

ABSTRACT
Ceramide synthase 6 (CerS6) is an enzyme that preferentially generates pro-apoptotic C16-ceramide in the sphingolipid metabolic pathway. Reduced expression of CerS6 has been associated with apoptosis resistance and recent studies point to a role for CerS6 in epithelial mesenchymal transition (EMT). Because cells that undergo EMT are also more resistant to apoptosis, we hypothesized that reduced expression of CerS6 could induce changes that are associated with EMT. We found that shRNA-mediated knockdown of CerS6 increases expression of the EMT transcription factor SNAI2 but not SNAI1 or TWIST. Treatment with C6-ceramide nanoliposomes (CNL) resulted in a preferential increase in C16-ceramide and suppressed SNAI2 transcriptional activation and protein expression. The increase in C16-ceramide following CNL treatment was dependent on CerS activity and occurred even when CerS6 shRNA was expressed. shRNA against CerS5, which like CerS6 preferentially generates C16-ceramide, also decreased transcriptional activation of SNAI2, suggesting a role for C16-ceramide rather than a specific enzyme in the regulation of this transcription factor. While loss of CerS6 has been associated with apoptosis resistance, we found that cells lacking this protein are more susceptible to the effects CNL. In summary, our study identifies SNAI2 as a novel target whose expression can be influenced by C16-ceramide levels. The potential of CNL to suppress SNAI2 expression has important clinical implications, since elevated expression of this transcription factor has been associated with an aggressive phenotype or poor outcomes in several types of solid tumors.
KEYWORDS: SNAI2, ceramide synthase, ceramide nanoliposome
Introduction
Sphingolipids have dual functions as structural components of cellular membranes and as signal transducers. Stress-induced stimulation of sphingolipid metabolism increases the generation of ceramide, which is known to play a role in anti-proliferative responses1. Ceramide is a central molecule in sphingolipid metabolism and exists in multiple species with distinct fatty acid chain lengths. Six members of the ceramide synthase family (CerS) with preferential substrate specificity are responsible for the variety of ceramide species that make up ceramide profiles in cells and tissues.
CerS6 preferentially generates C16-ceramide, which has been associated with cell death in response to radiation, chemotherapy, and death ligands in cancer cells2. The enzyme is highly responsive to various stressors and was identified as a direct transcriptional target of p533-5. CerS6 is required for the toxic effects of celecoxib and increases in response to stichoposide D, which leads to activation of the mitochondria–dependent apoptotic pathway6,7. Overexpression of the tumor suppressor gene MDA-7/IL24 also increases CerS6 expression5. Although the tumor suppressive effects of CerS6 have been demonstrated in multiple models, CerS6 function is still relatively unknown.
A recent study exploring the role of sphingolipids in epithelial mesenchymal transition (EMT) identified a signature of 35 sphingolipid genes associated with a significant reduction in ceramide or an increase in the pro-survival lipid sphingosine-1-phosphate (S1P)8. Among the ceramide generating enzymes, CerS6 mRNA was significantly decreased8. In a separate study by Edmond et al. cancer cells within the NCI-60 panel were grouped into “epithelial” or “mesenchymal” phenotypes based on E-cadherin/vimentin ratios, which revealed that mesenchymal cells had significantly reduced levels of CerS6 mRNA expression9. Analysis of CerS6 expression at the protein level in a molecularly defined model of EMT confirmed that reduced CerS6 expression coincides with the “cadherin switch”9. Cadherin switching is a process during which cells switch expression of cadherin transmembrane proteins that form cellular junctions, which has a profound effect on cell phenotype and behavior10. Since cells that undergo EMT acquire apoptosis resistance, we hypothesized that loss of CerS6 contributes to this process. Here we show that the EMT transcription factor SNAI2 is regulated in a C16-ceramide-dependent manner.
Results
Loss of Cers6 increases expression of the SNAI2 transcription factor
Recent studies have investigated potential links between EMT and sphingolipids8,9. EMT is a tightly regulated process that depends on altered expression of cell adhesion molecules such as cadherins, which mediate calcium-dependent cell-cell adhesion. As cells undergo EMT expression of the calcium-dependent adhesion protein E-cadherin decreases and vimentin, an intermediate filament of mesenchymal origin increases. We previously generated SW480 cells that stably express an inducible shRNA against CerS611. Given that CerS6 mRNA decreases with induction of EMT and that cells with a mesenchymal phenotype have significantly lower levels CerS6 mRNA than cells with an epithelial phenotype, we asked whether CerS6 knockdown differentially affects proteins associated with EMT8,9. shRNA-mediated targeting of CerS6 decreased E-cadherin and increased vimentin expression (Figure 1(a,c)). EMT is a multi-step process that is mediated by several families of transcription factors. Analysis of EMT transcription factors revealed that expression of SNAI2 but not SNAI1 or Twist expression was increased in cells with reduced CerS6 (Figure 1(b,c)). CerS6 knockdown in PPC1 prostate cancer cells using an adenoviral expression system also increased expression of SNAI2, suggesting the effect of CerS6 shRNA-mediated knockdown was not cell line specific or dependent on the expression system used (Figure 1(d)).
Figure 1.
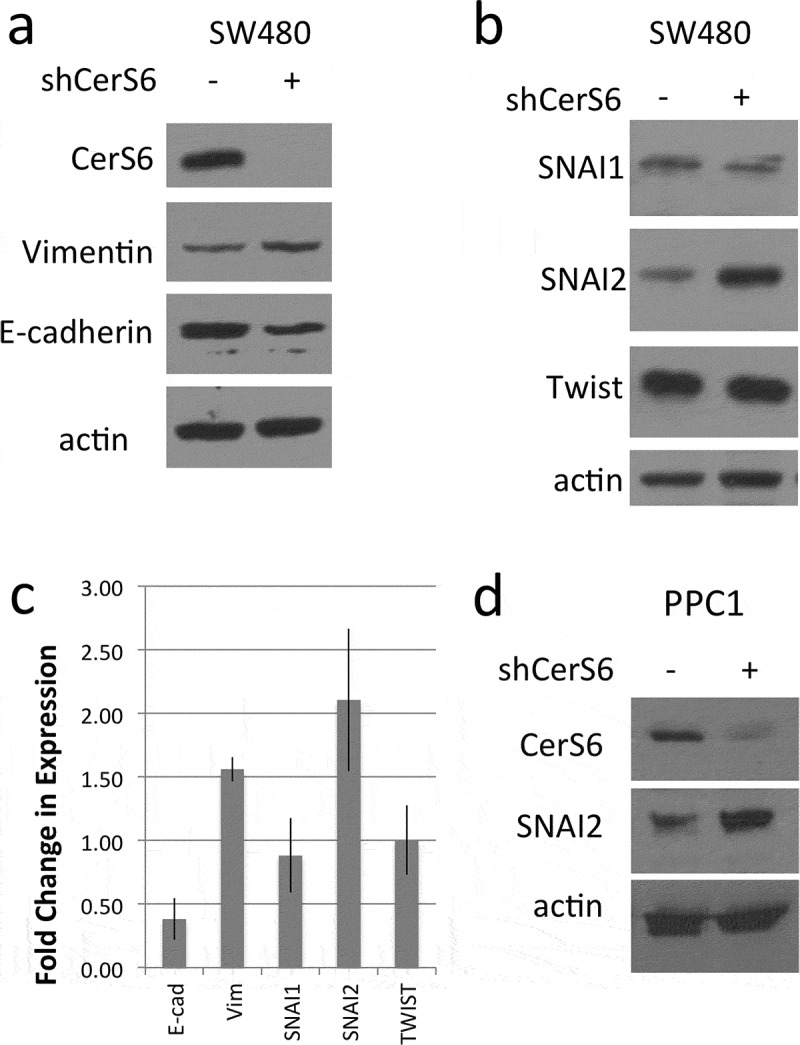
Protein analysis in cells with different CerS6 expression. (a, b) SW480 cells stably transfected with an inducible shRNA against CerS6 were analyzed for protein expression. (c) Quantification of the results from at least three experiments. (d) PPC1 with adenovirally expressed CerS6 shRNA. Whole cell lysates were analyzed by Western blot for protein expression. Actin served as loading control.
