

Abstract
The diverse chemistry possible with flavin cofactors positions flavin-dependent enzymes as versatile synthetic tools. This focused review highlights applications of flavin-dependent enzymes in organic synthesis. Select examples of monoamine oxidases, ene-reductases, monooxygenases and halogenases in target-oriented synthesis are presented.
Introduction:
Flavin-dependent enzymes are essential in primary metabolism in all kingdoms of life, performing a plethora of chemical reactions.1 The versatile flavin cofactor that defines this enzyme family is capable of mediating both oxidative and reductive processes depending on the form of the cofactor employed. Further diversifying the chemistry possible with flavoproteins, Nature has evolved flavin-dependent enzymes to carry out specialized chemistry within secondary metabolic pathways.2 Chemists have begun to tap into this incredible set of catalysts, exploring the chemistry possible with various classes of flavin-dependent enzymes. These endeavors capture the advantages and challenges in biocatalytic synthesis.
Selectivity, tunability, safety and sustainability can motivate chemists to choose biocatalytic methods over traditional chemical catalysts and reagents.3 For flavin-dependent enzymes, chemo-, site- and stereoselectivity can often be achieved at a higher level or with orthogonal selectivity to that attainable with small molecule reagents.4 Mediating oxidation reactions with flavin-dependent enzymes presents the opportunity to use molecular oxygen as the stoichiometric oxidant and similarly economical and abundant stoichiometric reductants in reductive transformations. Additionally, the ability to carry out reactions in water without the need for extreme temperatures or a controlled atmosphere adds to the procedural simplicity of biocatalytic chemistry. Despite these advantages, synthetic chemists have been slow to fully embrace biocatalysis in preparative-scale synthesis. The barriers to incorporating flavin-dependent biocatalysts in a synthetic route can include access to the biocatalyst, limited or unknown substrate scope of an enzyme, or catalyst stability. As the biocatalysis community expands, these obstacles are disappearing. More enzymes are commercially available than ever, and the technology for evolving proteins to access variants with an altered substrate scope and increased stability has been established.5
Decades of experimental and computational investigation of flavin-dependent enzyme mechanisms have provided a framework for understanding the reactions possible from each flavin state. Figure 1 illustrates a portion of the flavin states relevant to flavoprotein chemistry.6 Starting from the oxidized form of the cofactor flavin-adenine dinucleotide (FAD, 1) or flavin mononucleotide (FMN, 2) addition of a single electron affords the anionic flavin semiquinone (3). Further one electron reduction of the semiquinone form (3) or two electron reduction of 1 or 2 provides access to FADH2 (5) or FMNH2 (6), respectively. NAD(P)H is the most common native cofactor used for the reduction of FAD, however this substrate is costly and unstable. As such, a number of NAD(P)H recycling systems have been developed for reactions which generate FAD from FADH2. This reduced form of the cofactor can engage molecular oxygen to form peroxyflavin 7, which upon protonation yields hydroperoxyflavin 9. The protein scaffold and reaction conditions dictate the forms of the cofactor accessible and the chemistry that can be carried out in a given system. Herein, select examples of flavin-dependent enzymes in organic synthesis are presented. This work is organized according to the form of the flavin cofactor involved in each reaction.
Figure 1.
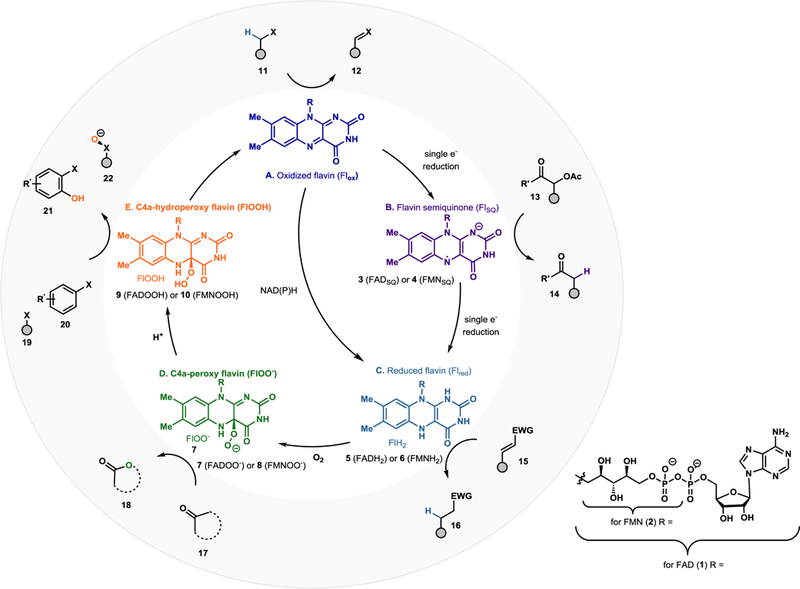
Catalytically active species of flavin (flavin-adenine dinucleotide and flavin mononucleotide) referenced in this review. (A) Flox can act as a one or two electron acceptor to afford the semiquinone or reduced flavin (Flred). (B) Flred in the context of ene-reductases can perform the two-electron reduction of activated olefins. Flred may also react with molecular oxygen to form the C4a-peroxy (D) or hydroperoxy flavin (E) which can be the source of an oxygen atom for incorporation into organic substrates. Elimination of peroxide affords Flox..
