

ABSTRACT
Patients with non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping can benefit from crizotinib treatment. Currently, the main resistance mechanisms to crizotinib are MET D1228N and Y1230C mutations. We reported a case of a Chinese NSCLC patient with MET exon 14 skipping detected by targeted next-generation sequencing (NGS) achieved clinical and imaging remission after crizotinib treatment. Then, amplification of multiple genes such as erb-b2 receptor tyrosine kinase 2 (HER2) was detected when disease progressed, indicating novel resistance mechanisms to crizotinib. Ultimately the patient died from cancer-related factors. This was the first NSCLC case with MET exon 14 skipping which reported the HER2 gene amplification at the time of progression during crizotinib treatment, indicating that bypass mechanisms contribute to the development of acquired resistance to MET inhibitors.
KEYWORDS: NSCLC, MET 14 skipping, crizotinib, resistance, HER2 amplification, NGS, targeted therapy
Background
As the research on the NSCLC genome and targeted therapy moves along, more and more molecular targets for anti-tumor therapy have been applied into clinical practice,1 including epidermal growth factor receptor (EGFR) gene,2-4 anaplastic lymphoma kinase (ALK) gene,5-7 and ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) gene,8 etc. MET proto-oncogene, receptor tyrosine kinase gene (MET) is another important driver gene in NSCLC.5,9,10 Activation of MET includes mutation, amplification, and protein overexpression, which is a potential target for NSCLC treatment, indicating its association with prognosis.11-13 Clinical evidence has demonstrated that MET is not only a driver proto-oncogene in lung cancer, but also one cause of acquired resistance to EGFR-targeted therapy.14
The MET exon 14-encoded portion of the juxtamembrane domain includes a binding site for Y1003 and c-Cbl E3 ubiquitin ligase. When MET exon 14 skipping mutations occur, the binding site for Y1003 and c-Cbl E3 ubiquitin ligase will be deleted, subsequently causing a decrease in receptor ubiquitination, a block in MET protein degradation, and continuously activated MET as a driver proto-oncogene.11 It was reported by previous studies that the incidence of MET exon 14 skipping mutation in pancreatic cancer was approximately 3%,15 much higher than the incidence of 0.9% in China shown by some domestic research.16 There are various types of MET exon 14 skipping mutations, mainly DNA mutations at 5’ splice site and 3’ splice site of MET exon 14.17 MET exon 14 skipping has been found in lung adenocarcinoma, lung squamous carcinoma, and pulmonary sarcomatoid carcinoma, for which clinical research indicated crizotinib treatment is effective.18 The currently reported resistance mechanisms to crizotinib in patients with MET exon 14 skipping are MET D1228N and Y1230C mutations.19,20 Currently known resistance mechanisms to kinase inhibitors can almost be categorized into the following types: second mutations, gene amplification, activation of bypass or downstream signaling pathways, histology and phenotypic transformation, etc.21 Using targeted NGS of cancer-related genes, we detected the response of MET 14 exon skipping in the tumor tissue of a patient with lung adenocarcinoma to crizotinib treatment. After treatment progress, we did not detect point mutations of the MET gene, but found other acquired resistance mechanisms such as HER2 gene amplification.
Pathological report
A 65-year-old woman without a prior history of smoking was hospitalized for cough and chest pain. In Feb. 2015, the patient was diagnosed with stage Ib (pT2aN0M0) invasive lung adenocarcinoma (Figure 1) with alveolar adenocarcinoma as the main type (ALK IHC, negative; EGFR ARMS-PCR, negative). The patient received adjuvant chemotherapy (gemcitabine + carboplatin) after operation with DFS 8 months. In Oct. 2015, tumor recurrence and metastasis was detected by the follow-up CT scan, and the patient had the chemotherapy (first-line pemetrexed + carboplatin + bevacizumab, pemetrexed maintenance chemotherapy + bevacizumab) with PFS 14 months. In Jan. 2017, the progression of pulmonary lesions was detected by the follow-up CT scan, and the second-line chemotherapy (paclitaxel liposome + carboplatin) was given.
Figure 1.
