transfection
The HSC-T6 cell line (Dr Scott Friedman, Division of Liver Diseases, Icahn School of Medicine at Mount Sinai, New York, NY, USA), an immortalized rat HSC line, has been described previously (22). The HSC-T6 cells were grown in Dulbecco's modified Eagle's medium (DMEM; HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; HyClone; GE Healthcare Life Sciences), 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2. For transient transfection, the HSC-T6 cells were plated at a density of 2×105 cells per well in six-well plates. A total of 18 h after seeding, the cells were transiently transfected with 2 µg pcDNA3.1 plasmids containing Rap1 (mock vector, M), pcDNA3.1 plasmid containing dominant-negative Rap1 (N17) and constitutively active Rap1 (V12; all Missouri S&T cDNA Resource Center, PA, USA) using Lipofectamine 2000 reagent (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol.
Expression, purification, and transduction of Tat-C3
The HIV-1 Tat transduction domain (amino acid residues 49-57, KKKRRQRRR) were fused to the N-terminus of the C3 exoenzyme containing a His tag (Tat-C3) for the purpose of efficiently introducing C3 exoenzymes into cells. Tat-C3 purification was performed following a previously described method (13,23). To analyze the transduction of Tat-C3, the cells were plated at a density of 2×105 cells per well in 6-well plates and were incubated with 1 µg/ml Tat-C3 at 37°C with 5% CO2. After 1 h, the Tat-C3 transduced cells were analyzed by western blotting using anti-RhoA antibody. The Tat-C3-transduced cells showed a mobility shift band, which was modified-RhoA (Arg 41) by Tat-C3.
Western blot analysis
The cells were collected, washed once with ice-cold phosphate-buffered saline (PBS) and lysed with RIPA buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 10 mM NaF, 1 mM Na3VO4, 2 mM EDTA, 1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml leupeptin and 1 µg/ml pepstatin]. The cell lysates were centrifuged at 13,000 × g for 20 min at 4°C, and protein concentrations in the supernatants were analyzed using a Bradford assay (Bio-Rad Laboratory, Inc., Hercules, CA, USA). Equal quantities of proteins (40 µg) were separated using SDS-PAGE on 10% gels for Col1a1 and fibronectin or 12% gels for all other proteins, transferred onto PVDF membranes, blocked with 5% skimmed milk or 5% BSA and probed with the appropriate antibodies (Table I). The immunoreactive bands were visualized on digital images captured with FUSION FX7 SPECTRA (Vilber Lourmat, Eberhardzell, Germany) using EzWestLumi plus Western blot detection reagent (ATTO Corporation, Tokyo, Japan). The band intensities were quantified using ImageJ software (version 1.50i; National Institutes of Health, Bethesda, MD, USA). Statistical analyses were performed using GraphPad Prism 4 (GraphPad Software, Inc., San Diego, CA, USA).
Filamentous actin (F-actin) staining
The HSC-T6 cells were treated with 10 ng/ml TGF-β1 in DMEM for 1 h at 37°C under 5% CO2. The cells were washed with PBS, fixed with 4% methanol-free formaldehyde and permeabilized with 0.2% Triton X-100 in PBS. F-actin (green) was then stained with Alexa Fluor 488-phalloidin (1 µm) for 30 min at room temperature. DAPI (blue) was used to counterstain the nuclei. The immunolabeled cells were examined using an LSM 700 laser confocal microscope (Carl Zeiss AG, Oberkochen, Germany).
Glutathione S-transferase (GST)-Rhotekin-Rho-binding domain (RBD) pull-down assays for activated RhoA
The cells were collected, washed with PBS and then lysed in binding/washing/lysis buffer [25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 1 mM DTT, 5% glycerol, 10 mM NaF, 1 mM Na3VO4, 1 mM EDTA and 1 mM EGTA] with a protease inhibitor cocktail tablet. The lysates were centrifuged at 13,000 × g for 10 min at 4°C. The supernatant was incubated with GST-Rhotekin-RBD for 2 h at 4°C to detect RhoA-GTP, as previously described (13). The beads were washed three times with binding/washing/lysis buffer. The bound proteins were eluted with 2X Laemmli sample buffer (Bio-Rad Laboratories, Inc.) by boiling. The samples were electrophoresed and analyzed by western blotting with an anti-RhoA antibody (1:1,000) for overnight at 4°C.