Oxidized flavin (Flox).
The oxidized form of the flavin cofactor, FAD or FMN (Flox see 1 and 2), can participate in reduction of substrates (Fig. 1A). In the context of complex molecule synthesis, the chemistry of Flox has been most widely exploited in the synthesis of imines from amine substrates by monoamine oxidases (MAOs).7 MAOs have been applied to the dynamic resolution of racemic amines as outlined in Fig. 2A.8,9 MAOs are often selective for a single enantiomer of the amine substrate, enriching for the enantiomer not oxidized by the biocatalysts. Coupling the oxidase with a reducing reagent leads to a dynamic resolution. For example, Turner and coworkers have engineered a MAO from Aspergillus niger to create a panel of MAO variants with complementary substrate scope.9 By applying both directed evolution and rational design strategies, useful catalysts for the resolution of alkyl, cyclic and benzylic amines have been generated.9–11
Figure 2.
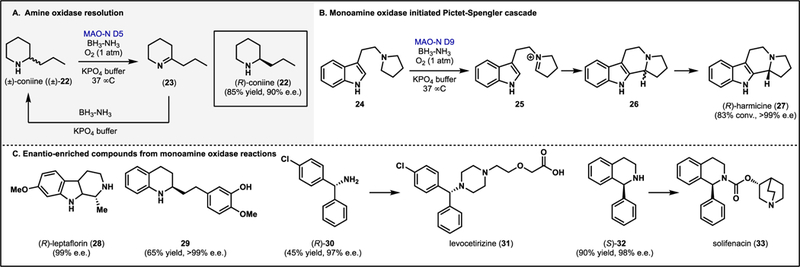
Monoamine oxidases (MAOs) in the synthesis of enantio-enriched amines. (A) Dynamic resolution of amines using MAO-N variants and a reducing agent. (B) Monoamine oxidase initiated Pictet-Spengler cascade for the synthesis of (R)-harmicine (27).9 (C) Enantio-enriched amines accessible through MAO resolution.9
The utility of these biocatalysts has been demonstrated through the synthesis of natural products as well as enantioenriched precursors to active pharmaceutical ingredients (APIs). Notably, a number of enantioenriched alkaloid natural products were obtained through this deracemization approach including the neurotoxin (R)-coniine (22), plant alkaloid (R)-leptaflorin (28) and anti-Leishmania compound (R)-harmicine (27) (Fig. 2).9 This panel of products demonstrate the stereochemical preference of MAO-N variants for the (S)-enantiomer of these amine substrates, thus enriching for the (R)-enantiomer of each amine. Further studies have demonstrated the potential for MAO-N variants to enrich for the (S)-enantiomer with specific substrate classes8 or through protein engineering.9 MAO-mediated deracemization of complex amines provides a valuable alternative to traditional approaches, which require asymmetric installation of the amine moiety at an earlier stage in the synthesis. In addition to the resolution of alkaloid natural products, MAO deracemization has been used to access enantioenriched intermediates in the synthesis of amine-containing drugs. Specifically, the MAO-D11 variant can discriminate between two similar aryl groups to afford 4-chlorobenzhydrylamine (30) in 97% ee, which can subsequently be elaborated to the antihistamine drug, levocetirizine (31).8 Similarly, solifenacin precursor (S)-tetrahydroisoquinoline 32 can be accessed in 98% ee. by the same MAO-N variant.8 Amine oxidases beyond MAO-N have also been explored for synthetic applications. For example, enantio-enriched tetrahyroquinolines such as 29 can be obtained through deracemization with cyclohexylamine oxidase (CHAO).12 MAOs can also be applied beyond simple deracemization reactions. For example, MAO-generated iminium ions such as 25 can be intercepted in a Pictet-Spengler type bond forming event (Fig. 2B),9 and cyclic amines can be aromatized using MAOs.13 These are just select examples of the synthetic potential of these enzymes, in an area ripe for innovation.
