

Abstract
A new area of visible light-induced Pd-catalysis has been emerging into the field. In contrast to the classical ground-state Pd-catalyzed reactions mostly proceeding via two-electron redox manifold, the mechanisms for these new Pd-photoexcited methods usually operate via a single electron transfer process, thus leading to putative Pd-radical hybrid species, which exhibit both radical and classical Pd-type reactivity. This minireview highlights the recent progress in this rapidly growing area.
Keywords: Palladium, Visible light, Pd-radical hybrid species, Excited-state Pd-complex
Graphical Abstract
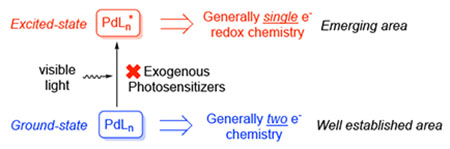
Photoexcited Pd-catalysis: This minireview highlights the recent emerging area of visible light-photoexcited Pd-catalysis. Its novel reactivities, scope and mechanistic aspects are discussed.
1. Introduction
Ground-state Pd-catalysis, one of the most efficient and versatile tools in modern organic synthesis,[1] generally relies on two-electron redox chemistry of Pd-complexes.[2] Recently, it was reported that upon excitation by visible light in the absence of exogenous photosensitizers,[3,4,5] the forming photoexcited Pd-complexes can undergo single electron transfer (SET) process, generating Pd-radical hybrid species[6] via a Pd0/PdI pathway (Scheme 1, Excited-state). This unique reactivity has led to the development of novel transformations that are difficult to achieve through traditional ground-state reactivity. Additionally, the photoexcited Pd-complexes are also capable of dramatic acceleration of known Pd-catalyzed transformations such as Heck and Negishi cross-coupling reactions of aryl halides. This minireview summarizes the recent progress in the emerging area of photoexcited Pd-catalysis without employment of exogenous photosensitizers.[7] The scope and mechanistic aspects of this new reactivity is discussed. It is systematized by the type of the Pd-radical hybrid intermediates produced. Since this new area is in its infancy, the detail mechanisms for these novel methods are still underdeveloped. Hence, the mechanisms discussed throughout the review are based on the best hypotheses by the authors. Related palladium-catalyzed reactions under UV-irradiation are not discussed herein.[8]
Scheme 1.
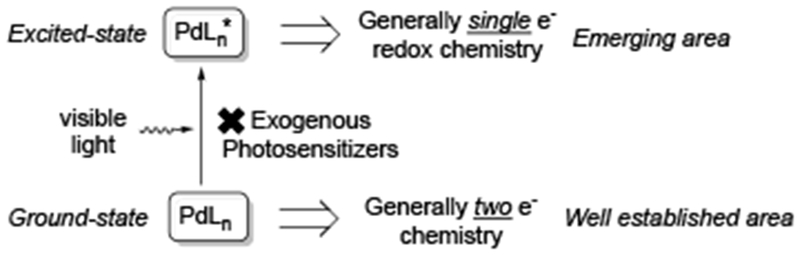
Ground-state- vs photoexcited Pd-catalysis.
2. Reactions Involving Aryl Electrophiles
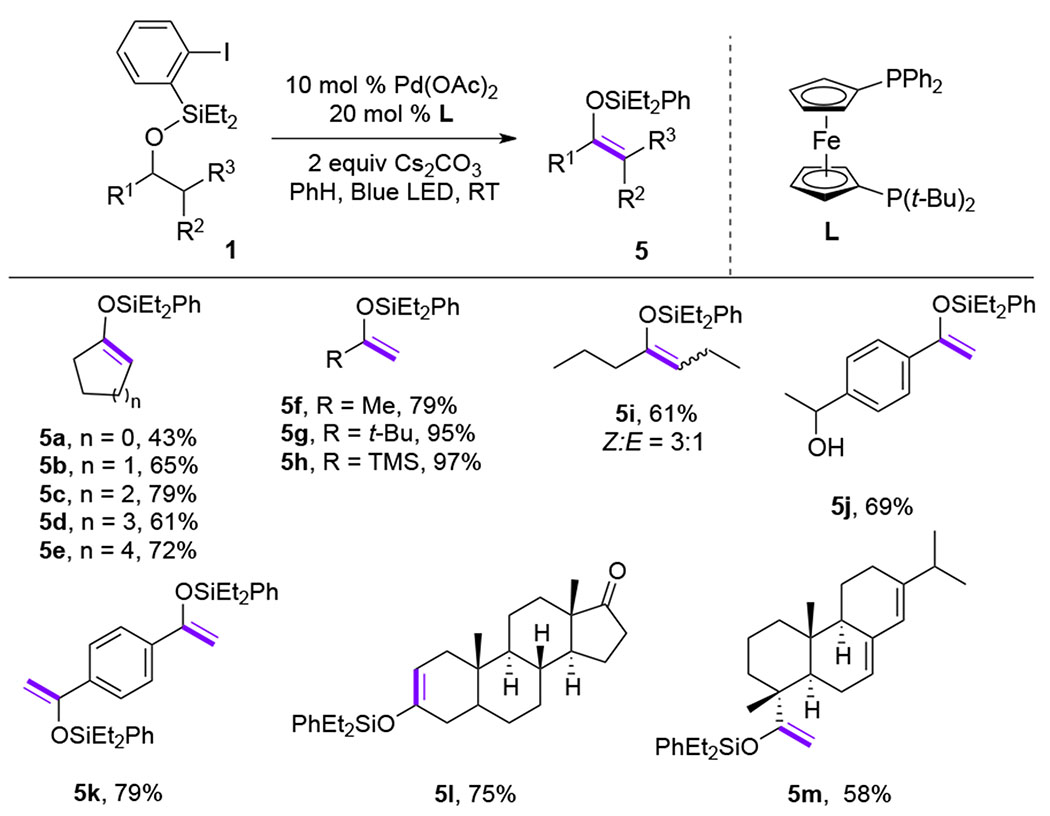
The first visible light-induced generation of Pd-radical hybrid species was reported by Gevorgyan group in 2016,[9] where the aryl hybrid Pd-radical species 2 was generated under mild photoinduced conditions from aryl iodide and the Pd0 complex. This key transfromation enabled room temperature exogeneous photosensitizer- and external oxidant-free dehydrogenation of silyl ethers 1 into silyl enol ethers 2. Both Pd-catalyst and visible light were found to be crucial components for this reaction. This method uncovers new reactivity of aryl iodides in the presence of Pd0 catalyst featuring unusual one-electron chemistry, distinct to the common two-electron oxidative addition of aryl halides. After generation of the aryl Pd-radical hybrid species, it undergoes unprecedented for Pd-catalyzed reactions 1,5-hydrogen atom transfer (HAT) process (2→3), to produce silyl enol ether 5 upon a subsequent β-H-elimination (Scheme 2). This novel method allows for a direct oxidation of silyl ethers into silyl enol ethers under mild conditions, with an improved selectivity and efficiency compared to the prior art using Rh- or Ir-catalytic systems.[10] The scope of this reaction was found to be relatively broad as cyclic-(5a-5e), acyclic- (5f-5k) and complex (5l, 5m) silyl ethers underwent efficient desaturation generating silyl enol ether products in high yields (Table 1). The mechanistic studies, including radical scavengers and radical clock experiments supported the radical nature of this transformation. Besides, isotope-labeling studies ruled out an alternative concerted metalation deprotonation (CMD) type mechanism. In summary, this work represents the synergistic combination of two unique reactivities, the radical HAT process, and the Pd-involved β-H-elimination event, which provided a new platform for development of novel remote C(sp3)-H functionalization methods.
