

Abstract
A female child with deafness was diagnosed to have neonatal diabetes mellitus at the age of 6 months, on routine evaluation prior to cochlear implant surgery. She presented to us at 11 months of age with diabetic ketoacidosis due to an intercurrent febrile illness. Her haematological parameters showed megaloblastic anaemia and thrombocytopenia. Therefore a possibility of Thiamine Responsive Megaloblastic Anaemia (TRMA) syndrome was considered. She was empirically treated with parenteral thiamine hydrochloride (Hcl). Subsequently, due to the unavailability of pharmacological preparation of oral thiamine Hcl in a recommended dose she was treated with benfotiamine. She had a sustained improvement in all her haematological parameters on oral benfotiamine. The insulin requirement progressively reduced and she is currently in remission for last 2 years. The genetic analysis confirmed the diagnosis of TRMA syndrome. Thus benfotiamine can be considered a new treatment option in management of TRMA syndrome.
Keywords: endocrine system, paediatrics (drugs and medicines), diabetes, haematology (incl blood transfusion), neonatal and paediatric intensive care
Background
Thiamine Responsive Megaloblastic Anaemia (TRMA) syndrome is a rare autosomal recessive disorder characterised by the cardinal triad of neonatal diabetes mellitus (NDM), megaloblastic anaemia and progressive sensorineural hearing loss.1 Thus every case of NDM requires screening for megaloblastic anaemia. Diabetes, in most of the patients with TRMA, remits with thiamine therapy. Although thiamine hydrochloride (Hcl) is the recommended pharmacological therapy, oral benfotiamine could be considered as an alternative treatment option for the management of TRMA syndrome.
Case presentation
An 11-month-old female child, diagnosed to have neonatal diabetes at the age of 6 months, was admitted to the paediatric services with diabetic ketoacidosis (DKA) (random plasma glucose: 24 mmol/L (432 mg/dL); venous pH: 7.29, HCO-3: 5.5 mEq/L, urine ketones: 3+) due to intercurrent febrile illness. On physical examination, she was irritable, had pallor with petechial rash over the body. Patient was tachypnoic (respiratory rate of 44/minute). She was managed with the institutional DKA management protocol. After she stabilised, the child was shifted to insulin glargine twice a day and injection lispro at meal times. Her total daily dose requirement was 1.5 U/kg/day.
The child was born to non-consanguineously married Muslim parents. Around the age of 5 months, the parents noticed that the child was not responding to sounds. On Brainstem Evoked Response Audiometry she had severe to profound sensorineural hearing loss for which cochlear implant surgery was considered. During the preoperative evaluation, she was detected to have a high fasting plasma glucose of 12.2 mmol/L (216 mg/dL) and an elevated HbA1C of 68 mmol/mol (8.4%). Further evaluation revealed that basal C-peptide was 0.47 ng/mL and Glutamate decarbxylase (GAD) antibody was negative. Prior to the current admission, she was initiated on insulin glargine. Detailed family history revealed that, one of her maternal uncles was diagnosed to have diabetes and deafness at the age of 8 years, expired soon after the diagnosis, due to DKA.
Investigations
Even after recovery from the acute febrile illness, she had persistent thrombocytopenia and macrocytic anaemia. Her vitamin B12 and folic acid levels were normal. Therefore a bone marrow examination was performed which showed megaloblastic bone marrow (figure 1). The above constellation of clinical (neonatal diabetes, deafness) and laboratory findings (megaloblastic anaemia) suggested the diagnosis of TRMA syndrome. Her blood sample was sent for genetic analysis.
Figure 1.
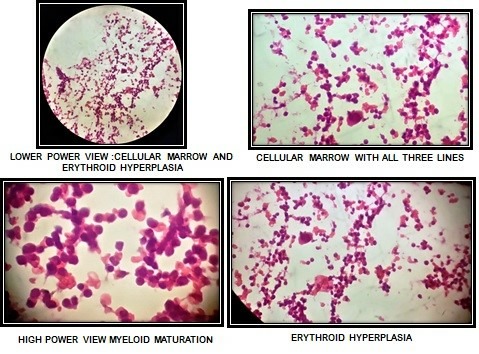
Bone marrow examination.