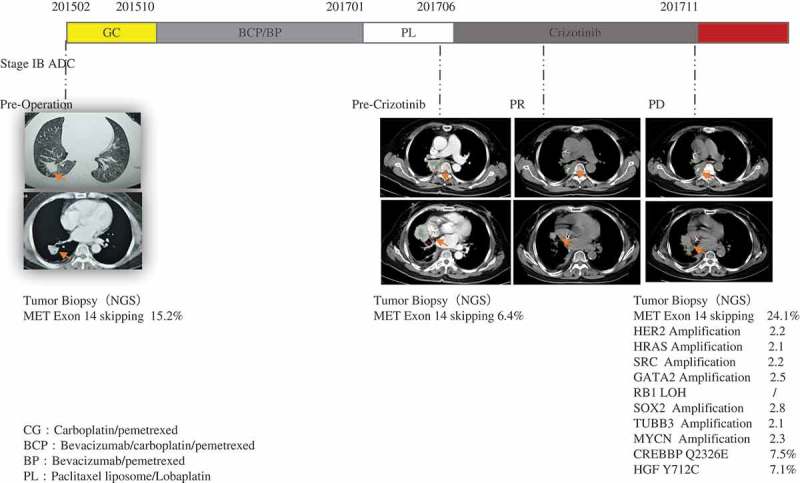
The patient can undergo a CT scan and have a tumor biopsy for NGS to ensure the efficacy of crizotinib and tumor progression during crizotinib treatment. MET: MET proto-oncogene, receptor tyrosine kinase; HER2: erb-b2 receptor tyrosine kinase 2; HRAS: HRas proto-oncogene, GTPase; SRC: SRC proto-oncogene, non-receptor tyrosine kinase; GATA2: GATA binding protein 2; RB1: RB transcriptional corepressor 1; SOX2: SRY-box 2; TUBB3: tubulin beta 3 class III; MYCN: MYCN proto-oncogene, bHLH transcription factor; CREBBP: CREB binding protein; HGF: hepatocyte growth factor.
In Jun. 2017, the patient progressed, and tumor biopsy (via bronchofiberscope) and plasma were taken for mutation profiling using targeted NGS for 422 cancer-relevant genes[Supplemental Materials, Table 1]. According to the instructions of KAPA Library Quantification Kit, fragmented DNA or ctDNA (>20 ng) were obtained for sequencing library construction and Hiseq 4000 NGS platforms (Illumina) were used for high-throughput sequencing. MET exon 14c.2942-17_2942-6del12 skipping mutation was detected in both tissue and plasma of the patient (tissue abundance 6.4%; plasma abundance 0.5%)(Figure 2) and other tumor-specific mutations were not detected. From Jun. 2017, the patient has undergone crizotinib treatment, and the tumor regressed after 42 days indicated by tumor imaging, showing partial response (PR).
Table 1.
The list of 422 genes panel.
| ABCB1 | ABCB4 | ABCC2 | ADH1A | ADH1B | ADH1C | AIP | AKT1 | AKT2 | AKT3 | ALDH2 | ALK |
| AMER1 | APC | AR | ARAF | ARID1A | ARID1B | ARID2 | ARID5B | ASCL4 | ASXL1 | ATF1 | ATIC |
| ATM | ATR | ATRX | AURKA | AURKB | AXIN2 | AXL | BAI3 | BAK1 | BAP1 | BARD1 | BCL2 |
| BCL2L11 | BCR | BIRC3 | BLM | BMPR1A | BRAF | BRCA1 | BRCA2 | BRD4 | BRIP1 | BTG2 | BTK |
| BUB1B | c11orf30 | CASP8 | CBL | CBLB | CC2D2B | CCND1 | CCNE1 | CD274 | CD74 | CDA | CDC73 |