Scheme 2.
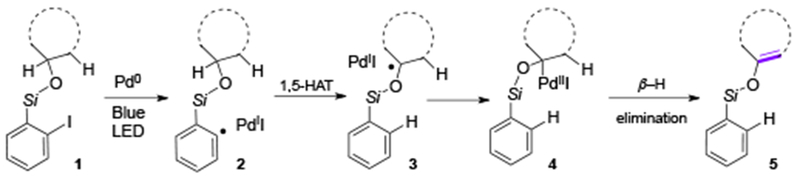
Reaction design for direct oxidation of silyl ethers into silyl enols.
Table 1.
Scope of Gevorgyan’s direct oxidation of silyl ethers into silyl enol ethers.
 |
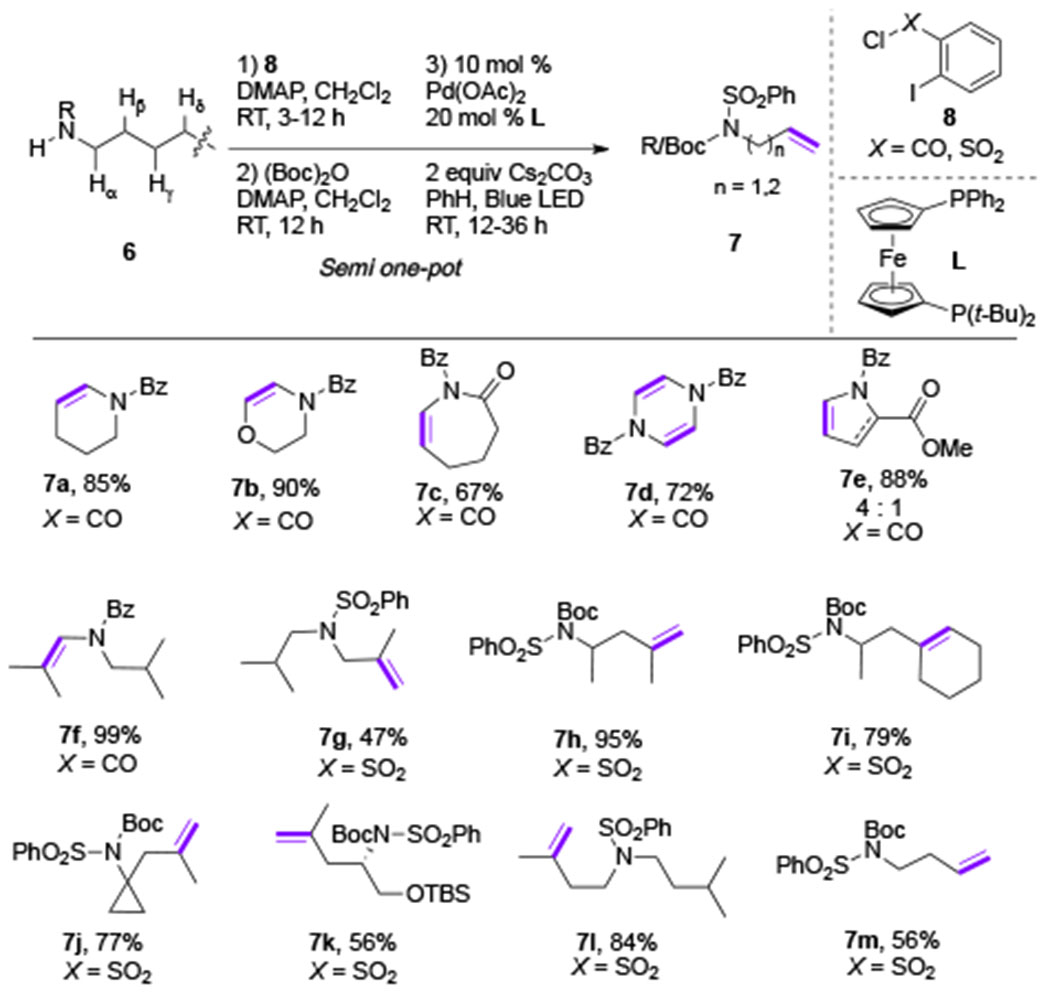
Thereafter, the same group expanded this strategy towards selective desaturation of aliphatic amines.[11] The authors showed that iodobenzamide (X = CO) and iodosulfonamide (X = SO2) groups were also capable donors towards aryl Pd-radical hybrid species under visible light-induced conditions. Interestingly, these two auxiliaries showed distinct selectivity for an HAT process: 1,5-HAT for iodobenzamides, and 1,7-HAT (also 1,6-HaT) for iodosulfonamides. Syntheses of enamines were accomplished in excellent yields (7a-7e) using the iodobenzamide tether (Table 2). Likewise, iodosulfonamide-tethered amines yielded allylic (7f) and homoallylic amines (7g-7m) in good yields (Table 2). Notably, all these unsaturated amines were synthesized directly from aliphatic amines via a semi one-pot procedure. Mechanistic studies of this work also supported the radical nature of this transformation. Deuterium-labeling study provided evidence for an HAT process. Moreover, photophysical studies revealed that the Pd-complex is a sole light-absorbing species among reactants, whose excited state is quenched by an aryl iodide.
