

ABSTRACT
Despite incessant research, colon cancer still is one of the most common causes of fatalities in both men and women worldwide. Also, nearly 50% of patients with colorectal cancer show tumor recurrence. Recent investigations have highlighted the involvement of colon cancer stem cells (CCSCs) in cancer relapse and chemoresistance. CCSCs deliver a significant protumorigenic niche through persistent overexpression of self-renewal capabilities. Moreover, CSCs cross network with stromal cells, immune infiltrates, and cyotokine-chemokine, which potentiate their aggressive proliferative potential. Targeting CCSCs through small molecule inhibitors, miRNAs, and monoclonal antibodies (mAbs) in in vivo studies has generated compelling evidence for the effectiveness of these various treatments. This review effectively compiles the role of CCSC surface markers and dysregulated and/or upregulated pathways in the pathogenesis of colorectal cancer that can be used to target CCSCs for effective colorectal cancer treatment.
KEYWORDS: Colon cancer stem cells, Colorectal cancer, Notch pathway, Hedgehog pathway, Wnt/beta-catenin pathway
1. Introduction
Although colorectal cancer has been studied for years, it is still one of the leading types of cancer that results in death worldwide. In 2012, colon cancer accounted for approximately 9.7% of the total cancer cases and 8.5% of total deaths worldwide1. A linear relationship exists between the risk and age of the people who develop colorectal cancer. Varying lifestyles throughout the world serve to reinforce how lifestyle modification can actually affect the incidence of colorectal cancer. In the last few decades, there has been a slow but steady improvement in the prognosis of patients with colorectal cancer in many countries.1,2
Smoking, excessive alcohol consumption, high consumption of red and processed meats, inflammatory bowel disease, obesity, diabetes, family history of colorectal cancer, age, and gender are considered some of the risk factors for colorectal cancer. Based on emerging data, it has been observed that infection with Helicobacter pylori, Fusobacterium spp, and other potential infectious agents can also increase the risk of colorectal cancer.3–8 Surgery, chemotherapy, and radiation therapy are some of the commonly used treatment strategies to treat colorectal cancer. The principal chemotherapeutic regimens include 5 fluorouracil (5FU), oxaliplatin and/or leucovorin or 5-FU, leucovorin, and irinotecan (FOLFIRI). Despite recent advancements in the field of medicine, nearly 50% of patients with colorectal cancer show tumor recurrence. Colon cancer prevention includes physical exercise, hormone replacement therapy, and aspirin, which accounts for a reduction of about 20–30%. Despite these positive dietary lifestyle modifications, they are only modestly effective in preventing colon cancer.9–15
Recurrence of cancer has proven to be a major problem, which renders the effect of current treatments temporary and incomplete. This can partially be explained, because current treatments primarily reduce tumor bulk rather than totally eradicating the tumor, as well as the potential for tumor metastasis and development of drug resistance by cancer cells. Captivating evidence from previous studies suggest that cancer stem cells (CSCs) possess various intrinsic resistant mechanisms largely responsible for metastasis, drug resistance, and relapse of the disease after initial therapy. Specific targeting of CSCs, combined with current therapies, could potentially prevent recurrence.16,17 This review effectively assembles current information on the role of CCSC surface markers and dysregulated and/or upregulated pathways in colorectal cancer that can be used to target CCSCs for more effective treatment. It also provides insights into the drugs/molecules that are either in preclinical or clinical testing and currently being used to target CCSCs.
2. Colon cancer stem cells
Neoplastic cells, supporting vascular cells, inflammatory cells, and fibroblasts comprise the cell types included in most solid tumors.18 The majority of the cells in the bulk tumor mass lack self-renewal capacity and are nontumorigenic. However, a small subpopulation of the cells in the tumor bulk known as cancer stem cells (CSCs) are immortal and, therefore, possess a capacity for self-renewal and the ability to “reform” the original tumor.19,20
Data from previous studies suggests their involvement in tumor growth, initiation, maintenance, survival, metastasis, and cancer recurrence. The property of pluripotency enables them to generate tumor cells with different phenotypes, which results in the growth of the primary tumor and emergence of new tumors.21,22 CSCs also have the ability to generate heterogeneous lineages of cancer cells that comprise the tumor.23–25 Interestingly, CSCs represent approximately 0.1–10% of all tumor cells and only some of them have the capacity to form a tumor. Because CSCs express antigens at lower levels, it makes them ‘difficult-to-target’. In fact, their identification is based on the presence of populations of cells that have stem cell-like properties and not on the overexpression of tumor antigens.26 For the expansion of a tumor, CSCs tend to undergo either a symmetrical, or asymmetrical, self-renewal process during cell division. Symmetrical cell division generates two identical daughter CSCs, whereas asymmetrical cell division generates one daughter CSC and one differentiated progenitor cell, which results in the expansion of the number of CSCs as the tumor grows.26
Rudolf Virchow, a German pathologist, was the first to propose the CSC hypothesis in 1855. Through his studies, he predicted that activation of dormant embryonic-like cancerous cells present in mature tissues leads to cancer.25 In 1994, Lapidot proved the CSC hypothesis by successfully producing leukemia in immunocompromised mice following transplantation of human acute myeloid lymphoma cells that manifested stem cell characteristics.26 Subsequent to this discovery, the presence of CSCs was explored in solid tumors.25 In 2007, O‘Brien and Vitiani, through independent investigations, discovered CCSCs.24
CCSCs are resistant to conventional chemotherapeutic drugs and radiotherapy due to a variety of known and unknown intrinsic mechanisms. Some of these proposed mechanisms include increased expression of ATP-binding cassette (ABC) drug transporters, activation of Wnt/beta catenin, Hedgehog and Notch signaling pathways, amplified activity of aldehyde dehydrogenase 1 (ALDH1), radiation-induced conversion of cancer cells to CCSCs, protection by microenvironment and niche networks, and metabolic alterations with a preference for hypoxia. CSC driven chemoresistance has been reported in human leukemia, malignant melanoma, and in brain, breast, pancreatic, and colorectal cancers.27
Treatment approaches that target CCSCs have shown increased efficacy and a reduced risk of tumor relapse, as well as metastasis, at preclinical stages. CCSCs are one of the major reasons for tumor redevelopment, therapeutic resistance, and malignant progression in cancer patients.
3. Origin of colon cancer stem cells
Stem cells play an important role in tumor development, growth, and metastasis and recurrence in many tumors including colon cancer. The colon is composed of various compartments; for example, the crypts of Lieberkuhn. These crypts are uniform test-tube shaped structures containing 2000–3000 cells. Stem cells are housed at the bottom of these crypts. In the human colon, these stem cells account for < 20 cells per crypt. However, these stem cells at the bottom of the crypt undergo self-renewal and produce new cells, which begin with proliferating cells that travel up the crypt as differentiating cells and ultimately replace the apoptotic cells at the top of the crypt. Despite the similarity of this process in healthy living tissue, it can result in tumor formation if it goes awry.28–31 The most widely accepted hypothesis for the origin of CCSC’s suggests the involvement of microenvironment; dedifferentiation of colon cancer cells and malignant transformation of colon cancer cells and normal stem cells.32–36 Colon CSCs show the presence of various populations of cells that are in different stages of development, which gives them the characteristic of heterogeneity.37 CSCs are normally in a quiescent state. The most vexing therapeutic challenge associated with disease relapse and treatment resistance is that current therapies target only the tumor bulk and, consequently, spare quiescent stem cells.38 Self-renewal, production of differentiated progeny, expression of specific surface markers and oncogenes, utilization of common signaling pathways, and the importance of the stem cell niche are the common characteristics shared by CSC and normal stem cells, but CSC are not identical to normal stem cells, because normal stem cells lack tumorigenic potential.18,24,39
4. Targeting colon cancer stem cells for the treatment of colorectal cancer
The current approved therapies against cancer have several limitations that frequently lead to treatment failure. As mentioned earlier, resistance to chemotherapy and radiotherapy is among the most common cause for treatment failure. Also, the current therapies fail to eliminate CCSC leading to metastasis and tumor reoccurrence. In order to avoid toxicity of the other healthy tissues and cells, specific CCSC targeting is necessary. Therefore, eliminating CCSC is crucial in order to treat malignant disorders. Recently, multiple targets are being identified and potential agents are being developed that could specifically target CCSC. These potential targets range from various cell surface markers to signaling pathways, the microenvironment niche that provides the necessary conditions for tumor growth, and the ABC transporters that are responsible for resistance to the gold standard anti-cancer drugs like doxorubicin, paclitaxel, and cisplatin. Currently, there are few therapeutic strategies that can specifically kill CCSC and many of them are undergoing preclinical and clinical evaluation. As mentioned above, CCSCs account for only 0.3–2.2% of the total cancer cells in colorectal cancer. They have the capacity for infinite proliferation and, therefore, specific treatments that would target the CCSC would help in potentially eliminating the tumor.40 However, the goal for any CCSC targeted therapy should be to potentially eradicate the entire CCSC population in order to avoid their progeny, as well as the occurrence of CCSC treatment-resistant cells. In order to achieve this therapeutic goal, more than one intrinsic pathway involved in CCSC maintenance, growth, and renewal needs to be targeted. Therefore, in order to increase the potential of a cure in colorectal cancer patients, it is necessary to combine CCSC targeted therapy with conventional chemotherapy and novel tumor-targeted drugs that can facilitate the elimination of CCSC, additional differentiated progeny, and lastly, the bulk tumor cell population.40
4.1. Targeting cell surface markers
Cell surface markers vary with the type of tumor. Specific targeting of these cell surface markers could potentially eliminate CCSC. In order to enhance the specificity of therapeutic strategies, researchers often choose ligands or antibodies against tumor cell surface makers. Monoclonal antibodies, which target CCSCs, have generated much interest in the cancer research community.41
In CCSC, EpCAM (high)/CD44+ acts as a specific biomarker. EpCAM is a 40 kDa glycoprotein that functions as an epithelial cell adhesion molecule. In normal tissues, EpCAM is only expressed basolaterally and is shielded by tight junctions that limit its accessibility. In contrast, in tumor cells, EpCAM is expressed on the entire cell surface, and therefore, becomes more accessible for binding.42 EpCAM plays an important role in cell proliferation and inhibits differentiation of cells. Approximately 85% of colorectal carcinomas express EpCAM. It is one of the earliest tumor markers that is involved in signal transduction, regeneration of tissue, and other biological functions and its expression level varies depending on the stage of the disease. EpCAM can induce acceleration of the cell cycle and promote proliferation by upregulating the expression of oncogene c-myc. Interestingly, the proliferative and invasive capacity of tumors is enhanced by increased expression of EpCAM and its downregulation by RNA interference inhibits these capacities.40,43,44 CD44 is involved in cell adhesion, migration, invasion, and angiogenesis and is located on the cell membrane. In a study conducted by Schulenburg et al., it was demonstrated that CD44+ cell isolates from colon cancer samples had stem cell-like properties and CD44- cells lacked stem cell-like properties. Also, CD44+ cells showed greater capacity for proliferation and invasion.45 The composite EpCAM(high)/CD44+ is a more specific colorectal stem cell marker than the individual cell adhesion molecule and the composite structure of EpCAM(high)/CD44+ can promote tumor invasion and metastasis. It was demonstrated that EpCAM(low)/CD44- cells lack the capacity for tumor initiation and formation. In a study conducted by Liu et al., 80 cases of colorectal cancer and liver metastasis were examined. It was observed that EpCAM(high)/CD44+ was not present in adjacent normal intestinal epithelium, but was visible in colorectal cancer tissue and corresponding metastatic liver specimens. The percentage of double-positive cells was 0.8–3.1% in colorectal cancer. The results from this study demonstrate that EpCAMhigh/CD44+ expression is significantly correlated with invasion and metastasis, and confirms that EpCAMhigh/CD44+ cells may serve as effective biomarkers for CCSCs.46 Based on the theory**47 that CCSCs are capable of tumor invasion and metastasis, specific targeting of the EpCAM(high)/CD44+ colorectal stem cell marker may help in eliminating the tumor.