Downregulation of Cers6 results in transcriptional activation of SNAI2
Next, we used reporter constructs to determine if knockdown of CerS6 increases expression of SNAI2 at the transcriptional level. HEK293 cells were co-transfected with plasmids expressing shRNAs against CerS6 (shCerS6) or a negative control (shScr) and luciferase reporter constructs in which reporter gene expression is either under the control of the SNAI1 or the SNAI2 promoter. Co-transfection of the shCerS6 plasmid with the SNAI1-luciferase reporter plasmid had no effect on luciferase reporter activity relative to the control (Figure 2(a)). In contrast, co-transfection of the shCerS6 plasmid and with the SNAI2-luciferase reporter plasmid increased the luciferase signal about 2–3-fold relative to the control (Figure 2(b)). We also investigated whether expression of SNAI2 affects CerS6 levels but did not detect any change in reporter gene activity or CerS6 protein expression (Figure 2(c,d)). These results suggest that loss of CerS6 is permissive for transcriptional activation of SNAI2 but not vice versa.
Figure 2.
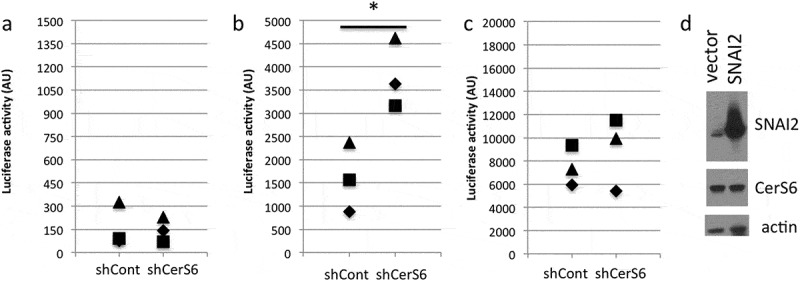
Transcriptional regulation by CerS6. HEK293 cells were co-transfected with 100ng of each plasmid and luciferase reporter activity quantified 24 hours post-transfection. (a) control and CerS6 shRNA co-transfected with SNAI1-luc reporter, (b) control and CerS6 shRNA co-transfected with SNAI2-luc reporter, (c) empty and SNAI2 expressing vector co-transfected with CerS6-luc reporter. Data are from 3 independent experiments each performed in triplicate. (d) Western blot of HEK293 cells transfected with SNAI2 showing lack of impact on CerS6 expression. Actin serves as loading control.
Restoration of C16-ceramide reverses increased SNAI2 expression due to loss in CerS6
To gain a better understanding on how CerS6 knockdown affects ceramide composition, we performed LC/MS profiling of SW480 cells with differential CerS6 expression. When analyzed by picomol sphingolipid per nanomol total phosphate, C16-ceramide as well as saturated very long chain ceramides (C22, C24, C26) were significantly decreased in cells with reduced CerS6 expression compared to control (Table 1). Hetero-dimerization of CerS6/CerS2 has been reported and is important for CerS activity12. Therefore, the decrease in total ceramides including very long chain ceramides may reflect reduced overall CerS activity due to differential dimer formation in cells with and without CerS6. Since CerS6 knockdown affected total ceramide levels, we calculated percentages of each ceramide species. Using this approach only C16-ceramide was significantly decreased (Table 1).
Table 1.
Sphingolipid composition in SW480 with and without CerS6 expression. SW480 cells were analyzed for sphingolipids by LC/MS. Data is the average and standard deviation from 2 independent experiments each performed in triplicate expressed as pmol sphingolipid per nmol total phosphate (pmol SL/nmol Pi). Other ceramides includes C14, C18, C18:1, C20, C22:1 and C26:1, which were each 1% of total ceramide or less.
| CerS6 expressed |
CerS6 knockdown |
|||
|---|---|---|---|---|
| Sphingolipid | pmol SL/ nmol Pi | % of total ceramide | pmol SL/ nmol Pi | % of total ceramide |
| C16-ceramide | 2.4078 ± 0.2954 | 28 ± 4 | 0.9959 ± 0.0146*** | 19 ± 5*** |
| C22-ceramide | 0.5706 ± 0.0362 | 7 ± 2 | 0.378.6 ± 0.0558*** | 7 ± 0 |
| C24-ceramide | 3.3829 ± 0.4618 | 39 ± 4 | 2.3480 ± 0.4853** | 43 ± 1 |
| C24:1 ceramide | 2.0166 ± 1.1361 | 22 ± 8 | 1.4731 ± 0.4265 | 27 ± 3 |
| C26-ceramide | 0.1093 ± 0.0127 | 1 ± 0 | 0.0451 ± 0.0152*** | 1 ± 0 |
| other ceramides | 0.0268 ± 0.0113 | 3 ± 1 | 0.0214 ± 0.0091 | 4 ± 1 |
| dhC16-ceramide | 0.3093 ± 0.0239 | 0.0804 ± 0.0158*** | ||
| sphingosine | 0.0794 ± 0.0099 | 0.0389 ± 0.0035*** | ||
| S1P | 0.0044 ± 0.0014 | 0.0031 ± 0.0008 | ||
| Total ceramide | 8.7549 ± 1.9267 | 5.4554 ± 0.9953** | ||
p < 0.01,* p < 0.001, ***p < 0.0001.
Next, we tested whether restoration of ceramide could reverse the increase in SNAI2 expression in cells lacking CerS6. The therapeutic utility of ceramide is greatly limited by its inherent hydrophobicity, which leads to precipitation, insolubility and limited membrane intercalation in biological fluids. Even though delivery platforms have been engineered for long chain ceramides, these formulations are still limited by bench top and biological shelf life issues. A possible solution is the use of C6-ceramide nanoliposomes (CNL), which have shown anti-tumor efficacy in numerous cancer models and are currently being evaluated in a Phase I clinical trial (NCT02834611)13–17. First, we investigated how the addition of CNL impacts ceramide composition. Ghost nanoliposomes (GNL), which are identical to CNL except that they lack C6-ceramide, were used as a negative control. As shown in Figure 3(a), treatment with CNL preferentially increased C16-ceramide. A CNL-driven increase in C16-ceramide was also observed in cells with CerS6 knockdown, although the magnitude was reduced (Figure 3(b)).