Table 2.
Scope of Gevorgyan’s desaturation of aliphatic amines (DMAP = 4-dimethylaminopyridine; Bz = benzoyl; Boc = tert-butyloxycarbonyl).
 |
Based on these studies, the authors proposed similar mechanism to that for the desaturation reactions of silyl ethers, which involves aryl Pd-radical hybrid species (Scheme 3). First, the excitation of the Pd-catalyst by visible light generates an excited Pd-complex, which undergoes SET with an aryl iodide. The formed radical anion then fragments into an aryl Pd-radical hybrid species 9 followed by a 1,n-HAT process to produce a transposed alkyl Pd-radical hybrid species 10. At this point, the end-game scenario (10→5 or 7), was unclear, thus, four β-H-elimination pathways were proposed: a radical recombination with PdIX species (11) followed by the β-H-elimination (A1), a direct radical abstraction (A2), an oxidation to carbocation 12, followed by a proton loss (A3); and a route involving an I-atom transfer intermediate 13 with a subsequent dehydroiodination (A4).
Scheme 3.
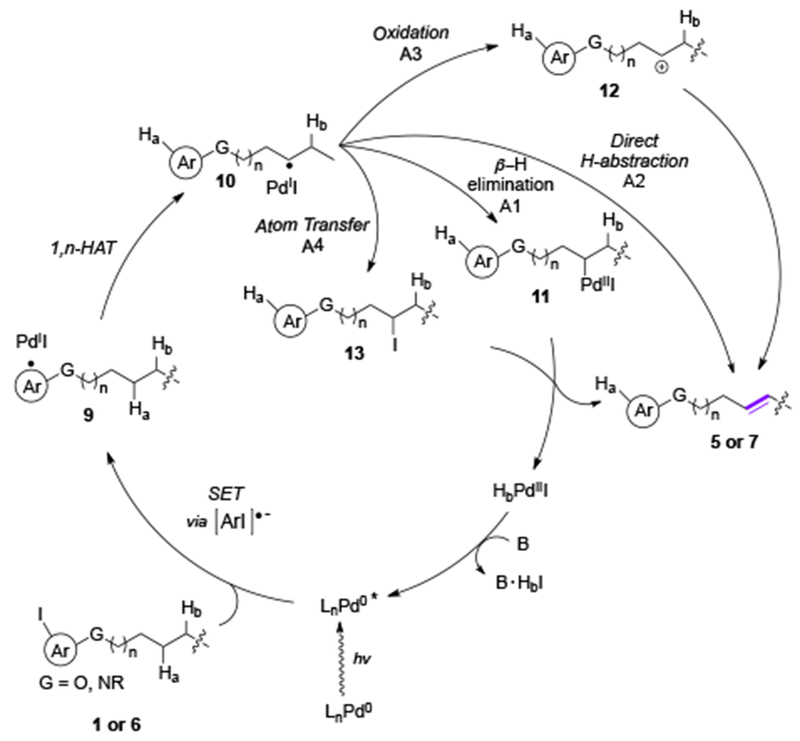
Mechanism for desaturation reactions involving aryl hybrid Pd-radial species.
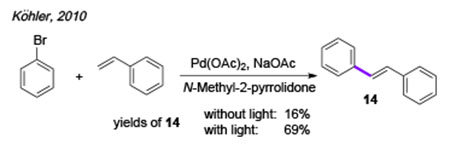
Visible light is also capable of dramatic acceleration of the classical Pd-catalyzed reactions. In 2010, Köhler group reported visible light-accelerated Heck reaction of bromobenzene with styrene (eq 1),[12] where the reaction under visible light irradiation proceeded much faster and resulted in higher yields of the stilbene product 14 compared to that under thermal conditions. However, based on control experiments and mechanistic studies the authors stated that the sole role of visible light in this transformation is to accelerate the off-cycle reduction of the PdII-precatalyst into the active Pd0-catalyst. No hybrid Pd-radical intermediates were proposed for this transformation (Scheme 4A).
 |
(eq 1) |
Scheme 4.
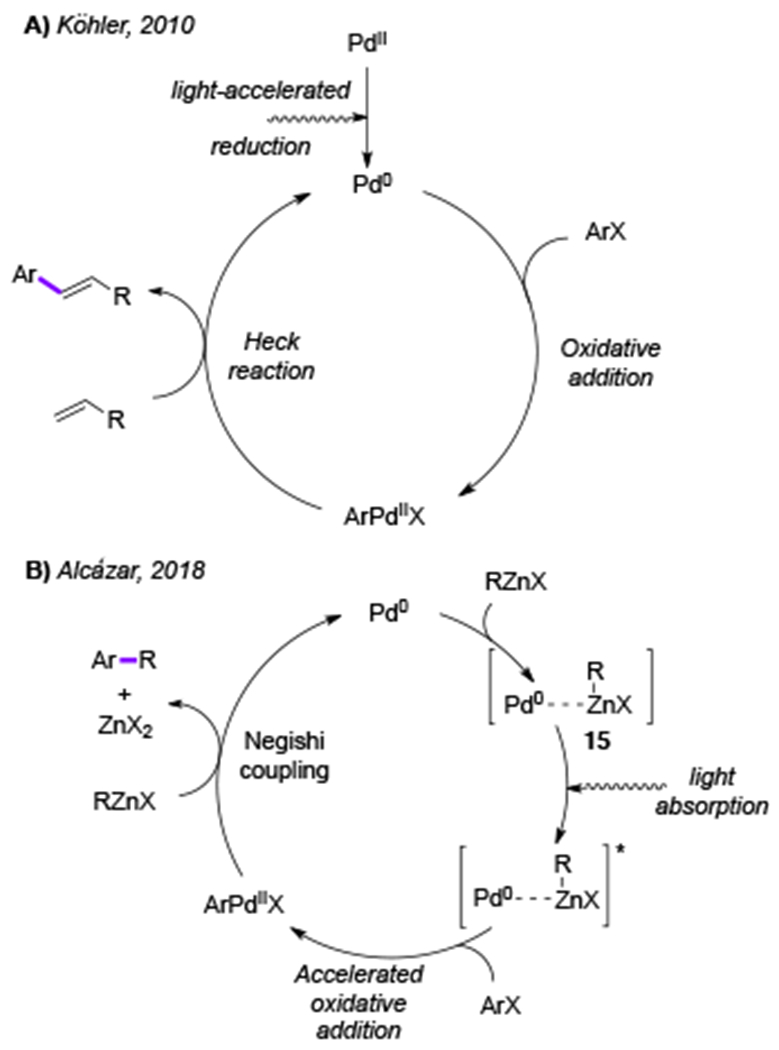
Mechanism for light-accelerated Pd-catalyzed reactions. A) Heck reaction. B) Negishi-coupling.