Catumaxomab is a specific targeted antibody that has been developed to act on the above mentioned stem cell marker. It obtained market approval in Europe in 2009 for the treatment of malignant ascites in cancer patients. Catumaxomab is a bispecific trifunctional antibody consisting of mouse IgG2a and rat IgG2b produced using quadroma technology. The antibody binds at three different sites; namely, T-cells via the CD3 marker, EpCAM, and the Fc region and exerts its cytotoxicity through T-cell mediated lysis, secretion of cytokines (e.g., IL-1, IL-2, IL-6, IL-12, or DC-CK1), and phagocytosis. Additionally, catumaxomab effectively eliminates CD133+/EpCAM+ CSCs from malignant ascites of patients with advanced ovarian, gastric, and pancreatic cancers, which suggests its potential therapeutic application in eradicating epithelial cancers expressing CSCs.48–50
A bispecific, bifunctional single-chain antibody MT110 is a construct of the bispecific T-cell engager (BiTE) class, which binds to EpCAM and CD3 and primes the resting human peripheral CD4+ and CD8 + T-cells. This subsequently leads to apoptosis of the EpCAM+ CSCs and the epithelial bulk tumor cells. Upon injecting EpCAM+ CSCs derived from patients with primary colon or pancreatic cancers into NOD/SCID mice, MT110 effectively eradicated CSCs. Interestingly, CSCs did not proliferate into fully grown tumors in MT110-treated NOD/SCID xenograft mice, which further strongly suggests the potential therapeutic benefits of using MT110 to eradicate EpCAM+ CSCs and bulk tumor cells.50–52 Statistical analysis on patients with human**53 adenocarcinoma and colorectal cancer frequently reveals EpCAM overexpression (~99.7) with protumorigenic capabilities.44,54 However, complete elucidation of the mechanisms involved in CCSC-driven tumor proliferation must be unraveled prior to an improvement in the overall prognosis.
4.2. Targeting signaling pathways
Self-renewal, programmed proliferation, differentiation, and signaling pathways are the hallmarks of stem cells; however, sudden dysregulation of these hallmarks and aberrant activation of dormant oncogenes may result in unexplained formation of CCSCs leading to tumorigenesis. Hedgehog (Hh), Notch, Wnt/b-catenin, high mobility group AT-hook 2 (HMGA2), Bcl-2, and Bmi-1 are ubiquitously dysregulated and are required for sustaining the hallmarks or features of stem cells in CCSCs. Hh, Notch, and Wnt/b-catenin potentially regulate tumorigenesis in CCSCs and, therefore, therapeutic interventions that modulate these signaling pathways may offer new strategies for cancer therapy.55,56
4.2.1. Targeting the notch pathway
The Notch pathway plays an important role in embryogenesis, cellular homeostasis, differentiation, EMT, and apoptosis.57 Notch signaling is initiated by ligand binding to the Notch receptor. Figure 1 depicts a schematic representation and function of the Notch canonical and non-canonical pathway. In CCSCs, Notch signaling inhibits apoptosis by repressing the cell cycle inhibitor p27 and maintaining its “stemness.” Additionally, a 10–30-fold higher expression of Notch signaling is observed in CCSCs leading to an unparalleled proliferative potential and chemoresistance as compared to the bulk tumor cells. However, Notch signaling can be inhibited by monoclonal antibodies directed against Notch receptors and gamma secretase inhibitors (GSIs).56,60
Figure 1.
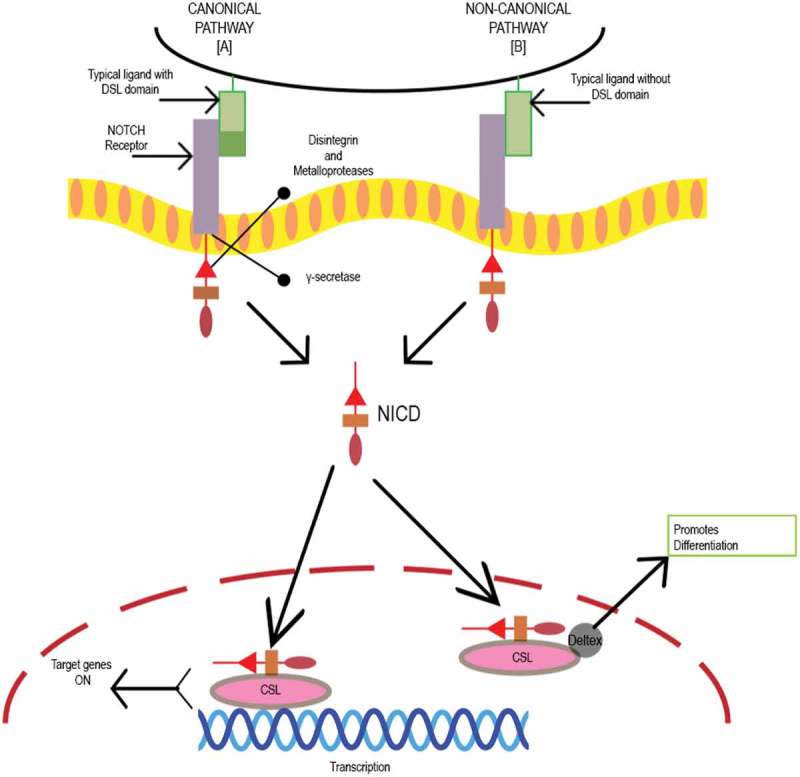
Schematic representation of Notch canonical and non-canonical pathway. Initiation of Notch signaling occurs on ligand binding to the receptor. There are five typical Notch ligands Delta like, 1, 3, and 4 and Jagged 1 and 2 with a Delta-Serrate-Lag 2 (DSL) domain, while atypical ligands include DNER, F3/Contactin and NB-3 without a DSL domain and four Notch receptors Notch 1–4.58 Proteolytic cleavage by disintegrin and metalloproteases family proteases (MMP), and gamma-secretase releases active Notch intracellular domain (NICD).59 The biological process is mediated by Notch via canonical and noncanonical pathways. In the canonical Notch pathway, NICD translocates to the nucleus and binds to the transcription factor CSL. Mastermind co-activators activate the CSL-NICD complex leading to suppression of differentiation and maintenance of stemness by activating transcriptional targets HES1 and HEY1. HES1 increases the stemness-related genes in CRC cells and CSC surface markers CD133, ALDH1, and ABCG2. In noncanonical pathway, atypical ligand interacts with receptor promoting differentiation by formation of CSL-NICD-Deltex complex.56,57,60.