Figure 3.
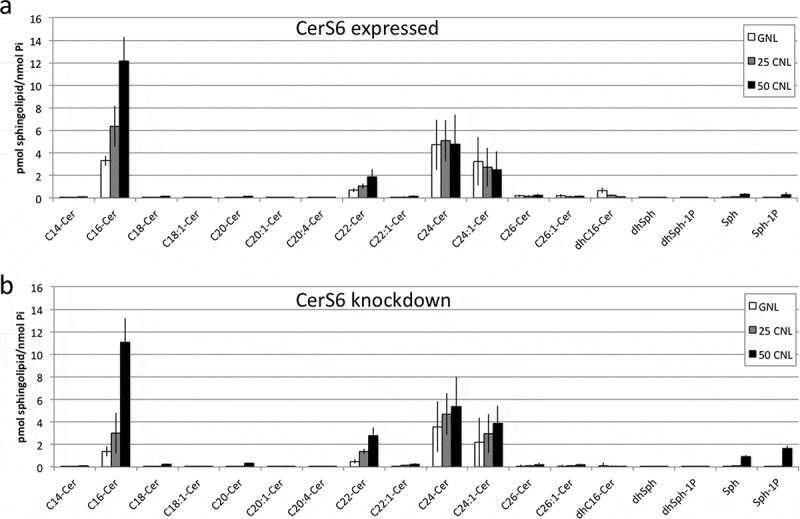
Sphingolipid profiles of SW480 cells following treatment with C6-ceramide nanoliposomes. SW480 cells expressing CerS6 (a) or with CerS6 knockdown (b) were incubated with 50μM ghost nanoliposomes or 25μM and 50μM C6-ceramide nanoliposomes. At 36 hours cells were collected for LC/MS analysis. Data shown are the average and standard deviation from two independent experiments each performed in triplicate.
We also evaluated the effect of CNL treatment on SNAI2 transcriptional induction. Treatment with CNL did not significantly impact SNAI2 promoter activity when the reporter construct was co-transfected with the control shRNA (Figure 4(a), column 1 vs. 2). Expression of the CerS6 shRNA approximately doubled luciferase reporter activity in cells treated with GNL (Figure 4(a), column 1 vs. 3). This is consistent with results shown in Figure 2 and indicates that the control liposomes do not interfere with the increase in reporter activity in response to CerS6 knockdown. The addition of 25μM or 50μM CNL diminished SNAI2 promoter activity in cells expressing CerS6 shRNA to levels comparable to controls (Figure 4(a), column 5/6 vs. 1/2). Similar results were observed at the protein level. Treatment with 25μM GNL did not affect SNAI2 expression in SW480 cells lacking CerS6 but the addition 25μM CNL decreased SNAI2 to 48% ± 5% relative to controls (n = 3) (Figure 4(b)).
Figure 4.
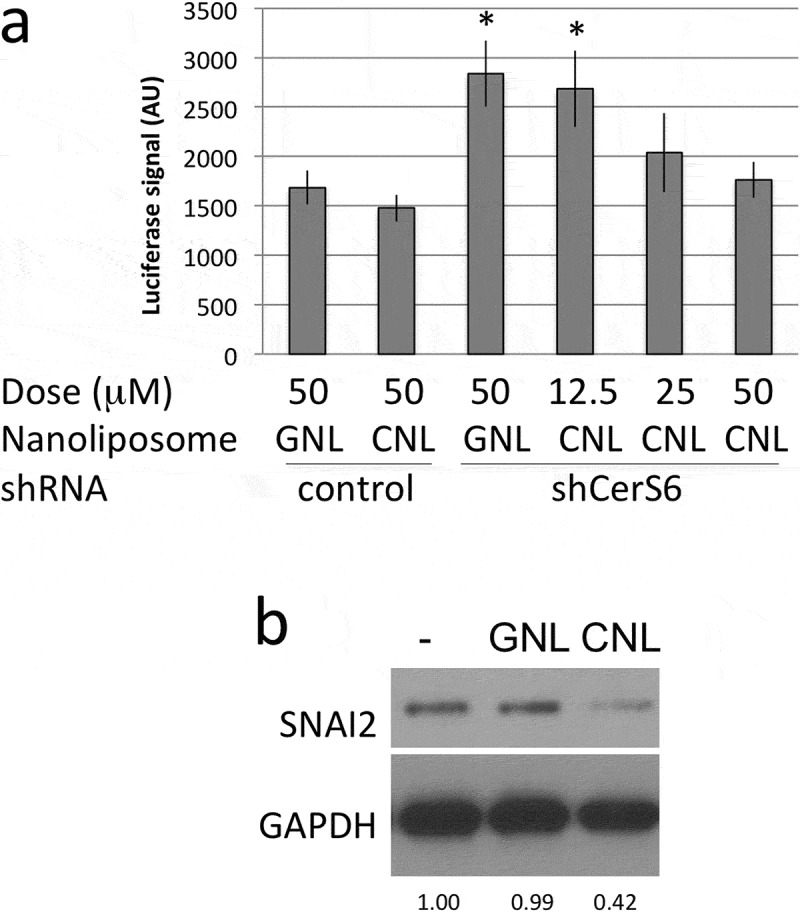
CNL reduce transcriptional activation and expression of SNAI2. (a) HEK293 cells co-transfected with control or CerS6 shRNA and the SNAI2-luciferase reporter were in parallel incubated with the indicated dose of ghost- or C6-ceramide nanoliposomes. Data shown are the average and standard deviation of the luciferase signal at 24h from two independent experiments each performed in triplicate. *p < 0.05. (b) SW480 cells expressing CerS6 shRNA were untreated (-) or incubated with 25μM GNL or CNL. A representative experiment is shown. Similar results were obtained in two independent experiments.
Our results suggested that loss of CerS6 decreases intracellular C16-ceramide, which is permissive for transcriptional activation of SNAI2 but can be reversed through treatment with CNL through a preferential increase C16-ceramide. The increase in C16-ceramide in cells expressing CerS6 shRNA suggested that the activity of another enzyme such as CerS5, which also preferentially generates C16-ceramide, compensates for the loss of CerS6. To investigate the role of CerS activity in the generation of C16-ceramide from C6-CNL, we pretreated cells with the pan-CerS inhibitor fumonison B1. In the absence of FB1, expression of CerS6 shRNA reduced C16-ceramide by about 50% (see GNL, -FB1, Figure 5(a)). In the presence of FB1, the C16-ceramide content was reduced equally regardless of CerS6 expression (see GNL, +FB1, Figure 5(a)). These results suggest that a very small pool of C16-ceramide is generated independent of CerS and that the amount of C16-ceramide in cells lacking CerS6 likely results from CerS5 activity. FB1 significantly reduced the amount of C16-ceramide upon treatment with CNL, indicating that CerS5/6 activity is required for incorporation of C6-ceramide from CNL into C16-ceramide (Figure 5(a), CNL, -FB1 vs. CNL, +FB1). In cells expressing the CerS6 shRNA, C16-ceramide was presumably generated by activity of CerS5. To further investigate the role of specific CerS, we performed experiments in which the SNAI2 promoter driven luciferase plasmid was co-transfected with plasmids targeting other CerS family members. CerS1 preferentially generates C18-ceramide and its knockdown did not significantly alter SNAI2-driven reporter activity (Figure 5(b)). Similarly shRNA against CerS4, which preferentially generates C18-ceramide and C20-ceramide, did not significantly impact on reporter gene activity. Knockdown of CerS2, which generates C24-ceramides, decreased SNAI2-driven reporter activity, although it fell short of reaching statistical significance. An increase in SNAI2 reporter activity was detected only when either CerS5 or CerS6 shRNA were expressed (Figure 5(b)). These results suggest that C16-ceramide levels rather than a specific enzyme transcriptionally regulates SNAI2 expression.