Treatment
The child was first initiated on an infusion of 100 mg of thiamine Hcl followed by intramuscular injection of 50 mg daily for 6 days. Thereafter, we administered oral benfotiamine 50 mg a day due to unavailability of oral preparation of thiamine. She maintained euglycemia for 48 hours without insulin after the first infusion of thiamine Hcl. However, she decompensated subsequently and required 2 units of inj. glargine a day (0.3 U/kg/day) to maintain euglycemia (5.5–8.3 mmol/L). Subsequently we could gradually taper and omit the insulin at the time of discharge.
Outcome and follow-up
Within a week, the haematological parameters also improved. This supported our clinical suspicion of TRMA syndrome(table 1).
Table 1.
Laboratory profile at baseline, on thiamine and benfotiamine therapy
| Parameters | Before starting thiamine | After 1 week on parenteral thiamine | After 6 months on benfotiamine |
| Haemoglobin (g/L) | 84 | 101 | 114 |
| Packed cell volume (PCV) (%) | 24 | 30 | 35.3 |
| Mean corpuscular volume (MCV) (fL) | 91.5 | 82 | 80 |
| Platelet (109/L) | 98 | 564 | 466 |
| HbA1C (mmol/mol) | 68 | – | 40 |
The molecular genetic analysis revealed a homozygous frameshift mutation in SLC19A2 (c262del) which codes for high affinity thiamine transporter in the proband. Both parents were heterozygous for the same mutation. She did not have cardiovascular or ocular abnormalities. She has been given a hearing aid, and is being scheduled for cochlear implant.
At 6 months follow-up, the haemoglobin is 11.4 gm% with MCV of 80 fL and plasma glucose is well maintained (4.1–8.3 mmol/L)with HbA1C of 40 mmol/mol without insulin (table 1).Currently she is off insulin for last 2 years.
Discussion
TRMA (OMIM 249270) syndrome is a rare autosomal recessive disorder characterised by the cardinal triad of diabetes mellitus, megaloblastic anaemia and progressive sensorineural hearing loss. The disease can manifest any time between infancy and adolescence. The TRMA syndrome is also known as Rogers Syndrome, after LE Rogers who first described it in 1969.1 The widespread consanguinity in some countries leads to increased incidence of this disease with autosomal recessive inheritance.
Thiamine uptake is thought to occur via two pathways: (1) Non-linear, active transport by a saturable, high-affinity/low-capacity carrier (thiamine transporter protein 1 (THTR1)); (2) Passive uptake by a low-affinity/high capacity carrier- Thiamine Transporter protein 2 (THTR2), both belonging to the solute carrier family of transporters. At physiological concentrations (<2 mmol/L) THTR1 transporter is crucial for the transport of thiamine intracellularly mainly in the peripheral tissues and intestinal lumen. Intracellularly, thiamine is converted into the active form, that is, thiamine pyrophosphate, which is incorporated into four mammalian enzymes. These are transketolase of the pentose phosphate cycle and another three enzymes involved in oxidative decarboxylation reaction that is, pyruvate dehydrogenase, ketoglutaratedehydrogenase and branched chain ketoacid dehydrogenase.2
The gene responsible for TRMA syndrome has been identified on chromosome 1q23.3 and the homozygous mutation was found in SLC19A2 which encodes for high-affinity THTR1.3 4 Because of the mutation in THTR1, thiamine is not transported normally at physiological concentrations (<2 mmol/L) and intracellular thiamine deficiency leads to decrease in the activity of enzymes associated with thiamine pyrophosphate.5
Stagg et al have documented a reduced expression of THRH1 transporter on fibroblasts of patients with TRMA syndrome and shown that the low intracellular thiamine concentration in the mutant cells causes biochemical abnormalities that lead to apoptotic cell death.2
Early onset diabetes mellitus, one of the cardinal components of TRMA syndrome is non-immune mediated and due to intracellular thiamine deficiency. Funk et al for the first time demonstrated the role of thiamine in glucose metabolism.6 Subsequently Oishi et al have demonstrated reduced insulin secretion in a mouse model of TRMA syndrome and shown that diabetes can be ameliorated with thiamine repletion.7 Most of the patients reported in the literature responded to thiamine treatment in the form of complete remission or significant reduction in the insulin requirement. As described in our patient, insulin requirement usually reduces by 30%–80% after starting thiamine. Glycemic control however may worsen during puberty, probably due to physiological decompensation.8
Megaloblastic anaemia, another cardinal feature of TRMA syndrome may manifest at any time from infancy to adolescence.9 Bone marrow shows megaloblastosis affecting the erythroid, myeloid and megakaryocyte lines. The classical haematological feature is megaloblastic anaemia with erythroblasts, which is often complicated with ringed sideroblasts, thrombocytopenia and neutropenia.10 Boros et al have shown that defective nucleic acid synthesis through impaired transketolase catalysis is the underlying biochemical disturbance. This induces cell cycle arrest or apoptosis in bone marrow cells leading to megaloblatosis.11Anaemia responds very quickly to the megadose of thiamine, but megaloblastic changes may remain persistent in bone marrow.12 13 In our patient, we documented persistent improvement in megaloblastic anaemia and thrombocytopenia with benfotiamine.