GSIs prevent the final proteolytic cleavage of Notch receptors that releases the active intracellular fragment. GSIs inhibit both Notch1 and Notch2 within the stem-progenitor compartment of the intestinal crypt. For instance, RO4929097, a small molecule GSI developed by Roche, is currently under clinical investigation as either monotherapy, or in combination with conventional chemotherapeutics. However, RO4929097 has been used in combination with cetuximab against CRCs, but resulted in significant toxicity such as activation of cyp450, which caused a decrease in its bioavailability. Furthermore, administration of RO4929097 during Phase II clinical trials demonstrated ineffectiveness as a monotherapeutic agent, because no significant response was observed in patients with CRC (NCT01116687).57,61 GSIs are also known to cause severe gastrointestinal toxicity due to goblet-cell metaplasia of the small-intestinal epithelium (a target-mediated effect due to inhibition of Notch1 and Notch2). Mechanistic evaluation of GSI-mediated toxicity revealed its involvement in abolishing the proliferative potential of crypt progenitors causing them to differentiate into postmitotic goblet cells. Additionally, systemic toxicity and off-target effects have been reported, which has limited its therapeutic potential. Interestingly, the efficacy of GSIs in a preclinical model yields a synergistic cytotoxicity with chemotherapeutic agents through both antiangiogenesis and anti-CSC effects. Nevertheless, the further design of GSIs to optimize their efficacy appears warranted.56,62
Suppression of activated Notch signaling in the presence of Honokiol (traditional Chinese medicine) increased the sensitivity of CCSCs to ionizing radiation (IR). Results from the combination of Honokiol and IR on cancer cell lines demonstrated its effectiveness by inducing apoptosis, reducing DCLK1, and activating Notch-1, Jagged-1, and Hes-1. These promising results were then extrapolated to a xenograft animal model, which led to a reduction in the expression of CSC marker and Notch signaling in xenograft tissues, as well as suppression of tumor growth.55,59
4.2.2. Targeting the hedgehog pathway
Hedgehog (Hh) pathways includes a wide variety of cellular and molecular mechanisms. For instance, protein trafficking, protein–protein interactions, feedback loops, and post-translational modifications to name but a few. These processes help in the tight regulation of Hh signaling in a temporally and spatially specific manner; a key requirement for tissue patterning, cell fate determination, and self-renewal.63 By orchestrating reciprocal communicative events between different cells and tissues, the Hh pathway plays a crucial role during organogenesis in the developing embryo. Based on the receiving cell type, Hh signaling varies and includes directing cell proliferation, cell fate determination, epithelial-to-mesenchymal transitions (EMT), and the rearrangement of cells by motility and adhesion changes.63,64 Therefore, it is not surprising that inappropriate activation of Hh signaling can contribute to the initiation, growth, and maintenance of cancer. Failure in aggressive and chemoresistant tumors is due to the overexpression in the Hh signaling cascade.63 In vivo studies have demonstrated the involvement of the Hedgehog-GLI (HH-GLI) pathway in the maintenance of self-renewal capacity of CCSCs and CD133+ colon CSCs 64–65. Figure 2 depicts a schematic representation of the Hedgehog pathway and its role in signaling in CCSC’s. When specific ligands, such as exo-secretion ligand Shh, bind to the trans-membrane receptor PATCHED1 (PTCH1), it initiates the HH-GLI pathway. Binding of this ligand to the receptor inhibits SMOOTHENED (SMO) protein, leading to its translocation to primary villi of the cell and inhibition of GLI repressors (mostly GLI3R) and activation of the intracellular signaling cascade. This cascade causes translocation and nuclear activation of the transcription factor Gli2. In parallel with this process, the transcription factor Gli2 undergoes up-regulation, which subsequently affects cell proliferation, regulation, and cell fate determination by ‘turning on’ the expression of specific genes. It also causes transcription of three GLI zinc finger transactivation factors PTCH1, GLI1, HIP. Targeted inhibition of SMO and GLI1 could possibly lead to cell death. Therefore, directly or indirectly inhibiting the HH-GLI pathway could potentially eradicate the bulk tumor and the CSC population within the tumor.58,63
Figure 2.
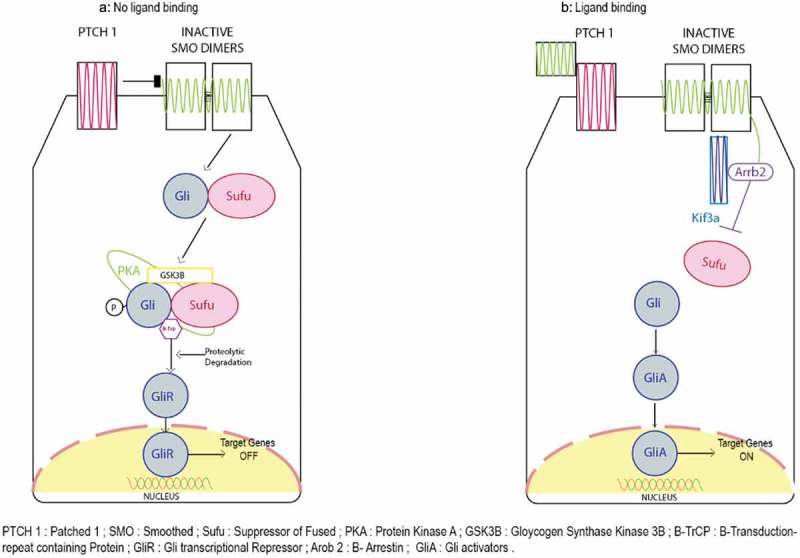
The process of Hedgehog pathway begins on binding of specific ligands such as exo-secretion ligand Shh bind to the trans-membrane receptor PATCHED1 (PTCH1) it initiates the HH-GLI pathway. Binding of this ligand to the receptor inhibits SMOOTHENED (SMO) protein leading to its translocation to primary villi of the cell and inhibition of GLI repressors (mostly GLI3R) and activation of intracellular signaling cascade. The cascade causes translocation and nuclear activation of the transcription factor Gli2. In parallel to this, the transcription factor Gli2 is upregulated playing an important role in cell proliferation, regulation and cell fate determination by turning on specific gene expression. It also causes transcription of three GLI zinc finger transactivation factors PTCH1, GLI1, HIP. Targeted inhibition of SMO and GLI1 could lead to cell death. Therefore, directly or indirectly inhibiting HH-GLI pathway could potentially eradicate the tumor and CSC population in the tumor.58,63.
Cyclopamine is a naturally occurring teratogenic alkaloid that is thought to be a strong inhibitor of the HH-GLI pathway. It inhibits cholesterol biosynthesis and interacts directly with SMO, which inhibits the HH-GLI pathway. These results have been demonstrated in HT-116 cell lines. HT-116 spheres were grown in serum-free, nonadherent culture and showed an increased expression of stem cell markers such as NANOG, POU5F1, CD-44, and EpCAM compared to cells grown in conventional culture medium. Treatment with cyclopamine decreased the level of stem cell markers NANOG, POU5F1, and CD-44. Hence, HT-116 cells with sphere formation capacity possessed CSC-like properties and, by using this model, it was demonstrated that cyclopamine downregulated the stem-cell associated markers and genes.65
GDC-0449 (Vismodegib), a SMO antagonist has been approved by US-FDA for treating basal cell skin carcinomas. The potency of GDC-0449 was demonstrated on Caco-2 and HT-29 cell lines by its downregulation of GLI1, inhibition of the HH-GLI pathway, and its anti-CSC effects, which were made evident by its capacity to down regulate the CSC surface markers CD44 and ALDH.65,66
Compelling evidence suggests that the characteristics normally supporting chemoresistance is limited by inhibition of Hh signaling in CSCs that promotes commitment or differentiation and a loss of stemness, as supported by a reduction in clonogenicity and pluripotency markers. In order to prevent tumor relapse and maximize patient outcomes, an attractive approach would be the combinatorial targeting of CSCs and tumor bulk with Hh inhibitors and conventional chemotherapeutic agents and/or radiation. However, further investigation into the sequencing of Hh inhibitors and conventional therapies is required to determine whether priming CSCs prior to cytotoxic treatment, coadministration, and/or use as maintenance therapy following tumor debulking will lead to optimal outcomes. Importantly, the type of Hh antagonist required for individual cancer subtypes must be carefully considered and based on the mode of Hh pathway activation. However, a greater understanding of Hh-mediated CSC maintenance and how to best combine Hh antagonists with conventional therapies in the clinic will be required before the full potential of this therapeutic strategy is realized.58
4.2.3. Targeting the WNT signaling pathway
The Wnt signaling pathway regulates stem cell self-renewal in epithelial cancers, including colon cancer. Wnt signals are either transduced to the canonical Wnt pathway for cell fate determination or to the noncanonical Wnt pathway for controlling tissue polarity and cell movement. Different Wnt ligands activate either the canonical or noncanonical Wnt pathway. The canonical Wnt signal regulates specific gene expression by activating the downstream target TCF/LEF. This activation is achieved by stabilization of β-catenin through inhibition of its phosphorylation-dependent degradation via Frizzled/LRP/6. High Wnt signaling activity defines human CCSCs, and these cells preferentially localize to a myofibroblast niche. This has been reported based on Wnt reporter constructs. Differentiated cells are relatively “Wnt-low,” whereas CSCs are relatively “Wnt‐high.” Wnt activity levels are dependent on extrinsic factors such as Hepatocyte Growth Factor (HGF) and RSPO3, and on various intrinsic factors such as mutation and expression levels of microRNAs (miRs). Since the CSC phenotype is not stable, differentiation and dedifferentiation are ongoing processes with CSC. Specific targeting of the Wnt pathway could potentially eliminate the CSC population.67
Figure 3 depicts a schematic representation of the Wnt pathway and also depicts its mechanism as it pertains to CCSC’s. β-catenin is a protein kept at a low cytoplasmic concentration by the destruction complex mainly regulating the Wnt pathway. The destruction complex consists of the tumor suppressor protein adenomatous polyposis coli (APC), casein kinase 1 (CK1) and glycogen synthase kinase 3b (GSK3-b), and Axin2, which scaffold the complex together. The membrane receptor complex is formed by frizzled (Fzd) and low-density lipoprotein receptor-related protein 5/6 (LRP5/6). In the absence of Wnt ligands, this membrane receptor complex is not engaged. Thereafter, CK1 and GSK3-b phosphorylate β-catenin at specific serine and threonine residues and leads to priming of its recognition by the U3 ubiquitin ligase β-transducin repeat-containing protein (β-TRCP). As a consequence, β-catenin is ubiquitinated and targeted for proteosomal degradation.67 Gene transcription is actively repressed in the nucleus as TCF transcription factors are bound to corepressor (Groucho). In the presence of Wnt ligands, they bind to Fzd and LRP5/6 coreceptors and trigger the formation of Dvl-Fzd complex. Also, this leads to phosphorylation of LRP by GSK3-b. The destruction complex is dissolved as this phosphorylation recruits the scaffolding protein Axin2 to the coreceptors.67 As a result, β-catenin stabilization occurs and can therefore accumulate in the cytosol. Subsequently, β-catenin translocates in the nucleus where it converts TCF into a transcriptional activator. This step is mediated by the displacement of the Groucho protein and recruitment of coactivators that include CBP, BCL9, and PYG. This recruitment ensures efficient transcription of genes that are important regulators of stem cell fate (LGR5, ASCL2), cell proliferation (C-MYC), and also, negative regulators of the pathway (Axin2). In CRC, truncating mutations in APC are frequently observed. In such mutations, there is inefficient targeting of β-catenin for degradation as the destruction complex is not properly formed. Therefore, even in the absence of external signals, β-catenin can accumulate and form active transcription factor complexes with TCF proteins in the nucleus.67
Figure 3.