Reverse transcription-quantitative-polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the HSC-T6 cells using an RNA purification kit (GeneAll, Seoul, Korea) according to the manufacturer's protocol. Complementary DNA (cDNA) was generated using Maxime™ RT-PCR PreMix (Intron Biotechnology, Inc., Seongnam, Korea) according to the instructions of the manufacturer. RT-qPCR was performed with the SYBR Green PCR mixture (Enzynomics, Daejeon, Korea) under the following conditions: One cycle at 95°C for 10 min and then 40 cycles at 95°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec. Real-time detection of the SYBR Green emission intensity was performed with a Thermo Scientific™ PikoReal™ PCR system (Thermo Fisher Scientific, Inc.). RT-qPCR was performed using primers specific for α-SMA (forward, 5′-AAG CCC AGC CAG TCG CTG TCA-3′ and reverse 5′-GAA GCC GGC CTT ACA GAG CCC-3′) and COL1A1 (forward 5′-GCT CCT TCT TAG GGG CCA CT-3′ and reverse 5′-ATT GGG GAC CCT TAG GCC AT-3′). The relative expression of each gene was calculated using the ∆∆Cq method with GAPDH (forward 5′-TGG CCT TCC GTG TTC CTA-3′ and reverse 5′-GAG TTG CTG TTG AAG TCG CA-3′). Quantification was performed by normalizing the quantification cycle (Cq) values with GAPDH Cq and analyzed with the 2−ΔΔCq method (24).
Cell migration assay
The migration of HSC-T6 cells was examined using a Transwell assay (Corning, Inc., New York, NY, USA). The cells (~1×105) were resuspended in 200 µl of DMEM and seeded on cell culture inserts with 8.0-µm pores, which were precoated with gelatin. The cells were pretreated with in the presence or absence of a RhoA specific inhibitor (Tat-C3, 1 µg/ml), a Rho associated coiled-coil containing protein kinase inhibitor (Y27632, 10 µM) and an inhibitor of NF-κB activation and phosphorylation of IκBα (BAY 11-7085, 1 µM) for 1 h, and then incubated with 10 ng/ml TGF-β1 added to the bottom chamber. The cells were allowed to migrate for 16 h at 37°C. The cells were fixed and stained with crystal violet, and the number of cells on the lower surface of the filters was counted under the microscope (Nikon TS100; Nikon, Tokyo, Japan). A total of four fields were counted for each Transwell filter.
Statistical analysis
Data were analyzed using Student's t-test, one-way analysis of variance (ANOVA) followed by Tukey's post hoc test and multiple comparison tests using GraphPad Prism 4 software (GraphPad Software, Inc., La Jolla, CA, USA). The data are presented as mean ± SEM. P<0.05 indicated that the difference between groups was statistically significant.
Results
TGF-β1 induces the activation of HSCs
TGF-β1 is known to induce the activation of HSCs. To confirm the activation of HSCs following TGF-β1 treatment (Fig. 1A-D), the major markers of activated HSCs, namely, α-SMA and Col1a1, were investigated. Compared with levels in the untreated HSC-T6 cells, the mRNA expression levels of α-SMA and Col1a1 were significantly increased in the TGF-β1-treated HSC-T6 cells (Fig. 1E and F), indicating that TGF-β1 induced the activation of HSCs.
Figure 1.

TGF-β1 induces the expression of Col1a1 and α-SMA in HSC-T6 cells. (A) HSC-T6 cells were treated with or without 10 ng/ml TGF-β1 for 16 h. Western blot analysis of the expression levels of collagen type 1. (B) Western blot analysis of the expression levels of Col1a1. (C) Western blot analysis of the expression levels of α-SMA. (D) Western blot analysis of the expression levels of α-SMA (mean ± SEM, n=3, *P<0.05, Student's t-test). (E and F) Reverse transcription-quantitative polymerase chain reaction analysis of the mRNA expression of α-SMA and Col1a1 (mean ± SEM, n=3, ***P<0.001, one-way ANOVA, Tukey's post hoc test). TGF-β1, transforming growth factor-β1; Col1a1, collagen type I; α-SMA, α-smooth muscle actin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CON, control.
TGF-β1 regulates the formation of F-actin through RhoA signaling
To examine whether TGF-β1 activates RhoA in HSC-T6 cells, a pull-down assay was performed with GST-Rhotekin-RBD in HSC-T6 cells with/without TGF-β1 treatment, as previously described (19). Compared with that in the untreated HSCs, the expression level of RhoA-GTP was significantly increased in the TGF-β1-treated cells (Fig. 2A and B).
Figure 2.

TGF-β1 regulates the RhoA signaling pathway. Cells were incubated with TGF-β1 for various durations (0, 0.5, 1 and 3 h). Detection of GTP-RhoA was performed via GST pull-down assays. (A) Total protein levels of RhoA were detected by western blotting and (B) quantified (mean ± SEM, n=3, *P<0.05, one-way ANOVA, Tukey's post hoc test). (C) Phosphorylation of cofilin was assessed in TGF-β1-stimulated HSC-T6 cells and (D) quantified (mean ± SEM, n=3, *P<0.05, one-way ANOVA, Tukey's post hoc test). (E) Immunocytochemical staining for F-actin formation in HSC-T6 cells using Alexa Fluor 488-phal-loidin (green). DAPI (blue) was used to counterstain the nuclei. All images are representative of multiple images from three independent experiments (scale bar=20 µm). TGF-β1, transforming growth factor-β1; p-, phosphorylated; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CON, control.
