

ABSTRACT
Glioblastoma is the most invasive form of brain tumor. Although temozolomide chemotherapy has been shown to significantly improve survival in patients with GBM, this increase is only trivial. The underlying cause is that many GBMs do not respond to temozolomide, and the rest produces resistance. In the past two decades, many attempts have been made to understand resistance mechanisms and to combine other treatments with temozolomide to maximize patient benefit. Unfortunately, it seems to be a red queen game, and the speed of disease development is as fast as the progress in the field. In order to win this game, a comprehensive approach is needed to decipher the details of the resistance mechanism and to transfer the basic research to the clinic. This article reviews the following: temozolomide discovery, chemistry, and mechanism of action, and mechanisms of resistance, as well as combination therapy with other strategies.
KEYWORDS: Temozolomide, glioma, glioblastoma, alkylating agents, MGMT, BER, MMR, DNA repair, chemoresistance
Introduction
Glioblastoma (GBM) is the most common form of primary brain tumor in adults. It is highly aggressive and has a median survival fewer than 2 years.1 Due to its heterogeneous and infiltrative nature, GBM has an unpredictable response to most treatment modalities. The complexity of this disease is a major obstacle to the development of effective treatment. Decades of research in the field of GBM have helped us to understand its biology extensively, but the translational transfer of most studies has not yet been achieved. Therefore, current standards of care include maximum safe resection, followed by simultaneous chemoradiation therapy and temozolomide adjuvant chemotherapy for newly diagnosed GBM, which remains the same for more than two decades.2
Temozolomide is a monofunctional DNA alkylating agent used in the clinical treatment of newly diagnosed GBM. The addition of temozolomide regimen improved the overall survival and progression-free survival in patients with GBM compared with radiotherapy alone.3,4 Temozolomide is a lipophilic molecule that crosses the blood-brain barrier and is stable at the acidic pH of the stomach and is therefore administered orally.5
The effect of temozolomide is highly scheduled dependent. A single higher dose is actually ineffective and a strict regimen must be followed to obtain a favorable result.6 In addition, due to genetic or acquired resistance to temozolomide, there is an inevitable recurrence. In many clinical trials, relapsed recurrent tumors have been studied using temozolomide as a single drug, as well as alternative therapies such as CCNU (lomustine), BCNU (carmustine) and bevacizumab, but no Long-term benefits.7,8 The only definitive marker predicting temozolomide resistance is the MGMT promoter methylation status.9 Although many other resistance mechanisms are reported in the literature, most antibody mechanisms either fail in clinical trials or have not been tried.
Thanks to the efforts of scientists and clinicians around the world, the understanding of GBM at the molecular level has increased significantly. A variety of important pathways and driving mutations have been identified and candidate targets for treatment have been provided.10-12 Although many of these targets showed promising results in animal models, most failed in the translation step and there was no further improvement in patient survival.13,14 A new GBM classification scheme was established by incorporating molecular markers into histopathological grades.15 However, this classification does not represent any strategy for stratifying patients, according to the temozolomide response, except for patients with IDH1/2 mutations who predict a good prognosis for low-grade gliomas and GBM.16,17
Therefore, GBM is still a frustrating disease that beats all efforts to cure success. Like the Red Queen, each intervention is overcome by GBM‘s adaptation, modification, and resistance, leaving the study in the same place it started (Box 1). In this article, we discuss how temozolomide was discovered and became popular in GBM. We also provide its mechanism of action and report on the reasons for resistance development. At the same time, we reviewed various clinical trials to improve the response to temozolomide in GBM patients. A complete overview of this GBM-preferred drug hopes to find the possibility for the scientific community, clinicians and young readers to win the Red Queen‘s race against GBM.
Box 1.
Red queen race conversation from Alice in the wonderland by Lewis Carroll.
| “Well, in our country,” Alice said, still panting. “You usually get to other places – if you run fast for a long time, as we have been doing.” |
| “A slow country!” said the Queen. “Now, here, you see, it requires all the runs you can do, stay in the same place. If you want to reach other places, you must run at least twice as fast as you have been doing!”. |
History and chemistry
The drug development process involves the synthesis of new chemical entities and evaluating their potential as a drug. The process is very lengthy and detailed, as the goal is to provide patients with the safest and most effective drugs. There are two types of drug development processes, first, where a series of compounds are formed and tested for their ability to capitulate various diseases, and the other is a known target, where the compound is pre-designed for development to abolish the activity of the desired target.18,19 However, for the development of temozolomide, neither option was followed. Therefore, after the first synthesis of precursors for patients in 1960, temozolomide has covered a strange but exciting journey that is completely different from consistent drug design. For example, the entire process of testing in the first phase was carried out by the efforts of a small group of academic pharmacists.