FlSQ.
Single electron reduction of Flox provides the flavin semiquinone (FlSQ, 3). FlSQ has been extensively studied in a number of flavin-dependent enzymes, including glucose oxidase14 and methanol oxidase.15 Despite the accessibility of the flavin radical, there have been few reports of synthetic methods that proceed via a single electron mechanism. Recently, Sandoval and coworkers reported the ene-reductase mediated radical dehalogenation of α-bromo esters.16 They propose a mechanism in which reduced flavin (FMNH2, 6) performs a single electron transfer to the substrate; the resulting flavin semiquinone serves as a hydrogen atom donor to afford the dehalogenated products. Mutation of Y177 in G. oxydans ene-reductase (GluER), which protonates alkenes in the native reaction of this enzyme, to alanine improved the yield and also enantioselectivity of the abiotic transformation. Flavin cofactors are typified by their ability to undergo both one and two electron redox transformations; however, this is the first application of a single-electron process to a synthetically relevant transformation. With the groundwork laid by this report we anticipate future biocatalytic methods will take advantage of radical flavin species.
Flred. Ene reductases.
Ene-reductases (ERs) are a large class of enzymes characterized by their capacity to reduce activated olefins including enones, enals, unsaturated carboxylic acids, esters and cyclic imides.17 Enantioenriched amino acid derivatives, important polymer precursors, and chiral building blocks have been synthesized using ERs as a key synthetic step.17 The Old Yellow Enzyme (OYE) family of flavoproteins belong to this class and have been employed in a large number of reaction cascades and industrially-relevant processes due to their substrate promiscuity and excellent stereoselectivity.18,19 The mechanism of OYEs in the asymmetric reduction of olefins has been well-studied,20 and is understood to involve the delivery of a hydride from flavin mononucleotide (FMNH2, 6) to the β-position of the substrate which is coordinated by a strictly conserved histidine/asparagine or histidine/histidine pair (Figure 3A). To arrive at the product, tyrosine protonates the alpha position on the face opposite to the flavin cofactor.20
Figure 3.
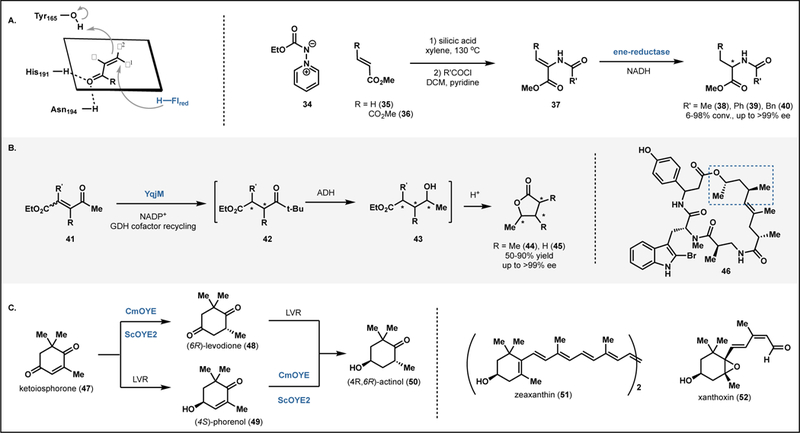
Reactions with ene-reductases. (A) Chemoenzymatic synthesis of amino acid derivatives via an ene-reductase-catalyzed hydrogenation.21 (B) One-pot three-step cascade to enantioenriched γ-butyrolactones.22 (C) One-pot, two-step cascade to generate actinol (50).23
Stueckler and coworkers devised an approach that highlights the utility of this class of flavoproteins in the synthesis of stereochemically dense small moleucles.21 They reported a chemoenzymatic route to N-acyl amino acid derivatives in which N-acetylamino acrylic esters were reduced in modest yields by OYE1–3, YqjM and OPR1, to afford products in 85–95% ee (Fig. 3A, see 38-40).21 Only terminal olefins (such as 35) were productive substrates, as compounds with additional substitution of the double bond gave no conversion to the corresponding saturated product with any of the enzymes tested. However, one exception to this rule was identified; diester 36 was converted quantitatively and with perfect stereoselectivity to afford the (S)-isomer of 38 by OYE1 and OYE3. Sterically bulky amides were also converted to products in good yields and enantioselectivities.