To further investigate the mechanism of TGF-β1-induced RhoA signaling, the present study assessed whether TGF-β1 modulates the RhoA-Rho-associated kinase (ROCK)-LIM-kinase (LIMK)-cofilin pathway. In a previous study, RhoA phosphorylated at Ser188 (p-RhoA) negatively regulated the activity of RhoA by promoting interactions with RhoGDI and the translocation of RhoA from the membrane to the cytosol (25) to enhance the levels of p-LIMK1/2 and p-cofilin. Similar to the above results, it was observed that TGF-β1 induced an increase in the expression of p-cofilin in activated HSCs (Fig. 2C and D).
Previous studies have shown that the activation of RhoA serves a role in the regulation of cytoskeletal reorganization through actin stress fiber formation and the induction of cell adhesion (26,27). Therefore, the present study investigated the effect of TGF-β1 on the formation of actin stress fibers in HSC-T6 cells. Stress fibers were observed to form F-actin, which was detected with FITC-conjugated phalloidin. As shown in Fig. 2E, the formation of F-actin was more marked in the TGF-β1-treated HSC-T6 cells than in the untreated HSC-T6 cells (Fig. 2E).
To investigate whether RhoA signaling regulates the activation of HSCs, the present study examined changes in the expression levels of α-SMA and Col1a1 following the inhibition of RhoA by Y27632 (a ROCK inhibitor) or Tat-C3 (a Rho-specific inhibitor). Treatment with Y27632 or Tat-C3 inhibited the expression of α-SMA and Col1a1 in TGF-β1-treated HSC-T6 cells. In addition, the inhibition of RhoA signaling by Y27632 and Tat-C3 significantly decreased the levels of p-cofilin (Fig. 3A and B).
Figure 3.

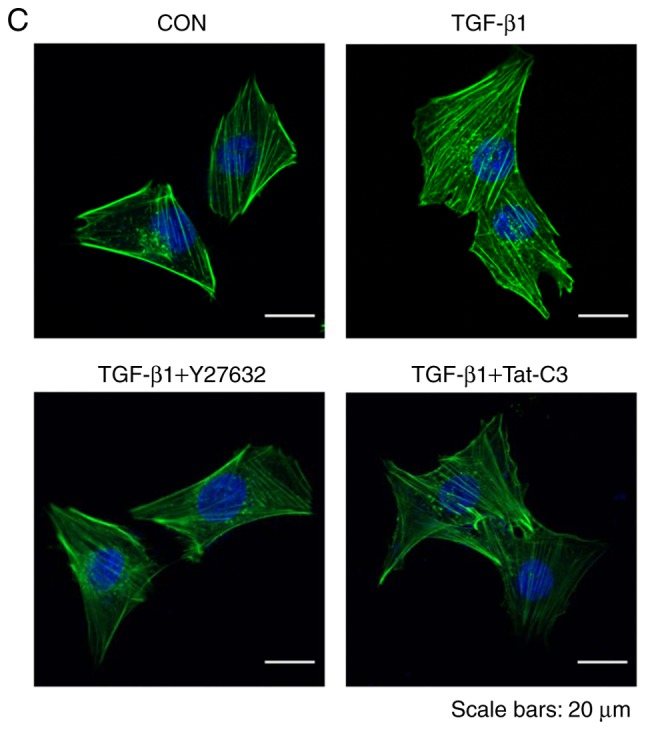
Inhibition of RhoA inhibits HSC activation and F-actin formation. HSC-T6 cells were pretreated with or without 10 µM Y27632 and 1 µg/ml Tat-C3 for 1 h, and 10 ng/ml TGF-β1 was added for 1 h. (A) Levels of α-SMA, p-cofilin and Col1a1 were assessed by western blotting and (B) quantified (mean ± SEM, n=3, *P<0.05, **P<0.01 and ***P<0.001, one-way ANOVA, Tukey's post hoc test). (C) Immunocytochemical staining for F-actin formation in HSC-T6 cells using Alexa Fluor 488-phalloidin (green). DAPI (blue) was used to counterstain the nuclei. All images are representative of multiple images from three independent experiments (scale bar=20 µm). TGF-β1, transforming growth factor-β1; Col1a1, collagen type 1; α-SMA, α-smooth muscle actin; p-, phosphorylated; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CON, control.
To assess the level of inhibition of F-actin formation by Y27632 or Tat-C3, the level of F-actin was examined in TGF-β1-treated HSC-T6 cells. The RhoA inhibitors also reduced F-actin formation in response to TGF-β1 (Fig. 3C), indicating that RhoA signaling regulates cytoskeletal reorganization, including F-actin formation, during the process of HSC activation.