This process begins with the establishment of imidazotetrazinone, a class of prodrugs when reacting diazonium with various isocyanates. The first synthesis of temozolomide was the result of diazo-IC and methyl isocyanate.20 Later, temozolomide was also made by using other precursors, but these two compounds proved to be effective for large-scale synthesis. In 1978, at Aston University, Pharmacy graduate Robert Stone joined Malcolm Stevens as part of the student program to join Plum and Baker Co., Ltd. Stone in collaboration with Eddy Lunt and Chris Newton, prepared a bicyclic compound having an imidazole ring bonded to 1,2,3,5-tetrazine ring.19 The laboratory name for this product is Azolastone (Aston+Stone; later called mitozolomide). Another Aston University student, Neil Gibson, showed anti-tumor activity against leukemia and lymphoma in a mouse tumor model. The pharmacist at Aston University used the drug in clinical trials in 1983, but due to its cross-linking properties, it ended in despair resulting in severe drug-induced thrombocytopenia. Later, studies showed that the replacement of a chloroethyl group with a methyl group in azolastone completely changed the toxicity characteristics, so a new molecule called temozolomide appeared. The Temozolomide project and the efforts of Aston University were originally funded by cancer charity of the Cancer Research Campaign, UK, and pharmaceutical company, May and Baker Ltd., After the failure of azolastone, the resource input of temozolomide may be easily reduced due to lack of funds, but it has been promoted and the world has obtained a successful anti-tumor drug that can treat patients with GBM. This success represents a spectacular blend of chemical and biological sciences against the GBM war[18].
Mechanism of action
DNA alkylating agents have been discovered almost a century ago. They were synthesized from mustard gas and used during World War I (1924–1928). Although these compounds have been used in the chemotherapy of 1946,21,22 it was not known that they are alkylating the DNA. In fact, the DNA structure itself was deciphered in 1953.23 The most common alkylating agents used to combat various types of cancer are dacarbazine (DTIC), BCNU, CCNU, and temozolomide. These drugs or their active forms produce methyldiazonium ions, which are electrophilic methylated species. Inside the cell, the DNA acts as a nucleophile and result in the formation of multiple adducts. The distribution, half-life, nature and cellular characteristics of these adducts determine the degree of cytotoxicity and the likelihood of resistance.5 There are two classes of alkylating agents: monofunctional, which form an adduct with DNA or which cause biphasic cross-linking of DNA. Temozolomide is a monofunctional DNA alkylating agent that acts as a prodrug. It is inactive and stable at acidic pH and is completely absorbed when administered orally. Hydrolysis of temozolomide occurs at physiological pH, resulting in the formation of 5-(3-methyltriazol-1-yl)imidazole-4-carboxamide (MTIC). MTIC is further hydrolyzed to 5-amino-imidazole-4-carboxamide (AIC) and methyldiazolium, which react with DNA and releases its methyl group (Figure 1). This process resulted in the formation of the following DNA adducts (Figure 2).
Figure 1.

Mechanism of action of temozolomide. Temozolomide is stable at acidic pH. At physiological pH, it is chemically converted to 5-(3-methyltriazol-1-yl)imidazole-4-carboxamide (MTIC, active compound. MTIC is further hydrolyzed to 5-amino-imidazole-4-methyl Amide (AIC) and methyldiazolium. Methyldiazo ions react with DNA and release its methyl group.
Figure 2.
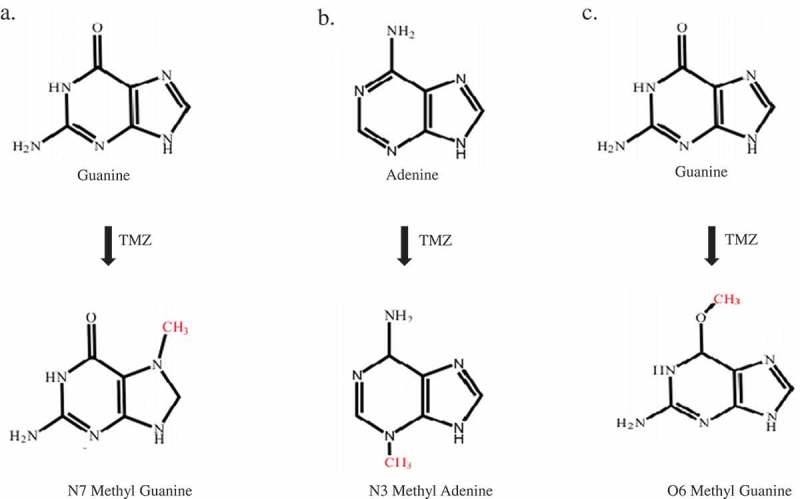
Adduct formation as a result of the reaction of DNA and temozolomide. A) N7 methylguanine (N7MG): The N7 position of guanine is methylated by temozolomide. The 70% adduct formed in the cells under temozolomide exposure was N7MG. B) N3 methyl adenine (N3MA): The N3 position of adenine is methylated by temozolomide. The 10% adduct formed in the cells under temozolomide exposure was N3MA. C) O6 methylguanine (O6MG): The O6 position of adenine is methylated by temozolomide. The 5% adduct formed in the cells upon exposure to temozolomide is O6MG.