| CDH1 | CDK10 | CDK12 | CDK4 | CDK6 | CDK8 | CDKN1A | CDKN1B | CDKN1C | CDKN2A | CDKN2B | CDKN2C |
| CEBPA | CEBPB | CEBPD | CEP57 | CHD4 | CHEK1 | CHEK2 | CLEC2D | CREBBP | CRKL | CSF1R | CTCF |
| CTLA4 | CTNNB1 | CUL3 | CUX1 | CXCR4 | CYLD | CYP19A1 | CYP2A13 | CYP2A6 | CYP2A7 | CYP2B6 | CYP2C19 |
| CYP2C9*3 | CYP2D6 | CYP3A4 | CYP3A5 | DAXX | DDR2 | DENND1A | DHFR | DHFRL1 | DICER1 | DNMT3A | DPYD |
| DUSP2 | EGFR | EML4 | EP300 | EPAS1 | EPCAM | EPHA2 | EPHA3 | EPHA5 | EPHB2 | ERBB2 | ERBB2IP |
| ERBB3 | ERBB4 | ERCC1 | ERCC2 | ERCC3 | ERCC4 | ERCC5 | ESR1 | ETV1 | ETV4 | EWSR1 | EXT1 |
| EXT2 | EZH2 | FANCA | FANCC | FANCD2 | FANCE | FANCF | FANCG | FANCL | FANCM | FAT1 | FBXW7 |
| FGF19 | FGFR1 | FGFR2 | FGFR3 | FGFR4 | FH | FLCN | FLT1 | FLT3 | FLT4 | FOXA1 | FOXP1 |
| FRG1 | GATA1 | GATA2 | GATA3 | GATA4 | GATA6 | GNA11 | GNA15 | GNAQ | GNAS | GRIN2A | GRM3 |
| GRM8 | GSTM1 | GSTM4 | GSTM5 | GSTP1 | GSTT1 | HDAC2 | HDAC9 | HGF | HLA-A | HNF1A | HNF1B |
| HRAS | HSD3B1 | IDH1 | IDH2 | IGF1R | IGF2 | IKBKE | IKZF1 | IL7R | INPP4B | IRF2 | JAK1 |
| JAK2 | JAK3 | JARID2 | JUN | KDM5A | KDM6A | KDR | KEAP1 | KIF1B | KIF5B | KIT | KITLG |
| KLLN | KMT2A | KMT2B | KMT2C | KMT2D | KRAS | LHCGR | LMO1 | LRP1B | LYN | LZTR1 | MAP2K1 |
| MAP2K2 | MAP2K4 | MAP3K1 | MAP3K4 | MAP4K3 | MAX | MCL1 | MDM2 | MDM4 | MECOM | MED12 | MEF2B |
| MEN1 | MET | MGMT | MITF | MLH1 | MLH3 | MLLT1 | MLLT3 | MLLT4 | MPL | MRE11A | MSH2 |
| MSH6 | MTHFR | MTOR | MUTYH | MYC | MYCL | MYCN | MYD88 | MYH9 | NAT1 | NAT2 | NBN |
| NCOR1 | NF1 | NF2 | NFE2L2 | NFKBIA | NKX2-1 | NKX2-2 | NKX2-4 | NOTCH1 | NOTCH2 | NOTCH3 | NPM1 |
| NQO1 | NRAS | NRG1 | NSD1 | NTRK1 | NTRK3 | PAK3 | PALB2 | PALLD | PARK2 | PARP1 | PARP2 |
| PAX5 | PBRM1 | PDCD1 | PDCD1LG2 | PDE11A | PDGFRA | PDGFRB | PDK1 | PGR | PHOX2B | PIK3C3 | PIK3CA |
| PIK3R1 | PIK3R2 | PKHD1 | PLAG1 | PLK1 | PMS1 | PMS2 | POLD1 | POLD3 | POLE | POLH | POT1 |
| PPP2R1A | PRDM1 | PRF1 | PRKACA | PRKACG | PRKAR1A | PRKCI | PRKDC | PRSS1 | PRSS3 | PTCH1 | PTEN |