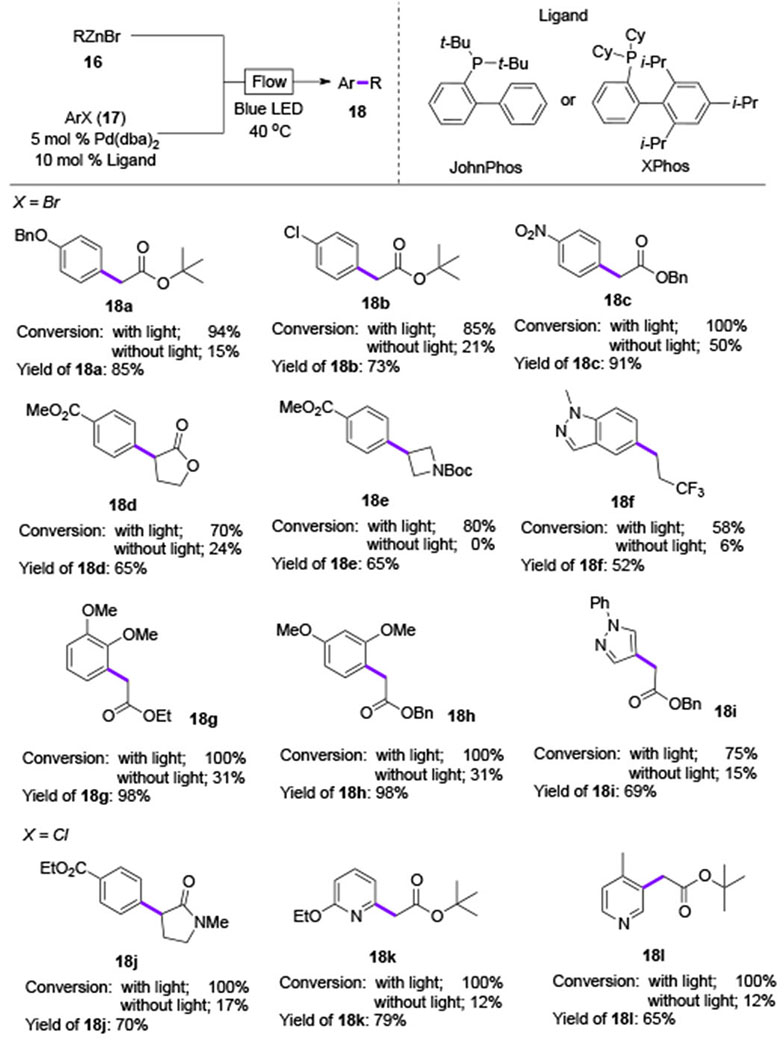
Recently, light-accelerated Pd-catalyzed Negishi cross-coupling reaction under flow conditions was reported by Alcazar and co-workers.[13] Absorption spectra revealed that the formed Pd0-Zn complex 15 absorbs light at 430 nm. Then, upon visible light irradiation, this complex was consumed rapidly in the presence of aryl bromide. Accordingly, the authors proposed that the photoexcited Pd0-Zn complex accelerates the oxidative addition of aryl halides, followed by the classical scenario (transmetallation–reductive elimination) of Negishi cross-coupling reaction (Scheme 4B). In this study on the light-accelerated Pd-catalyzed reaction, no formation of radical intermediates was observed.
Importantly, the scope of this light-accelerated Negishi cross-coupling reaction was broader compared to that employing standard thermal conditions (Table 3). In all cases, coupling reactions of aryl bromides with different alkyl zinc reagents were greatly accelerated by visible light irradiation (18a-18i). Notably, the typically sluggish coupling partners, such as electron-rich aryl bromides, or aryl chlorides were also capable substrates in this method, producing the coupling products 18g-18l in good yields.
Table 3.
Scope of Alcazar’s visible light-promoted Negishi coupling.
 |
In 2018, the groups of Wang, Jurca, and Ong applied palladium carbodicarbene complex 21 for mediating a visible light-induced directed C–H arylation and a coupling of aldehydes with amines (Scheme 5).[14] This Pd-complex showing a strong absoprion peak at 499 nm acted as both photoredox catalyst and cross-coupling catalyst. With this new capability, the directed C–H arylation with aryl diazonium salts previously described by Sanford,[7a] can now be accomplished with a single Pd-catalyst 21, that plays a double duty in this transformation (Scheme 5). The key step of the mechanism is the reduction of aryl diazonium species by the photoexcited PdII complex towards aryl radical species that couples with PdII-Ar1-DG 23 to eventually generate the 22a-22e biaryl products through a convential cross-coupling cycle. The estimated reduction potential (E) of the excited state (PdIII/PdII*) was found to be around −1.31 V, which indicates its greater reductive ability compared to the RuII(bpy)32+-catalyst (E for RuIII(bpy)33+/RuII(bpy)32+ = −0.81 V). Due to its strong reductive properties, the excited Pd-complex 21 was also able to catalyze oxidation of amine (26) to imine (27) under air generating hydrogen peroxide, that is known to promote amide synthesis from benzaldehydes 28 and secondary amines 26 (Scheme 6).
Scheme 5.
