

ABSTRACT
Cdc42 is a member of the Rho family of small GTPases that are at the crossroads of major oncogenic signaling pathways involved in both lung and prostate cancers. However, the therapeutic potential of Cdc42 regulation is still unclear due to the lack of pharmacological tools. Herein, we report that ZCL367 is a bona fide Cdc42 inhibitor that suppressed cancer development and ZCL278 can act as a partial Cdc42 agonist. In lung cancer cell lines with varying EGFR and Ras mutations as well as both androgen-independent and androgen-dependent prostate cancer cell lines, ZCL367 impeded cell cycle progression, reduced proliferation, and suppressed migration. ZCL367 decreased Cdc42–intersectin interactions and reduced Cdc42-mediated filopodia formation. ZCL367 showed increased potency and selectivity for Cdc42 when compared to Rac1 and RhoA. ZCL367 reduced A549 tumorigenesis in a xenograft mouse model. Altogether, ZCL367 is a selective Cdc42 inhibitor and an excellent candidate for lead compound optimization for further anticancer studies.
KEYWORDS: Cdc42, Rho GTPase, intersectin, lung cancer, prostate cancer, drug development
1. Introduction
Cell division cycle 42 (Cdc42) is a member of the Rho family of Ras-related GTPases, which includes Rac1 and RhoA, that influences cytoskeleton organization, cell cycle progression, cell survival, and cell migration.1 Although mutations of the Cdc42 gene are uncommon in cancers,2 Cdc42 is overexpressed in lung3 and prostate4 cancer. Moreover, Cdc42 becomes activated upon expression of oncogenic Ras5,6 and is involved in Ras-related transformation.7 Thus, Cdc42 plays a vital role in Ras-related cancers and its inhibition is a viable anticancer approach. We have recently reviewed the therapeutic potential of the Cdc42 signaling pathway in Ras-related cancers and summarized the pharmacological tools currently available to target the Cdc42 signaling pathway.8
Cdc42 is upregulated in several human lung cancer cell lines and its expression was directly correlated with tumor stage, lymph node metastasis, and patient survival.3 Effects of genetically silencing Cdc42 include inhibiting cell cycle progression and tumorigenesis.9,10 Inhibition of Cdc42 in the RNA level decreased A549 lung cancer cell proliferation but was restored upon upregulation of Cdc42.10 In prostate cancer (CaP), androgen-independence (AI) is a major concern in the development of resistance to current CaP chemotherapies, and several downstream effectors of Cdc42, such as activated Cdc42-associated kinase 1 (Ack1), is activated in CaP.11 Taken together, Cdc42 is a putative anticancer therapeutic target that affects a variety of cancers such as lung and prostate cancer.
Cdc42 function is dependent on its GDP- and GTP-bound state. Activity is intricately controlled by its interactions with the following regulators. GTPase activating proteins (GAPs) promote the hydrolysis of GTP, guanine nucleotide exchange factors (GEFs) stimulate the release of GDP, and guanine nucleotide dissociation inhibitors (GDIs) prevent the nucleotide exchange. When bound to GTP, Cdc42 interacts with over 20 downstream effectors12 involved in a variety of cellular signals. For example, GTP-bound Cdc42 and its effector, PAK, affect the PI3K-Akt13 and Raf-MEK-ERK pathways.14 Attempts to target small GTPases have focused on the inhibition of GEF interactions thereby preventing the prerequisite nucleotide exchange.15,16 We previously reported the discovery of a number of novel small molecule modulators of Cdc42–intersectin (ITSN) interaction, which we called the ZCL compounds.17 Herein, we report that ZCL278 can act as an ITSN/GEF-like agonist of Cdc42. We also identified ZCL367 as a bona fide Cdc42 inhibitor and potential anticancer therapeutic agent after screening the ZCL compounds for their activity against inhibiting cancer cell migration, cell proliferation, and cell cycle progression in several lung and prostate cancer cell lines. We further evaluate the anticancer potential of Cdc42 inhibition via ZCL367 in vivo using an A549 lung cancer tumor xenograft mouse model as well as validate the proposed mechanism of action of the ZCL367 as a Cdc42-GEF inhibitor.
2. Results
2.1. ZCL367 inhibits cancer cell migration without reducing cell viability
Our previous study applied high throughput in silico screening to identify a library of small molecules that modulate Cdc42–ITSN interactions.17 To evaluate their therapeutic potential as anticancer agents, these leads (ZCL compounds) were further screened against A549 lung and PC3 prostate cancer cell lines. The A549 lung cancer cells overexpress Cdc423 and the PC3 prostate cancer cells were used given the role of Cdc42 in the development of androgen-independence in prostate cancer.18 Our initial screen of the ZCL compounds focused on wound healing/migration given the function of Cdc42 in cell motility. ZCL367 significantly inhibited A549 and PC3 cancer cell migration in a time-dependent manner (Figure 1(a)). ZCL367 was more potent when compared to ZCL193, ZCL278, ZCL251, ZCL254, ZCL257, and ZCL269, which were previously reported to inhibit microspike formation of Swiss 3T3 cells. For comparison, cells were treated with AZA1 (EC/IC50 = 5–10 μM), a nonselective Rac1/Cdc42 inhibitor.19 Treatment with 10 μM AZA1 resulted in a significant decrease of wound closure, but also caused a morphological change in cells indicating potential toxicity (Figure S1).
Figure 1.