N7 methyl guanine (N7MG)
The N7 position of guanine is the most nucleophilic site in DNA. This property, due to its nature and its accessibility in the main groove, makes it the most preferred methylation site for temozolomide. Therefore, N7MG accounts for about 70% of the total temozolomide adduct. When present in DNA, the adduct will neither interfere with the replication process nor cause a mismatch. Therefore, despite its high abundance, the biological significance of this adduct remains questionable. Thus, the N7MG adducts are inherently harmless and their spontaneous depurination or enzymatic removal can result in the generation of apurinic sites that are highly toxic to cells.24
N3 methyl adenine (N3MA)
The N3 position of adenine in DNA is also a strong nucleophilic site. Temozolomide produced a second frequency of N3MA lesions, accounting for 10% of the total adduct. N3MA also has no mismatch, but has been shown to block DNA polymerization in vitro. However, it has been reported that this activity is N3MA induced by other alkylating agents, not temozolomide. It has been shown that N3MA hydrolyzes DNA at neutral pH and physiological temperature, resulting in abasic sites, which may result in cytotoxicity.24
O6 methyl guanine (O6MG)
O6 methylguanine is a weak nucleophile with a frequency of at least 5%. However, this lesion is the main cause of the cytotoxic effect of temozolomide. O6MG mismatches with thymine, as a result, are recognized by the mismatch repair machinery (MMR). MMR corrects the O6MG-thymidine pair by removing thymine from the undamaged strand instead of removing O6MG, which causes additional thymidine incorporation during replication.
Other alkylating agents have also been reported for other methyl groups such as N1MA, N7MA, N2MG, and N3MG, but their relevance or correlation to temozolomide toxicity has not been determined.24
Resistance mechanisms
Chemical resistance to drugs may be the result of activation of anti-apoptotic factors, DNA repair pathways or drug efflux mechanisms. Depending on the strength of DNA damage by chemotherapy, the cells undergo DNA repair, cell cycle arrest, apoptosis or senescence. The development of chemoresistance to temozolomide is a major obstacle to GBM management. Resistance may be inherent or acquired during treatment.25,26 In the next section, the effects of various DNA repair pathways and other signaling pathways reported in the regulation of temozolomide resistance are discussed (Figure 3).
Figure 3.
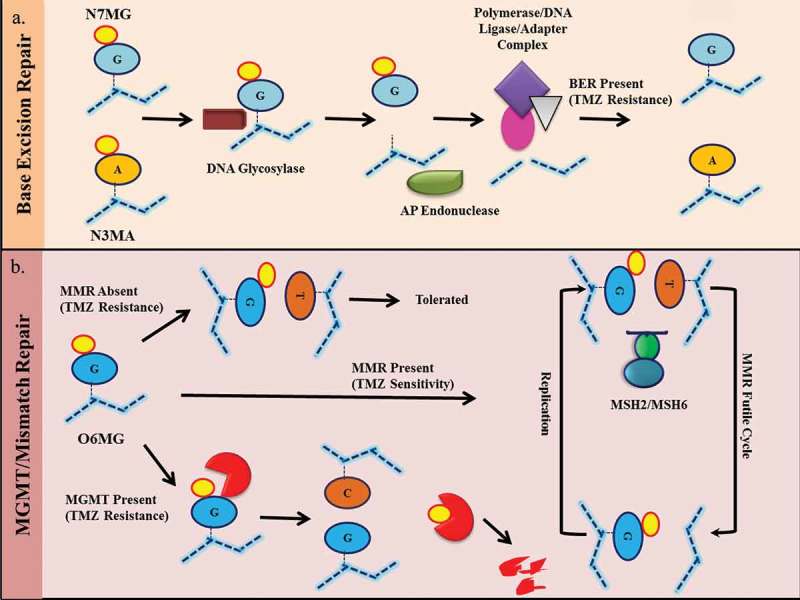
Resistance mechanism of temozolomide. A) N7MG and N3MA induced base excision repair (BER) pathways leading to recognition of modified bases by DNA glycosylation enzymes followed by base excision by AP endonuclease. This results in the recruitment of complexes containing DNA polymerase, DNA ligase, and adaptor molecules. This complex replaces the damaged site with the correct foundation. An effective BER results in resistance to temozolomide. B) O6MG on DNA results in an O6MG-thymidine mismatch. In the presence of active mismatch repair (MMR), the incorporated thymine was removed from the undamaged strand and incorporated again during the next replication cycle. This results in an ineffective loop of MMR which results in cytotoxicity. In the absence of an active MMR mechanism, the O6MG-thymidine mismatch is tolerated and cells survive. In the presence of MGMT, the O6MG is repaired, resulting in resistance.