Figure 5.
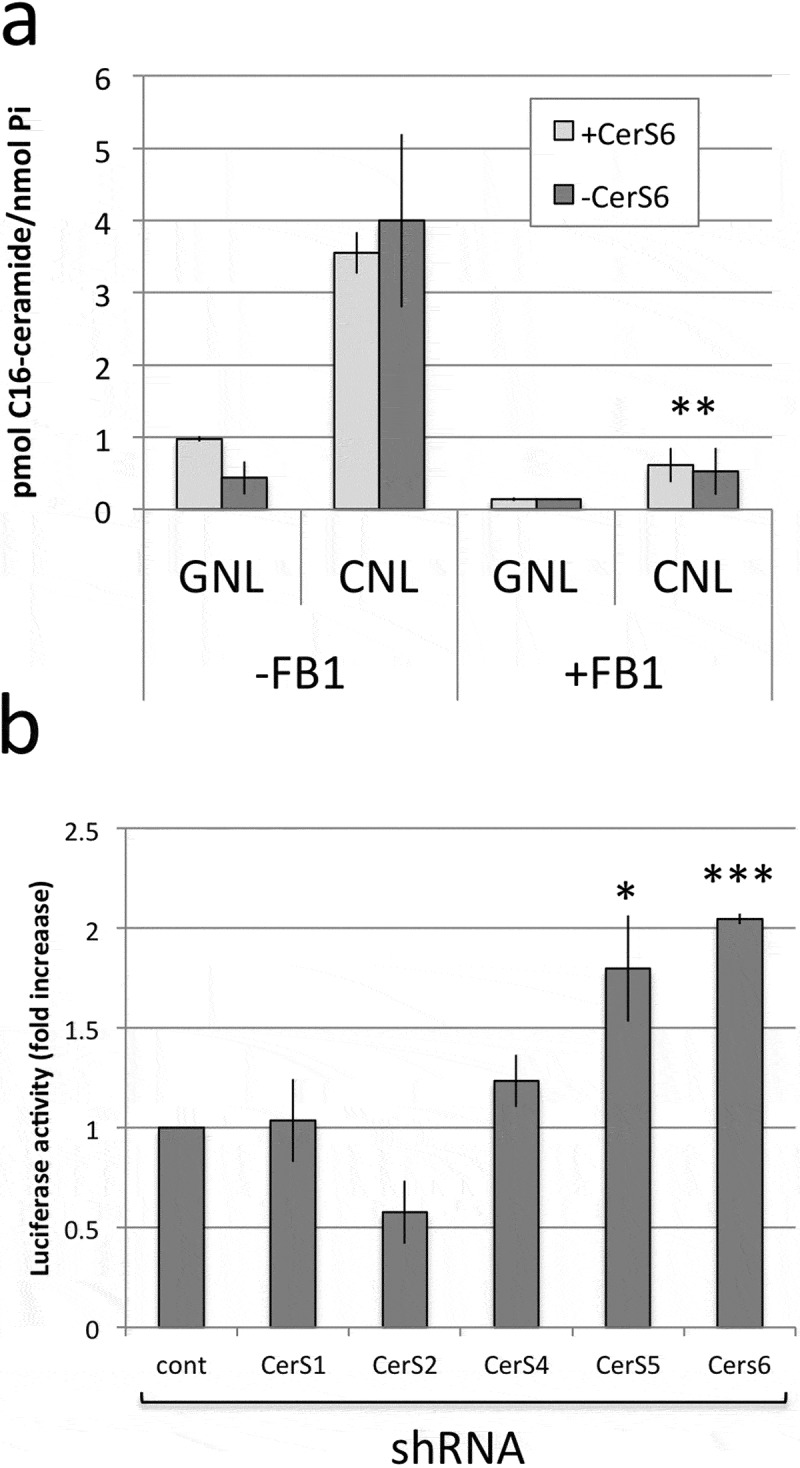
C16-ceramide generation from CNL occurs in a CerS-dependent manner and its loss is sufficient for transcriptional activation of SNAI2. (a) SW480 cells were pretreated with 25μM FB1 for one hour before adding 25μM GNL or CNL for 18 hours. Data shown are the mean and standard deviation from two experiments performed in duplicate. (b) Control and CerS shRNAs were co-transfected with the SNAI2-luc reporter to determine luciferase activity. Data are from two independent experiments each performed in triplicate. * p < 0.05, *** p < 0.005.
Since CNL have recently entered the clinic, we evaluated their effect on viability in cells expressing and lacking CerS6. Cells expressing CerS6 tolerated dosages up to 50 μM CNL with minimal loss in viability as determined by mitochondrial activity, although cells assumed a rounded morphology at the higher concentrations (Figure 6). Interestingly, cells lacking CerS6 were more susceptible to CNL with an approximately 50% decrease in mitochondrial activity at 50 μM CNL and appearance of the rounded cell morphology at 30 μM CNL (Figure 6). These results suggest that cells lacking CerS6 may be preferentially eliminated by CNL.
Figure 6.