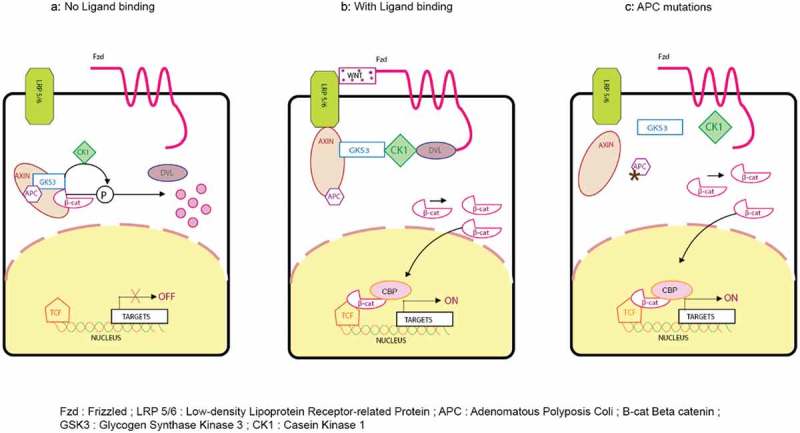
Wnt signaling pathway with and without ligand binding and APC mutations. β-catenin is a protein kept under low cytoplasmic concentration by the destruction complex mainly regulating the Wnt pathway. The destruction complex consists of the tumor suppressor protein adenomatous polyposis coli (APC); casein kinase 1 (CK1) and glycogen synthase kinase 3b (GSK3-b); and Axin2, which scaffold the complex together. The membrane receptor complex is formed by frizzled (Fzd) and low-density lipoprotein receptor–related protein 5/6 (LRP5/6). In the absence of Wnt ligands, this membrane receptor complex is not engaged. Thereafter, CK1 and GSK3-b phosphorylate β-catenin at specific serine and threonine residues and leads to priming of its recognition by the U3 ubiquitin ligase β-transducin repeat-containing protein (β-TRCP). As a consequence, β-catenin is ubiquitinated and targeted for proteosomal degradation.67 Gene transcription is actively repressed in the nucleus as TCF transcription factors are bound to corepressor (Groucho).67 In the presence of Wnt ligands, they bind to Fzd and LRP5/6 coreceptors and trigger the formation of Dvl-Fzd complex. Also, it leads to phosphorylation of LRP by GSK3-b. The destruction complex is dissolved as this phosphorylation recruits the scaffolding protein Axin2 to the coreceptors.68 As a result, β-catenin stabilization occurs and can therefore accumulate in the cytosol. Subsequently, β-catenin translocates in the nucleus where it converts TCF into a transcriptional activator. This step is mediated by the displacement of the Groucho protein and recruitment of coactivators that include CBP, BCL9, and PYG.69 This recruitment ensures efficient transcription of genes that are important regulators of stem cell fate (LGR5, ASCL2), cell proliferation (C-MYC), and also, negative regulators of the pathway (Axin2). In CRC, truncating mutations in APC are frequently observed. In such mutations, there is inefficient targeting of β-catenin for degradation as the destruction complex is not properly formed. Therefore, even in the absence of external signal, β-catenin can accumulate and form active transcription factor complexes with TCF proteins in the nucleus.70.
Traf2- and Nck-interacting kinase (TNIK) are essential regulatory components of the T-cell factor-4 and β-catenin transcriptional complex. TNIK is required for the tumor-initiating function of CCSCs. NCB-0846 has been shown to block TNIK/Wnt signaling and demonstrated not only marked anti-tumor and anti-CSC activity, but also inhibited the expression of mesenchymal marker proteins. Since TNIK is a multifunctional protein, it also regulates other pathways in addition to Wnt. Based on the results of this study, it can be inferred that specific TNIK inhibition could be a potential therapeutic approach for eradicating CCSCs.71
The R-spondin (RSPO) pathway plays a pivotal role in regulating stem cell maintenance and renewal. WNT signaling is activated by RSPO with the help of Wnt ligands. In CRC, translocation of RSPO occurs. OMP-131R10 is a novel IgG1 that inhibits the binding of RSPO3 to leucine-rich repeat-containing G-coupled receptors (LGRs) and the activation of the RSPO-LGR pathway. This may result in an inhibition of both CSC survival and proliferation. Anti-tumor effects have been demonstrated in patient-derived xenograft animal models as a single agent and in combination with traditional chemotherapeutic agents. A phase Ia/Ib study is ongoing with this agent (NCT02482441; sponsored by Onco Med Pharmaceuticals).72
4.2.4. Targeting the hippo pathway
It was not more than a decade ago that one of the most important pathways driving “stemness” to cancer stem cells was revealed. This pathway, known as Hippo pathway, not only plays an important role in organ development, tumorigenesis, and tissue regeneration, but also, recent studies have highlighted its role in driving cancer stem cell biology that includes EMT, drug resistance, and self-renewal.73 Its identification and role in tumor suppression can be traced to Drosophila melanogaster, which was later followed by its recapitulation in transgenic mouse models.74,75
The cytoplasmic kinase module and nuclear transcription module are the two major components of the Hippo pathway. The oncogenic transcriptional module is composed of yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ). YAP/TAZ, together with the TEA domain family member (TEAD), function as transcriptional coactivators. Mammalian STE20-like protein kinase 1 (MST1) and (MST2) activate large tumor suppressor 1 (LATS1) and LATS2 by phosphorylating them and thus, forming the kinase module. However, recent studies provide evidence that supports the MAP4K family as also being a part of the kinase family along with MST1 and MST2. The biological function of paramount importance that is performed by the kinase cascade is the inhibition of the oncogenic transcriptional module.76,77
The Hippo pathway in mammals functions as a tumor suppressor pathway under normal circumstances. Figure 4 depicts a schematic representation of the Hippo pathway. There has been considerable evidence supporting the role of the Hippo pathway in regulating cell-cell, contact-induced growth inhibitory signals. Under normal physiological conditions, such as wound healing and embryonic development, activation of the Hippo pathway occurs due to release of E-cadherin, alpha-catenin, and Crumbs during cell-cell contact. However, loss of cell-cell contact inhibition facilitates tumorigenesis during EMT due to hyperactivation of YAP/TAZ.79–81 Importantly, there are also additional potent regulators of YAP/TAZ, which include mechanical cues such as extracellular matrix (ECM) stiffness, cell attachment or detachment, cell geometry, and cytoskeleton tension. These additional factors support YAP/TAZ as being a mediator of mechano-transduction, thereby validating its significant role in a myriad of pathological conditions.82 Nutrient stress also regulates the activation of YAP/TAZ, since it has long been known that extracellular nutrients such as glucose and amino acids regulate cell metabolism and proliferation. Furthermore, intracellular and environmental stresses also regulate the Hippo pathway due to the fact that this pathway also plays an important role in combating cellular stress signals.83
Figure 4.
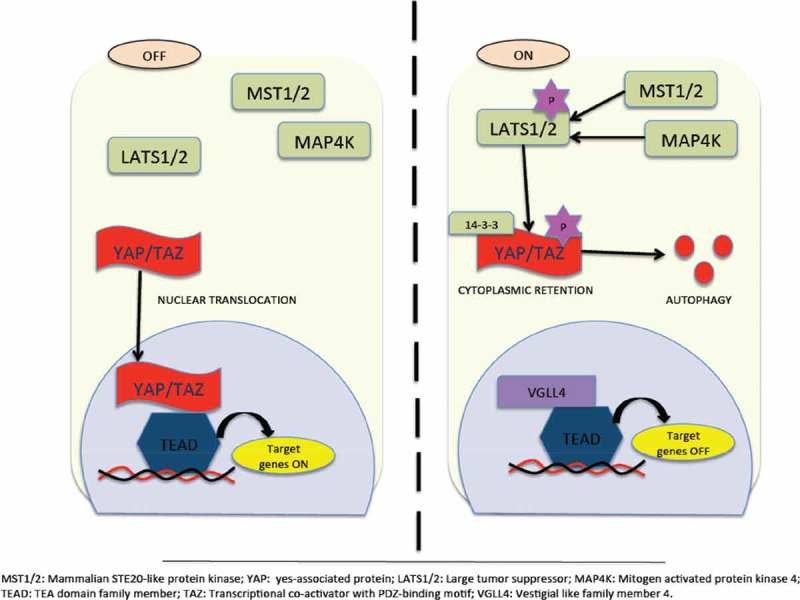
Hippo Pathway for tumor suppression. Inactivation of YAP/TAZ leads to oncogenic transcriptional module. Its inactivation is due to activation of hippo kinases MST1/2 that facilitates activation of LATS1/2 thereby phosporylating and retaining YAP/TAZ in the cytoplasm via 14–3-3 or being subjected to proteasomal or autophagy-induced degradation. Followed by this, suppression of TEAD-mediated gene transcription occurs. On the other hand, inactivation of hippo kinases occurs due to myriad reasons. Inactivation of hippo kinases leads to dephosphorylation of YAP/TAZ and translocates inside the nucleus inducing TEAD target gene expression. However, recent studies highlight Hippo-YAP-independent activation of TEAD too.77,78.
Various studies have demonstrated the involvement of YAP in the development of CRC. In fact, it has been shown that increased expression of YAP is co-related to high histological grade, enrichment of colon stem cell signatures, metastasis characteristics, and cancer progression. Other studies have also demonstrated resistance to cetuximab therapy due to upregulation of YAP. Moreover, increased expression of YAP and PGE2 has been associated with CRC, which substantiates the premise that YAP inhibitors may act in synergism with NSAIDs.84,85 Despite a significant body of evidence supporting the association of YAP with poor prognosis and development of CRC, there have been other conflicting studies that suggest YAP to be a tumor-suppressor gene. Therefore, the function of YAP appears to depend significantly on the tissue involved. In the case of CRC, the enhancement in the function of the p73 transcription factor in the promoter region of apoptotic genes leads to induction of apoptosis as a response to DNA damage.86 The contrasting results from various investigations as it pertains to YAP appear to suggest that if YAP can reduce cell proliferation and induce cell apoptosis and death in CRC, then YAP activation could be beneficial in the treatment of CRC. Therefore, further in-depth study is urgently needed regarding the specific functions of YAP.