DNA repair pathways
Depending on the nature of the DNA damage, different types of DNA repair pathways are activated by exposure to chemotherapy4. DNA repair pathways involve direct repair (DR), mismatch repair (MMR) and base excision repair (BER). Each of these has been reported to contribute to cellular responses to temozolomide exposure, although at different levels.
Direct repair by MGMT
O6-methylguanine-DNA methyltransferase (MGMT, also known as ATase, AGT, AGAT) reverses O6MG damage caused by alkylating agents in a one-step transalkylation reaction. It transfers an alkyl substituent (alkyl or chloroethyl) from the oxygen to the cysteine residue of its catalytic pocket (Cys 145). This results in inactivation of MGMT and its subsequent degradation by proteasome mechanisms. Thus, one MGMT molecule can remove only one adduct, such that repair is dependent on continuous MGMT expression. Due to the high lethality of O6MG, this is one of the main mechanisms of anti-temozolomide. MGMT is an evolutionarily conserved enzyme that is regulated by a variety of methods, such as promoter methylation, altered expression, histone modifications, post-transcriptional modifications, and miRNA regulation of transcription levels.27,28 It is a cytoplasmic protein that shuttles through the nucleus after DNA alkylation damage. MGMT promoter methylation has been found to be viable in GBM and is associated with a good clinical response to temozolomide.9 However, the association of MGMT protein levels with GBM survival remains unclear.29,30 Many reports have shown changes in MGMT protein in response to temozolomide treatment by activating various signaling pathways. For example, the Wnt pathway has been shown to modulate MGMT expression in response to temozolomide because its inhibition results in a decrease in MGMT expression and chemical sensitization.31 It has been reported that 0.4% of MGMT mutations in gliomas indicate that genetic inactivation is not the primary mechanism of resistance development. The use of drugs that inhibit MGMT, such as O6-benzylguanine (O6BG), is limited in clinical applications due to its hematological cytotoxicity.32 On the other hand, MGMT methylation is associated with a high mutation phenotype at relapse.33 Due to its predictive significance, MGMT promoter methylation is used clinically for GBM (Box 2).
Mismatch repair
Mismatch repair pathways are used to prevent mismatches caused by replication errors and incorrect insertions. In mammals, MSH2 and MSH6 heterodimer recognize base mismatches.25 Next, another heterodimeric complex consisting of MLH1 and PMS2 was recruited to the repair site to modulate the process. As previously described, unrepaired O6MG pairs with thymine instead of cytosine. This O6MG-thymidine pair is recognized by MMR, and as a result, thymine is removed from the newly synthesized strand, leaving O6MG intact. In the next replication cycle, a mismatch occurs again and the repair cycle is repeated. This ineffective loop of MMR causes the replication fork to stall because the cells attempt to divide and cause the production of double-strand breaks. These double-strand breaks are cytotoxic to cells, so O6MG-induced cytotoxicity requires active MMR.34 In the absence of MMR, O6MG lesions are tolerated, resulting in chemical resistance. Very few mutations are reported in the MMR component of GBM. However, in recurrent GBM, MSH6 mutations have been found with no mutations in matched primary tumors. This mutation in MSH6 resulted in a decrease in its expression, further confirming the resistance induced by temozolomide by inactivating MMR.