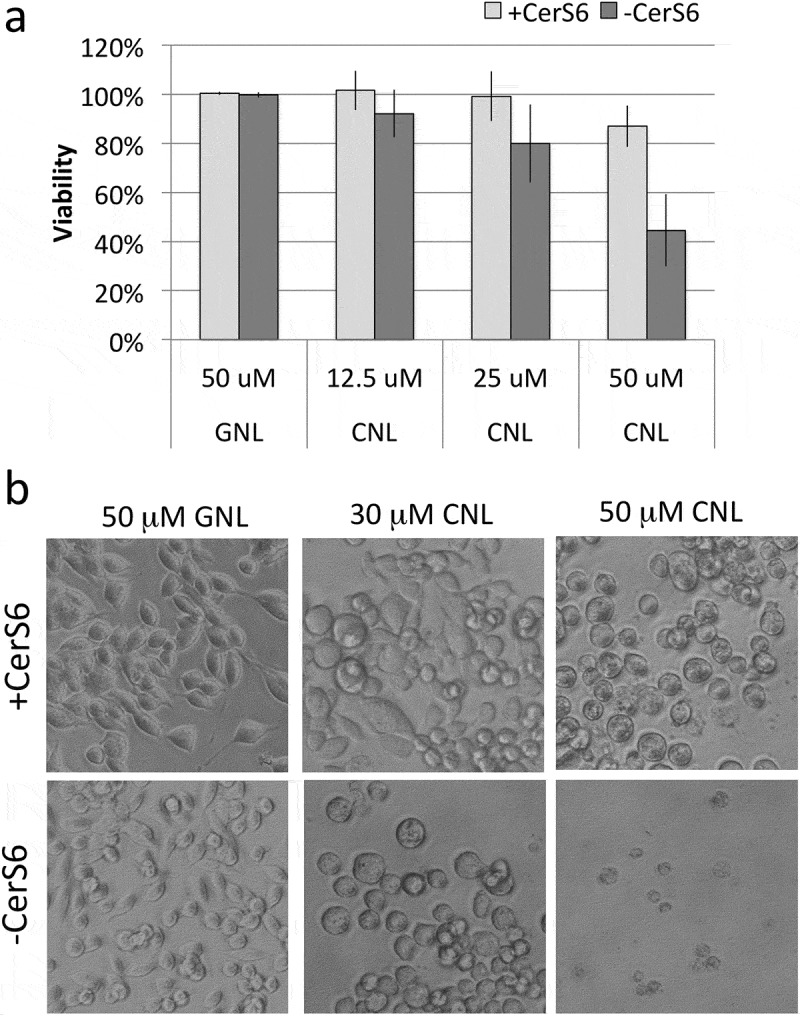
Loss of CerS6 increases susceptibility to CNL. SW480 cells were incubated with 50 μM GNL or increasing concentrations of CNL for 72 hours. (a) Data shown are the mean ± standard deviation from 5 experiments. (b) Representative images of SW480 cells treated with GNL or CNL (200x magnification).
Discussion
In solid tumors cancer progression involves loss of polarization and adhesion among epithelial cells, which allows them to migrate and invade other tissues and/or to acquire pluripotency18. This process is similar to EMT during embryonic development and is driven by several families of transcription factors that directly or indirectly suppress the expression of E-cadherin resulting in a so-called “cadherin switch” during which E-cadherin is lost and expression of the mesenchymal filament protein vimentin is gained. A recent study identified a 35-gene signature that associated EMT with alterations that lead to a significant reduction in ceramide and an increase in S1P8. One of the genes with reduced expression was CerS6, which has previously been linked to apoptosis resistance1,19. Since apoptosis resistance is also observed in EMT, we investigated how loss of CerS6 impacts on EMT markers and transcription factors. CerS6 knockdown led to reduced E-cadherin and increased vimentin expression, which is consistent with previous findings that associate low CerS6 levels with a mesenchymal phenotype (Figure 1(a))9. Our results also indicated that these changes were accompanied by increased expression of the SNAI2 but not SNAI1 or TWIST transcription factors, suggesting the possibility that loss of CerS6 expression (or reduced C16-ceramide) is specifically associated with a decrease in E-cadherin and increased SNAI2. Clinical studies appear to support this possibility. A recent TMA analysis of 251 samples of primary colorectal cancer failed to detect a correlation between decreased E-cadherin and increased SNAI1 expression20. SNAI1 expression, which was detected in 76% of samples, also did not correlate with nodal stage20. In contrast, low E-cadherin and high SNAI2 expression closely correlated with lymph node metastasis, tumor-node-metatastasis stage, and lymphatic vessel metastasis in breast cancer and in colorectal cancer patients high SNAI2 and low E-cadherin expression were associated with worst overall prognosis21,22. Lymph node progression in early stages of non-small cell lung cancer has also been associated with SNAI2 but not SNAI123,24. Similar results were found in a meta-analysis of 11 studies with 1817 patients with ovarian cancers25. In breast cancer, PLD2 expression, which is highly increased in larger tumors and associated with poor prognosis, is positively regulated by SNAI2 but not SNAI126. These results underscore important differences between closely related SNAI1 and SNAI2 family members.
The specific effect of CerS6 knockdown on increased SNAI2 (and not SNAI1) expression was also demonstrated at the transcriptional level in co-transfection experiments of HEK293 cells. To determine whether increased SNAI2 promoter activity was specifically dependent on loss of CerS6, we evaluated the effect of shRNAs against other CerS family members and found a similar effect on SNAI2 promoter activity occurred with CerS5 shRNA. Both of these CerS family members preferentially generate C16-ceramide, suggesting that levels of this specific ceramide species play an important role in SNAI2 regulation. Interestingly, expression of CerS2 shRNA trended towards decreasing SNAI2 promoter activity. CerS2 preferentially generates C24-ceramide and studies from CerS2-deficient mice have shown that loss of this very long chain ceramide is compensated through increased C16-ceramide to maintain total ceramide levels27,28. Thus decreasing CerS2 expression may also lead to an increase in C16-ceramide levels, which may decrease SNAI2.
Clinically, a significant correlation has been found between SNAI2 expression levels and Dukes stage, distant metastasis and overall survival in colorectal cancer patients22. The study identified SNAI2 as an independent prognostic factor associated with poor outcome22. SNAI2 has also been found to be a critical effector of EMT in head and neck squamous cell carcinoma29. The correlation between elevated SNAI2 expression and poor clinical outcome coupled with our finding that SNAI2 expression increases when C16-ceramide levels decrease, suggests that restoration of ceramide could emerge as an important therapeutic strategy. C16-ceramide has specifically been associated with anti-tumor effects but delivery of long chain ceramides is difficult due to hydrophobicity. Recently, C6-ceramide containing ceramide nanoliposomes (CNL) have entered the clinic as part of a Phase I trial (NCT02834611). CNL enter cells via a rapid bilayer exchange mechanism and it has been proposed that they subsequently accumulate in caveoli30,31. We found that treatment with CNL results in a preferential increase in C16-ceramide in the presence or absence of CerS6 expression. Since CerS5 and CerS6 preferentially generate C16-ceramide, these results suggest that CerS5 likely compensates for loss of CerS6 activity. Experiments using the pan-CerS inhibitor FB1 indicate the C6-ceramide from CNL is converted to C16-ceramide via the salvage pathway. Only a small fraction of C16-ceramide was generated independent of CerS activity. Hydrolysis of complex sphingolipids, such as C16-sphingomyelin, is likely responsible for this FB1-insensitve pool of C16-ceramide.
Treatment with CNL reduced SNAI2 promoter activity as well as SNAI2 protein expression indicating that SNAI2 is regulated in a C16-ceramide-dependent manner. While CNL treatment resulted in comparable levels of ceramide regardless of CerS6 expression, cells lacking CerS6 were more susceptible to the effects of CNL. We previously studied SW620 cells, an isogenic metastatic derivative of SW480 cells that has reduced CerS6 expression. Interestingly, treatment of SW620 cells with 30 μM C6-ceramide resulted in a similar rounded morphology as treatment with 30 μM CNL in SW480 cells expressing CerS6 shRNA32. It remains to be investigated why CerS6 knockdown renders cells more susceptible to CNL when intracellular C16-ceramide levels appear comparable. This prompts the question of compartmentalization as the CerS5-generated C16-ceramide could occur in a different subcellular location than CerS6-derived C16-ceramide. Regardless of mechanism, the potential therapeutic implication is that CNL have the potential to preferentially target cells with low or no CerS6 expression, which are more difficult to eliminate through radiation, chemotherapy, or death ligand stimulation2.