In 2014, Classen and coworkers reported a chemoenzymatic cascade which afforded access to γ-butyrolactones.22 Employing YqjM, a homolog of OYE involved in E. coli response to oxidative stress, substrates such as 41 were reduced to generate a substrate which could be intercepted by a second enzyme (Fig. 3B). For example, in a first step, α,β-unsaturated 4-oxo-esters (41), which can be accessed through a Wittig olefination of commercially available starting materials, undergo a formal anti-addition of hydrogen by enolate reductase to afford anti- or syn-γ-ketoester products (see 42). While the (E)-isomer typically gives rise to the anti-product and the (Z)-isomer gives rise to syn-product, this trend did not extend to all cases. Although a predictive model for YqjM stereoselectivity has not been reported to date, the authors employed deuterium-labeling and docking studies to understand the irregularities in stereoselectivity. Substrate activation occurs though hydrogen bonding to the most electron-withdrawing substituent. Umpolung selectivity was observed for substrates with particularly sterically demanding substituents proximal to the most electron-withdrawing substituent. Following YqjM reduction, intermediate 42 can be intercepted by an alcohol dehydrogenase (ADH) in a chemo- and stereoselective reduction of the ketone group and subsequent acid-catalyzed cyclization afford the lactone product 44 with high ee (>98%) and moderate to good yields. This methodology provides access to trisubstituted lactones with three contiguous stereocenters. Classical synthetic routes to this motif require multiple steps, and a general method for accessing various substitution patterns and stereoisomers has yet to be reported, as most methods require chiral pool reagents as a starting point.
A similar strategy was employed by Horita in the chemoenzymatic synthesis of enantioenriched actinol (50) from ketoisophorone (47).23 (4R,6R)-actinol (50) is a valuable chiral building block that serves as an intermediate in the synthesis of carotenoids such as xanthoxin, zeaxathin. The biocatalytic cascade begins with the Candida macedoniensis OYE (CmOYE) reduction of ketoisophorone (47) to (6R)-levodione (48) followed by the reduction to actinol (50) by (6R)-levodione reductase. However, when these enzymes were implemented in a one-pot system, the authors noted 67% conversion to the final product with the balance of the material accumulating as an intermediate, (4S)-phorenol (49). This was attributed to the low activity of CmOYE on this reduced species. By obtaining crystal structures with and without substrate mimic p-hydroxybenzaldehyde bound, the authors identified a flexible loop that appeared to act as a lid on the active site when substrate was present. Modeling of (4S)-phorenol in the active site showed potential steric clashes with the gem-dimethyl group at C6 and Phe250, Pro295, and Phe296. These residues were individually mutated to Gly and each of these variants exhibited higher catalytic activity with phorenol, although substitution of these Phe residues also led to lower activity with ketoisophorone (47). The CmOYE(P295G) variant was used in the one-pot, two-step biocatalytic reaction to afford actinol (50) in 90% yield.
Innovations in the application of ene-reductases continue to be reported. For example, Hartwig recently disclosed a ‘cooperative’ chemoenzymatic reaction in which a mixture of E/Z olefins are isomerized photocatalytically to the (E)-isomer preceding a stereoselective ene-reductase mediated reduction.24
C4a-peroxy flavin (FlOO-):
Flavin-dependent monooxygenases are a large family of enzymes that can be grouped into classes A-H based on structure and activity.25 Monooxygenases access the reactive flavin intermediate from reactions of FADH2 with molecular oxygen. The reaction between FADH2 and O2 can form peroxyflavin (7), providing a nucleophilic source of oxygen (Fig. 4A). In reactions, one atom of oxygen is incorporated into the organic substrate, whereas, the second atom of oxygen from O2 is lost as water. Within this family, Baeyer-Villiger monooxygenases (BVMOs)26 have received the most attention from the synthetic community. In addition to their canonical capacity to perform stereoselective oxidation of carbonyl containing compounds, BVMOs are also able to perform epoxidations27, sulfoxidations28,29 and amine oxidations30 with excellent stereocontrol. BVMOs have been employed in the chemoenzymatic synthesis of important pharmaceutical agents, such as modafinil31 and esomeprazole,32 and in the dynamic kinetic resolution of chiral building blocks employed in the synthesis of epothilones33 and isocoumarins.34 The process relevance of these transformations was highlighted in a 200 L-scale whole-cell biotransformation.35
Figure 4.