| PTK2 | PTPN11 | PTPN13 | PTPRD | QKI | RAC1 | RAC3 | RAD50 | RAD51 | RAD51C | RAD51D | RAF1 |
| RARA | RARG | RASGEF1A | RB1 | RECQL4 | RELN | RET | RHOA | RICTOR | RNF43 | ROS1 | RPTOR |
| RRM1 | RUNX1 | RUNX1T1 | RUNX3 | SBDS | SDC4 | SDHA | SDHB | SDHC | SDHD | SEPT9 | SETBP1 |
| SETD2 | SF3B1 | SGK1 | SLC34A2 | SLC7A8 | SMAD2 | SMAD3 | SMAD4 | SMAD7 | SMARCA4 | SMARCB1 | SMO |
| SOS1 | SOX1 | SOX14 | SOX2 | SOX21 | SOX3 | SPOP | SPRY4 | SRC | SRY | STAG2 | STAT3 |
| STK11 | STMN1 | STT3A | SUFU | TEK | TEKT4 | TERC | TERT | TET2 | TGFBR2 | THADA | TMEM127 |
| TMPRSS2 | TNFAIP3 | TNFRSF11A | TNFRSF14 | TNFRSF19 | TNFSF11 | TOP1 | TOP2A | TP53 | TP63 | TPMT | TSC1 |
| TSC2 | TSHR | TTF1 | TUBB | TUBB2A | TUBB2B | TUBB3 | TUBB4A | TUBB4B | TUBB6 | TYMS | U2AF1 |
| UGT1A1 | VEGFA | VHL | WAS | WISP3 | WRN | WT1 | XPA | XPC | XRCC1 | YAP1 | ZNF2 |
| ZNF217 | ZNF703 |
Figure 2.
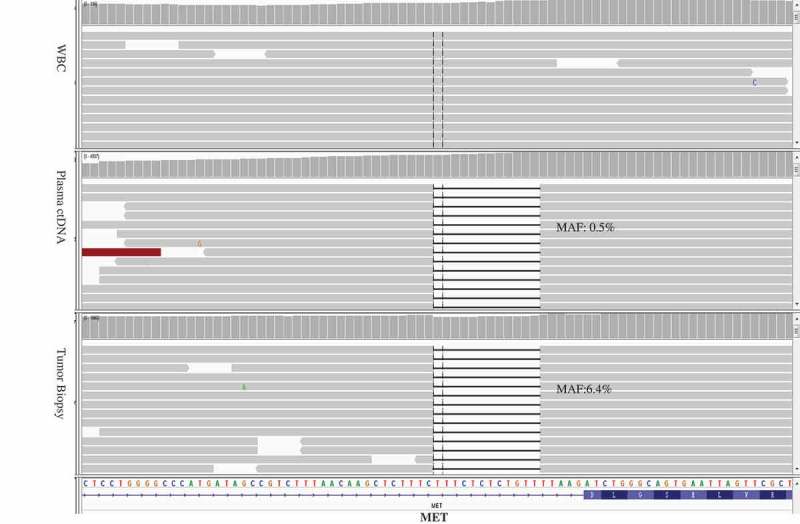
MET exon 14 skipping mutation identified in the pre-crizotinib tumor biopsy and plasma ctDNA.
In Nov. 2017, as the tumor progressed, tumor biopsy (via bronchofiberscope) and plasma were taken again for NGS. MET exon 14c.2942-17_2942-6del12 skipping mutation was detected with plasma abundance 1.7% and tissue abundance 24.1%. In the biopsy, we also detected that almost 2.2-fold amplification of the ERBB2 gene, almost 2.1-fold amplification of the HRAS gene, almost 2.2-fold amplification of the SRC gene, almost 2.5-fold amplification of the GATA2 gene, a single copy number loss of the RB1 gene, almost 2.8-fold amplification of the SOX2 gene, almost 2.1-fold amplification of the TUBB3 gene, almost 2.3-fold amplification of the MYCN gene, Q2326E mutation of the CREBBP gene with abundance 7.5%, and Y712C mutation of HGF gene with abundance 7.1%. IHC was used to measure HER2 protein expression in tumor biopsy, emphasizing the fact that HER2 over-expressed (Figure 3).
Figure 3.