TGF-β1 regulates RhoA via Rap1
Previous studies have reported that Rap1 inhibits cell migration by suppressing the activity of RhoA in response to TGF-β1 in macrophages (19). Therefore, the present study investigated whether TGF-β1 regulates the activity of Rap1 in HSC-T6 cells using a pull-down assay with the His-RalGDS-Rap-binding domain. The activity of Rap1 was inhibited by TGF-β1 until 1 h after treatment, but increased thereafter (Fig. 4A and B). By contrast, the levels of GTP-RhoA and GTP-Rap1 showed opposite patterns of time-dependent change following TGF-β1 stimulation (Figs. 2A and 4A). In addition, the inhibition of Rap1 (using Rap1 N17, a dominant-negative form of Rap1) induced the activation of RhoA, and the activation of Rap1 (using Rap1 V12, a constitutively active form of Rap1) inhibited the activation of RhoA. These results indicated that Rap1 regulates the activity of RhoA in TGF-β1-treated HSC-T6 cells (Fig. 4C and D).
Figure 4.

Rap1 downregulates the TGF-β1-induced activation of RhoA. Cells were incubated with 10 ng/ml TGF-β1 for various durations (0, 0.5, 1 and 3 h). (A) GTP-Rap1 was selectively precipitated from cell lysates using the His-RalGDS-RBD fusion protein and Ni-NTA His-Bind resin, and detected by immu-noblotting with an anti-Rap1 antibody. (B) Quantification of the protein expression of GTP-Rap1 (mean ± SEM, n=3, **P<0.01 and ***P<0.001, one-way ANOVA, Tukey's post hoc test). Mock vector, pcDNA-Rap1 N17 (encoding the dominant negative form of Rap1) and pcDNA-Rap1 V12 (encoding the consti-tutively active form of Rap1) were transfected into HSC-T6 cells, and the cells were stimulated with or without 10 ng/ml TGF-β1 for 1 h. GTP-RhoA levels were determined using a pull-down assay with GST-Rhotekin-RBD fusion protein and Glutathione-Sepharose beads. (C) Expression of RhoA was determined by western blotting using an anti-RhoA antibody and (D) quantified (mean ± SEM, n=3, *P<0.05, **P<0.01, and ***P<0.001, one-way ANOVA, Tukey's post hoc test). TGF-β1, transforming growth factor-β1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; M, mock vector.
TGF-β1 promotes cell migration via the inhibition of Rap1 and activation of RhoA
To confirm the molecular mechanism by which TGF-β1 induces the activation of NF-κB, the phosphorylation of NF-κB signaling components were measured. Compared with those in untreated cells, the expression levels of p-IKKα/β, p-IκBα and p-p65 in TGF-β1-treated HSC-T6 cells were significantly increased (Fig. 5A and B). The expression of ECM proteins and specific cytokines were also measured in the TGF-β1-treated HSC-T6 cells. Treatment with TGF-β1 induced an increase in the expression of fibronectin and TGF-β1, whereas the expression of interleukin (IL)-1β did not differ between the untreated cells and TGF-treated cells (Fig. 5C and D). Activated HSCs migrate and accumulate at tissue repair sites, regulating ECM production and secretion (28,29). To confirm this behavior, the migration of activated HSCs were measured using Transwell assays. Compared with that of the untreated cells, the migration of TGF-β1-treated HSC-T6 cells was significantly increased (Fig. 6A and B). RhoA inhibitors reduced cell migration in the TGF-β1-treated HSC-T6 cells (Fig. 6D), and the activation of Rap1 inhibited the migration of HSCs (Fig. 6C). To confirm that the migration of TGF-β1-treated HSC-T6 cells was regulated via RhoA and NF-κB signaling, the migration indices of HSC-T6 cells pretreated with Y27632, Tat-C3 and BAY 11-7085 were measured. The RhoA inhibitors and the NF-κB inhibitor reduced cell migration in the TGF-β1-treated HSC-T6 cells (Fig. S1). Taken together, these findings suggest that TGF-β1 modulates cell migration by regulating ECM protein and chemokine expression through Rap1, RhoA and NF-κB signaling.
Figure 5.


TGF-β1 regulates the NF-κB signaling pathway. HSC-T6 cells were treated with 10 ng/ml TGF-β1 for various durations (0, 10, 20, 30, 60 and 120 min). (A) Phosphorylation of IκB, IKKα/β and p65 was analyzed by western blotting and (B) quantified (mean ± SEM, n=3, *P<0.05 and **P<0.01 one-way ANOVA, Tukey's post hoc test). Cells were treated with or without 10 ng/ml TGF-β1 for 16 h. (C) Western blot analysis and (D) quantification of the expression levels of fibronectin, TGF-β1 and IL-β1 (mean ± SEM, n=3, *P<0.05, **P<0.01, Student's t-test). TGF-β1, transforming growth factor-β1; NF-κB, nuclear factor-κB; IκB, inhibitor of NF-κB; IKK, IκB kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; p-, phosphorylated; IL-1β1, interleukin-β1; CON, control.