Base excision repair
BER repair single nucleotide modification.35 As described above, temozolomide produces various types of adducts. The BER that primarily recognizes N7MG and N3MA is activated to resolve these adducts. Since the major lesion caused by temozolomide is N7MG alkylation, active BER plays a key role in temozolomide resistance. The BER pathway is initiated when a modified base is recognized by a DNA glycosylase. A DNA glycosylase scans DNA helices to recognize and excise modified or damaged bases. In eukaryotes, different types of glycosylation enzymes are activated by different underlying lesions. In the case of the temozolomide adduct, the alkyl adenine DNA glycosylase (AAG) plays a major role. Once the damaged base is removed, an apurinic site is created. At this stage, the depurination/depyrimidine endonuclease (APE1) cleaves the damaged end, and DNA polymerase β (DNA POLB) is synthesized thereon and filled with a single nucleotide gap. Finally, the nick sealing step is carried out by DNA ligase I or a complex of XRCC1 and DNA ligase III. In the absence of an active BER mechanism, temozolomide-induced N7MG adducts are cytotoxic to cells.36 It has been shown that BER intermediates that retain unrepaired single nucleotide gaps or gaps are more toxic than the initial damage of DNA. Therefore, inhibition of this repair process is fatal to the cells in an intermediate step after base excision. Consistent with this, up-regulation of AAG and silencing of DNA POLB have been shown to result in hypersensitivity of GBM cells to temozolomide. Furthermore, silencing AAG alone can disrupt the overall initiation of the BER mechanism and has therefore been shown to increase the chemosensitivity of cells to temozolomide.37
Other mechanisms
In addition to MGMT as part of the DNA repair mechanism that confers resistance to temozolomide, many other alternative mechanisms have been proposed. The role of gap junction intracellular communication (GJIC) has been shown to be important for the development of temozolomide resistance. Increased RNA and protein levels of connexin 43 (a GJIC component) were observed upon exposure to temozolomide. This increase was found to be dependent on the EGFR/JNK/ERK/AP1 axis, as it was found that binding of AP1 to the 5‘ regulatory region of connexin 43 is important for its upregulation.38,39 The functional role of connexin 43 was established by dye transfer, and increased metastasis in resistant cells, suggesting that intercellular transfer may be an important factor in temozolomide resistance In another study, inhibition of junction 43 by peptidomimetic was shown to result in inhibition of mTOR and activation of AMPK, resulting in increased cell death following temozolomide exposure. Similarly, another protein, p-Gp (p-glycoprotein), which is an ABC transporter, has been shown to efflux from temozolomide to help resist.40 In addition, protein homeostasis in response to temozolomide damage have been shown to be important for cell survival. In addition to DNA damage responses, chemical resistance is also dependent on the endoplasmic reticulum stress response. The ER stress inducer, JLK 1486, has been shown to increase the efficacy of temozolomide, suggesting that prolonged ER stress can play a synergistic role in temozolomide-induced cell death.41 In addition to protein homeostasis, an imbalance in metabolic response can also increase the effect of temozolomide. In a similar series, it has been reported that AMPK is induced after temozolomide exposure and promotes apoptosis by inhibiting mTORC1.42 In another study, activation of β-catenin and AKT signaling has been shown to confer chemical resistance to the U87 glioma cell line. The combination of a GSK3β inhibitor cocktail with temozolomide has been shown to be beneficial in a mouse model.43
Clinical trials and combination therapy
A landmark study to establish current standards of care called the “Stupp regimen,” indicates that newly diagnosed gliomas are synchronized with radiation and adjuvant chemotherapy (75 mg/m2 body surface area daily for six weeks, followed by six cycles of temozolomide at 150–200 mg/m2 for 5 days during each 28 day cycle) is given compared to radiation alone. The median overall survival of the Stupp regimen branch was 14.6 months, compared to 12.1 for RT alone. At the same time, the importance of the methylation status of the MGMT promoter to the patient‘s chemotherapy response was established. In this study, it was found that MGMT promoter methylation increased the survival rate of GBM patients regardless of treatment, making it a prognostic marker. In addition, GBM patients with promoter methylation when receiving RT and temozolomide had improved median survival compared to RT alone. This underscores the role of MGMT in temozolomide resistance.9 Based on this, it has been suggested that increasing the dose of temozolomide should increase its cytotoxic effect as it will overcome the ability to repair. However, this trial failed to provide a survival difference between the two arms.44,45 Several driving gene changes have been described in GBM10. Based on this knowledge, the efficacy of many targeted therapies alone or in combination with temozolomide has been tested. Unfortunately, these efforts to date have not led to the emergence of any successful therapy that can benefit patients. The EGFR pathway has been reported to be altered in a variety of ways in most GBM, such as EGFR amplification or mutation. EGFR inhibitors (erlotinib and gefitinib) and monoclonal antibodies (cetuximab and nimotuzumab) have undergone multiple phases II and III clinical trials, but they survive or The benefits of the combined use of temozolomide could not be established.46 Similarly, inhibition of the PI3K/mTOR signaling pathway with temsirolimus (an mTOR inhibitor) failed to provide a survival benefit compared to temozolomide.47 In addition to active GBM signaling, tumor-gene angiogenesis is essential for disease progression. Bevacizumab is a humanized antibody against vascular endothelial growth factor A (VEGFA), combined with standard therapies for newly diagnosed patients in two independent clinical trials, both confirming the lack of benefit in the overall survival of GBM patients.17,47,49
Conclusion: can the red queen race be won?