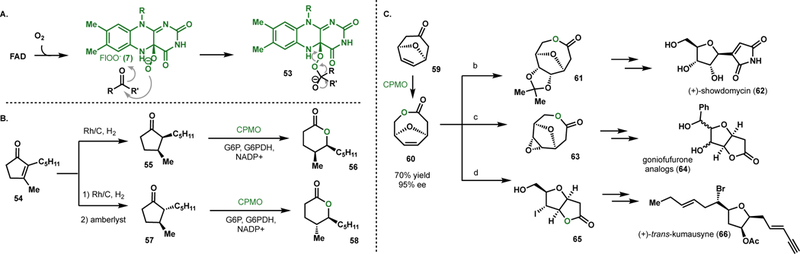
Reactions with BVMOs. (A) General mechanism for BVMOs. (B) Aerangis lactone synthesis featuring a stereoselective BV oxidation by cyclododecanone monooxygenase (CDMO) or cycopentanone monooxygenase (CPMO). (C) Chemoenzymatic synthesis of tetrahydrofuran-based natural products via a chiral intermediate generated by CPMO-mediated Baeyer-Villager oxidation a) OsO4, NMO, DCM, rt, then acetone–AlCl3, 0 °C to 40 °C, 47%; b) mCPBA, DCM, reflux, 98%; c) MeCN, H2O, KOH, rt, then I2–KI, 40 °C, dark, 75%.
BVMOs have been employed in chemoenzymatic cascades to generate enantioenriched products. Fink reported the chemoenzymatic synthesis of the aroma compound Aerangis lactone.36 Rh/C hydrogenation in continuous flow of cyclopentenone 54 afforded selectively the cis-cyclopentanone 55 (Fig. 4B). Acid-mediated isomerization gave rise to the theromodynamically favored trans-isomer. Crude cell extract of cyclododecanone monooxygenase (CDMO) catalyzed the kinetic resolution of 56 with >99% ee. Cyclopentanone monooxygenases (CPMO) afforded the trans-lactone 58 in >99% ee. Using 2% w/v biocatalyst at 20 mM or 15 mM substrate concentration afforded lactone product 58 in 27% or 28% yield, respectively.
The utility of BVMOs to generate chiral building blocks for multistep synthesis of valuable complex molecules has been explored by a number of groups. Mihovilovic et al. reported the formal chemoenzymatic synthesis of tetrahydrofuran-based natural products using BVMOs as a key synthetic step (Fig. 4C).37 [4+3] cycloaddition of furan and tetrabromoacetone followed by reductive dehalogenation afforded prochiral oxabicycloketone 59. This bicycle was subjected to fermentation with cyclopentanone monooxygenase (CPMO)-expressing E. coli. Using substrate feeding and product removal (SFPR) allowed for a 70% isolated yield of ester 60 in 95% ee. Subsequent chemical steps allowed the authors to intercept pivotal intermediates in the total syntheses of showdomycin (62), trans-kumausyne (66), and goniofufurone (64) analogs. Upjohn dihydroxylation of intermediate 60 and subsequent protection afforded acetal 61. In a separate synthesis, the chiral intermediate also underwent epoxidation to afford the goniofufurone precursor 63. The hydrolysis of the lactone followed by halolactonization gave access to the trans-kumausyne precursor 65. This work demonstrated complexity-generating potential of the biooxidative desymmetrization of the simple ketone 59.
BVMOs have also been employed for in vivo enzymatic cascades. Engineered E. coli harboring three biocatalytic enzymes allowed for the in vivo cascade starting from 2-cyclohexenol to caprolactone, featuring CHMO catalyzed Baeyer-Villager oxidation as the final step.38 In addition to this canonical reactivity, CHMO has been reported to perform hydroxylation of boronic acids, and the stereoselective epoxidation of dimethyl and diethyl vinyl phosphonate.27 Interestingly, CHMO has also been demonstrated to perform heteroatom oxidation, presumably from the hydroperoxy flavin.27
C4a-Hydroperoxy flavin (FlOOH):
While BVMOs use peroxyflavin (7) to oxidize organic substrates, group A flavin-dependent monooxygenases perform hydroxylation on electron-rich substrates using the hydroperoxyflavin form of the cofactor (8). The prototypical group A member, p-hydroxybenzoate 3-hydroxylase (PHBH), has been well-studied.39 This work has led to an understanding of the mechanism of substrate recognition, cofactor binding and flavin reduction. Recently, group A members from biosynthetic pathways have received attention for their synthetic potential.40,41
Sorbicilin natural products have been of interest to the synthetic community for nearly two decades due to their dense structural complexity and biological activity.42 The shortest linear sequences to these natural products have employed an oxidative dearomatization as a key synthetic step; however, due to the lack of a general asymmetric method for this transformation, only racemic syntheses have been achieved via this route.42 Recently, Sib and coworkers employed SorbC, a flavin-dependent monooxygenase from the sorbicillactone biosynthetic pathway,43 as key synthetic step in the chemoenzymatic synthesis of bisorbicillinol (72), trichodimerol (73) and sorbiquinol 71.40 Sorbicillin (67) was transformed in a SorbC-mediated oxidative dearomatization to afford sorbicillinol (68) in >99.5% ee. In the presence of different organic cosolvents, sorbicillinol (68) could be selectively dimerized to afford the natural products 71, 72, or 73 (Fig. 5A).