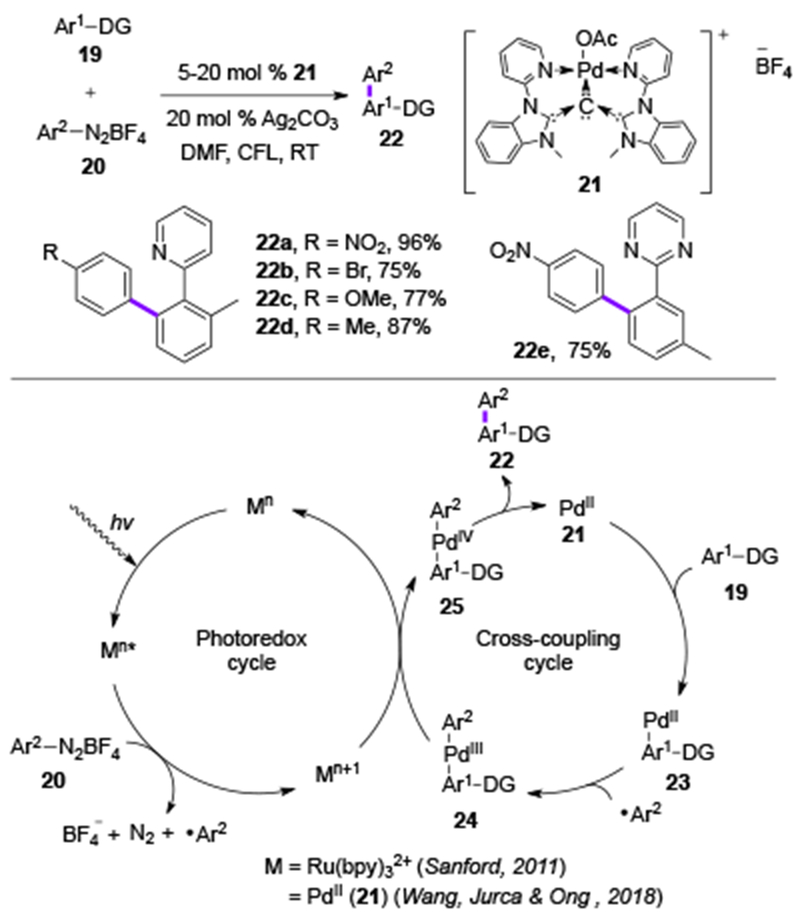
Scope and mechanism of Wang, Jurca & Ong’s Pd-catalyzed directed C-H arylation (DMF = N, N-dimethylformamide, bpy = 2,2’-bipyridine, DG = directing group).
Scheme 6.
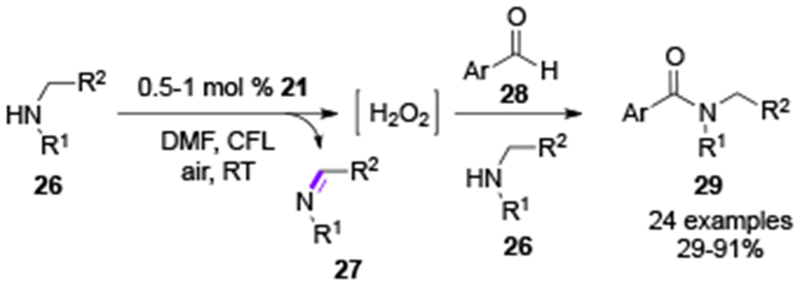
Wang, Jurca & Ong’s amide synthesis via photoinduced generation of hydrogen peroxide.
3. Reaction Involving Vinyl Halides
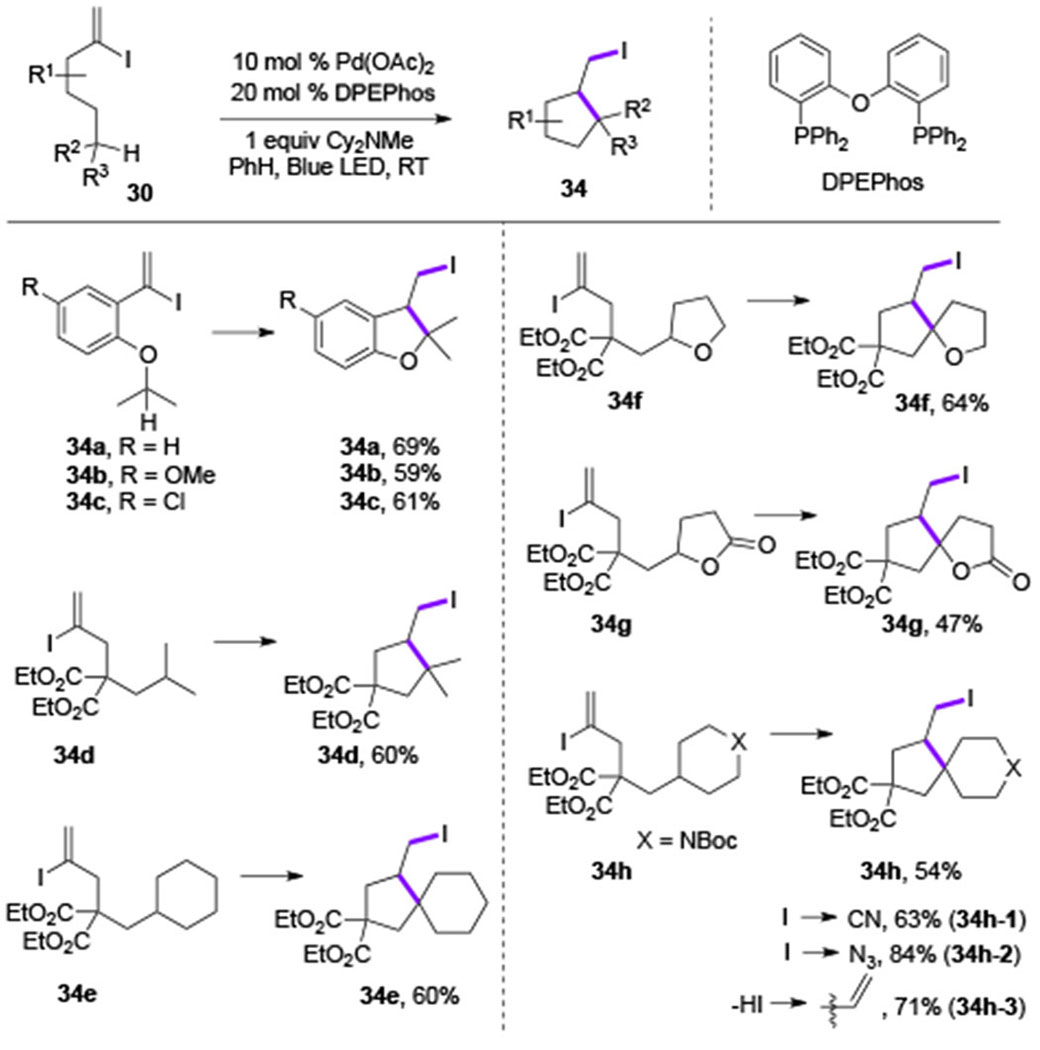
In 2018, Gevorgyan group reported generation of vinyl Pd-radical hybrid species from vinyl iodide under visible light-induced Pd-catalyzed conditions.[15] This oxidative radical cyclization of vinyl iodide 30 via the 1,5-HAT/oxidative Heck-type coupling was designed towards methylenecyclopentane 35 (Scheme 7). That process would represent an oxidative version of the classical Curran’s reductive cascade cyclization into methylcyclopentane 36, proceeding under standard radical conditions (AIBN/Bu3SnH).[16] Interestingly, the targeted 35 was not observed in the reaction, instead, an atom transfer radical cyclization product 34 was obtained selectively. This protocol was found to be effective for synthesis of iodomethyl benzofurans (34a-34c) and iodomethyl cyclopentane derivatives (34d-34h) (Table 4). Naturally, the obtained iodomethyl products could be easily converted into other spirocyclic derivatives via nucleophilic substitution (34h-1, 34h-2) or elimination (34h-3) reactions. Based on the radical trapping experiments, deuterium labeling-, and photophysical studies, a Pd-radical hybrid mechanism was proposed for this transformation (Scheme 8).