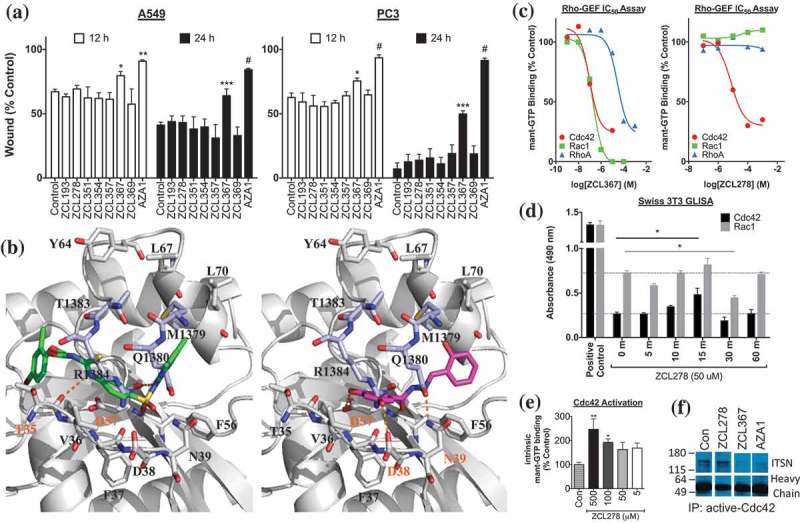
ZCL compound screening and validation as Cdc42-ITSN regulators. (a) ZCL367 inhibits migration/wound healing of A549 and PC3 cells. Confluent cells were scratched and treated with mitomycin C before treating with ZCL compound. The percentage of the wound closure was quantified from three scratches from two independent experiments and is expressed as mean±SEM. (b) Molecular docking of ZCL278 (green) and ZCL367 (pink) into the Cdc42-ITSN binding site. ZCL278 and ZCL367 show different poses with a certain extent of overlap with each other. ZCL278 formed two hydrogen bonds with Thr35 and Asp57, and hydrophobic interactions with Val36 and Thr35 of Cdc42. ZCL367 formed three hydrogen bonds with Asp38, Asn39, and Asp57, and hydrophobic interactions with Phe56 and Val36 of Cdc42. Gray: Cdc42, light purple: ITSN, orange dots: hydrogen bond. (c) ZCL compounds inhibit GEF-mediated Rho GTPase activation. Both ZCL278 (IC50 = 7.5 μM) and ZCL367 (IC50 = 0.098 μM) inhibit DH domain-mediated mant-GTP binding/Cdc42 activation. ZCL367 is more potent against Cdc42 than RhoA (IC50 = 29.7 μM) and Rac1 (IC50 = 0.19 μM). (d) ZCL278 activates Cdc42. Serum-starved Swiss 3T3 cells were treated with ZCL278 and analyzed for Cdc42 and Rac1 activation via GLISA. (e) ZCL278 increases intrinsic GTP binding of Cdc42. In the absence of GEF/DH domain, treatment with ZCL278 increased binding of GTP to Cdc42. (f) Cdc42 regulators inhibit Cdc42-ITSN. A co-immunoprecipitation of ITSN with active-Cdc42 revealed that ZCL367 (50 μM) and AZA1 (10 μM) was able to dislodge ITSN from active-Cdc42. At the same concentration, ZCL278 was not effective. All data are presented as mean±SEM from duplicates or triplicates from two independent experiments. ANOVA compared treatments to their respective control (* p < 0.05, ** p < 0.01, *** p < 0.001, # p < 0.0001).
2.2. ZCL compounds regulate Cdc42-GEF and Cdc42–GTP interactions
To elucidate the potential interactions of the ZCL compounds with Cdc42, we performed in silico analysis (Figure 1(b)) as previously described.17 Residues Gln1380 and Arg1384 of ITSN were observed to form hydrogen bonds with Asn39 and Phe37 of Cdc42, respectively. Two clusters of hydrophobic interactions were found between Leu1376, Met1379, and Thr1383 of ITSN and Phe56, Tyr64, Leu67, and Leu70 of Cdc42. Both ZCL367 and ZCL278 were found to bind to Cdc42 at its ITSN-interacting interface. However, the two compounds adopted different binding poses with certain extent of overlap. ZCL367 was found to form three hydrogen bonds with Asp38, Asn39, and Asp57, and hydrophobic interactions with Phe56 and Val36 of Cdc42. ZCL278 was found to form two hydrogen bonds with Thr35 and Asp57, and hydrophobic interactions with Val36 and Thr35 of Cdc42. Thus, ZCL367, with an extra hydrogen bond, showed more favorable interactions with Cdc42 than ZCL278, which is consistent with its observed elevated activity.
To evaluate the direct effects of the ZCL compounds on Cdc42 activation, we utilized a Cdc42-GEF assay. The Cdc42-GEF assay employs a fluorescent mant-GTP reporter to monitor Cdc42-GTP binding. In the presence of GEF (DH domain), both ZCL367 and ZCL278 inhibited mant-GTP binding (Figure 1(c)). The estimated EC50 value of ZCL367 and ZCL278 for Cdc42 is 0.098 μM and and 7.5 μM, respectively. Moreover, ZCL367 showed a 2- and 303-fold increase in activity for Cdc42 (0.098 μM) when compared to Rac1 (0.19 μM) and RhoA (29.7 μM), respectively. ZCL278 was ineffective at inhibiting Rac1 and RhoA. Interestingly, we observed an increase in mant-GTP binding to Cdc42 in the presence of ZCL278 before the addition of the GEF (DH domain) (Figure S2). Therefore, we further investigated if treatment with ZCL278 could activate Cdc42. Indeed, ZCL278 activated Cdc42 in serum-starved Swiss 3T3 cells in a time-dependent manner (Figure 1(d)) and promoted the binding of mant-GTP to Cdc42 in a concentration-dependent manner (Figure 1(e)).
The agonistic effect of ZCL278 in the absence of GEF implies that binding of ZCL278 may be able to induce a conformational change of Cdc42 into a state that resembles the GEF-induced change. Docking of ZCL278 into free Cdc42 (glide score = −4.55) and Cdc42 complexed with GEF (glide score = −5.33) showed that ZCL278 has significantly increased affinity with Cdc42 in its GEF-complexed conformation, which supports the implication that ZCL278 may have induced conformational change of Cdc42 into its GEF-induced state that has higher binding affinity to GTP.
To determine if ZCL367 could inhibit Cdc42-GEF/ITSN interactions in vitro, we performed a co-immunoprecipitation assay that examined ITSN bound to active-Cdc42 (Figure 1(f)). In A549 lysates, ZCL367 (50 μM) and AZA1 (10 μM) decreased the amount of bound ITSN to active-Cdc42 while ZCL278 (50 μM) did not appear to effective.
2.3. ZCL compounds regulate filopodia formation and alter GM130 localization
Given the established roles of Rho GTPases in the actin cytoskeletal organization, we determined the effects of the ZCL compounds on Swiss 3T3 cells. Swiss 3T3 cells activated with bradykinin were used to examine the formation/localization of GM130, filopodia, lamellipodia, and stress fibers (Figures 2 & 3). Control cells displayed (1) a crescent shape staining for GM130 indicating organization/localization in the Golgi, (2) little/no filopodia/microspikes and lamellipodia/membrane ruffles, and (3) well-defined stress fibers (Figure 2). Activation with bradykinin resulted in the formation of filopodia and lamellipodia as well as a morphological change (Figures 2 & 3) in the overall shape of the cell, as previously reported.20 Treatment with ZCL367 before bradykinin-stimulation resulted in a decreased number of fully activated cells. Microspike formation/elongation was inhibited while membrane ruffling was apparent in a large number of cells. Treatment with ZCL367 alone showed no adverse effects. Meanwhile, treatment with ZCL278 resulted in cellular changes similar to that of treatment with bradykinin. ZCL278 altered cellular morphology in a time-dependent manner. Diminished stress fibers and diffused actin staining were prominent (>80% of cells) 2 h after treatment and complete change in cell morphology was observed 6 h after treatment.