Figure 5.

Reactions of group A flavin-dependent monooxygenases. (A) Access to sorbicilin natural products via oxidative dearomatization. (B) Biocatalytic route to azaphilone natural products. (C) Enzymatic cascade to form stipitaldehyde (79).
We have employed SorbC in a chemoenzymatic route to urea sorbicillin 70 by employing a non-sorbicilinol cycloaddition partner (Fig. 5A).41 Using hydantoin derivative 69, the Diels-Alder reaction with sorbicilinol (68) proceeded at 80 °C. Subsequent removal of the acetate groups afforded urea sorbicillin 70. A homologous monooxygenase, AzaH, provides orthogonal site and facial selectivity to SorbC on the resorcinol scaffold, providing access to C3-hydroxylated products such as 75. Using AzaH, the azaphilone natural product scaffold was obtained through oxidative dearomatization of ketone 74 followed by spontaneous cyclization to afford natural product 75 in 52% yield. A third group A flavin-dependent monooxygenase, TropB, provides the same site selectivity as AzaH but differs in substrate scope. TropB-generated ortho-quinol 77 was enzymatically elaborated to the tropolone natural product stipitaldehyde (79) through selective hydroxylation and ring-expansion mediated by α-ketoglutarate-dependent non-heme iron(II)-dependent dioxygenase TropC.
Halogenases:
Group F flavin-dependent monooxygenases are structurally related to class A monooxygenases and follow the same catalytic sequence up to the formation of the C4a-hydroperoxide (FlOOH, 8). Halogenases bind halide ions which are oxidized by the C4a-hydroperoxide to form hypochlorous or hypobromous acid (Figure 6A, 80).44 As the substrate binding site is distant from the flavin binding site, researchers hypothesize that the acid traverses a tunnel to the substrate binding site where it reacts with a lysine residue to form a covalent chloramine adduct (81), in the case of chlorination.44 This electrophilic chlorine species is thought to undergo nucleophilic attack by an aromatic substrate (83). The Wheland intermediate is then deprotonated by a conserved Glu residue to form the chlorinated product (84).
Figure 6.

Reactions of halogenases. (A) General reaction mechanism for halogenase-mediated halogenation (EAS = Electrophilic aromatic substitution). (B) Halogenation, Pd-catalyzed Suzuki coupling cascade for late-stage functionalization of indole natural product scaffolds.45 (C) Evolution of RebH variants using substrate walking from simple indole derivatives to pentacyclic alkaloids.46
Select flavin-dependent halogenases have received significant attention from the synthetic community for their ability to halogenate aromatic systems under mild reaction conditions with excellent site selectivity. For example, Durak and coworkers demonstrated the application of enzymatic halogenation of indole derivatives in conjunction with Pd-catalyzed cross coupling reactions on crude extracts of reaction mixtures to afford further functionalized products (Fig. 6B, see 85 to 87).45 The parent enzyme, RebH, was evolved using a substrate-walking directed evolution strategy to obtain variants capable of halogenating a broad range of large indole and carbazole substrates (Fig. 6C, 88-91).46 Recently, Fraley and coworkers characterized halogenase MalA from the malbrancheamide biosynthetic pathway, which performs iterative halogenation of the bicyclo[2.2.2]diazaoctane core.47 These enzymes hold promise for future use in the selective halogenation of indoles and other electron-rich arenes.
The breadth of chemistry accessible with flavin-dependent biocatalysts coupled with the demonstrated scalability of these reactions positions this enzyme class to make major contributions to synthetic organic chemistry. Current applications have largely been constrained to native reaction mechanisms; however, with an ever-increasing understanding of the structural and function of these enzymes, synthetic chemists have the opportunity to employ these catalysts in novel reactions and integrate them into complexity-generating cascade sequences.