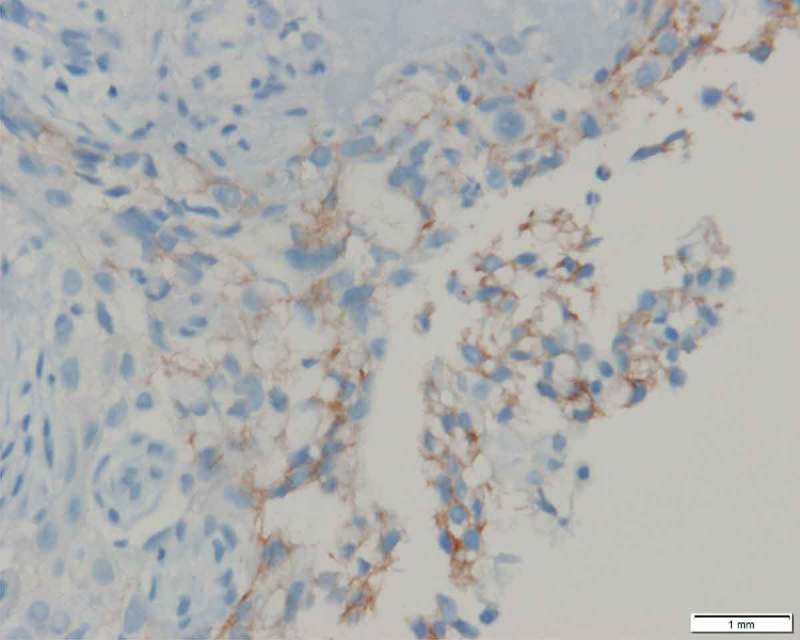
HER2 over-expression by IHC at 400× magnification.
In Dec. 2017, a retrospective NGS analysis of the patient’s paraffin-embedded tissue after operation was performed and it detected MET exon 14c.2942-17_2942-6del12 skipping mutation (MAF 15.2%).
Discussion
We observed a case of alveolar adenocarcinoma in a patient with MET exon 14 skipping. The patient had response to crizotinib with a PFS of nearly 5 months. Liu et al. reported the incidence of MET exon 14 skipping is 0.9% among the Chinese patients with NSCLC, of whom 83% are with lung adenocarcinoma, 0.8% with lung squamous carcinoma, and 0.8% with adenosquamous carcinoma of the lung.16 Currently, the histological type of patients with lung adenocarcinoma harboring MET exon 14 skipping has not been reported yet.
Profile 1001 analyzed the efficacy of crizotinib in treating NSCLC with MET exon 14 skipping. This research obtained an OR of 44% and found crizotinib was generally well tolerated. The median duration of therapy in Profile 1001 was 5.3 months, which was consistent with the PFS of nearly 5 months in this report.
Several case reports have explored the resistance mechanisms to crizotinib in patients with MET exon 14 skipping mutations.19,20,22 Those patients at first received first-line or second-line chemotherapy and local radiotherapy. When the disease progressed, they received crizotinib therapy and achieved partial response (PR) until the disease progressed again. Then they accepted gene detection again. A patients was de tedetected new MET D1228N mutation and the pre-existing MET exon 14 skipping mutation, D1010H; in another case, the level of pre-existing MET exon 14 skipping mutation D1010H has decreased to 10.9%, while that of MET Y1230C mutation has increased to 3.5%; in another case, there emerged new MET exon 19 D1228N/H and Y1230H mutations on the basis of the pre-existing MET exon 14 skipping mutation, which meant the co-occurrence of three mutations. The above cases indicated that D1228 and Y1230 mutations might be the main reason why patients with MET exon 14 skipping mutations resisted to crizotinib. MET Y1230 and D1228 mutations were certificated to be the mechanisms of acquired resistance to MET inhibitors in vivo. When the patient in our report had partial relief after crizotinib treatment, we conducted a biopsy and ctDNA detection. HER2 gene amplification was detected in the biopsy. HER2, also known as ERBB2, is a transmembrane glycoprotein with receptor tyrosine kinase (RTK) activity and is a member of the epidermal growth factor receptor (EGFR) family. HER2 gene amplification and protein overexpression, without the need of ligand activation, can directly induce the formation of HER2 homodimers or heterodimers, activate receptor tyrosine kinase (RTK) and downstream signaling pathways, and promote proliferation and metastasis of tumor cells. HER2 gene amplification can elevate HER2 protein expression and participate in tumor initiation and progression and affect prognosis through promoting proliferation, invasion and metastasis of tumor cells, and may reduce sensitivity of tumors to EGFR-TKIs, serving as one of the resistance mechanisms of first-generation EGFR-TKIs.23 Crizotinib inhibited continuous MET activation caused by MET exon 14 skipping, whereas elevated HER2 protein expression also promoted the proliferation of tumor cells, suggesting maybe bypass activation can also be a resistance mechanism to crizotinib in the treatment of NSCLC with MET exon 14 skipping.