Figure 2.
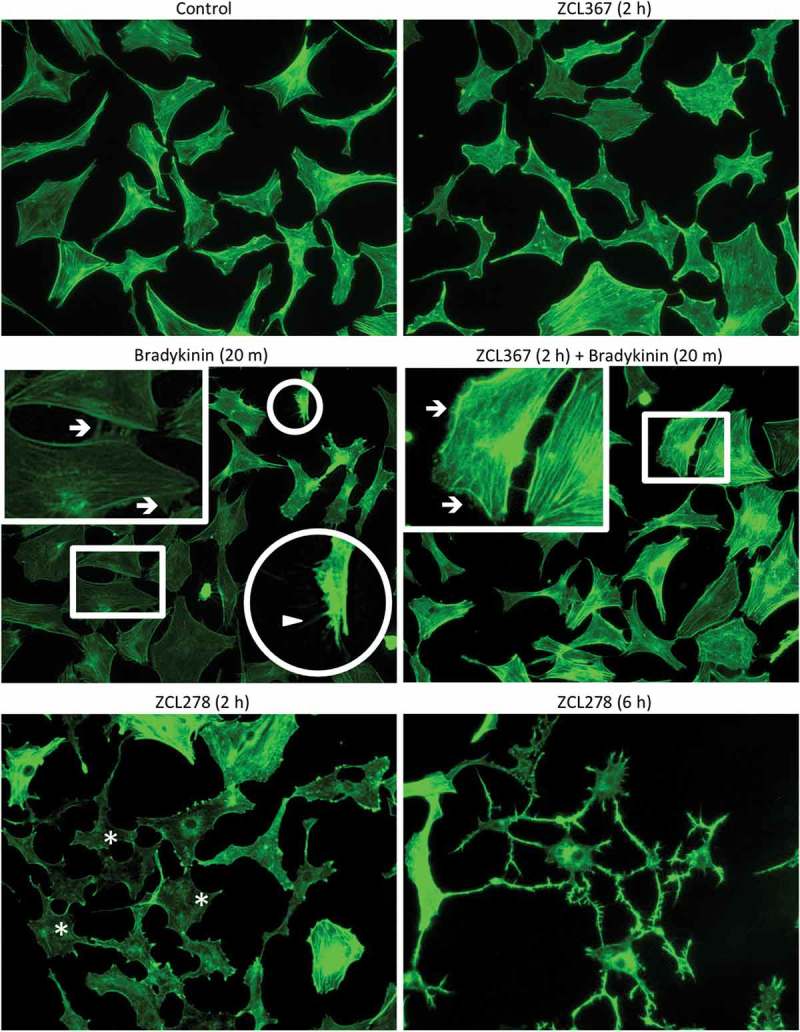
ZCL compounds regulate microspike formation. Swiss 3T3 cells were starved ON then preincubated with ZCL367 (50 μM) for 2 h. Bradykinin (100 ng/mL) was used to activate Cdc42/Rac1 for 20 m. Cells were also treated with ZCL278 (50 μM) for comparison. Cells were fixed and stained with phalloidin. Boxes are magnifications of membrane ruffling and highlighted by →. Circles are magnifications of microspikes and highlighted by ▲. Asterisks (*) highlight diminished stress fibers.
Figure 3.
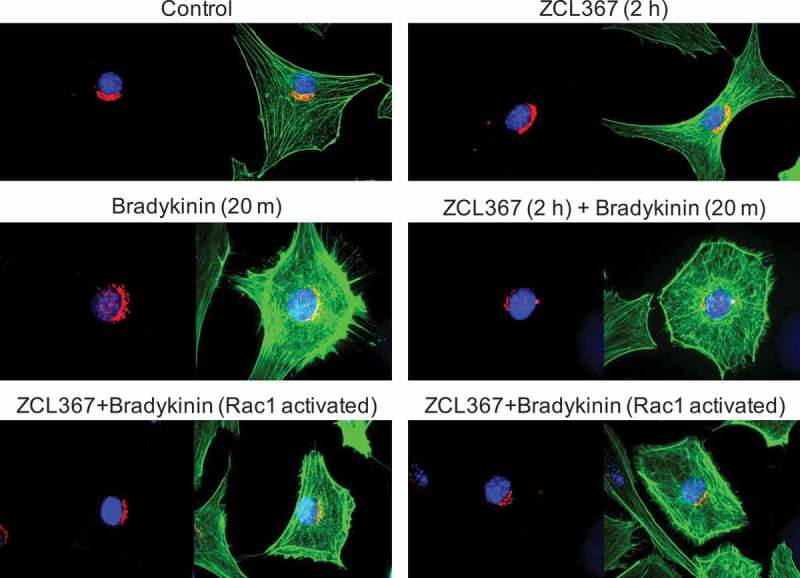
ZCL367 inhibits Cdc42 activation downstream of GM130. Swiss 3T3 cells were starved ON then preincubated with ZCL367 (50 μM) for 2 h. Bradykinin (100 ng/mL) was used to activate Cdc42/Rac1 for 20 m. Cells were fixed and stained for GM130 (red), nucleus (blue), and phalloidin (green). GM130 organization was conserved in ZCL367 treated cells. Activation with bradykinin resulted in altered distribution of GM130. Treatment with ZCL367 inhibited filopodia formation but did not affect GM130 distribution. Cells that showed strong Rac1 activation in the presence of ZCL367 showed a GM130 staining similar to control.
Given that ZCL367 appeared to inhibit Cdc42 activation, we determined its effects on GM130 translocation. GM130 staining in both control and ZCL367 treated cells showed a crescent shape staining localized in the Golgi (Figure 3). Activation of Swiss 3T3 cells with bradykinin led to the redistribution of GM130 corresponding to the formation of filopodia and lamellipodia. Treatment with ZCL367 prior to bradykinin-stimulation resulted in a slight decrease in GM130 redistribution with decreased filopodia formation but did not appear to affect lamellipodia formation (Figure 3). These results are consistent with previous studies that reported Cdc42 function downstream of GM130 and altered Golgi organization.17,21,22 Interestingly, cells with prominent membrane ruffling showed a GM130 staining similar to control.