Temozolomide is the drug of choice for GBM treatment in the clinic. Although it is beneficial to the survival of patients, the recurrence and prognosis of GBM remain frustrating. The temozolomide response lacks robust predictive markers. To date, combination therapies that have been tried have failed to provide additional benefits to patients. This highlights the gap between the positive results of the in vitro and in vivo models, but the human trials fail. Therefore, any improvement in this area will allow us to re-enter the same place we started, which is the overall survival of 15 months. The question is when will the Red Queen race be won? The need for hours is to understand drug resistance from a new perspective so that other tumor susceptibility can be utilized and targeted improvements in survival can be achieved. An understanding of the mechanisms by which tumor cells play an important role in combating temozolomide will enable the addition of other agents to the regimen, making it possible for a double sword to make tumor recurrence difficult.
Box 2.
Methods for MGMT measurement.
| The MGMT promoter methylation status is an important requirement test for gliomas for clinical decision making. The value of this test is that the state can be proven to be prognostic and predictive of the response to temozolomide. The MGMT gene consists of five exons and is located on chromosome 10q26. Most of the promoter and first exon consist of a stretch of CpG dinucleotide.50 The following tests were performed to measure the methylation status of the MGMT promoter: |
Methylation-specific PCR (MSP)
Methylation-specific PCR is most commonly used for MGMT promoter methylation diagnostic tests. This is a simple and cost-effective test that can be performed on a small number of stereotactic biopsy samples in the case of unresectable GBM and formalin-fixed, paraffin-embedded (FFPE) clinical samples. Extraction of genomic DNA from the sample and bisulfite treatment results in the conversion of the cytosine residue to uracil. The methylated cytosine residue in the form of 5-methylcytosine remains unchanged after this treatment. Although the methylation-specific primer pair contains a sequence complementary to the untransformed 5-methylcytosine, on the other hand, the non-methylation-specific primer pair contains a sequence complementary to the unmethylated cytosine that has been transformed. For thymine. After the completion of the PCR, gel electrophoresis was performed to visualize the bands. Therefore, one of the above two primer pairs obtains the amplification product as determined by the MGMT26.
Quantitative methylation-specific PCR
The method also utilizes bisulfite conversion of genomic DNA and amplification using methylation-specific primers such as an MSP. However, instead of semi-quantitative analysis based on gel electrophoresis, RT-qPCR values were used to report methylation status. The Ct value obtained for each sample was used to calculate the copy number based on a linear regression of the values plotted on the copy number equivalent standard curve. A standard curve was obtained using a plasmid containing the sequence of interest after bisulfite conversion. Data normalization in terms of sample processing, volume, DNA separation, etc. is obtained by the ratio of the MGMT copy value obtained from the standard curve to the internal control copy value (for example, ACTB). A cutoff value is applied to the ratio to define the methylation status.28
Pyrosequencing
Pyrosequencing is based on the principle of “synthesis sequencing”, which is used to determine the sequence of methylated CpG sites on the MGMT promoter. This technique relies on the measurement of light signals produced by bioluminescence, which can be detected when pyrophosphate is released during DNA synthesis. Previously, bisulfite conversion of the genomic DNA to be analyzed was performed, which resulted in the conversion of unmethylated CpG to TpG, keeping the methylated CpGs the same. The transformed genomic DNA was used as a sequencing template, primers, specific enzymes and enzyme substrates for synthesis. The enzymes used include the Klenow fragment of DNA polymerase I, ATP sulfurylase, luciferase, apyrase, and the enzyme substrates include adenosine phosphate sulfate and D-luciferin. Four nucleotides were added in a continuous cycle in the sequence, and the light-emitting recording recorded the incorporated nucleotides. Based on this data, the DNA template sequence and its methylation status in the initial sample can be derived. This method is highly analytical and is recommended for use in current clinical settings.50
Acknowledgments
KS acknowledges the research grant by the Indian government CSIR and DBT. The DST-FIST, Indo-French Centre for the Promotion of Advanced Research (Grant no: 5603-1), DBT grants and UGC (Advanced Research Center for Molecular Microbiology) have been recognized for their infrastructure support. AA acknowledges the Indian Institute of Science for the fellowship. KS is a JC Bose Fellow in the Department of Science and Technology.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Abbreviations