Figure 6.

TGF-β1 regulates HSC-T6 cell migration via RhoA and Rap1 signaling. (A) Serum-starved HSC-T6 cells were added to the upper chamber, and 10 ng/ml TGF-β1 was added to the lower chamber (magnification, ×40). The cells in the upper chamber were then allowed to migrate for 16 h and (B) quantified (mean ± SEM, n=3, **P<0.01, Student's t-test). (C) Mock vector, pcDNA-Rap1 N17 (encoding the dominant negative form of Rap1) and pcDNA-Rap1 V12 (encoding the constitutively active form of Rap1) were transfected into HSC-T6 cells, and the cells were allowed to migrate for 16 h (mean ± SEM, n=3, *P<0.05 and **P<0.01, one-way ANOVA, Tukey's post hoc test). (D) HSC-T6 cells were incubated with 10 µM Y27632 and 1 µg/ml Tat-C3 and allowed to migrate for 16 h (mean ± SEM, n=3, ***P<0.001, one-way ANOVA, Tukey's post hoc test). TGF-β1, transforming growth factor-β1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CON, control; M, mock.
Discussion
NF-κB modulates various essential functions of hepatocytes, HSCs and Kupffer cells (30-32). The crucial role of NF-κB in the liver is emphasized by the fact that the deletion of NF-κB signaling components in mouse models leads to spontaneous liver damage, fibrosis and hepatocellular carcinoma (33). Hepatic fibrosis is a reversible wound-healing response characterized by the accumulation of fibrillar matrix in the injured liver. The activation of HSCs during hepatic fibrosis is considered a major factor in the progression of chronic liver disease (3,34,35). HSCs are an important source of growth factors in the liver, and in addition to HSCs increasing in number, ECM production per cell increases, leading to fibrosis (36). Activated HSCs are a key source of cytokines and establish an autocrine stimulation loop. HSCs are activated by TGF-β1 (37), and the activation of HSCs increases the expression of TGF-β1. The present study also demonstrated that TGF-β1 increased the protein and mRNA levels of α-SMA and Col1a1, consistent with previous studies (Fig. 1) (38,39).
Cell migration serves a pivotal role in numerous biological processes, including differentiation and immune responses. Cell migration is a multistep process involving changes in the cytoskeleton and can be divided into the following four steps: i) lamellipodium extension, ii) new adhesion, iii) cell body contraction and iv) tail detachment (40). Rho GTPases regulate several aspects of cell migration and affect other components of the cytoskeleton, in addition to cell-substrate adhesion and matrix reconstitution. Actin filament reorganization through cofilin activity is observed at the leading edge of migrating cells (41). The activation of RhoA induces the phosphorylation of cofilin into its inactivate form, promoting actin polymerization (42). The present study found that TGF-β1 significantly increased the level of p-cofilin, inducing the activation of cofilin and facilitating F-actin formation. In addition, the inhibition of RhoA signaling by Y27632 and Tat-C3 significantly decreased F-actin formation (Fig. 3). Iwamoto et al (43) reported that Y27632 consistently suppressed cell spread and proliferation, and that Y27632 also decreased the gene expression and protein accumulation of collagen type I in primary cultured HSCs. Kato et al also demonstrated that Tat-C3 treatment in activated HSCs distorted cell shape, and decreased stress fiber formation and the level of Col1a1 (44). These results were consistent with findings in a rat model of dimethyl nitrosamine-induced hepatic fibrosis (45). The results of the present study also demonstrated that the inhibition of RhoA signaling suppressed the phosphorylation of cofilin and subsequently decreased F-actin formation in activated HSCs, suggesting that RhoA signaling is an important mechanism underlying the phenotypic changes in HSCs during the initiation of hepatic fibrosis.
Rap1 is a member of the Ras GTPase family, which is inactive in its GDP-bound form and becomes active following binding to GTP. GAPs and GEFs regulate Rap1, with GAPs promoting the GDP-bound (inactive) form and GEFs promoting the GTP-bound (active) form. GTP-binding Rap1 binds to effector molecules, including Raf-1, B-Raf, RalGDS and AF-6 (46,47). The activation of Rap1 is associated with CD31-dependent adhesion to intercellular adhesion molecule (ICAM) and vascular cell adhesion molecule, thereby mediating the interaction of platelets and leukocytes with vascular endothelial cells and facilitating the transmigration and extravasation of leukocytes (48). In addition, lymphocyte function-associated antigen-1 binds to ICAM-1 following the introduction of constitutively active Rap1, and increases Rap-dependent adhesion and migration in pro-B cells (49,50). Regarding the stimulation of cell migration by Rap1, our previous study showed that Rap1 inhibits the activity of RhoA and inhibits cell migration in mouse macrophages (19). Although the mechanism by which Rap1 signaling modulates HSC migration is unknown, our data suggest that Rap1 can regulate the ability of cells to produce F-actin and express cytokines by inhibiting the activity of RhoA.