Scheme 7.
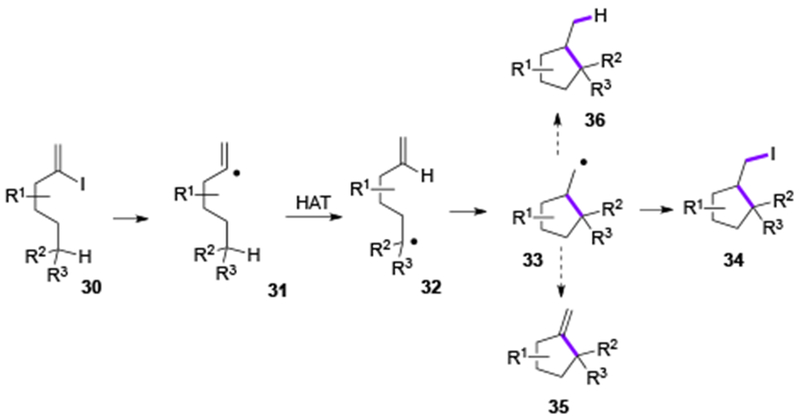
Gevorgyan’s HAT/atom transfer radical cyclization reaction.
Table 4.
Scope of Gevorgyan’s HAT/atom transfer radical cyclization reaction.
 |
Scheme 8.
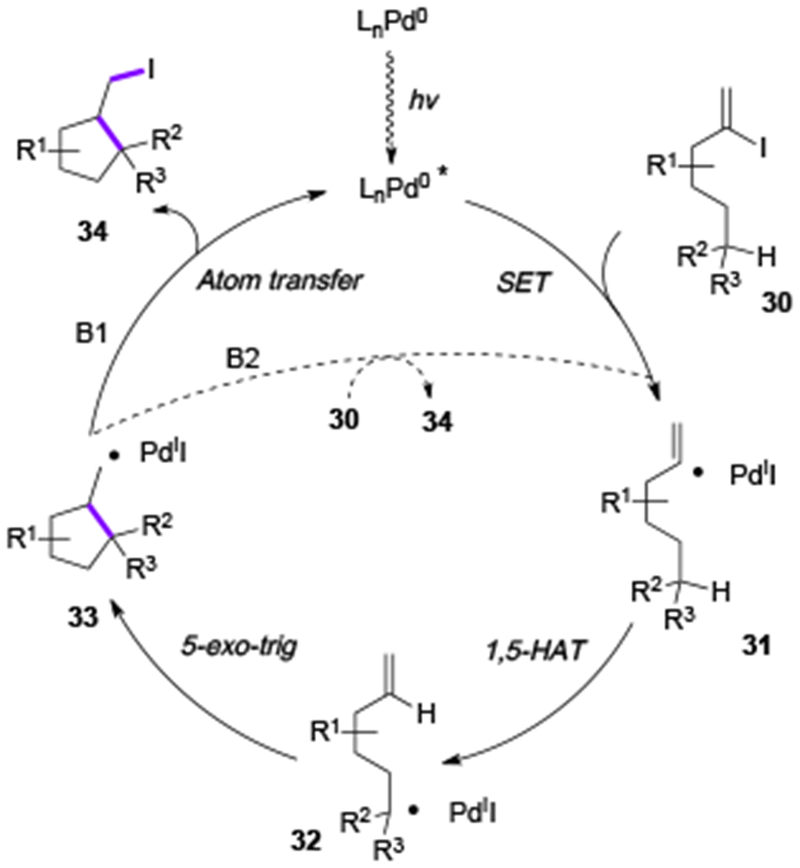
Mechanism of HAT/atom transfer radical cyclization reaction.
First, upon visible light irradiation, the excited Pd-complex species is produced, which is engaged in an SET event with the vinyl iodide 30. The formed vinyl Pd-radical hybrid species 31 undergoes a 1,5-HAT process producing an alkyl hybrid Pd-radical species 32. A subsequent 5-exo-trig radical cyclization followed by an iodine atom transfer from the PdII species by the formed primary radical 33 furnishes the product 34 and regenerates the Pd-catalyst (B1). Apparently, an alternative radical chain mechanism[17] (B2) is unlikely due to the unfavorable for this process C(sp3)–I vs C(sp2)–I bond energies.
4. Reactions Involving Alkyl Electrophiles
Until groundbreaking works by Fu,[18] Alexanian,[19] and Zhou,[20] the Pd-catalyzed reactions of alkyl electrophiles have been problematic due to the slower rates of oxidative addition and undesired premature β-H-elimination process.[21] Recently, an employment of photoexcited palladium catalysis has provided a remarkable further progress in this area. In 2017, Gevorgyan group reported the first visible light-induced Heck reaction of alkyl halides at room temperature (Table 5).[22] The reaction of α-heteroatom-substituted methyl iodides and -bromides with vinyl arenes proceeded smoothly, generating a diverse functionalized allylic systems in good yields (39a-39m, 39q). Notably, unactivated primary (37n, 37o) and secondary (37p) alkyl iodides were found to be competent substrates for this reaction as well. Later, the same group applied these developed photoinduced conditions towards efficient Heck reaction of activated and unactivated tertiary alkyl iodides with styrene derivatives (39r-39z, 39ab, 39ac) and acrylonitrile (39aa).[23] Interestingly, light was not necessary for the reaction of activated tertiary alkyl halides (40→41) (eq 2).