| GBM | glioblastoma |
|---|---|
| MGMT | O6-methylguanine-DNA methyltransferase |
| MMR | mismatch repair |
| BER | base excision repair |
| N7MG | N7 methylguanine |
| N3MG | N3 methylguanine |
| O6BG | O6-benzylguanine |
References
- 1.Thakkar J. P. 2014. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. 23:1985–1996. doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O‘Reilly SM, Newlands ES, Glaser MG, Brampton M, Rice-Edwards JM, Illingworth RD, Richards PG, Kennard C, Colquhoun IR, Lewis P.. Temozolomide: a new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumors. Eur J Cancer. 1993;29A:940–942. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, van Den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. 2009. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomized phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van Den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C. Temozolomide: a review of its discovery, chemical properties, pre-clinical development, and clinical trials. Cancer Treat Rev. 1997;23:35–61. [DOI] [PubMed] [Google Scholar]
- 6.Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, Baig G, Goddard C, Gibson NW, Slack JA. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987;47:5846–5852. [PubMed] [Google Scholar]
- 7.Brada M, Stenning S, Gabe R, Thompson LC, Levy D, Rampling R, Erridge S, Saran F, Gattamaneni R, Hopkins K, et al. 2010. Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J Clin Oncol. 28:4601–4608. doi: 10.1200/JCO.2009.27.1932. [DOI] [PubMed] [Google Scholar]
- 8.Yung WK, Albright RE, Olson J, Fredericks R, Fink K, Prados MD, Brada M, Spence A, Hohl RJ, Shapiro W, et al. 2000. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 83:588–593. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. 2005. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 10.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. 2013. The somatic genomic landscape of glioblastoma. Cell. 155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research, N Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al. 2017. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomized, double-blind, international phase 3 trial. Lancet Oncol. 18:1373–1385. doi: 10.1016/S1470-2045(17)30517-X. [DOI] [PubMed] [Google Scholar]
- 14.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, et al. 2014. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 343:72–76. doi: 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. 2016. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 16.Leu S, von Felten S, Frank S, Vassella E, Vajtai I, Taylor E, Schulz M, Hutter G, Hench J, Schucht P, et al. 2013. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro-oncol. 15:469–479. doi: 10.1093/neuonc/nos317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. 2014. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 18.Lipinski C, Hopkins A. 2004. Navigating chemical space for biology and medicine. Nature. 432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 19.Collins I, Workman P. New approaches to molecular cancer therapeutics. Nat Chem Biol. 2006. 689–700. doi: 10.1038/nchembio840. [DOI] [PubMed] [Google Scholar]
- 20.Stevens MF, Newlands ES. From triazines and triazenes to temozolomide. Eur J Cancer. 1993;29A:1045–1047. [DOI] [PubMed] [Google Scholar]
- 21.Goodman LS, Wintrobe MM. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin‘s disease, lymphosarcoma, leukemia, and certain allied and miscellaneous disorders. J Am Med Assoc. 1946;132:126–132. [DOI] [PubMed] [Google Scholar]
- 22.Wintrobe MM. Nitrogen mustard therapy. Am J Med. 1948;4:313. [DOI] [PubMed] [Google Scholar]
- 23.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair, and resistance. Curr Mol Pharmacol. 2012;5:102–114. [DOI] [PubMed] [Google Scholar]
- 25.Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, Wick W. 2008. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin Cancer Res. 14:2900–2908. doi: 10.1158/1078-0432.CCR-07-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wick W, Platten M. 2014. Understanding and targeting alkylator resistance in glioblastoma. Cancer Discov. 4:1120–1122. doi: 10.1158/2159-8290.CD-14-0918. [DOI] [PubMed] [Google Scholar]
- 27.Kaina B, Christmann M, Naumann S, Roos WP. 2007. MGMT: a key node in the battle against genotoxicity, carcinogenicity, and apoptosis induced by alkylating agents. DNA Repair (Amst). 6:1079–1099. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Wick W, Weller M, van Den Bent M, Sanson M, Weiler M, von Deimling A, Plass C, Hegi M, Platten M, Reifenberger G. 2014. MGMT testing–the challenges for biomarker-based glioma treatment. Nat Rev Neurol. 10:372–385. doi: 10.1038/nrneurol.2014.100. [DOI] [PubMed] [Google Scholar]