Sphingolipids have been studied extensively in the context of proliferation and apoptosis but also play important roles in gene regulation33. The Chalfant group has shown that ceramide impacts SR proteins, which direct alternative splicing of pre-mRNA of caspase-9 and Bcl-XL thereby impacting apoptosis susceptibility34-36. More recently ceramide was shown to impact on p53 splicing37. The Ogretmen group has identified inhibitor 2 of protein phosphatase 2A (I2PP2A) as a ceramide-binding protein38. In addition, this group has shown that exogenous C6-ceramide or endogenous C16-ceramide decrease the expression of human telomerase reverse transcriptase (hTERT), an enzyme that is involved in cancer cell immortality through the addition of telomeres39,40. Our results suggest that SNAI2 is a novel addition to genes whose expression is regulated by intracellular C16-ceramide levels. The acyl chain of C16-ceramide has recently been shown to specifically impact cellular processes. For example, C16-ceramide directly binds to RIPK1, which leads to the formation of ceramidosomes during necroptosis41. C16-ceramide also directly binds to p53, thereby disrupting the interaction of the E3 ligase MDM2, which results in p53 accumulation42. A potential mechanism for SNAI2 regulation could involve direct binding of C16-ceramide to a SNAI2 transcriptional regulator. Using the Champion ChiP transcription factor search portal based on SABiosciences database known as DECODE, several transcription factors, including Sox9, Sp1, YY1, MyoD, C/EBPα, CREB, ΔCREB, HNF-4α1, and HNF-4α2, were identified. Increasing endogenous ceramide levels has been shown to inhibit the expression of C/EBPα but whether C/EBPα directly binds C16-ceramide or whether a reduction in C16-ceramide increases the expression of C/EBPα thereby inducing SNAI2 transcription remains to be determined43.
In conclusion, our results demonstrate that loss of C16-ceramide as a consequence of decreased CerS6 expression is permissive for transcriptional activation of the EMT transcription factor SNAI2, which has been associated with poor clinical outcomes. We also show that treatment with CNL that increase C16-ceramide can reverse the transcriptional induction and expression of SNAI2. Analysis of SNAI2 levels in cancer patients treated with CNL maybe worthwhile to determine if SNAI2 expression changes in response to therapy and whether an inverse relationship between C16-ceramide levels and SNAI2 expression can be detected in a clinical setting.
Materials and methods
Reagents
Antibodies were purchased commercially as follows: Epithelial Mesenchymal Transition Antibody Sampler Kit (Cell Signaling Technologies, #9782), anti-CerS6 (Abcam, #ab56582), anti-GAPDH (SantaCruz Biotechnologies, #sc32233), anti-actin (Sigma, #A2068). Plasmids expressing the control and CerS6 shRNA (B10, target sequence GAACTGCTTCTGGTCTTACTT) were purchased from OpenBiosystems. Additional shRNA plasmids were provided by Besim Ogretmen. shRNA sequences were as follows: shCerS1, gene id: 10715, TRCN0000168211:CCGGCTACTTCTTCTTCAATGCGCTCTCGAGAGCGCATTGAAGAAGAAGTAGTTTTTTG; shCers2, gene id: 29956, TRCN000276376: CCGGCCTGCCTTCTTTGGCTATTACCTCGAGGTAATAGCCAAAGAAGGCAGGTTTTTG; shCerS4, gene id: 204219, TRCN0000016986: CCGGGTTACATGATTCCTCTGACTACTCGAGTAGTCAGAGGAATCATGTAACTTTTT; shCerS5, gene id: 91012, TRCN0000022221: CCGGGTGGGCTTTATCACTATTATACTCGAGTATAATAGTGATAAAGCCCACTTTTT; shCerS6, gene id: 253782, TRCN0000344342: CCGGGAACTGCTTCTGGTCTTACTTCTCGAGAAGTAAGACCAGAAGCAGTTCTTTTTG. The CerS6-driven luciferase reporter plasmid was also provided by Besim Ogretmen44. SNAI1 and SNAI2-driven luciferase reporter plasmids (Snail_pGL2, Addgene plasmid #31694 and Slug_pGL2, Addgene plasmid #31695) were a gift from Paul Wade45. Mark Kester kindly supplied the Ghost NanoLiposomes (GNL) and C6-ceramide NanoLiposomes (CNL)31.
Cell culture
The cell lines SW480 (CRL-228) and HEK293A (CRL-1573) were purchased from ATCC. PPC1 prostate cancer cells were a gift from Dean Tang (Roswell Park Comprehensive Cancer Center). Cells were maintained in DMEM (SW480, HEK293A) or RPMI1640 (PPC1) medium supplemented with 10% heat-inactivated FBS (Hyclone,#SH30088-03), 1% Antibiotic-Antimycotic (Gibco Life Technologies, #15240–062) and 0.1% Gentamycin (Lonza, #17-519Z) and cultured at 37°C, 5% CO2 in humidified air. Cell lines were not carried more than 30 consecutive passages and were periodically tested for mycoplasma.
Western blot analysis
Western blot analysis was performed as previously described32. Following electrophoresis and transfer of proteins, nitrocellulose membranes were probed with primary antibodies overnight at 4°C followed by HRP-labeled secondary anti-mouse or anti-rabbit antibodies for 1 hour at room temperature (Santacruz Biotechnologies, #sc-2204 and sc-2005). Signals were detected using the Immobilon Western chemiluminescence HRP substrate (Immobilon #WBKL50100, Millipore). For Figure 1(c), signals were quantified using ImageJ software, normalized to the loading control, and expressed as fold change relative to cells expressing CerS6.
Transfections and transductions
SW480 cells expressing shRNA against CerS6 have previously been described11. Adenoviral vectors expressing scrambled or CerS6 shRNA were purchased from Vector Biolabs (#1122 and shADV-213700). For transductions, PPC1 cells were plated and allowed to adhere before adding adenoviral vector (MOI 50) for 72 hours. Transient transfections of HEK293 cells were performed in 96-well plates using 200 ng DNA (100 ng per plasmid) and 0.5μl lipofectamine (Invitrogen, #11668–019) per well according to manufacturer’s instructions.
Luciferase activity assay
Luciferase activity was determined at 24 hours post-transfection using the Steady-Glo kit (Promega, E2520). Signals were quantified using a BMG Optima plate reader. Background values from untransfected cells were subtracted and values from triplicate wells averaged. The fold-change in luminescence was calculated relative to cells that were co-transfected with control shRNA and the relevant luciferase reporter.