Table 5.
Scope of Gevorgyan’s Heck reaction of alkyl halides (TMS = trimethylsilyl, Bpin = bis(pinacolato)diboronyl, Piv = pivaloyl, Ts = tosyl, NPhth = phthalimidyl).
 |
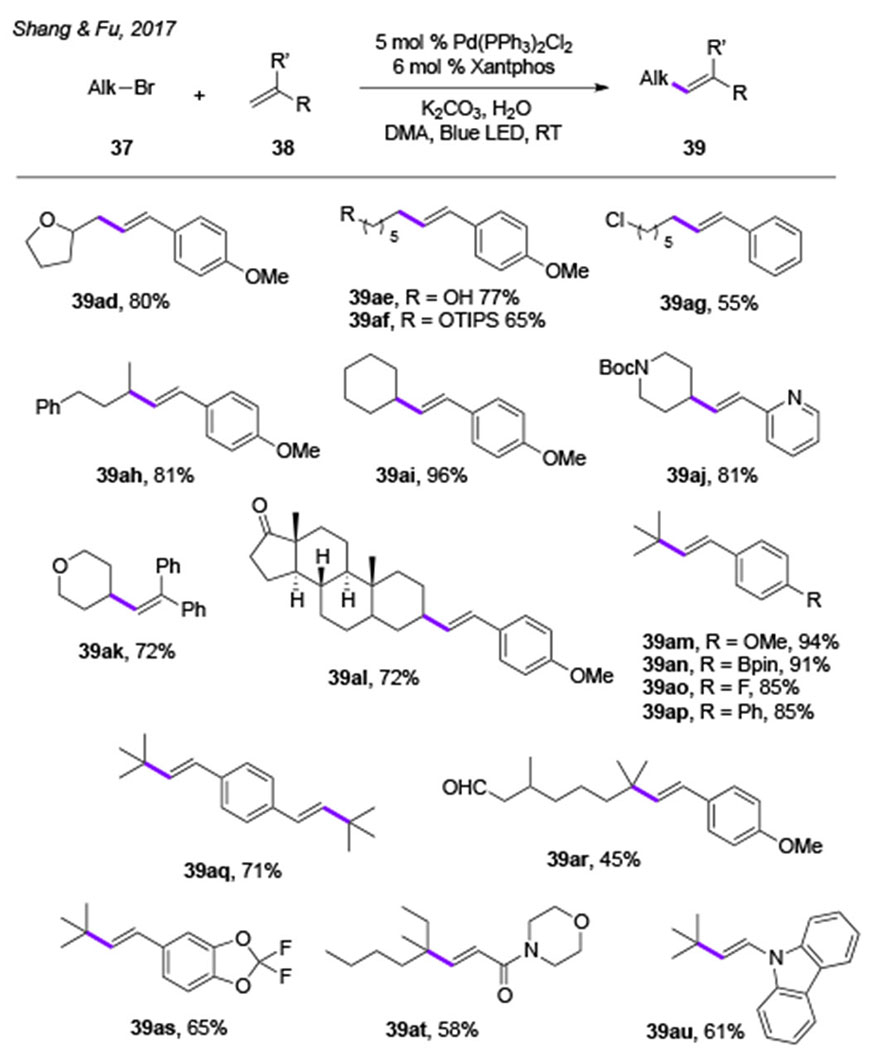
Nearly at the same time, independently, Shang and Fu groups reported the Pd-catalyzed Heck reaction of alkyl bromides under visible light irradiation.[24] The reaction features a fairly general scope (Table 6), so that primary, secondary, as well as unactivated tertiary alkyl halides, which were previously considered as incapable substrates for the Heck reaction, all reacted well producing Heck products (39ad-39au) in good to excellent yields. Moreover, the extensive mechanistic studies of this work involving X-ray photoelectron spectroscopy (XPS) measurements revealed the presence of three oxidation states of palladium in the reaction mixture, Pd0, PdI, and PdII, thus supporting the Pd0-PdI-PdII mechanistic scenario. Radical clock- and radical trapping experiments, described in all these reports on alkyl Heck reactions,[22–24] support radical-type mechanism. Photophysical studies confirmed that Pd0Ln complexes absorb light in the visible region, and that the formed photoexcited Pd0Ln* species is quenched by an alkyl halide presumably via an SET event. Accordingly, a Pd-radical hybrid mechanism was proposed for these Heck reactions of alkyl halides (Scheme 9).[19,20] The alkyl Pd-radical hybrid species 42, generated from photoexcited Pd0-Ln complex and alkyl halide, add to an alkene moeity producing a new alkyl Pd-radical hybrid species 43. β-H-Elimination from the latter delivers the product 39 and regenerates the Pd0 catalyst.
Table 6.
Scope of Shang’s and Fu’s Heck reaction of alkyl halides (DMA = N, N-dimethylacetamide).
 |
Scheme 9.
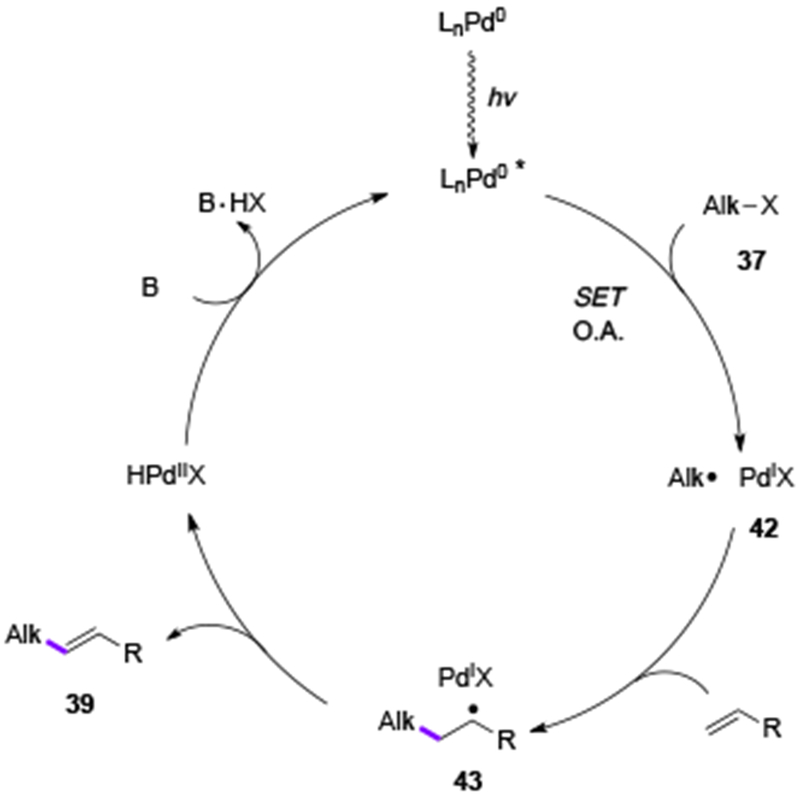
Mechanism for Heck reaction of alkyl halides.