When the patient in our report had partial relief after crizotinib treatment, a biopsy was performed, in which we detected SOX2 gene amplification and a single copy number loss of the RB1 gene. SOX2 is a stem cell transcription factor which plays a critical role in embryogenesis. It belongs to a set of genes (Oct4, SOX2, NANOG) which can reprogram human somatic cells to pluripotent stem cells.24,25 SOX2 is overexpressed in various histologic types of lung cancers, such as small-cell lung cancer (SCLC), lung squamous carcinoma and lung adenocarcinoma.26 The spectrum of genomic variants in small-cell lung cancer has already identified SOX2 as a potential target for therapeutic intervention.27 RB1 gene plays a regulatory role in the G1 phase of the cell cycle, and therefore RB1 gene deletions will lead to loss of G1 control. The gene has a critical role in SCLC transformation.28 Molecular sequencing confirmed that 100% of SCLC cells transformed from resistant EGFR mutant NSCLC had RB1 gene deletions. This gene change didn’t exist in NSCLC before SCLC transformation or the remaining NSCLC after SCLC transformation, indicating RB1 gene deletions played a key role in SCLC transformation.29 What’s more, SOX2 gene amplification and a single copy number loss of the RB1 gene detected in the biopsy suggested there might exist transformation to SCLC after crizotinib resistance. In 2013, NCCN Guidelines for NSCLC pointed out that one of the resistance mechanisms to EGFR-TKIs is transformation from NSCLC to SCLC, accounting for approximately 3%–15%.25 The case in our report indicated that SCLC transformation might be one of the resistance mechanisms in crizotinib treatment of MET exon 14 skipping, which still needs more evidence.
The resistance machanisms is wide-ranging for cancer’s ability, second-site mutations in the target gene and bypass tract signaling both can develop in response to target drugs. For NSCLC with MET exon 14 skipping, although second-site mutations in MET gene had been reported and demonstrated to be mechanisms of resistance to MET inhibitors, to our knowledge, this is the first clinical report of Her2 amplification arising in a patient with MET exon 14 skipping. The bypass tract signaling was emphasized to be resistance mechanisms to MET inhibitors and the range of resistance mechanisms observed may provide further insight into clinical decision.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, Varella-Garcia M, Franklin WA, Aronson SL, Su PF, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama. 2014;311:1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 3.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 4.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 5.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou S-HI, Kim D-W, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–1019. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J Usari T.. 2014. First-line crizotinib versus chemotherapy in alk-positive lung cancer. N Engl J Med. 371(23):2167–2177. [DOI] [PubMed] [Google Scholar]
- 7.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 8.Shaw AT, Solomon BJ.. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2015;372:683–684. doi: 10.1056/NEJMc1415359. [DOI] [PubMed] [Google Scholar]
- 9.Waqar SN, Morgensztern D, Sehn J. MET Mutation associated with responsiveness to crizotinib. J Thorac Oncol. 2015;10:e29–31. doi: 10.1097/JTO.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camidge DR, Ou SHI, Shapiro G, Otterson GA, Villaruz LC, Villalona-Calero MA. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J Clin Oncol. 2013;32(Suppl 15):8001. [Google Scholar]
- 11.Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009;4:5–11. doi: 10.1097/JTO.0b013e3181913e0e. [DOI] [PubMed] [Google Scholar]