2.4. ZCL367 inhibits cell proliferation and cell cycle progression
To determine the benefits of Cdc42 inhibition in lung and prostate cancers, we evaluated several lung and prostate cancer cell lines with varying backgrounds. We utilized A549 (EGFR WT, KRasG12S), NCI-H522 (EGFR WT, KRas WT), and HCC827 (EGFRexon19del E746–A750, KRas WT) lung cancer cells to define the differential effects of the ZCL compounds on EGFR-KRas signaling. Additionally, we utilized the androgen-independent prostate cancer (AICaP) cells PC3, DU145, and CWR-22Rv1 and the androgen-dependent prostate cancer cell (ADCaP) LNCaP for comparison. Cancer cell proliferation was determined after treatment with ZCL278, ZCL367, or AZA1 (Figure 4(a)). Treatment of lung cancer cells with ZCL278 or ZCL367 (50 μM) inhibited cell proliferation 48, 96, and 144 h after treatment. Similarly, AZA1 (10 μM) significantly inhibited lung cancer cell proliferation, but also appeared to have affected cell viability. Due to the increased sensitivity of PC3 cells to Cdc42 inhibition, the prostate cancer cell lines were treated with 20 μM ZCL278 and ZCL367 or 5 μM AZA1. Treatment of AICaP and ADCaP cells with ZCL367 or AZA1 significantly decreased cell proliferation after 48, 96, and 144 h. Meanwhile, ZCL278 was effective in only the PC3 and LNCaP cell lines.
Figure 4.
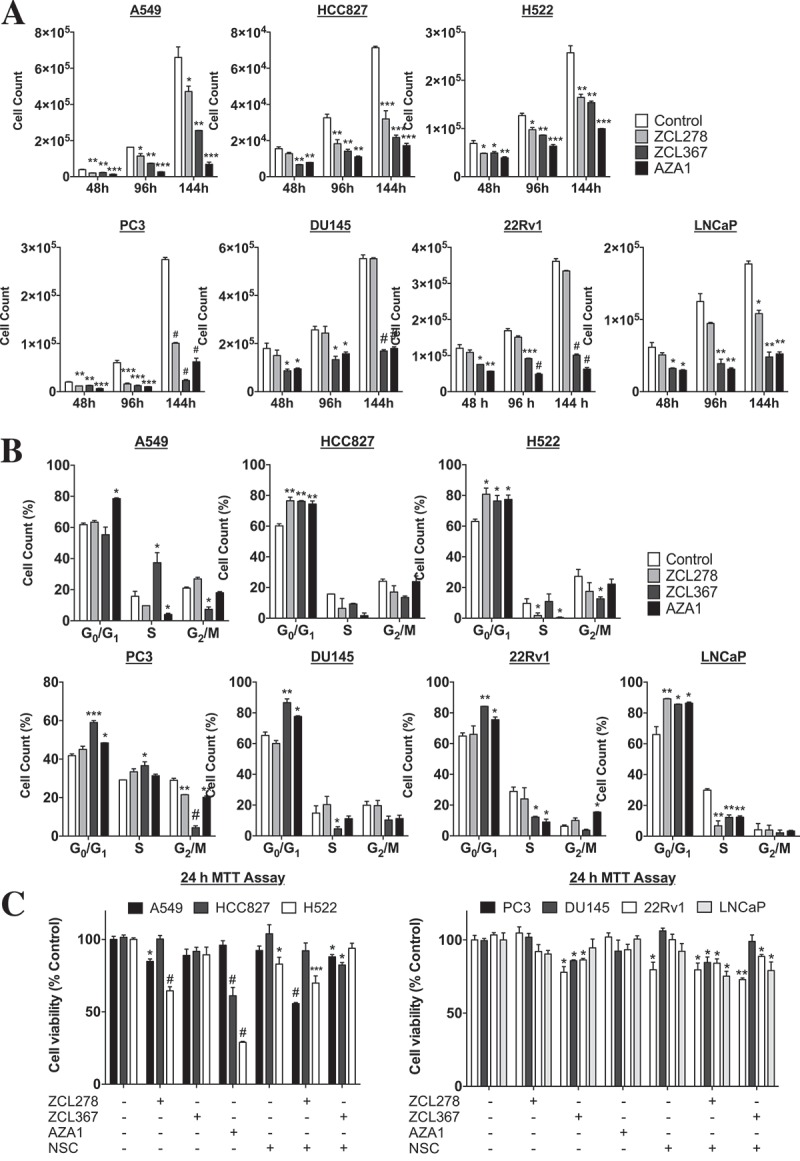
Cdc42 inhibition alters critical cellular processes. (a) Cdc42 regulators decrease cell proliferation. Relative cellular density of lung and prostate cancer cells up to 144 h after treatment with ZCL278, ZCL367, or AZA1. (b) Cdc42 inhibitors alter FBS-stimulated cell cycle progression. Relative cell cycle distribution of lung and prostate cancer cells after 24 h of starvation followed by 24 h FBS-stimulation with or without treatment (A549, HCC827, H522 = 50 μM ZCL367 or 10 μM AZA1; PC3, DU145, 22Rv1, LNCaP = 20 μM ZCL367 or 5 μM AZA1). (c) Effect of Cdc42 inhibitors on cell viability. Cells (5 × 104) were seeded and, after 24 h, treated with compound for 24 h. A solution of MTT was added and the reading was taken after 3 h. 50 μM NSC23766 (NSC), a Rac1 inhibitor, and 50 μM NSC + 50 μM ZCL367 is not as toxic as AZA1 alone. The absorbance was measured at 562 nm. All data are presented as mean±SEM from duplicates or triplicates from two independent experiments. ANOVA compared treatments to their respective control (* p < 0.05, ** p < 0.01, *** p < 0.001, # p < 0.0001).
Given the decrease in cell proliferation, we determined the effects of the drugs on cell cycle progression (Figure 4(b)). Treatment of AICaP and ADCaP with ZCL367 or AZA1 resulted in an increase in G0/G1 cell cycle arrest. Treatment of H522 and HCC827 lung cancer cells also resulted in an increase in G0/G1 cell cycle arrest. Alternatively, A549 cells treated with ZCL367 showed an increase in S phase arrest while AZA1 (5 μM) halted cells in the G0/G1 phase. While ZCL367 and AZA1 inhibited cell cycle progression of all the lung and prostate cancer cell lines, ZCL278 was most effective against the lung cancer cell lines and the PC3 and LNCaP prostate cancer cell lines.