The maintenance of “stemness” and tissue homeostasis involves one more key regulator, and that regulator is the Hippo pathway.87 There is evidence from previous studies that depict localization of the YAP protein to the crypt base, as well as its absence from villi. This fact supports the possibility that YAP maintains the lack of differentiation of stem cells by binding to the TEAD transcription factor. It is for this reason that recent studies have suggested a dual role for YAP in the regulation of intestinal stem cells (ISCs). Interestingly, it has been observed that YAP hyperactivation expands intestinal progenitor/stem cells, whereas YAP deletion impairs the regeneration of intestinal cells that have been experimentally damaged with dextran sodium acetate.88,89
The potential of YAP as a target for small-molecule modulators has been explored in a plethora of cancers and is classified into three main categories. These three categories are comprised of small-molecule modulators that (1) regulate the upstream molecules of YAP and the effects of YAP-TEAD transcriptional activity, (2) modulate YAP phosphorylation and block YAP nuclear translocation, and (3) inhibit YAP to subsequently inhibit YAP’s interaction with TEAD1.
Recently, oligomeric proanthocyanidins (OPCs) have been tested against CCSCs by Toden and colleagues.90 Proanthocyanidins are particularly found in fruits and vegetables and are a group of heterogeneous flavon-3-ol or flavon-3,4-diol oligomers. Their study demonstrated antitumorigenic potential of OPCs, as well as anti-CCSC properties. The main findings of the study not only demonstrated decreased expression of CCSC markers like LGR5, CD44, and CD133, but also verified OPCs as YAP/TAZ inhibitors, thereby inhibiting the Hippo pathway.90
Verteporfin is well known for enhancing phototherapy against neovascular macular degradation by inhibiting YAP. However, a recent study demonstrated its antitumor effects independent of its action on YAP in the context of CRC.91 Additionally, another study demonstrated the ability of verteporfin to reverse paclitaxel resistance due to YAP over-expression in HCT-8/T cells by inhibiting the expression of YAP.92
The major hurdle in the development of small-molecule modulators of YAP is its dual role of being both oncogenic and functioning as a tumor suppressor. The difficulty arises in determining whether inhibiting or stimulating the expression of YAP represents a more suitable strategy against CCSCs. Therefore, the role of YAP in relation to CCSCs requires further investigation and in-depth analysis.
4.2.5. Cross-talk between the hippo pathway and other signaling pathways
The interplay between the Hippo pathway and other signaling pathways was first studied in Drosophila. Thereafter, their connection in mammals was also determined. It was observed in a YAP-transgenic mouse model that YAP expression led to a rapid stimulation of HES expression, which was an indication that Notch signaling was activated. Additionally, there has been a considerable body of evidence that supports a positive feedback loop between the Hippo and Notch signaling pathways, since overexpression of NCID has been correlated to increased expression of YAP and TEAD. Moreover, the physical interaction of YAP with NICD facilitates Notch signaling output. Importantly, several studies have also identified Notch2 as a direct YAP/TAZ/TEAD target gene.93–95
In contrast to the Notch signaling pathway, there exists a negative feedback loop between the Hedgehog (Hh) and Hippo pathways, since studies have shown that protein levels of YAP1 increase due to an increase in Hh signaling. Coupled with this finding is the fact that there is suppression of Hh target genes when YAP binds to GLI transcription factors.96–98
The interplay between Wnt signaling and the Hippo pathway involves several opposing views and is less than straightforward with regard to regulatory mechanisms. To begin, the Hippo pathway limits a positive Wnt signaling regulator called DVL due to the cytoplasmic expression of its core kinase component, YAP1/TAZ, and thus, serves to repress the activity of β-catenin.99 As mentioned earlier with regard to the Wnt signaling pathway, the β-catenin degradation complex, which includes YAP, undergoes phosphorylation and results in β-catenin being retained in the cytoplasm. Subsequently, β-catenin is subjected to ubiquitination and degradation by the proteasome. As it pertains to the binding of the canonical Wnt ligand, activation of β-catenin and YAP1/TAZ targets genes occurs after binding to the transcriptional activators TCF and LEF or TEAD, respectively, which causes translocation of β-catenin and YAP1/TAZ to the nucleus. On the other hand, binding of the noncanonical Wnt ligand activates the G-protein-coupled receptor through Gα12/13. Importantly, inhibition of LATS1/2 results in activation of YAP1/TAZ due to subsequent activation of Rho GTPases (RHO). A final significant fact worth noting is that the interplay between APC and YAP/TAZ can occur independent of Wnt/β-catenin signaling. One of the most common observations with CRC is deletion or mutation of APC, which has been observed to lead to an inhibition of Hippo kinases and, in turn, facilitates an increase in YAP1 activity.85,100–104
4.3. Targeting miRNA expression
Recently, a new class of small noncoding, single-stranded RNA molecules (i.e., miRNAs) have been discovered, which regulate gene expression and lead to either degradation of mRNA, or inhibition of protein synthesis, by binding to the 3ʹ-untranslated region of the complementary mRNA. It has been discovered that alterations in several miRNAs are involved in the etiology and clinical outcome of many cancers, including CRC. Recent findings suggest that a single miRNA can regulate many mRNAs and that a single mRNA can be the target of many miRNAs. The tumor location and mutation status of p53 and K-Ras exemplifies the miRNA status on different tumor types. Apart from their potential role in carcinogenesis, they also play an important role in biological functions like cell development, cell proliferation, cell differentiation, intracellular metabolism, and signal transduction.105–107
To date, there has been a plethora of studies describing the role of miRNA in normal stem cells, but studies that clearly elucidate their exact role as it pertains to CSCs are rare. The research community has now adopted a heightened interest in identifying their precise role in the regulation of CCSCs. miRNA could be a potential target in eradicating CSCs. Different miRNAs are up- or downregulated by different tumors. The therapeutic strategy involving miRNAs to treat CRSCs likely consists of two mechanisms. The first strategy involves upregulation of genes that would be silenced by deregulated miRNAs by inhibiting the oncogenic miRNAs using miRNA antagonists (anti-miRNAs). The second strategy involves the use of miRNA mimics that would help to restore the normal expression levels of tumor suppressor microRNA. It has been observed that miRNA mimics and anti-miRNAs might help to re-establish dysregulated gene expression, thereby suggesting their potential role in anticancer therapy.105 The delivery of antisense oligonucleotides, locked nucleic acid constructs, miRNA sponge constructs, and miRNA masking antisense oligonucleotide delivery are just some of the approaches that have been reported for inhibiting oncogenic miRNA. Using inhibitors of an oncogenic pathway represents an additional approach that has been reported to inhibit oncogenic miRNA. Additionally, mature miRNA mimics, pre-miRNA mimics, and mature miRNA precursors are all viable approaches that could be used for restoring tumor suppressor miRNAs.108
The main challenges associated with targeting miRNA is the delivery system, its stability, safety, and possible off-target effects. The two main categories for delivery systems are viral and nonviral vectors. Viral vectors are further classified into adenoviral and lentiviral vectors. Lipids (cationic liposomes, neural lipid emulsions, stable nucleic acid lipid particles) and polymer-based [polyethylenimine (PEI)), poly (lactic-co-glycolic acid) (PGLA), and polyamidoamine (PAMAM)] nanoparticles encompass the nonviral vectors.108 Table 1 describes different types of miRNA and their role in CCSC’s.
Table 1.
Type of miRNA and their role in CCSC.
| Sr.no | miRNA | Role in CCSC’s | Reference |
|---|---|---|---|
| 1. | miRNA143 and 145 | Downregulation in CRC tissues as compared to normal tissues. | 105,109,110 |
| 2. | miRNA140 | On targeting Histone deactylases4 (HDAC4), miRNA140 inhibited cell proliferation conferring methotrexate and 5-FU resistant phenotype in CCSC. | 109,111 |
| 3. | miRNA215 | miRNA215 enhances chemo-resistance of CCSC to methotrexate and tomudex by inducing G2 arrest through suppression of DTL expression | 109,111 |
| 4. | miRNA19b and miRNA21 | There is up-regulation of miRNA19b and miRNA21 in 5-FU resistant colon cancer cells | 108,111 |
| 5. | miRNA21 and 145 | (a)Colon cancer cells highly enriched in CSCs shows an upregulation of miRNA21 and downregulation of miRN145, suggesting their role in regulating CSCs. (b)P53 gene regulates the expression of miRNA145 via RAS signaling pathway, whereas miRNA21 increases the RAS signaling activity leading to subsequent repression of miRNA143/145 cluster complex. |
110 |
A polyethylenimine (PEI)/miRNA145 complex administered in a xenograft SCID mouse model showed suppression of tumor along with a reduction in CSC biomarkers such as CD44, β-catenin, and SOX2, as well as an increase in the expression of PDCD4 and CK20. Based on these results, it can be concluded that administration of miR-145 decreases CSC proliferation and induces cell differentiation, which leads to suppression of tumor growth in SCID mice.109
Roy et al. demonstrated that colonospheres highly enriched in CSCs show an inverse relationship between miRNA21 and PTEN, a target of miRNA21. Decreased expression of PTEN leads to activation of Akt signaling, which, in turn, mediates acceleration of tumor outgrowth in SCID xenografts. Difluorinated curcumin (CDF), an analog of dietary curcumin with a high bioavailability, modulates the miRNA21-PTEN-Akt axis in both p53-positive and p53-negative colon cancer cells. CDF is also effective in restoring PTEN levels in the metastatic colon cancer cell line known as SW620. Taken together, these results suggest that CDF could be an effective therapeutic agent for different stages of colon cancer.110 Also, it has been observed that CDF caused modulation of the miR-21-PTEN-Akt axis in chemo-resistant colon cancer cells that are highly enriched in CSCs, suggesting that this novel analog of curcumin could also be therapeutically effective for recurrent cancer, which is known to be resistant to conventional chemotherapy.110,112,113
Through recent findings, it has been observed that miRNA plays an important role in regulating stemness of CSCs, but its role in CCSCs remains poorly understood and more research in this field is desperately needed.