- 12.Soh J, Okumura N, Lockwood WW, Yamamoto H, Shigematsu H, Zhang W, Chari R, Shames DS, Tang X, MacAulay C, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4:e7464. doi: 10.1371/journal.pone.0007464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeung SF, Tong JH, Law PP, Chung LY, Lung RW, Tong CY, Chow C, Chan AWH, Wan IYP, Mok TSK, et al. Profiling of oncogenic driver events in lung adenocarcinoma revealed MET mutation as independent prognostic factor. J Thorac Oncol. 2015;10:1292–1300. doi: 10.1097/JTO.0000000000000620. [DOI] [PubMed] [Google Scholar]
- 14.Drilon A, Cappuzzo F, Ou SHI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET?. J Thorac Oncol. 2016; 12:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrock AB, Frampton GM, Suh J, Chalmers ZR, Rosenzweig M, Erlich RL, Halmos B, Goldman J, Forde P, Leuenberger K, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol. 2016;11:1493–1502. doi: 10.1016/j.jtho.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Liu SY, Gou LY, Li AN, Lou NN, Gao HF, Su J, Yang -J-J, Zhang X-C, Shao Y, Dong Z-Y, et al. The unique characteristics of MET exon 14 mutation in Chinese non-small cell lung cancer patients. J Thorac Oncol. 2016;11:1503–1510. doi: 10.1016/j.jtho.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 17.Paik PK, Drilon A, Fan PD, Yu H, Rekhtman N, Ginsberg MS, Borsu L, Schultz N, Berger MF, Rudin CM, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–849. doi: 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drilon AE, Camidge DR, Ou SHI, Clark JW, Socinski MA, Weiss J, Riely GJ, Winter M, Wang SC, Monti K, et al. Efficacy and safety of crizotinib in patients (pts) with advanced MET exon 14-altered non-small cell lung cancer (NSCLC). J Clin Oncol. 2016;34:108. doi: 10.1200/JCO.2016.34.15_suppl.108. [DOI] [Google Scholar]
- 19.Ou SI, Young L, Schrock AB, Johnson A, Klempner SJ, Zhu VW, Miller VA, Ali SM. Emergence of preexisting MET Y1230C mutation as a resistance mechanism to crizotinib in NSCLC with MET exon 14 skipping. J Thorac Oncol. 2017;12:137–140. doi: 10.1016/j.jtho.2016.09.119. [DOI] [PubMed] [Google Scholar]
- 20.Heist RS, Sequist LV, Borger D, Gainor JF, Arellano RS, Le LP, Dias-Santagata D, Clark JW, Engelman JA, Shaw AT, et al. Acquired resistance to crizotinib in NSCLC with MET exon 14 skipping. J Thorac Oncol. 2016;11:1242–1245. doi: 10.1016/j.jtho.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Qi J, McTigue MA, Rogers A, Lifshits E, Christensen JG, Janne PA, Engelman JA. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011;71:1081–1091. doi: 10.1158/0008-5472.CAN-10-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong HJ, Li P, Wu CL, Zhou XY, Lu HJ, Zhou T. Response and acquired resistance to crizotinib in Chinese patients with lung adenocarcinomas harboring MET exon 14 splicing alternations. Lung Cancer. 2016;102:118–121. doi: 10.1016/j.lungcan.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okumura-Nakanishi S, Saito M, Niwa H, Ishikawa F. Oct-3/4 and Sox2 regulate Oct-3/4 gene in embryonic stem cells. J Biol Chem. 2005;280:5307–5317. doi: 10.1074/jbc.M410015200. [DOI] [PubMed] [Google Scholar]
- 25.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karachaliou N, Rosell R, Viteri S. The role of SOX2 in small cell lung cancer, lung adenocarcinoma and squamous cell carcinoma of the lung. Transl Lung Cancer Res. 2013;2:172–179. doi: 10.3978/j.issn.2218-6751.2013.01.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, Margenthaler JA, Chatterton RT, Jovanovic B, Dunn BK, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890. doi: 10.1158/1078-0432 CCR-13-1982. https://www.researchgate.net/publication/259254447_Rb_Loss_Is_Characteristic_of_Prostatic_Small_Cell_Neuroendocrine_Carcinoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]