An MTT assay confirmed the cytotoxicity of AZA1 (>20 μM) on the lung and prostate cancer cells lines and the increased sensitivity of PC3 cells to Cdc42 inhibition when compared to other prostate cancer cell lines (Figures 4(c) and S3). Alternatively, lung cancer cell lines tolerated treatment with ZCL367 (50 μM). H522 lung cancer cells appeared to be more sensitive to treatment with ZCL278 (50 μM), when compared to other lung cancer cell lines. AZA1 (>20 μM) sharply decreased the viability of all the lung and prostate cancer cell lines. To determine if the increased toxicity of AZA1 was due to the dual inhibition of Cdc42 and Rac1, cells were treated with NSC23766, a specific Rac1 inhibitor, with and without ZCL367 (Figure 4(c)). NSC23766 (50 μM) alone and NSC23766+ZCL278 or NSC23766+ZCL367 (50 and 50 μM, respectively) decreased cell viability but did not reach the same toxicity of AZA1. Similar results were obtained in Swiss 3T3 cells treated with ZCL278, ZCL367, or AZA1 (Figure S3).
2.5. ZCL367 inhibits A549 tumor growth
To evaluate the anticancer effects of ZCL367 in vivo, we utilized an A549 tumor xenograft mouse model. Mice treated with vehicle control showed a steady increase in tumor volume whereas ZCL367-treated mice showed a marked inhibition (~80%, p < 0.05) in overall tumor growth. After 4 weeks, the tumor volumes were 443.87 ± 228.85 mm3 and 93.41 ± 13.77 mm3 for control and ZCL367 group mice, respectively (Figure 5). ZCL367 treatment also decreased tumor mass (~85%, p < 0.05). The mass for the control tumors was 168.20 ± 93.37 mg and 26.00 ± 10.03 mg for ZCL367-treated tumors. Treatment with ZCL367 did not adversely affect the weight of the mice (data not shown).
Figure 5.
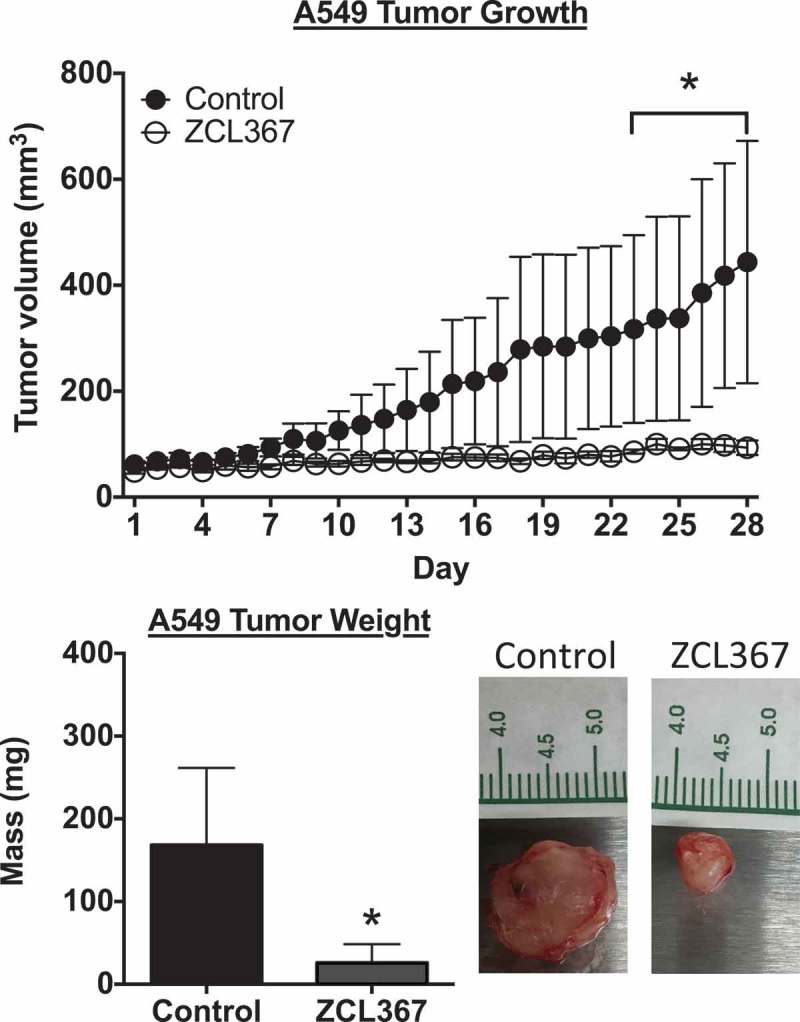
ZCL367 inhibits A549 tumor growth in mice. Mice with palpable A549 tumors were treated with ZCL367 (20 μg/g) or vehicle control (10% DMSO in sesame oil) every other day for 28 days. Tumor volume was calculated using the formula (length x width2)/2. After 28 days, the tumor was weighed. Data are presented as mean±SEM from groups of n = 6. ANOVA followed by Matt–Whitney test compared ZCL367 treatment to control (* p < 0.05).
3. Discussion
Cdc42 is gaining momentum as a putative target for cancers as well as in Alzheimer’s disease and other neurodegenerative diseases.8,23 Due to the limitations of currently available pharmacological agents that target Cdc42, the therapeutic potential of Cdc42 has not been determined. We previously identified a series of Cdc42 regulators (ZCL compounds) via high-throughput in silico analysis and biological assays.17 Therefore, we set out to evaluate the potential of the ZCL compounds as anticancer agents. Given the role of Cdc42 in cell motility, we evaluated the effect of the ZCL compounds on cancer cell migration using A549 and PC3 cells. Both the A549 and PC3 cell lines implicate Cdc42 as a viable target.3,18,24 We observed ZCL367 to be the most potent ZCL compound under the current screening condition. Interestingly, we observed that although ZCL278 functions as an inhibitor, ZCL278 may also function as an agonist in certain conditions.
We have presented data to support the function of ZCL278 as an agonist in certain conditions (i.e. in the absence of GEF (DH domain)). Our in vitro Cdc42-GEF assay and immunofluorescence experiments highlight the effects of Cdc42 activation with ZCL278. First, we observed that ZCL278 promotes the binding of GTP to Cdc42. Second, we observed that treatment of Swiss 3T3 cells with ZCL278 induced microspike formation and other morphological changes similar to that of treatment with bradykinin. Third, treatment of serum-starved Swiss 3T3 cells with ZCL278 induced the activation of Cdc42 via GLISA analysis. Last, we propose that ZCL278 acts as an ITSN/GEF-like Fagonist by interacting with Cdc42 in the corresponding ITSN/GEF groove via our in silico model. Altogether, these results provide the foundation for further studies to examine the functions of ZCL278 as a dual agonist and antagonist of Cdc42.