4.4. Targeting the tumor microenvironment
Persistent hypoxia in bulk tumors forms the basis of existence and maintenance of tumor stem cells, which confers resistance to conventional chemotherapy. The tumor stem cell niche plays a key role in regulating stem cell maintenance and self-renewal by either secreting paracrine factors, or by direct cell-to-cell interactions that potentiates self-renewal and acceleration during tumor proliferation. The tumor microenvironment (TME) consists of stromal cells, immune cells, networks of cytokines, chemokines, and growth factors, hypoxic regions, and the extracellular matrix (ECM). The TME modulates the Wnt/β-catenin, Notch, and Hedgehog signaling pathways and/or interrupts the master transcriptional regulators like NANOG, OCT-4, and SOX-2 to maintain the stemness of CSCs. Under specific micro-environmental stimuli, certain cancer cells exhibit plasticity, which enables them to resume proliferation through epithelial–mesenchymal transition EMT. The interaction between CCSCs and the microenvironment plays a key role in regulating CCSC self-renewal.38 Investigational studies on therapeutic interventions of stromal cells such as adipocytes, cancer-associated fibroblasts, and tumor-associated macrophages highlights the significance of targeting the TME. The major components of the TME that are known to play an important role in CSC and, CCSC in particular, have been described in Table 2. Furthermore, Table 3 lists all of the potential therapeutic agents that are undergoing clinical trials together with their specific target.
Table 2.
Stromal cells and their role in cancer and CSC.
| Sr.no | Name of the stromal cells | Role in cancer | Role in CSC | Pathway regulated | Reference |
|---|---|---|---|---|---|
| 1. | Adipocytes | Secretes adipokines and cytokines like leptin, adiponectin, IL-6, MCP-1, and TNF-α. | The leptin receptor maintains a self-reinforcing signaling cascade to expand the CSC population and tumor growth. | - | 114–116 |
| 2. | Cancer Associated Fibroblasts (CAFs) | Promotes tumor progression, invasion and guides structural changes by secreting mitogenic factors like hepatocyte growth factor (HGF), EGF family members, CCL12, fibroblast growth factors and stanniocalcin-1 | Supports CSC properties and invasive metastatic potential of CSCs. | Induces Wnt/β-catenin signaling by secreting HGF, matrix metalloproteases, cytokines like tumor necrosis factor-α (TNF-α). | 116–119 |
| 3. | Myeloid-Derived Suppressor Cells (MDSCs) | They secrete arginase 1, ROS, and inducible nitric oxide synthase (iNOS) to inhibit the anticancer function of NK cells and T cells. It helps in escaping immunosurveillance and promotes tumor initiation and progression. | Their infiltration to the intestinal mucosa is associated with CXCR2 expression on CRC endothelial cells and immunocytes. | - | 119–122 |
| 4. | Tumor Associated Macrophages (TAMs) | Pro-tumorgenic through induction of T cell anergy, driving ECM components, repairing the damaged tissues and neoangiogenesis. | Supports CSC growth through Milk-fat globule EGF-8 (MFG-E8) | Activates STAT3 and Hedgehog pathways triggering tumorigenicity and drug resistance in CSCs | 123–125 |
| 5. | T-regulatory cells (Treg cells) | Suppresses the immune system through the production of various cytokines like IL-10, IL-35, and TGF-β. Also suppresses cytotoxic T cells and NK cells. | Under hypoxia, FOXP3+ Treg cells express IL-17 and expands CCSC population as evidenced by an in increase in expression of CD133, CD44s and EpCAM. It also regulates CSC properties by secreting prostaglandin (PGE2) through NF-ҡB pathway. | IL-17 causes activation of Akt and MAPK pathway | 126–130 |
| 6. | HIF | HIF-1α and HIF-2α can bind to hypoxia response elements (HRE) aggravating tumor growth, invasion and metastasis. | HIF-1α can increase the transcriptional activity of β-catenin in canonical Wnt pathway. By maintaining CSCs in a quiescent state, hypoxia also contributes to drug resistance. | Wnt/β-catenin | 131,132 |
Table 3.
Colon Cancer Stem Cells Targeted Therapies.
| Sr. no | Name of the drug | Target | Preclinical/clinical | Reference |
|---|---|---|---|---|
| 1. | Catumaxomab | CD133+/FpCAM+ | Phase I-III in solid tumors. (NCT00836654) | 48,49,52 |
| 2. | MT110 | EpCAM | Phase I in solid tumors (NCT00635596) | 50,51 |
| 3. | NCB-0846 | TNIK | Preclinical | 71 |
| 4. | OMP-131R10 | Wnt pathway | Phase Ia/Ib (NCT02482441) | 72 |
| 5. | ICG-001 | Wnt pathway | Preclinical | 133 |
| 6. | PRI-724 | Wnt pathway | Phase I/II in solid tumors but withdrawn for Colon cancer. (NCT02413853) | 134 |
| 7. | EGCG | Wnt pathway | Preclinical | 135 |
| 8. | RO4929097 | Notch pathway | Phase II (NCT01116687) | 61 |
| 9. | OMP-21M18 | Anti-DLL4 antibody | Phase I/Ib (NCT01189942) | 136 |
| 10. | Honokiol | Notch pathway | Preclinical | 59 |
| 11. | Cyclopamine | Hedgehog pathway | Preclinical | 65 |
| 12. | GDC-0449 | Hedgehog pathway | Preclinical Phase II (in combination with standard drug) |
66 64 |
| 13. | LY2940002 | PI3K/AKT pathway | Preclinical | 137 |
| 14. | Polyethylenimine/miRNA145 | miRNA 145 | Preclinical | 108,110 |
| 15. | Difluorinated curcumin | miRNA21, ABCG2 | Preclinical | 109,112,113 |
| 16. | Grape seed extract | Browning of adipocytes | Preclinical | 115 |
| 17. | Acriflavine | HIF transcription inhibitor | Preclinical | 138 |
| 18. | Salinomycin | ABC transporters | Preclinical | 137,139,140 |
| 19. | Verteporfin | YAP | Preclinical | 91,92 |
| 20. | OPCs | Hippo pathway | Preclinical | 90 |
5. Conclusion
CCSCs have provided new avenues for the treatment of colorectal cancer that need to be further investigated. Chemoresistance and treatment failure are among the major drawbacks of current therapy for CRC, and it is indeed necessary to figure out how CCSCs escape current treatment. Also, to decrease the systemic and local toxicity associated with conventional chemotherapy, formulations need to be developed that can specifically target CCSCs to improve patient survival. Eradication of CCSCs has the potential to radically revolutionize the clinical outcome in CRC patients. The precise targeting of CCSCs with various therapeutic agents used alone, or in combination with conventional chemotherapeutic drugs, is currently being explored. However, since CCSCs share common characteristics with normal stem cells, the biggest challenge is to identify specific markers and techniques that exclusively target CCSCs and spare healthy normal stem cells. Therapeutic intervention via several pathways such as Wnt, HH, and Notch using various compounds has yielded positive results in several preclinical studies. However, there is a need for further reflection concerning potential targets and the generation of significant in vivo scientific evidence to better elucidate the mechanism of action of these therapeutic strategies on their targets at both the cellular and molecular levels.
Funding Statement
None.
Conflict of Interest
The authors declare no potential conflicts of interest.
References
- 1.Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F.. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683-691. doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J.. Lortet-Tieulent J and Jemal A. Global cancer statistics. CA Cancer J Clin. 2012;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Taylor DP, Burt RW, Williams MS, Haug PJ, Cannon-Albright LA. Population-based family history-specific risks for colorectal cancer: a constellation approach. Gastroenterology. 2010;138:877–885. doi: 10.1053/j.gastro.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fedirko V, Tramacere I, Bagnardi V, Rota M, Scotti L, Islami F, Negri E, Straif K, Romieu I, La Vecchia C, et al. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol. 2011;22:1958–1972. doi: 10.1093/annonc/mdq653. [DOI] [PubMed] [Google Scholar]
- 5.Chan DS, Lau R, Aune D, Vieira R, Greenwood DC, Kampman E, Norat T, Tomé D. Red and processed meat and colorectal cancer incidence: meta-analysis of prospective studies. PLoS One. 2011;6:e20456. doi: 10.1371/journal.pone.0020456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma Y, Yang Y, Wang F, Zhang P, Shi C, Zou Y, Qin H, Gorlova OY. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One. 2013;8:e53916. doi: 10.1371/journal.pone.0053916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang Y, Ben Q, Shen H, Lu W, Zhang Y, Zhu J. Diabetes mellitus and incidence and mortality of colorectal cancer: a systematic review and meta-analysis of cohort studies. Eur J Epidemiol. 2011;26:863–876. doi: 10.1007/s10654-011-9617-y. [DOI] [PubMed] [Google Scholar]
- 8.Sonnenberg A, Genta RM. Helicobacter pylori is a risk factor for colonic neoplasms. Am J Gastroenterol. 2013;108:208–215. doi: 10.1038/ajg.2012.407. [DOI] [PubMed] [Google Scholar]
- 9.Boyle T, Keegel T, Bull F, Heyworth J, Fritschi L. Physical activity and risks of proximal and distal colon cancers: a systematic review and meta-analysis. J Natl Cancer Inst. 2012;104:1548–1561. doi: 10.1093/jnci/djs354. [DOI] [PubMed] [Google Scholar]
- 10.Lin KJ, Cheung WY, Lai JY, Giovannucci EL. The effect of estrogen vs. combined estrogen-progestogen therapy on the risk of colorectal cancer. Int J Cancer. 2012;130:419–430. [DOI] [PubMed] [Google Scholar]
- 11.Bosetti C, Rosato V, Gallus S, Cuzick J, La Vecchia C. Aspirin and cancer risk: a quantitative review to 2011. Ann Oncol. 2012;23:1403–1415. doi: 10.1093/annonc/mds113. [DOI] [PubMed] [Google Scholar]
- 12.Aune D, Lau R, Chan DS. Nonlinear reduction in risk for colorectal cancer by fruit and vegetable intake based on meta-analysis of prospective studies. Gastroenterology. 2011;141:106–118. doi: 10.1053/j.gastro.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Aune D, Chan DS, Lau R, Vieira R, Greenwood DC, Kampman E, Norat T. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. BMJ. 2011;343:d6617. doi: 10.1136/bmj.d6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aune D, Lau R, Chan DS, Vieira R, Greenwood DC, Kampman E, Norat T. Dairy products and colorectal cancer risk: a systematic review and meta-analysis of cohort studies. Ann Oncol. 2012;23:37–45. doi: 10.1093/annonc/mdr269. [DOI] [PubMed] [Google Scholar]
- 15.Lochhead P, Chan AT. Statins and colorectal cancer. Clin Gastroenterol Hepatol. 2013;11:109–118. doi: 10.1016/j.cgh.2012.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han L, Shi S, Gong T, Zhang Z, Sun X. Cancer stem cells: therapeutic implications and perspectives in cancer therapy. Acta Pharm Sin B. 2013;3(2):65–75. doi: 10.1016/j.apsb.2013.02.006. [DOI] [Google Scholar]
- 17.Rich JN, Bao S. Chemotherapy and cancer stem cells. Cell Stem Cell. 2007;1:353–355. doi: 10.1016/j.stem.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Dou J, Gu N. Emerging strategies for the identification and targeting of cancer stem cells. Tumor Biol. 2010;31:243–253. doi: 10.1007/s13277-010-0023-y. [DOI] [PubMed] [Google Scholar]