In the present study, it was found that TGF-β1 regulated the activity of Rap1 and thus the RhoA-mediated signal transduction pathway. TGF-β1 increased the activity of RhoA following short-term treatment but reduced the activity of Rap1 (Figs. 2 and 3). In a previous report, Rap1-dependent RhoGAP (ARAP3) was activated by Rap1 and induced the inactivation of RhoA in PC12 cells with neurite outgrowth in response to nerve growth factor (51). ARAP3 is a potent GAP for RhoA and Rap1, and phosphatidylinositol (3,4,5)-triphosphate activates the Rho GAP activity of ARAP in vitro and in vivo (52). Similarly, dominant-negative Rap1 (Rap1 N17) increased the activity of RhoA, whereas constitutively active Rap1 (Rap1 V12) decreased the activity of RhoA (Fig. 3).
TGF-β is a multifunctional cytokine that is present in most cells. The TGF-β1-induced polyubiquitination of TAK1 lysine 158 is required for TAK1-mediated Smad-independent IKK, JNK and p38 activation (53,54). Several mitogen-activated protein kinase kinase kinases, including TAK1, serve a pivotal role in the activation of NF-κB. The overexpression of TAK1 and TAK1-binding protein 1 induces nuclear translocation of the NF-κB p50/p65 heterodimer, with the degradation of IκBα and IκBβ (55). Activated TAK1, which was induced via Ubc13-Uev1A-mediated K63-linked polyubiquitination, has been found to specifically phosphorylate IKKβ at S177 and S181 (56). IKKγ binds to the RhoA-RhoGDI complex and promotes the activation of RhoA by destroying the RhoA-RhoGDI complex in response to TGF-β1. Active RhoA-GTP and ROCK phosphorylate IKKβ, followed by the phosphorylation of IκB and p65 to induce the activation of NF-κB (17). In the present study, it was also found that TGF-β1 increased the phosphorylation of TAK1, IKKα/β, IκBα and p65 (Fig. 5) in activated HSCs. The decreased expression of TGF-β1 reduces the DNA-binding activity of NF-κB and decreases the production of fibronectin and chemokines, including IL-8 and monocyte chemoattractant protein 1, in the kidney (57). The present study also demonstrated that TGF-β1 increased the expression of fibronectin and TGF-β1 and formation of F-actin, ultimately enhancing the migration of cells. Additionally, the results demonstrated that TGF-β1-treated HSC-T6 cell migration was regulated via RhoA and NF-κB signaling (Fig. S1).
Taken together, the results of the present study demonstrated that Rap1 contributes to the inactivation of RhoA, thereby inhibiting F-actin formation and NF-κB activity, ultimately regulating HSC migration. These results are important for understanding the activity of TGF-mediated HSCs, the accumulation of ECM and the mechanisms associated with cell migration.
Supplementary Data
Acknowledgments
Not applicable.
Abbreviations
- HSC
hepatic stellate cell
- TGF
transforming growth factor
- SMA
smooth muscle actin
- Col1a1
collagen type I
- GST-Rhotekin-RBD
glutathione S-transferase-Rhotekin-Rho-binding domain
- HIS-RalGDS-RBD
His-Ral guanine nucleotide dissociation stimulator-Rap binding domain
Funding
This study was supported by a grant from the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (grant no. 2017R1C1B1005996), an NRF of Korea grant funded by the Korean Government (grant no. NRF-2014R1A1A3052139) and the Hallym University Research Fund (grant no. HURF-2015-56).
Availability of data and materials
The datasets used and/or analyzed in the present study are available from the corresponding author on reasonable request.