ZCL367 showed favorable selectivity for Cdc42 in vitro. ZCL367 inhibited bradykinin-activated Cdc42-mediated microspike/filopodia formation in Swiss 3T3 cells but not Rac1-mediated membrane ruffling/lamellipodia, without altering GM130 distribution indicating that ZCL367 functions further downstream. ZCL367 inhibited the interaction between Cdc42 and ITSN, a Cdc42-specific GEF, in A549 cell lysate. ZCL367 inhibited GEF-dependent binding of GTP to Cdc42 (IC50 = 98 nM) and showed a 2- and 303-fold increase in potency for Cdc42 when compared to Rac1 and RhoA, respectively. Since neighboring domains (SH3 and PH) contribute to the specificity of ITSN for Cdc42,25 this estimate of selectivity is limited by the sole use of the GEF (DH) domain. Thus, ZCL367 may be more selective for Cdc42 than initially reported. Overall, these results highlight the mechanism of ZCL367 as a competitive inhibitor of Cdc42 by inhibiting GEF binding and, unlike ZCL278, does not activate/promote GTP binding to Cdc42 in the absence of GEF. Taken together, we have demonstrated the potency and selectivity of ZCL367 for Cdc42.
There has been a prior attempt of targeting Cdc42/Rac1 in CaP with a nonspecific small molecule inhibitor called AZA1.19 Our results with several AICaP cell lines and an ADCaP cell line treated with ZCL367 and AZA1 provide some insight to the benefits of inhibiting Cdc42 in CaP. ZCL367 and AZA1 were effective at inhibiting several cancer cell processes. Although AZA1 generally performed better than ZCL367, we observed a change in cell morphology and cytotoxicity following treatment with AZA1. Given the dual inhibitory nature of AZA1, we performed dual treatment of ZCL367 and NSC23766, a specific Rac1 inhibitor. A cell viability assay indicated that the dual treatment was not as toxic as AZA1 thus implicating the nonspecific interactions of AZA1 leading to increased cytotoxicity unrelated to Cdc42/Rac1 inhibition. Cytotoxicity assays estimate the in vitro LD50 of AZA1 to be 15–20 μM while the EC/IC50 values range from 5 to 10 μM.19 We also observed increased sensitivity of PC3 cells to Cdc42 inhibition that was not present in other AICaP cell lines (CWR-22Rv1 and DU145). Androgen-independence is a major obstacle in prostate cancer treatment due to the development of resistance to current therapy. Thus, Cdc42 may represent a novel potential anticancer target for AICaP given the increased expression of Cdc42 in CaP and the critical role of Ack1, a Cdc42-specific effector, in progression to androgen-independence.18,26
Similarly, there have been attempts to inhibit Cdc42 in lung cancer with natural products such as TDB and curcumin. Our results with A549, H522, and HCC827 wound healing, cell proliferation, and cell cycle progression with ZCL367 highlighted the therapeutic potential of inhibiting Cdc42 in lung cancer. Thus, ZCL367 is an improvement on previously reported nonspecific, natural products. On the other hand, AZA1 showed a similar toxicity profile for lung cancer cells as observed in CaP cells. Cdc42 represents a putative therapeutic target in EGFR-Ras-related lung cancers as observed with A549 cells, given that Cdc42 is upregulated upon expression of EGFR and oncogenic Ras.5,27,28 Indeed, our in vitro and in vivo studies with the various lung cancer cell lines highlight the therapeutic potential of inhibiting Cdc42 and the potential of ZCL367 as an anticancer agent in lung cancer.
Our studies pointed to the ability of ZCL278 to function as an agonist of Cdc42. The validation of ZCL278 as an agonist would classify ZCL278 as the first small-molecule activator of Cdc42 that promotes the binding of GTP. Unlike AZA1, treatment with ZCL367 did not adversely affect cell viability. Treatment with both ZCL367 and NSC23766, a Rac1 inhibitor, did not exhibit the same cytotoxicity as that of AZA1 in any of the cell lines used. These results indicate that the toxicity of AZA1 may most likely be the result of nonspecific, off-target interactions. Altogether, our results underscore the potential of targeting Cdc42 in vivo with ZCL367 given its relatively low toxicity and high specificity/selectivity for Cdc42. Our results highlight the benefits and feasibility of inhibiting Cdc42 as an anticancer target due to the effects of Cdc42 inhibition in several lung and prostate cancer cell lines. ZCL367 is a promising small molecule primed for lead development/optimization that is distinct from and improves upon previously reported Cdc42 inhibitors.
4. Materials and methods
4.1. Cell lines and cell culture
A549 (CCL-185), HCC827 (CRL-2868), and NCI-H522 (CRL-5810) lung cancer cells, 22Rv1 (CRL-2505), DU145 (HTB-81), LNCaP (CRL-1740), and PC3 (CRL-7934) prostate cancer, and Swiss 3T3 (CCL-92) fibroblast cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured following ATCC guidelines. A549, DU145, PC3, and 22Rv1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (22400, Invitrogen, Carlsbad, CA, USA). HCC827, NCI-H522, LNCaP cell were culture in RMPI 1640 medium (A10491). Swiss 3T3 cells were cultured in Dubelco’s Modified Eagle Medium (DMEM; Invitrogen, Carlsbad, CA, USA). Medium was supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 1% penicillin-streptomycin. The cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
4.2. Wound healing assay
A549 or PC3 cells were seeded into a 3.5 cm plate and allowed to reach 90% confluency in complete medium. The monolayer was then wounded three times with a 10 μL pipette tip and rinsed to remove debris. Cells were then incubated with Mitomycin C (10 μg/mL) for 1 h at 37°C to inhibit cell proliferation. Phase-contrast images of a representative of each scratch were taken at 0, 12, and 24 h of treatment with an Axiovert S100 microscope (Carl Zeiss). Wound healing/migration was determined by measuring the distance between the leading edges of the migrating cells by using MetaMorph software. Data are representative of mean±SEM from triplicates conducted at least twice.
4.3. Immunofluorescence
Serum-starved Swiss 3T3 cells were pretreated with compound for 2 h before the addition of bradykinin (100 ng/mL) for 20 min. Cells were fixed in 4% paraformaldehyde for 15 min at RT and stored in PBS. Fixed cells were treated with 0.5% Triton for 30 min at RT, 100 mM Glycine 30 min at RT, and 10% BSA for 30 min at 37°C. Primary antibody for GM130 (1:1000, BD Biosciences) was used to probe for 1 h at RT followed by CY3-conjugated secondary antibody (1:1000) for 1 h at RT in the dark. Actin was stained with fluorescein-conjugated phalloidin (1:300). Hoechst (1:2500) was used to stain the nuclei. Coverslips were mounted onto slides using antifade medium from Life Technologies (Carlsbad, CA). Images were taken with a Zeiss Axiovert S100 microscope (Carl Zeiss).