- 19.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2009;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 20.Chen K, Huang Y, Chen J. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–740. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koch U, Krause M. Baumann M Cancer stem cells at the crossroads of current cancer therapy failures—radiation oncology perspective. Sem Cancer Biol. 2010;20:116. doi: 10.1016/j.semcancer.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs. 2009;1(1):12–25. doi: 10.4161/mabs.1.1.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 24.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-04-4557. [DOI] [PubMed] [Google Scholar]
- 25.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Julio C, Minden M, Paterson B, Caliguiri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 26.Abdullah LN, Chow EK. Mechanisms of chemo-resistance in cancer stem cells. Clin Transl Med. 2013;2:3. doi: 10.1186/2001-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine: III. Entero-endocrine cells. Am J Anat. 1974;141:503–519. [DOI] [PubMed] [Google Scholar]
- 28.Winton DJ, Ponder BA. Stem-cell organization in mouse small intestine. Proc Roy Soc Lond B Biol Sci. 1990;241:13–18. doi: 10.1098/rspb.1990.0059. [DOI] [PubMed] [Google Scholar]
- 29.Brittan M, Wright NA. Stem cell in gastrointestinal structure and neoplastic development. Gut. 2004;53:899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Koutsilieris M and Papavassiliou AG. Colorectal cancer stem cells. Stem Cells. 2012;30:363–371. doi: 10.1002/stem.1031. [DOI] [PubMed] [Google Scholar]
- 31.Pardal R, Clarke MF. Morrison SJ Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 32.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van Den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 33.Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P, et al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci USA. 1996;93:9049–9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castets M, Broutier L, Molin Y, Brevet M, Chazot G, Gadot N. DCC constrains tumour progression via its dependence receptor activity. Nature. 2011;482:534–537. doi: 10.1038/nature10708. [DOI] [PubMed] [Google Scholar]
- 35.Schwitalla S, Fingerle A A, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. Normal and neoplastic non stem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Sousa E, Melo F, Vermeulen L, Fessler E, Medema JP. Cancer heterogeneity - a multifaceted view. EMBO Rep. 2013;14:686–695. doi: 10.1038/embor.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan T, Xu J, Zhu Y. Self-renewal molecular mechanisms of colorectal cancer stem cells. Int J Mol Med. 2017;39:9–20. doi: 10.3892/ijmm.2016.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 40.Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naujokat C, Laufer S. Targeting cancer stem cells with defined compounds and drugs. J Can Res Updates. 2013;2:36–67. [Google Scholar]
- 42.Gaiser MR, Lämmermann T, Feng X, Igyarto BZ, Kaplan DH, Tessarollo. 2012. Cancer associated epithelial cell adhesion molecule (EpCAM; CD326) enables epidermal Langerhans cell motility and migration in vivo. Proc Natl Acad Sci USA. 109(15):E889–E897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Litvinov SV, Bakker HAM, Gourevitch MM, Velders M, Warnaar SO. Evidence for a role of the epithelial glycoprotein 40 (Ep-CAM) in epithelial cell-cell adhesion. Cell Adhes Commun. 1994;2:417–428. [DOI] [PubMed] [Google Scholar]
- 44.Munz M, Baeuerle PA, Gires O. The Emerging Role of EpCAM in Cancer and Stem Cell Signalling. Cancer Res. 2009;69:5627–5629. doi: 10.1158/0008-5472.CAN-08-3660. [DOI] [PubMed] [Google Scholar]
- 45.Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW, Zoeller M. CD44 and EpCAM: cancer-initiating cell markers. Curr Mol Med. 2008;8:784–804. [DOI] [PubMed] [Google Scholar]
- 46.Liu D, Sun J, Zhu J, Zhou H, Zhang X, Zhang Y. Expression and Clinical Significance of colorectal stem cell marker EpCAMHigh/CD44+ in Colorectal Cancer. Oncol Lett. 2014;7(5):1544–1548. doi: 10.3892/ol.2014.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin CW, Liao MY, Lin WW, Wang YP, Lu TY, Wu HC. Epithelial cell adhesion molecule regulates tumor initiation and tumorigenesis via activating reprogramming factors and epithelial-mesenchymal transition gene expression in colon cancer. J Biol Chem. 2012;287:39449–39459. doi: 10.1074/jbc.M112.386235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bezan A, Hohla F, Meissnitzer T. Systemic effect of catumaxomab in a patient with metastasized colorectal cancer: a case report. BMC Cancer. 2013;13:618. doi: 10.1186/1471-2407-13-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linke R, Klein A, Seimetz D. Catumaxomab-Clinical development and future directions. mAbs. 2010;2(2):129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T, Raum T, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–1143. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 51.Herrmann I, Baeuerle PA, Friedrich M, Murr A, Filusch S, Rüttinger D, Majdoub MW, Sharma S, Kufer P, Raum T, et al. Highly efficient elimination of colorectal tumor-initiating cells by an EpCAM/CD3-bispecific antibody engaging human T cells. PLoS One. 2010;5:e13474. doi: 10.1371/journal.pone.0013474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strohlein MA, Lordick F, Ruttinger D, Grutzner KU, Schemanski OC, Jager M. Immunotherapy of peritoneal carcinomatosis with the antibody catumaxomab in colon, gastric, or pancreatic cancer: an open-label, multicenter, phase I/II trial. Onkologie. 2011;34(3):101–108. doi: 10.1159/000324667. [DOI] [PubMed] [Google Scholar]
- 53.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Went P, Vasei M, Bubendorf L, Terracciano L, Tornillo L, Riede U, Kononen J, Simon R, Sauter G, Baeuerle PA. Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br J Cancer. 2006;94:128–135. doi: 10.1038/sj.bjc.6602924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merchant AA, Matsui W. Targeting Hedgehog—a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–3140. doi: 10.1158/1078-0432.CCR-10-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX, Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445–464. doi: 10.1038/nrclinonc.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, Wu GS, Wu K. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20–27. doi: 10.1016/j.canlet.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 58.Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1:338–351. doi: 10.1002/emmm.200900027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ponnurangam S, Mammen J.M., Ramalingam S., He Z., Zhang Y., Umar S., Subramaniam D, Anant S. Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol Cancer Ther. 2012;11(4):963–972. doi:10.1158/1535-7163.MCT-11-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013;139:95–110. doi: 10.1016/j.pharmthera.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, Almhanna K, Kim R, Valone T, Jump H, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012;48:997–1003. doi: 10.1016/j.ejca.2012.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling—are we there yet? Nat. Rev Drug Discov. 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 63.Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of cancer stem cells. Cancers. 2015;7:1554–1585. doi: 10.3390/cancers7030851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC, Mulcahy MF, Zalupski MM, Mackey HM, Yauch RL, et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin Cancer Res. 2013;19:258–267. doi: 10.1158/1078-0432.CCR-12-1800. [DOI] [PubMed] [Google Scholar]
- 65.Batsaikhan B, Yoshikawa K, Kurita N, Iwata T, Takasu C, Kashihara H. Cyclopamine decreased the expression of sonic hedgehog and its downstream genes in colon cancer stem cells. Anti-Cancer Res. 2014;34:6339–6344. [PubMed] [Google Scholar]
- 66.Wu C, Hu S, Cheng J, Wang G, Tao K. Smoothened antagonist GDC‐0449 (Vismodegib) inhibits proliferation and triggers apoptosis in colon cancer cell lines. Exp Ther Med. 2017;13:2529–2536. doi: 10.3892/etm.2017.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Melo F, Vermeulen L. Wnt signaling in cancer stem cell biology. Cancers (Basel). 2016;8(7). pii: E60. doi: 10.3390/cancers8070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci USA. 1999;96:6273–6278. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–1622. doi: 10.1126/science.1142109. [DOI] [PubMed] [Google Scholar]
- 70.Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol. 2005;12:364–371. doi: 10.1038/nsmb912. [DOI] [PubMed] [Google Scholar]
- 71.Masuda M, Uno Y, Ohbayashi N, Ohata H, Mimata A, Kukimoto-Niino M. TNIK inhibition abrogates colorectal cancer stemness. Nat Commun. 2016;7:12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bendell J, Eckhardt G.S, Hochster H.S, Morris V.K, Strickler J, Kapoun A.M, Wang M, Xu L, McGuire K, Dupont J, et al. Initial results from a phase 1a/b study of OMP-131R10, a first-in-class anti-RSPO3 antibody, in advanced solid tumors and previously treated metastatic colorectal cancer (CRC). Eur J Cancer. 2016;69(1):S2–S177. doi: 10.1016/S0959-8049(16)32668-5. [DOI] [Google Scholar]
- 73.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29(6):783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114(4):457–467. [DOI] [PubMed] [Google Scholar]
- 76.Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun. 2015;6:8357. doi: 10.1038/ncomms9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30(1):1–17. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin KC, Moroishi T, Meng Z, Jeong HS, Plouffe SW, Sekido Y, Han J, Park HW, Guan KL. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat Cell Biol. 2017;19(8):996–1002. doi: 10.1038/ncb3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108(29):11930–11935. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell. 2011;144(5):782–795. doi: 10.1016/j.cell.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 83.Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17(4):500–510. doi: 10.1038/ncb3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007;14:743–751. doi: 10.1038/sj.cdd.4402063. [DOI] [PubMed] [Google Scholar]