References
- 1 ).MacHeroux P; Kappes B; Ealick SE FEBS J. 2011, 278 (15), 2625–2634. [DOI] [PubMed] [Google Scholar]
- 2 ) A).Mansoorabadi SO; Thibodeaux CJ; Liu HW J. Org. Chem. 2007, 72 (17), 6329–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]; B) Fagan RL and Palfey BA In Comprehensive Natural Products II; Begley TP, Ed.; Elsevier, 2010; Vol 7, pp. 37–114. [Google Scholar]
- 3 ).Sheldon RA; Woodley JM Chem. Rev. 2018, 118 (2), 801–838. [DOI] [PubMed] [Google Scholar]
- 4 ).Ceccoli RD; Bianchi DA; Rial DV Front. Microbiol. 2014, 5, 1–0]14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5 ).Arnold FH Angew. Chem., Int. Ed. 2018, 57 (16), 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6 ).Palfey BA; and Massey V In Comprehensive Biological Catalysis; Sinnott M, ed.; Academic Press, 1998; Vol. 3, pp. 83–154 [Google Scholar]
- 7 ).Scrutton NS Nat. Prod. Rep. 2004, 21 (17), 722–730. [DOI] [PubMed] [Google Scholar]
- 8 ).Ghislieri D; Houghton D; Green AP; Willies SC; Turner NJ ACS Catal. 2013, 3 (12), 2869–2872. [Google Scholar]
- 9 ).Ghislieri D; Green AP; Pontini M; Willies SC; Rowles I; Frank A; Grogan G; Turner NJ J. Am. Chem. Soc. 2013, 135 (29), 10863–10869. [DOI] [PubMed] [Google Scholar]
- 10 ).Heath RS; Pontini M; Bechi B; Turner NJ ChemCatChem 2014, 6 (4), 996–1002. [Google Scholar]
- 11 ).Li GY; Yao PY; Gong R; Li JL; Liu P; Lonsdale R; Wu QQ; Lin JP; Zhu DM; Reetz MT Chem. Sci. 2017, 8 (5), 4093–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12 ).Cosgrove SC; Hussain S; Turner NJ; Marsden SP ACS Catal. 2018, 8 (6), 5570–5573. [Google Scholar]
- 13 ).Scalacci N; Black GW; Mattedi G; Brown NL; Turner NJ; Castagnolo D ACS Catal. 2017, 7 (2), 1295–1300. [Google Scholar]
- 14 ).Stankovich MT; Schopfer LM; Massay VJ Biol. Chem. 1978, 253 (14), 4971–4979. [PubMed] [Google Scholar]
- 15 ).Mincey T; Tayrien G; Mildvan a S.; Abeles RH Proc. Natl. Acad. Sci. U. S. A. 1980, 77 (12), 7099–7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16 ).Sandoval BA; Meichan AJ; Hyster TK J. Am. Chem. Soc. 2017, 139 (33), 11313–11316. [DOI] [PubMed] [Google Scholar]
- 17 ).Knaus T; Toogood HS; Scrutton NS In Green Biocatalysis; Patel RN, Ed.; John Wiley & Sons: Hoboken, NJ, 2016; pp 473–488. [Google Scholar]
- 18 ).Winkler CK; Faber K; Hall M Curr. Opin. Chem. Biol. 2018, 43, 97–105. [DOI] [PubMed] [Google Scholar]
- 19 ).Durchschein K; Hall M; Faber K Green Chem. 2013, 15 (7), 1764–1772. [Google Scholar]
- 20 ).Kohli RM; Massey VJ Biol. Chem. 1998, 273 (49), 32763–32770. [DOI] [PubMed] [Google Scholar]
- 21 ).Stueckler C; Winkler CK; Hall M; Hauer B; Bonnekessel M; Zangger K; Faber K Adv. Synth. Catal. 2011, 353 (7), 1169–1173. [Google Scholar]
- 22 ).Classen T; Korpak M; Schölzel M; Pietruszka J ACS Catal. 2014, 4 (5), 1321–1331. [Google Scholar]
- 23 ).Horita S; Kataoka M; Kitamura N; Nakagawa T ChemBioChem 2015, 8502, 440–445. [DOI] [PubMed] [Google Scholar]
- 24 ).Litman ZC; Wang Y; Zhao H; Hartwig JF Nature 2018, 560 (7718), 355–359. [DOI] [PubMed] [Google Scholar]
- 25 ).Huijbers MME; Montersino S; Westphal AH; Tischler D; Van Berkel WJ H. Arch. Biochem. Biophys. 2014, 544, 2–17. [DOI] [PubMed] [Google Scholar]
- 26 ).Leisch H; Morley K; Lau PCK Chem. Rev. 2011, 111 (7), 4165–4222. [DOI] [PubMed] [Google Scholar]
- 27 ).Colonna S; Gaggero N; Carrea G; Ottolina G 2002, 43, 1797–1799. [Google Scholar]
- 28 ).De Gonzalo G; Torres Pazmiño DE; Ottolina G; Fraaije MW; Carrea G Tetrahedron Asymmetry 2005, 16 (18), 3077–3083. [Google Scholar]
- 29 ).Sheldon RA; Woodley JM Chem. Rev. 2018, 118 (2), 801–838. [DOI] [PubMed] [Google Scholar]
- 30 ).Branchaud BP; Walsh CT J. Am. Chem. Soc. 1985, 107 (7), 2153–2161. [Google Scholar]
- 31 ).Riva S, Fassi P, Allegrini P & Razzetti G European Patent Application, EP 1777295 (2007).