4.4. MTT assay
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was conducted as previously described.29 Briefly, cells were treated for 24 h followed by incubation with MTT. The absorbance was measured at 562 nm. Data are representative of mean±SEM from triplicates conducted at least twice.
4.5. Synthesis of ZCL367
4.5.1. General procedure
Reagents were purchased from Sigma–Aldrich (Milwaukee, WI), Acros (Morris Plains, NJ), TCI America (Portland, OR), or Alfa Aesar (Ward Hill, MA) and used without further purification. AZA1 was purchased from EMD Millipore (Billerica, MA). NMR spectra were obtained from a Bruker 400 spectrometer in CDCl3 or DMSO-d6. Chemical shifts (δ) are given in ppm relative to the signal for the deuterated solvent and are reported consecutively as position (dH), relative integral, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, m = multiplet and br = broad), coupling constant (J/Hz) and assignment.
4.5.2. Synthesis of ZCL367
Salicylaldehyde (0.85 g, 7 mmol) was added to an ethanol solution (25 mL) of 3, 5-diphenolbenzoichydrazide (1 g, 6 mmol) and the solution refluxed for 6 h. After cooling to room temperature, the precipitated solid was filtered, washed with ethanol and recrystallized from absolute ethanol to give the title product as a light brown solid (0.87 g, 53.2%). 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 11.37 (s, 1H), 9.62 (s, 2H), 8.63 (s, 1H), 7.50 (m, 1H), 7.30 (m, 1H), 6.93 (m, 2H), 6.78 (d, J = 2.1 Hz, 2H), 6.45 (t, J = 2.1 Hz, 1H).
4.6. Cell proliferation assay
Cells (~5 x 104) were seeded in 2.2 cm plates and treated with a compound in growth medium after 24 h. Media was changed 48 h after treatment. Cell proliferation and viability were determined 48, 96, and 144 h after treatment with a Countess Automated Cell Counter (Invitrogen) per manufacturer’s instructions. Data are representative of mean±SEM from duplicates from experiments conducted at least twice.
4.7. Cell cycle progression assay
Cells were seeded in 3.5 cm plates and allowed to reach roughly 70% confluency. After starving 24 h, cells were treated in FBS-supplemented medium. After 24 h, cells were trypsinized, washed in PBS, fixed in 70% ethanol at −20°C for 24 h, washed with PBS, and stained with a solution of 50 μg/mL propidium iodide and 50 mg/mL RNase. The solution was incubated at 37°C for 1 h and allowed to cool for 30 min at RT. 104 events were analyzed on a FACScan flow cytometer. Data are representative of mean±SEM from duplicates conducted at least twice.
4.8. Rho-GEF assay
Rho-GEF assay (Cytoskeleton) was conducted per manufacturer’s instructions. Briefly, a solution (50 μL) of purified Rho GTPase (1 μM) and mant-GTP (1.5 μM) with or without compound (0.001–500 μM) was prepared and the absorbance monitored for 10–30 min to obtain the background/baseline. The addition of purified GEF/DH domain (0.08–0.2 μM) was carried out simultaneously and the absorbance was recorded for 30–45 min. The linear slope following the addition of the GEF was calculated and expressed as the percentage of the control and plotted on an IC50 curve.
4.9. GLISA
Serum-starved Swiss 3T3 cells (50% confluent) were treated with ZCL278 (50 μM) for 0, 5, 10, 15, 30, and 60 m. Cells were lysed and ~15 μg protein was used for GLISA analysis per manufacturer’s (Cytoskeleton) instructions. Readings were taken with a Synergy HT plate reader (BioTek).
4.10. Co-immunoprecipitation
A549 lysate (240 μg) was precleared for 30 min on a nutator at 4°C with protein G. Protein G was removed by centrifugation at 1,000 rpm for 5 min. Anti-active Cdc42 mouse monoclonal antibody (New East) was added to the lysates and incubated ON on a nutator at 4°C with or without Cdc42 inhibitor. Protein G was then added to the cell lysates then incubated for 2 h at 4°C on a nutator. Protein G was washed three times by centrifugation at 1,000 rpm for 5 min, removal of the supernatant, addition of buffer, and gently vortexing for 15 s. The Protein G pellet was used for Western blot analysis for ITSN (Santa Cruz).
4.11. A549 tumor xenograft mouse model
The Institutional Animal Care and Use Committee of East Carolina University approved the experiments performed in this study. Six-week-old male or female nude BALB/C mice (CAnN.Cg-Foxn1nu/Crl, Charles River) were sedated with isoflurane and inoculated with 1 × 106 human A549 lung cancer cells suspended in PBS via subcutaneous injection in the right flank. Mice bearing palpable tumors (~50 mm3) were divided into control and treatment group (n = 6/group). ZCL367, 20 μg/g, or vehicle control (10% DMSO in sesame oil) was administered every other day for 4 weeks via intraperitoneal (IP) injection. Tumors were measured with a caliper daily, and tumor volume was determined using the formula (length x width2)/2. After 4 weeks of treatment, the mice were euthanized and the tumors isolated and weighed.
4.12. Statistical analysis
The in vitro cell data are compared by nonparametric one-way analysis of variance (ANOVA) with Greenhouse-Geisser correction followed by Dunnett’s multiple comparisons test calculated with GraphPad Prism 6 (La Jolla, CA). Data are presented as mean±SEM from replicates and are representative of results from experiments conducted at least twice. Tumor volume and weight were analyzed via ANOVA followed by Matt–Whitney test. Data are presented as mean±SEM.
Funding Statement
This work was supported in part by National Cancer Institute Grants [CA111891 and CA165202] (to Q.L.), the Harriet and John Wooten Laboratory for Alzheimer’s and Neurodegenerative Disease Research (to Q.L.), National Natural Science Foundation of China Grant [81573264 (to H.Z.) and 81603029 (to Y.Z.)], and by a grant from the Alzheimer’s Association [AARFD-17-533421] (to B.J.A.).