- 85.Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature. 2013;493:106–110. doi: 10.1038/nature11818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Strano S, Monti O, Pediconi N, Baccarini A, Fontemaggi G, Lapi E. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol Cell. 2005;18:447–459. doi: 10.1016/j.molcel.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 87.Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010;24:2383–2388. doi: 10.1101/gad.1978810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–2060. doi: 10.1016/j.cub.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 89.Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LSB, Abujarour R, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24:1106–1118. doi: 10.1101/gad.1903310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toden S, Ravindranathan P, Gu J, Cardenas J, Yuchang M, Goel A. Oligomeric proanthocyanidins (OPCs) target cancer stem-like cells and suppress tumor organoid formation in colorectal cancer. Sci Rep. 2018;8:3335. doi: 10.1038/s41598-018-21478-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Győrffy B, Sebolt-Leopold JS, Dame MK, Varani J, Brenner DE, et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8(397):ra98. doi: 10.1126/scisignal.aac5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pan W, Wang Q, Zhang Y, Zhang N, Qin J, Li W, Wang J, Wu F, Cao L, Xu G. Verteporfin can Reverse the Paclitaxel Resistance Induced by YAP Over-Expression in HCT-8/T Cells without Photoactivation through Inhibiting YAP Expression. Cell Physiol Biochem. 2016;39:481–490. doi: 10.1159/000445640. [DOI] [PubMed] [Google Scholar]
- 93.Li Y, Hibbs MA, Gard AL, Shylo NA, Yun K. Genome-wide analysis of N1ICD/RBPJ targets in vivo reveals direct transcriptional regulation of Wnt, SHH, and hippo pathway effectors by Notch1. Stem Cells. 2012;30(4):741–752. doi: 10.1002/stem.1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B. Hippo pathway activity influences liver cell fate. Cell. 2014;157(6):1324–1338. doi: 10.1016/j.cell.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu FX, Karin M, Pan D, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015;29(12):1271–1284. doi: 10.1101/gad.262816.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12(6):393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 97.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–2472. doi: 10.1101/gad.1613608. [DOI] [PubMed] [Google Scholar]
- 98.Tariki M, Dhanyamraju PK, Fendrich V, Borggrefe T, Feldmann G, Lauth M. The Yes-associated protein controls the cell density regulation of Hedgehog signaling. Oncogene. 2014;3:e112. doi: 10.1038/oncsis.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, Dawson D, Willis JE, Markowitz SD, Camargo FD, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci USA. 2011;108(49):E1312–20. doi: 10.1073/pnas.1110428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Varelas X, Miller BW, Sopko R, Song S, Gregorieff A, Fellouse FA, Sakuma R, Pawson T, Hunziker W, McNeill H, et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev Cell. 2010;18(4):579–591. doi: 10.1016/j.devcel.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 101.Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, et al. YAP/TAZ incorporation in the beta-catenin destruction complex or- chestrates the Wnt response. Cell. 2014;158(1):157–170. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 102.Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, Piccolo S. Role of TAZ as mediator of Wnt signaling. Cell. 2012;151(7):1443–1456. doi: 10.1016/j.cell.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 103.Cai J, Maitra A, Anders RA, Taketo MM, Pan D. Beta-catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015;29(14):1493–1506. doi: 10.1101/gad.264515.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, Meng Z, Lin K, Yu F-X, Alexander C, et al. Alternative Wnt signaling activates YAP/TAZ. Cell. 2015;162(4):780–794. doi: 10.1016/j.cell.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caruso S, Bazan V, Rolfo C, Insalaco L, Fanale D, Bronte G, Corsini LR, Rizzo S, Cicero G, Russo A. MicroRNAs in colorectal cancer stem cells: new regulators of cancer stemness? Oncogenesis. 2012;1:e32. doi: 10.1038/oncsis.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang B, Pan X, Cobb GP, Anderson TA. MicroRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 107.Papagiannakopoulos T, Kosik KS. MicroRNAs: regulators of oncogenesis and stemness. BMC Med. 2008;6:15. doi: 10.1186/1741-7015-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ramalingam S, Subramaniam D, Anant S. Manipulating miRNA Expression: A Novel Approach for Colon Cancer Prevention and Chemotherapy. Curr Pharmacol Rep. 2015;1(3):141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fanale D, Caruso S, Bazan V, Bronte G, Piazza FD. MicroRNAs in Colorectal Cancer Drug Resistance: shooters become Targets. J Carcinogene Mutagene. 2013;4:136. [Google Scholar]
- 110.Yu Y, Nangia-Makker P, Farhana L, G Rajendra S, Levi E, Majumdar AP. miR-21 and miR-145 cooperation in regulation of colon cancer stem cells. Mol Cancer. 2015;14:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kurokawa K, Tanahashi T, Iima T, Yamamoto Y, Akaike Y, Nishida K, Masuda K, Kuwano Y, Murakami Y, Fukushima M, et al. Role of miR-19b and its target mRNAs in 5-fluorouracil resistance in colon cancer cells. J Gastroenterol. 2012;47:883–895. doi: 10.1007/s00535-012-0547-6. [DOI] [PubMed] [Google Scholar]
- 112.Roy S, Yu Y, Padhye SB, Sarkar FH, Majumdar APN, Goel A. Difluorinated-Curcumin (CDF) Restores PTEN Expression in Colon Cancer Cells by Down- Regulating miR-21. PLoS One. 2013;8(7):e68543. doi: 10.1371/journal.pone.0068543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Padhye S, Yang H, Jamadar A, Cui QC, Chavan D. New difluoro Knoevenagel condensates of curcumin, their Schiff bases and copper complexes as proteasome inhibitors and apoptosis inducers in cancer cells. Pharm Res. 2009;26:1874–1880. doi: 10.1007/s11095-008-9767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115(5):911–920. doi: 10.1016/j.jaci.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 115.Kumar S, Kumar D, Raina K, Agarwal R, Agarwal C. Functional modification of adipocytes by grape seed extract impairs their pro-tumorigenic signaling on colon cancer stem cells and the daughter cancer cells. Oncotarget. 2014;5(20):10151–10169. doi: 10.18632/oncotarget.v5i20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen S, Huang EH. The colon cancer stem cell microenvironment holds keys to future cancer therapy. J Gastrointest Surg. 2014;18:1040–1048. doi: 10.1007/s11605-014-2497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumor cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012;31:195–208. doi: 10.1007/s10555-011-9340-x. [DOI] [PubMed] [Google Scholar]
- 118.Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Céspedes MV. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. 2013;24:631–644. doi: 10.1016/j.ccr.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 121.Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. 2011;11:702–711. doi: 10.1038/nri3064. [DOI] [PubMed] [Google Scholar]
- 122.Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunolo. 2015;15(2):73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour- associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42(6):717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 124.Jinushi M, Sato M, Kanamoto A, Itoh A, Nagai S, Koyasu S, Dranoff G, Tahara H. Milk fat globule epidermal growth factor-8 blockade triggers tumor destruction through coordinated cell-autonomous and immune-mediated mechanisms. J Exp Med. 2009;206(6):1317–1326. doi: 10.1084/jem.20082614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, Yagita H, Takaoka A, Tahara H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108(30):12425–12430. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3 + regulatory T cells in the human immune system. Nat Rev Immunolo. 2010;10(7):490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 127.Facciabene A, Peng X, Hagemannetal IS, Balint K, Barchetti A, Wang L-P, Gimotty PA, Gilks CB, Lal P, Zhang L, et al. Tumor hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 128.Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3 regulatory T cells in gastric cancer. Int J Cancer. 2008;122(22):2286–2293. doi: 10.1002/ijc.23392. [DOI] [PubMed] [Google Scholar]
- 129.Yang S, Wang B, Guan C, Wu B, Cai C, Wang M, Zhang B, Liu T, Yang P. Foxp3 IL-17 T cells promote development of cancer-initiating cells in colorectal cancer. J Leukoc Biol. 2011;89(1):85–91. doi: 10.1189/jlb.0910506. [DOI] [PubMed] [Google Scholar]
- 130.Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology. 2015;149(7):1884–1895. doi: 10.1053/j.gastro.2015.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McIntyre A, Harris AL. Metabolic and hypoxic adaptation to anti-angiogenic therapy: A target for induced essentiality. EMBO Mol Med. 2015;7:368–379. doi: 10.15252/emmm.201404271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Santoyo-Ramos P, Likhatcheva M, García-Zepeda EA, Castañeda-Patlán MC, Robles-Flores M. Hypoxia-inducible factors modulate the stemness and malignancy of colon cancer cells by playing opposite roles in canonical Wnt signaling. PLoS One. 2014;9:e112580. doi: 10.1371/journal.pone.0112580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo J-L, Oh SW, Kim HY, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc Natl Acad Sci U S A. 2004;101(34):12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.El-Khoueiry A. B, Ning Y, Yang D, Cole S, Kahn M, Berg MZ, Fujimori M, Inada T, Kouji H, Lenz HJ. A phase I first- in-human study of PRI-724 in patients with advanced solid tumors. J Clin Oncol. 2013;31: supplement abstract 2501. doi: 10.1200/JCO.2013.49.0219. [DOI] [Google Scholar]
- 135.Chen Y, Wang X.Q, Zhang Q, Zhu J.Y, Li Y, Xie C.F, Li XT, Wu JS, Geng SS, Zhong CY, et al. (−)-Epigallocatechin-3-gallate inhibits colorectal cancer stem cells by suppressing Wnt/β-catenin pathway. Nutrients. 2017;9:572. doi: 10.3390/nu9060572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA, Lagasse E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008;68:6932–6941. doi: 10.1158/0008-5472.CAN-07-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen S, Fisher R.C, Signs S, Molina L.A, Shenoy A.K, Lopez M.C. Inhibition of PI3K/Akt/mTOR signaling in PI3KR2- overexpressing colon cancer stem cells reduces tumor growth due to apoptosis. Oncotarget. 2017;8(31):50476–50488. doi: 10.18632/oncotarget.9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shay JE, Imtiyaz HZ, Sivanand S, Durham AC, Skuli N, Hsu S. Inhibition of hypoxia-inducible factors limits tumor progression in a mouse model of colorectal cancer. Carcinogenesis. 2014;35:1067–1077. doi: 10.1093/carcin/bgu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene. 2007;26:1357–1360. doi: 10.1038/sj.onc.1210200. [DOI] [PubMed] [Google Scholar]
- 140.Zhang C, Tian Y, Song F, Fu C, Han B, Wang Y. Salinomycin inhibits the growth of colorectal carcinoma by targeting tumor stem cells. Oncol Rep. 2015;34:2469–2476. doi: 10.3892/or.2015.4253. [DOI] [PubMed] [Google Scholar]