Authors' contributions
SK planned and conceptualized the study; MM, HK and SK designed the study; MM and SK drafted the manuscript; MM, HK and MK performed experiments; MM, HK, MK, JP, KS, JP and SK performed data analysis and interpretation. All authors reviewed the manuscript and approved the final version.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arriazu E, Ruiz de Galarreta M, Cubero FJ, Varela-Rey M, Perez de Obanos MP, Leung TM, Lopategi A, Benedicto A, Abraham-Enachescu I, Nieto N. Extracellular matrix and liver disease. Antioxid Redox Signal. 2014;21:1078–1097. doi: 10.1089/ars.2013.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang C, Zeisberg M, Mosterman B, Sudhakar A, Yerramalla U, Holthaus K, Xu L, Eng F, Afdhal N, Kalluri R. Liver fibrosis: Insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology. 2003;124:147–159. doi: 10.1053/gast.2003.50012. [DOI] [PubMed] [Google Scholar]
- 4.Wang XM, Yu DM, McCaughan GW, Gorrell MD. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. Hepatology. 2005;42:935–945. doi: 10.1002/hep.20853. [DOI] [PubMed] [Google Scholar]
- 5.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 7.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 8.Nobes CD, Hawkins P, Stephens L, Hall A. Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J Cell Sci. 1995;108:225–233. doi: 10.1242/jcs.108.1.225. [DOI] [PubMed] [Google Scholar]
- 9.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 10.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 11.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edlund S, Landstrom M, Heldin CH, Aspenstrom P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Boil Cell. 2002;13:902–914. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JS, Kim JG, Moon MY, Jeon CY, Won HY, Kim HJ, Jeon YJ, Seo JY, Kim JI, Kim J, et al. Transforming growth factor-beta1 regulates macrophage migration via RhoA. Blood. 2006;108:1821–1829. doi: 10.1182/blood-2005-10-009191. [DOI] [PubMed] [Google Scholar]
- 14.Papadimitriou E, Kardassis D, Moustakas A, Stournaras C. TGFβ-induced early activation of the small GTPase RhoA is Smad2/3-independent and involves Src and the guanine nucleotide exchange factor Vav2. Cell Physiol Biochem. 2011;28:229–238. doi: 10.1159/000331734. [DOI] [PubMed] [Google Scholar]
- 15.Li L, Wang JY, Yang CQ, Jiang W. Effect of RhoA on transforming growth factor β1-induced rat hepatic stellate cell migration. Liver Int. 2012;32:1093–1102. doi: 10.1111/j.1478-3231.2012.02809.x. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto Y, Gaynor RB. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Kim HJ, Kim JG, Moon MY, Park SH, Park JB. IκB kinase γ/nuclear factor-κB-essential modulator (IKKγ/NEMO) facilitates RhoA GTPase activation, which, in turn, activates Rho-associated KINASE (ROCK) to phosphorylate IKKβ in response to transforming growth factor (TGF)-β1. J Boil Chem. 2014;289:1429–1440. doi: 10.1074/jbc.M113.520130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bos JL, de Bruyn K, Enserink J, Kuiperij B, Rangarajan S, Rehmann H, Riedl J, de Rooij J, van Mansfeld F, Zwartkruis F. The role of Rap1 in integrin-mediated cell adhesion. Biochem Soc Trans. 2003;31:83–86. doi: 10.1042/bst0310083. [DOI] [PubMed] [Google Scholar]
- 19.Moon MY, Kim HJ, Kim JG, Lee JY, Kim J, Kim SC, Choi IG, Kim PH, Park JB. Small GTPase Rap1 regulates cell migration through regulation of small GTPase RhoA activity in response to transforming growth factor-β1. J Cell Physiol. 2013;228:2119–2126. doi: 10.1002/jcp.24383. [DOI] [PubMed] [Google Scholar]
- 20.Teo H, Ghosh S, Luesch H, Ghosh A, Wong ET, Malik N, Orth A, de Jesus P, Perry AS, Oliver JD, et al. Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat Cell Biol. 2010;12:758–767. doi: 10.1038/ncb2080. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh AS, Tergaonkar V. Telomeres and inflammation: Rap1 joins the ends? Cell Cycle. 2010;9:3834–3835. doi: 10.4161/cc.9.19.13383. [DOI] [PubMed] [Google Scholar]
- 22.Vogel S, Piantedosi R, Frank J, Lalazar A, Rockey DC, Friedman SL, Blaner WS. An immortalized rat liver stellate cell line (HSC-T6): A new cell model for the study of retinoid metabolism in vitro. J Lipid Res. 2000;41:882–893. [PubMed] [Google Scholar]
- 23.Park J, Kim JS, Jung KC, Lee HJ, Kim JI, Kim J, Lee JY, Park JB, Choi SY. Exoenzyme Tat-C3 inhibits association of zymosan particles, phagocytosis, adhesion, and complement binding in macrophage cells. Mol Cells. 2003;16:216–223. [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Ellerbroek SM, Wennerberg K, Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem. 2003;278:19023–19031. doi: 10.1074/jbc.M213066200. [DOI] [PubMed] [Google Scholar]
- 26.Sit ST, Manser E. Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci. 2011;124:679–683. doi: 10.1242/jcs.064964. [DOI] [PubMed] [Google Scholar]
- 27.Spiering D, Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr. 2011;5:170–180. doi: 10.4161/cam.5.2.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marra F, Arrighi MC, Fazi M, Caligiuri A, Pinzani M, Romanelli RG, Efsen E, Laffi G, Gentilini P. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor's actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology. 1999;30:951–958. doi: 10.1002/hep.510300406. [DOI] [PubMed] [Google Scholar]