- 32 ).Bong YK; Song S; Nazor J; Vogel M; Widegren M; Smith D; Collier SJ; Wilson R; Palanivel SM; Narayanaswamy K; Mijts B; Clay MD; Fong R; Colbeck J; Appaswami A; Muley S; Zhu J; Zhang X; Liang J; Entwistle DJ Org. Chem. 2018, 83 (14), 7453–7458. [DOI] [PubMed] [Google Scholar]
- 33 ).Ödman P; Wessjohann LA; Bornscheuer UT J. Org. Chem. 2005, 70 (23), 9551–9555. [DOI] [PubMed] [Google Scholar]
- 34 ).Rioz-Martínez A; De Gonzalo; Torres Pazmiño DE; Fraaije MW; Gotor V J. Org. Chem. 2010, 75 (6), 2073–2076. [DOI] [PubMed] [Google Scholar]
- 35 ).Baldwin CVF; Wohlgemuth R; Woodley JM Org. Process Res. Dev. 2008, 8 (7), 660–665. [Google Scholar]
- 36 ).Fink MJ; Schön M; Rudroff F; Schnürch M; Mihovilovic MD ChemCatChem 2013, 5 (3), 724–727. [Google Scholar]
- 37 ).Mihovilovic MD; Bianchi DA; Rudroff F Chem. Commun. 2006, 8 (30), 3214–3216. [DOI] [PubMed] [Google Scholar]
- 38 ).Oberleitner N; Peters C; Muschiol J; Kadow M; Saß S; Bayer T; Schaaf P; Iqbal N; Rudroff F; Mihovilovic MD; Bornscheuer UT ChemCatChem 2013, 5 (12), 3524–3528. [Google Scholar]
- 39 ).Entsch B; Palfey BA; Ballou DP; Massey VJ Biol. Chem. 1991, 266, 17341. [PubMed] [Google Scholar]
- 40 ).Sib A; Gulder TA M. Angew. Chem. Int. Ed. 2017, 56 (42), 12888–12891. [DOI] [PubMed] [Google Scholar]
- 41 ).Baker Dockrey SA; Lukowski AL; Becker MR; Narayan AR H. Nat. Chem. 2018, 10 (2), 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42 ).Nicolaou KC; Vassilikogiannakis G; Simonsen KB; Baran PS; Zhong YL; Vidali VP; Pitsinos EN; Couladouros EA J. Am. Chem. Soc. 2000, 122 (13), 3071–3079. [Google Scholar]
- 43 ).Al Fahad A; Abood A; Fisch KM; Osipow A; Davison J; Avramović M; Butts CP; Piel J; Simpson TJ; Cox RJ Chem. Sci. 2014, 5 (2), 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44 ).Dong C, Flecks S, Unversucht S, Haupt C, Van Pee KH, & Naismith JH 2012, 309 (5744), 2216–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45 ).Durak LJ; Payne JT; Lewis JC ACS Catal. 2016, 6 (3), 1451–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46 ).Payne JT; Poor CB; Lewis JC Angew. Chem., Int. Ed. 2015, 54 (14), 4226–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47 ).Fraley AE; Garcia-Borràs M; Tripathi A; Khare D; Mercado-Marin EV; Tran H; Dan Q; Webb GP; Watts KR; Crews P; Sarpong R; Williams RM; Smith JL; Houk KN; Sherman DH J. Am. Chem. Soc. 2017, 139 (34), 12060–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]