Sphingolipid analysis
Sphingolipid analysis was performed as described previously32. Briefly, cell pellets were stored at −80°C until processing for sphingolipid analysis by liquid chromatography/mass spectrometry (LC/MS) in the MUSC Lipidomics facility46. An aliquot of the lipid extract was used to carry out lipid phosphate estimation using Bligh Dyer extraction and a colorimetric assay47.
Viability assays and microscopy
Viability was determined at 72 hours using the CellTiterBlue kit according to manufacturer’s instructions (Promega). Images were captured at 200x using a Zeiss Axiovert 200 microscope equipped with an Axiocam MRC digital camera. Images were captured using the Axiovision Rel.6 software.
Statistical analysis- Linear regression was used to model log of fold change of expression in luciferase reporter assays, adjusting for other factors including well and repeats. Student’s t test was performed using GraphPad software.
Funding Statement
This work was supported by the HHS | NIH | National Cancer Institute (NCI) [P01 CA203628.];HHS | NIH | National Cancer Institute (NCI) [P01 CA171983].
Acknowledgments
The authors would like to thanks Dr. Besim Ogretmen for CerS shRNA and CerS6-reporter plasmids, Dr. Dean Tang for the PPC1 cells, the Lipidomics Shared Resource for excellent technical support and the Biostatistics Shared Resource for the linear regression analysis. This project was in part supported by P01 CA203628 (CVJ) and P01 CA171983 (MK).
Disclosure of Potential Conflicts of Interest
MK—Penn State Research Foundation (PSRF) has licensed ceramide nanoliposomes to Keystone Nano, Inc. MK is co-founder and chief medical officer of Keystone Nano, Inc, State College, PA. The remaining authors declare no competing financial interests.
References
- 1.Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18:33–50. doi: 10.1038/nrc.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tirodkar TS, Voelkel-Johnson C. Sphingolipids in apoptosis. Exp Oncol. 2012;34:231–242. [PubMed] [Google Scholar]
- 3.Fekry B, Jeffries KA, Esmaeilniakooshkghazi A, Ogretmen B, Krupenko SA, Krupenko NI. CerS6 is a novel transcriptional target of p53 protein activated by non-genotoxic stress. J Biol Chem. 2016;291:16586–16596. doi: 10.1074/jbc.M116.716902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoeferlin LA, Fekry B, Ogretmen B, Krupenko SA, Krupenko NI. Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6. J Biol Chem. 2013;288:12880–12890. doi: 10.1074/jbc.M113.461798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wegner MS, Schiffmann S, Parnham MJ, Geisslinger G, Grosch S. The enigma of ceramide synthase regulation in mammalian cells. Prog Lipid Res. 2016;63:93–119. doi: 10.1016/j.plipres.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Schiffmann S, Sandner J, Schmidt R, Birod K, Wobst I, Schmidt H, Angioni C, Geisslinger G, Grösch S. The selective COX-2 inhibitor celecoxib modulates sphingolipid synthesis. J Lipid Res. 2009;50:32–40. doi: 10.1194/jlr.M800122-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Schiffmann S, Ziebell S, Sandner J, Birod K, Deckmann K, Hartmann D, Rode S, Schmidt H, Angioni C, Geisslinger G, et al. Activation of ceramide synthase 6 by celecoxib leads to a selective induction of C16:0-ceramide. Biochem Pharmacol. 2010;80:1632–1640. doi: 10.1016/j.bcp.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Meshcheryakova A, Svoboda M, Tahir A, Kofeler HC, Triebl A, Mungenast F, Heinze G, Gerner C, Zimmermann P, Jaritz M, et al. Exploring the role of sphingolipid machinery during the epithelial to mesenchymal transition program using an integrative approach. Oncotarget. 2016;7:22295–22323. doi: 10.18632/oncotarget.7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edmond V, Dufour F, Poiroux G, Shoji K, Malleter M, Fouque A, Tauzin S, Rimokh R, Sergent O, Penna A, et al. Downregulation of ceramide synthase-6 during epithelial-to-mesenchymal transition reduces plasma membrane fluidity and cancer cell motility. Oncogene. 2015;34:996–1005. doi: 10.1038/onc.2014.55. [DOI] [PubMed] [Google Scholar]
- 10.Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. 2008;121:727–735. doi: 10.1242/jcs.000455. [DOI] [PubMed] [Google Scholar]
- 11.Tirodkar TS, Lu P, Bai A, Scheffel MJ, Gencer S, Garrett-Mayer E, Bielawska A, Ogretmen B, Voelkel-Johnson C. Expression of ceramide synthase 6 transcriptionally activates acid ceramidase in a c-jun n-terminal kinase (JNK)-dependent manner. J Biol Chem. 2015;290:13157–13167. doi: 10.1074/jbc.M114.631325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laviad EL, Kelly S, Merrill AH Jr., Futerman AH. Modulation of ceramide synthase activity via dimerization. J Biol Chem. 2012;287:21025–21033. doi: 10.1074/jbc.M112.363580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doshi UA, Shaw J, Fox TE, Claxton DF, Loughran TP, Kester M. STAT3 mediates C6-ceramide-induced cell death in chronic lymphocytic leukemia. Signal Transduct Target Ther. 2017;2:17051. doi: 10.1038/sigtrans.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Y, DiVittore NA, Kaiser JM, Shanmugavelandy SS, Fritz JL, Heakal Y, Tagaram HRS, Cheng H, Cabot MC, Staveley-O’Carroll KF, et al. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol Ther. 2011;12:574–585. doi: 10.4161/cbt.12.7.15971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryland LK, Doshi UA, Shanmugavelandy SS, Fox TE, Aliaga C, Broeg K, Baab KT, Young M, Khan O, Haakenson JK, et al. C6-ceramide nanoliposomes target the Warburg effect in chronic lymphocytic leukemia. PLoS One. 2013;8:e84648. doi: 10.1371/journal.pone.0084648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tagaram HRS, Divittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET, Jiang Y, Isom HC, Kester M, Staveley-O’Carroll KF. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut. 2011;60:695–701. doi: 10.1136/gut.2010.216671. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Kitatani K, Toyoshima M, Ishibashi M, Usui T, Minato J, Egiz M, Shigeta S, Fox T, Deering T, et al. Ceramide nanoliposomes as a MLKL-dependent, necroptosis-inducing, chemotherapeutic reagent in ovarian cancer. Mol Cancer Ther. 2018;17:50–59. doi: 10.1158/1535-7163.MCT-17-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nieto MA, Huang RY-J, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 19.White-Gilbertson S, Mullen T, Senkal C, Lu P, Ogretmen B, Obeid L, Voelkel-Johnson C. Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene. 2009;28:1132–1141. doi: 10.1038/onc.2008.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroepil F, Fluegen G, Vallböhmer D, Baldus SE, Dizdar L, Raffel AM, Hafner D, Stoecklein NH, Knoefel WT. Snail1 expression in colorectal cancer and its correlation with clinical and pathological parameters. BMC Cancer. 2013;13:145. doi: 10.1186/1471-2407-13-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T, Zhang X, Shang M, Zhang Y, Xia B, Niu M, Liu Y, Pang D. Dysregulated expression of Slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J Surg Oncol. 2013;107:188–194. doi: 10.1002/jso.23240. [DOI] [PubMed] [Google Scholar]