- 29.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, Heymann F, Weber A, Weiskirchen R, Liedtke C, Gassler N, et al. TAK1 suppresses a NEMO-dependent but NF-kappaB- independent pathway to liver cancer. Cancer Cell. 2010;17:481–496. doi: 10.1016/j.ccr.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 31.Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci USA. 2010;107:844–849. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 33.Luedde T, Schwabe RF. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 35.Benyon RC, Arthur MJ. Mechanisms of hepatic fibrosis. J Pediatr Gastroenterol Nutr. 1998;27:75–85. doi: 10.1097/00005176-199807000-00013. [DOI] [PubMed] [Google Scholar]
- 36.Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 38.Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S. TGF-beta/Smad signaling in the injured liver. Z Gastroenterol. 2006;44:57–66. doi: 10.1055/s-2005-858989. [DOI] [PubMed] [Google Scholar]
- 39.Friedman SL. Liver fibrosis: From mechanisms to treatment. Gastroenterol Clin Biol. 2007;31:812–814. doi: 10.1016/S0399-8320(07)73970-2. [DOI] [PubMed] [Google Scholar]
- 40.Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer. 2007;7:429–440. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DesMarais V, Ghosh M, Eddy R, Condeelis J. Cofilin takes the lead. J Cell Sci. 2005;118:19–26. doi: 10.1242/jcs.01631. [DOI] [PubMed] [Google Scholar]
- 43.Iwamoto H, Nakamuta M, Tada S, Sugimoto R, Enjoji M, Nawata H. A p160ROCK-specific inhibitor, Y-27632, attenuates rat hepatic stellate cell growth. J Hepatol. 2000;32:762–770. doi: 10.1016/S0168-8278(00)80245-7. [DOI] [PubMed] [Google Scholar]
- 44.Kato M, Iwamoto H, Higashi N, Sugimoto R, Uchimura K, Tada S, Sakai H, Nakamuta M, Nawata H. Role of Rho small GTP binding protein in the regulation of actin cytoskeleton in hepatic stellate cells. J Hepatol. 1999;31:91–99. doi: 10.1016/S0168-8278(99)80168-8. [DOI] [PubMed] [Google Scholar]
- 45.Tada S, Iwamoto H, Nakamuta M, Sugimoto R, Enjoji M, Nakashima Y, Nawata H. A selective ROCK inhibitor, Y27632, prevents dimethylnitrosamine-induced hepatic fibrosis in rats. J Hepatol. 2001;34:529–536. doi: 10.1016/S0168-8278(00)00059-3. [DOI] [PubMed] [Google Scholar]
- 46.Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: Adhering to new models. Nat Rev Mol Cell Boil. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 47.Fujita H, Fukuhara S, Sakurai A, Yamagishi A, Kamioka Y, Nakaoka Y, Masuda M, Mochizuki N. Local activation of Rap1 contributes to directional vascular endothelial cell migration accompanied by extension of microtubules on which RAPL, a Rap1-associating molecule, localizes. J Biol Chem. 2005;280:5022–5031. doi: 10.1074/jbc.M409701200. [DOI] [PubMed] [Google Scholar]
- 48.Reedquist KA, Ross E, Koop EA, Wolthuis RM, Zwartkruis FJ, van Kooyk Y, Salmon M, Buckley CD, Bos JL. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J Cell Biol. 2000;148:1151–1158. doi: 10.1083/jcb.148.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katagiri K, Hattori M, Minato N, Irie SK, Takatsu K, Kinashi T. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol. 2000;20:1956–1969. doi: 10.1128/MCB.20.6.1956-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tohyama Y, Katagiri K, Pardi R, Lu C, Springer TA, Kinashi T. The critical cytoplasmic regions of the alphaL/beta2 integrin in Rap1-induced adhesion and migration. Mol Boil Cell. 2003;14:2570–2582. doi: 10.1091/mbc.e02-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jeon CY, Kim HJ, Lee JY, Kim JB, Kim SC, Park JB. p190RhoGAP and Rap-dependent RhoGAP (ARAP3) inactivate RhoA in response to nerve growth factor leading to neurite outgrowth from PC12 cells. Exp Mol Med. 2010;42:335–344. doi: 10.3858/emm.2010.42.5.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krugmann S, Williams R, Stephens L, Hawkins PT. ARAP3 is a PI3K- and rap-regulated GAP for RhoA. Curr Boil. 2004;14:1380–1384. doi: 10.1016/j.cub.2004.07.058. [DOI] [PubMed] [Google Scholar]
- 53.Mao R, Fan Y, Mou Y, Zhang H, Fu S, Yang J. TAK1 lysine 158 is required for TGF-β-induced TRAF6-mediated Smad-independent IKK/NF-κB and JNK/AP-1 activation. Cell Signal. 2011;23:222–227. doi: 10.1016/j.cellsig.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Boil. 2008;10:1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 55.Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB. TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Boil. 2003;326:105–115. doi: 10.1016/S0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 56.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 57.Qi W, Chen X, Holian J, Mreich E, Twigg S, Gilbert RE, Pollock CA. Transforming growth factor-beta1 differentially mediates fibronectin and inflammatory cytokine expression in kidney tubular cells. Am J Physiol Renal Physiol. 2006;291:F1070–F1077. doi: 10.1152/ajprenal.00013.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed in the present study are available from the corresponding author on reasonable request.