Abbreviations
- Ack1
activated Cdc42-associated kinase 1
- Cdc42
cell division cycle 42
- DH
Dbl homology
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinases
- GAP
GTPase-activating protein
- GDI
guanine nucleotide dissociation inhibitor
- GDP
Guanosine-5'-diphosphate
- GEF
Guanine nucleotide exchange factor
- GTP
Guanosine-5'-triphosphate
- IP
intraperitoneal
- ITSN
intersectin
- PAK
p21-activated kinase
- PH
pleckstrin homology
- PI3K
Phosphoinositide 3-kinases
- Rac1
Ras-related C3 botulinum toxin substrate 1
- Raf
Rapidly Accelerated Fibrosarcoma
- SH3
SRC homology 3
- TDB
4,5,4’-trihydroxy-3,3’-dimethoxy-bibenzyl
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Yang L, Wang L, Zheng Y.. Gene targeting of Cdc42 and Cdc42GAP affirms the critical involvement of Cdc42 in filopodia induction, directed migration, and proliferation in primary mouse embryonic fibroblasts. Mol Biol Cell. 2006;17:4675–4685. doi: 10.1091/mbc.e06-04-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rihet S, Vielh P, Camonis J, Goud B, Chevillard S, de Gunzburg J. Mutation status of genes encoding RhoA, Rac1, and Cdc42 GTPases in a panel of invasive human colorectal and breast tumors. J Cancer Res Clin Oncol. 2001;127:733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Q-Y, Jiao D-M, Yao Q-H, Yan J, Song J, Chen F-Y, Lu G-H, Zhou J-Y. Expression analysis of Cdc42 in lung cancer and modulation of its expression by curcumin in lung cancer cell lines. Int J Oncol. 2012;40:1561–1568. doi: 10.3892/ijo.2012.1336. [DOI] [PubMed] [Google Scholar]
- 4.Knyazev YP, Cheburkin YV, Spikermann K, Peter S, Jenster G, Bangma KH, Karelin MI, Shkolnik MI, Urbanskii AI, Evtushenko VI. The cDNA microarray profiling of protein kinases and phosphatases: molecular portrait of human prostate carcinomas. Mol Biol [Internet] 2003;37:89–101. http://link.springer.com/article/10.1023/A:1022341015018 [PubMed] [Google Scholar]
- 5.Beeser A, Jaffer ZM, Hofmann C, Chernoff J. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J Biol Chem. 2005;280:36609–36615. doi: 10.1074/jbc.M502306200. [DOI] [PubMed] [Google Scholar]
- 6.Bagrodia S, Laudano AP, Shalloway D. Accessibility of the c-Src SH2-domain for binding is increased during mitosis. J Biol Chem. 1994;269:10247–10251. [PubMed] [Google Scholar]
- 7.Qiu RG, Abo A, McCormick F, Symons M. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol Cell Biol. 1997;17:3449–3458. doi: 10.1128/MCB.17.6.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguilar BJ, Zhou H, Lu Q. Cdc42 signaling pathway inhibition as a therapeutic target in Ras- related cancers. Curr Med Chem. 2017;24:3485–3507. doi: 10.2174/0929867324666170602082956. [DOI] [PubMed] [Google Scholar]
- 9.Stengel KR, Zheng Y. Essential role of Cdc42 in Ras-induced transformation revealed by gene targeting. PLoS One. 2012;7:e37317. doi: 10.1371/journal.pone.0037317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang T, Chen T, Li Y, Gao L, Zhang S, Wang T, Chen M. Downregulation of miR-25 modulates non-small cell lung cancer cells by targeting CDC42. Tumour Biol. 2015;36:1903–1911. doi: 10.1007/s13277-014-2793-0. [DOI] [PubMed] [Google Scholar]
- 11.Manser E, Leung T, Salihuddin H, Tan L, Lim L. A non-receptor tyrosine kinase that inhibits the GTPase activity of p21cdc42. Nature. 1993;363:364–367. doi: 10.1038/363364a0. [DOI] [PubMed] [Google Scholar]
- 12.Bishop A, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241. [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Y, Bagrodia S, Cerione RA. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J Biol Chem. 1994;269:18727–18730. [PubMed] [Google Scholar]
- 14.King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, Marshall MS. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 1998;396:180–183. doi: 10.1038/24184. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang X, Marchioni F, Evelyn CR, Sipes N, Zhou X, Seibel W, Wortman M, Zheng Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc Natl Acad Sci USA. 2013;110:3155–3160. doi: 10.1073/pnas.1212324110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friesland A, Zhao Y, Chen Y-H-H, Wang L, Zhou H, Lu Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc Natl Acad Sci USA. 2013;110:1261–1266. doi: 10.1073/pnas.1116051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, Earp HS, Whang YE. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci USA. 2007;104:8438–8443. doi: 10.1073/pnas.0700420104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zins K, Lucas T, Reichl P, Abraham D, Aharinejad S. A Rac1/Cdc42 GTPase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS One. 2013;8:e74924. doi: 10.1371/journal.pone.0074924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–1952. doi: 10.1128/MCB.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baschieri F, Confalonieri S, Bertalot G, Di Fiore PP, Dietmaier W, Leist M, Crespo P, Macara IG, Farhan H. Spatial control of Cdc42 signalling by a GM130-RasGRF complex regulates polarity and tumorigenesis. Nat Commun. 2014;5:4839. doi: 10.1038/ncomms5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kodani A, Kristensen I, Huang L, Sütterlin C. GM130-dependent control of Cdc42 activity at the Golgi regulates centrosome organization. Mol Biol Cell. 2009;20:1192–1200. doi: 10.1091/mbc.e08-08-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguilar BJ, Zhu Y, Lu Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res Ther. 2017;9:97. doi: 10.1186/s13195-017-0320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang D, Zhu Z, Wang W, Shen P, Wei Z, Wang C, Cai Q. Expression profiles analysis of pancreatic cancer. Eur Rev Med Pharmacol Sci. 2013;17:311–317. [PubMed] [Google Scholar]
- 25.Zamanian JL, Kelly RB. Intersectin 1L guanine nucleotide exchange activity is regulated by adjacent src homology 3 domains that are also involved in endocytosis. Mol Biol Cell. 2003;14:1624–1637. doi: 10.1091/mbc.e02-08-0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1091/mbc.e06-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng C-M-M, Li H, Gasman S, Huang J, Schiff R, Chang EC. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol Cell Biol. 2011;31:983–997. doi: 10.1128/MCB.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng Y, Xia Y, Hawke D, Halle M, Tremblay M, Gao X, Zhou X, Aldape K, Cobb M, Xie K, et al. FAK phosphorylation by ERK primes Ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell. 2009;35:11–25. doi: 10.1016/j.molcel.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boykin C, Zhang G, Chen Y-H-H, Zhang R-W-W, Fan X-E-E, Yang W-M-M, Lu Q. Cucurbitacin IIa: a novel class of anti-cancer drug inducing non-reversible actin aggregation and inhibiting survivin independent of JAK2/STAT3 phosphorylation. Br J Cancer. 2011;104:781–789. doi: 10.1038/bjc.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.