- 22.Shioiri M, Shida T, Koda K, Oda K, Seike K, Nishimura M, Takano S, Miyazaki M. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br J Cancer. 2006;94:1816–1822. doi: 10.1038/sj.bjc.6603193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emprou C, Le Van Quyen P, Jegu J, Prim N, Weingertner N, Guerin E, Pencreach E, Legrain M, Voegeli A-C, Leduc C, et al. SNAI2 and TWIST1 in lymph node progression in early stages of NSCLC patients. Cancer Med. 2018. doi: 10.1002/cam4.2018.7.issue-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atmaca A, Wirtz RW, Werner D, Steinmetz K, Claas S, Brueckl WM, Rauh C, Fasching PA, Beckmann MW, Thiel FC. SNAI2/SLUG and estrogen receptor mRNA expression are inversely correlated and prognostic of patient outcome in metastatic non-small cell lung cancer. BMC Cancer. 2015;15:300. doi: 10.1186/s12885-015-1584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Li S, Chen R, Lu X. Increased risk of poor survival in ovarian cancer patients with high expression of SNAI2 and lymphovascular space invasion. Oncotarget. 2017;8:9672–9685. doi: 10.18632/oncotarget.14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganesan R, Mallets E, Gomez-Cambronero J. The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol Oncol. 2016;10:663–676. doi: 10.1016/j.molonc.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pewzner-Jung Y, Brenner O, Braun S, Laviad EL, Ben-Dor S, Feldmesser E, Horn-Saban S, Amann-Zalcenstein D, Raanan C, Berkutzki T, et al. A critical role for ceramide synthase 2 in liver homeostasis: II. J Biol Chem. 2010;285:10911–10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pewzner-Jung Y, Park H, Laviad EL, Silva LC, Lahiri S, Stiban J, Erez-Roman R, Brügger B, Sachsenheimer T, Wieland F, et al. A critical role for ceramide synthase 2 in liver homeostasis: I. J Biol Chem. 2010;285:10902–10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srivastava K, Pickard A, Craig SG, Quinn GP, Lambe SM, James JA, et al. DeltaNp63gamma/SRC/slug signaling axis promotes epithelial-to-mesenchymal transition in squamous cancers. Clin Cancer Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watters RJ, Kester M, Tran MA, Loughran TP Jr., Liu X. Development and use of ceramide nanoliposomes in cancer. Methods Enzymol. 2012;508:89–108. doi: 10.1016/B978-0-12-391860-4.00005-7. [DOI] [PubMed] [Google Scholar]
- 31.Zolnik BS, Stern ST, Kaiser JM, Heakal Y, Clogston JD, Kester M, McNeil SE. Rapid distribution of liposomal short-chain ceramide in vitro and in vivo. Drug Metab Dispos. 2008;36:1709–1715. doi: 10.1124/dmd.107.019679. [DOI] [PubMed] [Google Scholar]
- 32.Voelkel-Johnson C, Hannun YA, El-Zawahry A. Resistance to TRAIL is associated with defects in ceramide signaling that can be overcome by exogenous C6-ceramide without requiring down-regulation of cellular FLICE inhibitory protein. Mol Cancer Ther. 2005;4:1320–1327. doi: 10.1158/1535-7163.MCT-05-0086. [DOI] [PubMed] [Google Scholar]
- 33.Patwardhan GA, Liu YY. Sphingolipids and expression regulation of genes in cancer. Prog Lipid Res. 2011;50:104–114. doi: 10.1016/j.plipres.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, Hannun YA. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells, Dependence on protein phosphatase-1. J Biol Chem. 2002;277:12587–12595. [DOI] [PubMed] [Google Scholar]
- 35.Massiello A, Chalfant CE. SRp30a (ASF/SF2) regulates the alternative splicing of caspase-9 pre-mRNA and is required for ceramide-responsiveness. J Lipid Res. 2006;47:892–897. doi: 10.1194/jlr.C600003-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Massiello A, Roesser JR, Chalfant CE. SAP155 Binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5ʹ splice site selection of Bcl-x pre-mRNA. FASEB J. 2006;20:1680–1682. doi: 10.1096/fj.05-5021fje. [DOI] [PubMed] [Google Scholar]
- 37.Patwardhan GA, Hosain SB, Liu DX, Khiste SK, Zhao Y, Bielawski J, Jazwinski SM, Liu -Y-Y. Ceramide modulates pre-mRNA splicing to restore the expression of wild-type tumor suppressor p53 in deletion-mutant cancer cells. Biochim Biophys Acta. 2014;1841:1571–1580. doi: 10.1016/j.bbalip.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, Snook CF, Arnold HK, Sears RC, Hannun YA, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009;23:751–763. doi: 10.1096/fj.08-120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wooten LG, Ogretmen B. Sp1/Sp3-dependent regulation of human telomerase reverse transcriptase promoter activity by the bioactive sphingolipid ceramide. J Biol Chem. 2005;280:28867–28876. doi: 10.1074/jbc.M413444200. [DOI] [PubMed] [Google Scholar]
- 40.Wooten-Blanks LG, Song P, Senkal CE, Ogretmen B. Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J. 2007;21:3386–3397. doi: 10.1096/fj.07-8621com. [DOI] [PubMed] [Google Scholar]
- 41.Nganga R, Oleinik N, Kim J, Selvam SP, De Palma R, Johnson KA, Parikh RY, Gangaraju V, Peterson Y, Dany M, et al. Receptor-interacting Ser/Thr kinase 1 (RIPK1) and myosin IIA-dependent ceramidosomes form membrane pores that mediate blebbing and necroptosis. J Biol Chem. 2019 Jan 11;294(2):502–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fekry B, Jeffries KA, Esmaeilniakooshkghazi A, Szulc ZM, Knagge KJ, Kirchner DR, Horita DA, Krupenko SA, Krupenko NI. C16-ceramide is a natural regulatory ligand of p53 in cellular stress response. Nat Commun. 2018;9:4149. doi: 10.1038/s41467-018-06650-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sprott KM, Chumley MJ, Hanson JM, Dobrowsky RT. Decreased activity and enhanced nuclear export of CCAAT-enhancer-binding protein beta during inhibition of adipogenesis by ceramide. Biochem J. 2002;365:181–191. doi: 10.1042/BJ20020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyers-Needham M, Ponnusamy S, Gencer S, Jiang W, Thomas RJ, Senkal CE, Ogretmen B. Concerted functions of HDAC1 and microRNA-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol Med. 2012;4:78–92. doi: 10.1002/emmm.201100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–219. [DOI] [PubMed] [Google Scholar]
- 46.Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Adv Exp Med Biol. 2010;688:46–59. [DOI] [PubMed] [Google Scholar]
- 47.Van Veldhoven PP, Bell RM. Effect of harvesting methods, growth conditions and growth phase on diacylglycerol levels in cultured human adherent cells. Biochim Biophys Acta. 1988;959:185–196. [DOI] [PubMed] [Google Scholar]