Patients usually have an early onset progressive, profound, high-frequency sensorineural deafness. The hearing loss may not be present at birth. It has been proposed that selective inner hair cell loss of the cochlea because of deficiency of THTR1 may be responsible for deafness. Progressive sensorineural deafness is irreversible and may not be prevented with thiamine treatment. However, in a recent case report, it was claimed that thiamine could prevent the development of hearing impairment when started in early infancy.8 Contrary to that, there are few case reports where hearing loss developed even after starting thiamine early in life.14 15
Apart from these cardinal features, other findings, reported in association with TRMA are cardiac abnormalities (arrhythmias, structural defects and cardiomyopathy), retinal abnormalities (optic atrophy, retinal degeneration and rod-cone dystrophy), situsinversus, stroke-like episodes, short stature and aminoaciduria.16 17
The treatment of TRMA syndrome is using thiamine Hcl in pharmacological doses (25–75 mg/day) life long. Due to unavailability of oral thiamine Hcl in the required strength to treat TRMA, we used benfotiamine in our patient. Benfotiamine (S-benzoylthiamine-o-monophosphate) is a lipophilic derivative of thiamine. After getting phosphorylated in the intestinal mucosa, benfotiamine enters into the blood stream as S-benzylthiamine that is converted to thiamine in erythrocytes and liver. As benfotiamine is lipophilic, its absorption is much better than that of water-soluble thiamine salts. The maximum plasma levels of thiamine are about five times higher with benfotiamine than thiamine Hcl. Its bioavailability is about 3.6 times as high as that of thiamine Hcl and is better than other lipophilic thiamine derivatives.18 Our patient showed sustained improvement in clinical features after starting benfotiamine. To the best of our knowledge, this is the first case report in literature where benfotiamine has been used as a treatment modality in TRMA syndrome.
In conclusion, TRMA syndrome is a rare genetic disease with clinical heterogeneity. We should always look for haematological parameters particularly megaloblastic anaemia, while evaluating cases of NDM. Though thiamine hydrochloride has been commonly described as treatment in the literature, oral benfotiamine can be considered as an alternative treatment option for the management of TRMA syndrome.
Learning points.
Thiamine Responsive Megaloblastic Anaemia (TRMA) is a rare form of autosomal recessively transmitted neonatal diabetes mellitus (NDM).
Screening for haematological parameters forms an important part of work up for NDM.
Treatment with water soluble salts of thiamine forms the crux of TRMA management.
Benfotiamine can be used as an alternative to water soluble salts of thiamine for the treatment of TRMA, which can better manage the diabetes that might go into remission on benfotiamine. Follow-up is mandatory to withhold insulin therapy.
As the different features of the syndrome may have different ages of presentation, meticulous follow-up makes an important part of TRMA syndrome management.
Acknowledgments
The authors would like to express gratitude to Dr Elisa De Franco, Dr Andrew Hattersley and their team for genetic analysis of TRMA syndrome. (Molecular Genetics Laboratory, Royal Devon & Exeter NHS Foundation Trust, University of Exeter Medical School, RILD-Level 3, Barrack Road, Exeter, EX2 5DW, UK.)
Footnotes
Contributors: PD and ASJ have conceptualised and designed the case report. DST and PD have contributed to acquisition and interpretation of data. DST, ASJ and NMB drafted the article. NMB finalised the article by revising and editing it critically.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Parental/guardian consent obtained.