- 29.van Nifterik KA, van Den Berg J, van der Meide WF, Ameziane N, Wedekind LE, Steenbergen RDM, Leenstra S, Lafleur MVM, Slotman BJ, Stalpers LJA, et al. 2010. Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide. Br J Cancer. 103:29–35. doi: 10.1038/sj.bjc.6605712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang K, Jin Q, Yan W, Zhang W, You G, Liu Y, Jiang T. 2012. Clinical correlation of MGMT protein expression and promoter methylation in Chinese glioblastoma patients. Med oncol. 29:1292–1296. doi: 10.1007/s12032-011-9901-4. [DOI] [PubMed] [Google Scholar]
- 31.Wickstrom M. 2015. Wnt/beta-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signaling prevents chemoresistance. Nat Commun. 6:8904. doi: 10.1038/ncomms9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Gerson SL. 2006. Targeted modulation of MGMT: clinical implications. Clin Cancer Res. 12:328–331. doi: 10.1158/1078-0432.CCR-05-2543. [DOI] [PubMed] [Google Scholar]
- 33.Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ, Grimsby J, Sougnez C, Muller F, Hu J, et al. 2015. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveal patterns of tumor evolution. Genome Res. 25:316–327. doi: 10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McFaline-Figueroa JL, Braun CJ, Stanciu M, Nagel ZD, Mazzucato P, Sangaraju D, Cerniauskas E, Barford K, Vargas A, Chen Y, et al. 2015. Minor changes in expression of the mismatch repair protein MSH2 exert a major impact on glioblastoma response to temozolomide. Cancer Res. 75:3127–3138. doi: 10.1158/0008-5472.CAN-14-3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krokan HE, Bjoras M. 2013. Base excision repair. Cold Spring Harb Perspect Biol. 5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. 2005. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 65:6394–6400. doi: 10.1158/0008-5472.CAN-05-0715. [DOI] [PubMed] [Google Scholar]
- 37.Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS, Kelly J, McGown G, Thorncroft M, Carlson BL, et al. 2012. Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J Clin Invest. 122:253–266. doi: 10.1172/JCI59334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy SF, Varghese RT, Lamouille S, Guo S, Pridham KJ, Kanabur P, Osimani AM, Sharma S, Jourdan J, Rodgers CM, et al. 2016. Connexin 43 inhibition sensitizes chemoresistant glioblastoma cells to temozolomide. Cancer Res. 76:139–149. doi: 10.1158/0008-5472.CAN-15-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munoz J. L. 2014. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 5:e1145. doi: 10.1038/cddis.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munoz JL, Walker ND, Scotto KW, Rameshwar P. 2015. Temozolomide competes for P-glycoprotein and contributes to chemoresistance in glioblastoma cells. Cancer Lett. 367:69–75. doi: 10.1016/j.canlet.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 41.Weatherbee JL, Kraus JL, Ross AH. 2016. ER stress in temozolomide-treated glioblastomas interferes with DNA repair and induces apoptosis. Oncotarget. 7:43820–43834. doi: 10.18632/oncotarget.9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang WB, Wang Z, Shu F, Jin Y-H, Liu H-Y, Wang Q-J, Yang Y. 2010. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem. 285:40461–40471. doi: 10.1074/jbc.M110.164046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi GZ, Liu Y-W, Xiang W, Wang H, Chen Z-Y, Xie S-D, Qi S-T. 2016. Akt and beta-catenin contribute to TMZ resistance and EMT of MGMT negative malignant glioma cell line. J Neurol Sci. 367:101–106. doi: 10.1016/j.jns.2016.05.054. [DOI] [PubMed] [Google Scholar]
- 44.Wick W, Platten M, New WM. 2009. (Alternative) temozolomide regimens for the treatment of glioma. Neuro-oncol. 11:69–79. doi: 10.1215/15228517-2008-078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT, et al. 2013. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 31:4085–4091. doi: 10.1200/JCO.2013.49.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown PD, Krishnan S, Sarkaria JN, Wu W, Jaeckle KA, Uhm JH, Geoffroy FJ, Arusell R, Kitange G, Jenkins RB, et al. 2008. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol. 26:5603–5609. doi: 10.1200/JCO.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wick W, Gorlia T, Bady P, Platten M, van Den Bent MJ, Taphoorn MJB, Steuve J, Brandes AA, Hamou M-F, Wick A, et al. 2016. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation (EORTC 26082). Clin Cancer Res. 22:4797–4806. doi: 10.1158/1078-0432.CCR-15-3153. [DOI] [PubMed] [Google Scholar]
- 48.Mason WP, Cairncross JG. 2005. Drug insight: temozolomide as a treatment for malignant glioma–impact of a recent trial. Nat Clin Pract Neurol. 1:88–95. doi: 10.1038/ncpneuro0045. [DOI] [PubMed] [Google Scholar]
- 49.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. 2014. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bienkowski M, Berghoff AS, Marosi C, Wöhrer A, Heinzl H, Hainfellner JA, Preusser M. Clinical Neuropathology practice guide 5-2015: MGMT methylation pyrosequencing in glioblastoma: unresolved issues and open questions. Clin Neuropathol. 2015;34:250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]