References
- 1. Porter FS, Rogers LE, Sidbury JB, et al. Thiamine-responsive megaloblastic anemia. J Pediatr 1969;74:494–504. 10.1016/S0022-3476(69)80031-4 [DOI] [PubMed] [Google Scholar]
- 2. Stagg AR, Fleming JC, Baker MA, et al. Defective high-affinity thiamine transporter leads to cell death in thiamine-responsive megaloblastic anemia syndrome fibroblasts. J Clin Invest 1999;103:723–9. 10.1172/JCI3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Labay V, Raz T, Baron D, et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet 1999;22:300–4. 10.1038/10372 [DOI] [PubMed] [Google Scholar]
- 4. Dutta B, Huang W, Molero M, et al. Cloning of the human thiamine transporter, a member of the folate transporter family. J Biol Chem 1999;274:31925–9. 10.1074/jbc.274.45.31925 [DOI] [PubMed] [Google Scholar]
- 5. Meire FM, Van Genderen MM, Lemmens K, et al. Thiamine-responsive megaloblastic anemia syndrome (TRMA) with cone-rod dystrophy. Ophthalmic Genet 2000;21:243–50. 10.1076/1381-6810(200012)2141-HFT243 [DOI] [PubMed] [Google Scholar]
- 6. Funk C. The Vitamins. Baltimore, MD: Williams & Wilkins, 1922:388. [Google Scholar]
- 7. Oishi K, Hofmann S, Diaz GA, et al. Targeted disruption of Slc19a2, the gene encoding the high-affinity thiamin transporter Thtr-1, causes diabetes mellitus, sensorineural deafness and megaloblastosis in mice. Hum Mol Genet 2002;11:2951–60. 10.1093/hmg/11.23.2951 [DOI] [PubMed] [Google Scholar]
- 8. Valerio G, Franzese A, Poggi V, et al. Long-term follow-up of diabetes in two patients with thiamine-responsive megaloblastic anemia syndrome. Diabetes Care 1998;21:38–41. 10.2337/diacare.21.1.38 [DOI] [PubMed] [Google Scholar]
- 9. Onal H, Bariş S, Ozdil M, et al. Thiamine-responsive megaloblastic anemia: early diagnosis may be effective in preventing deafness. Turk J Pediatr 2009;51:301–4. [PubMed] [Google Scholar]
- 10. Ganesh R, Ezhilarasi S, Vasanthi T, et al. Thiamine responsive megaloblastic anemia syndrome. Indian J Pediatr 2009;76:313–4. 10.1007/s12098-009-0058-5 [DOI] [PubMed] [Google Scholar]
- 11. Boros LG, Steinkamp MP, Fleming JC, et al. Defective RNA ribose synthesis in fibroblasts from patients with thiamine-responsive megaloblastic anemia (TRMA). Blood 2003;102:3556–61. 10.1182/blood-2003-05-1537 [DOI] [PubMed] [Google Scholar]
- 12. Haworth C, Evans DI, Mitra J, et al. Thiamine responsive anaemia: a study of two further cases. Br J Haematol 1982;50:549–61. 10.1111/j.1365-2141.1982.tb01955.x [DOI] [PubMed] [Google Scholar]
- 13. Neufeld EJ, Mandel H, Raz T, et al. Localization of the gene for thiamine-responsive megaloblastic anemia syndrome, on the long arm of chromosome 1, by homozygosity mapping. Am J Hum Genet 1997;61:1335–41. 10.1086/301642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akın L, Kurtoğlu S, Kendirci M, et al. Does early treatment prevent deafness in thiamine-responsive megaloblastic anaemia syndrome? J Clin Res Pediatr Endocrinol 2011;3:36–9. 10.4274/jcrpe.v3i1.08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Borgna-Pignatti C, Azzalli M, Pedretti S. Thiamine-responsive megaloblastic anemia syndrome: long term follow-up. J Pediatr 2009;155:295–7. 10.1016/j.jpeds.2009.01.062 [DOI] [PubMed] [Google Scholar]
- 16. Lorber A, Gazit AZ, Khoury A, et al. Cardiac manifestations in thiamine-responsive megaloblastic anemia syndrome. Pediatr Cardiol 2003;24:476–81. 10.1007/s00246-002-0215-3 [DOI] [PubMed] [Google Scholar]
- 17. Scharfe C, Hauschild M, Klopstock T, et al. A novel mutation in the thiamine responsive megaloblastic anaemia gene SLC19A2 in a patient with deficiency of respiratory chain complex I. J Med Genet 2000;37:669–73. 10.1136/jmg.37.9.669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loew D. Pharmacokinetics of thiamine derivatives especially of benfotiamine. Int J Clin Pharmacol Ther 1996;34:47–50. [PubMed] [Google Scholar]