Recently, Rueping group reported the decarboxylative cross-coupling reaction of alkyl halides with vinyl carboxylic acids 44 (Scheme 10)[25] to provide the Heck products (45a-d) in good yields. Mechanistic studies and DFT calculations supported the Pd-radical hybrid mechanism, where a single electron oxidative addition of alkyl bromides 37 with the photoexcited PdL3 to form alkyl radical 42 was proved to be “barrierless”. The subsequent radical addition at alkene followed by the decarboxylative desaturation create the reaction product 45 and regenerates the Pd-catalyst.
Scheme 10.
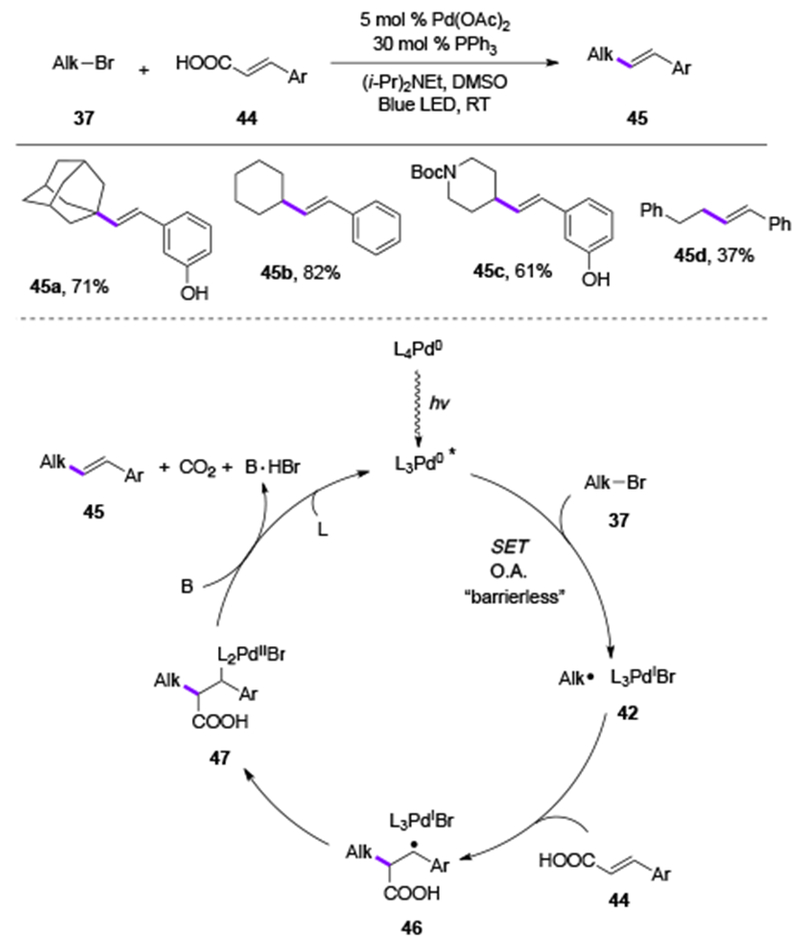
Scope and mechanism of Rueping’s decarboxylative coupling (DMSO = dimethyl sulfoxide).
In 2017, Gevorgyan and co-workers disclosed the ability of an alkyl Pd-radical hybrid species to undergo an HAT process at unactivated C(sp3)–H sites (Scheme 11).[26] The reaction proceeds under the similar sequence of steps as that in the desaturation protocols using aryl Pd-radical hybrid species (Scheme 3). An alkyl Pd-radical hybrid species 49, generated under visible light irradiation in the presence of Pd-catalyst, undergoes a selective 1,n-HAT process to produce new alkyl Pd-radical hybrid species 50 (n = 5-7) (Scheme 11). The subsequent β-H-elimination produces unsaturated alcohols 51, selectively. Mechanistic studies evidently supported Pd-radical hybrid mechanism, where a Pd-catalyst is needed for the generation of radical, and is also engaged in the β-H-elimination step. The control over selective β-/γ-, γ-/δ-, and δ-/ϵ-desaturation sites was achieved by an employment of easily installable/removable iodomethyl silyl tethers on alcohols (48). The activation of tertiary and secondary C(sp3)–H bonds proceeded smoothly at room temperature producing allylic- (51a, 51b), homoallylic-(51c-51i), and bishomoallylic- (51j, 51k) alcohols (Table 7). Based on the regiochemistry trend for desaturation of alcohols containing competitive sites of abstraction (51l-51n) and the kinetic studies, the selectivity preferences for this system were found: 1,6-HAT(γ) > 1,5-HAY(β) > 1,7-HAT(δ). Notably, the level of regiocontrol presented in this work is superior to previous metal-free methods for remote desaturation of aliphatic alcohols[27] due to high regioselectivity of the HAT step achieved by the silyl tether, and highly selective Pd-involved β-H-elimination step.
Scheme 11.
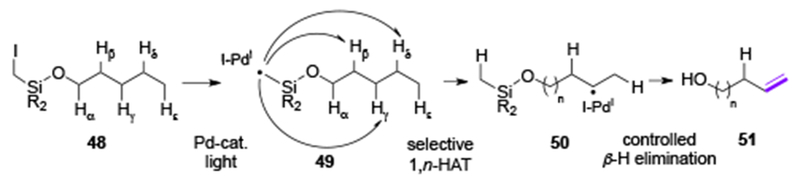
Remote desaturation of aliphatic alcohols via alkyl Pd-radical hybrid species.
Table 7.
Scope of Gevorgyan’s remote desaturation of aliphatic alcohols (TBAF = tetra-n-butylammonium fluoride).
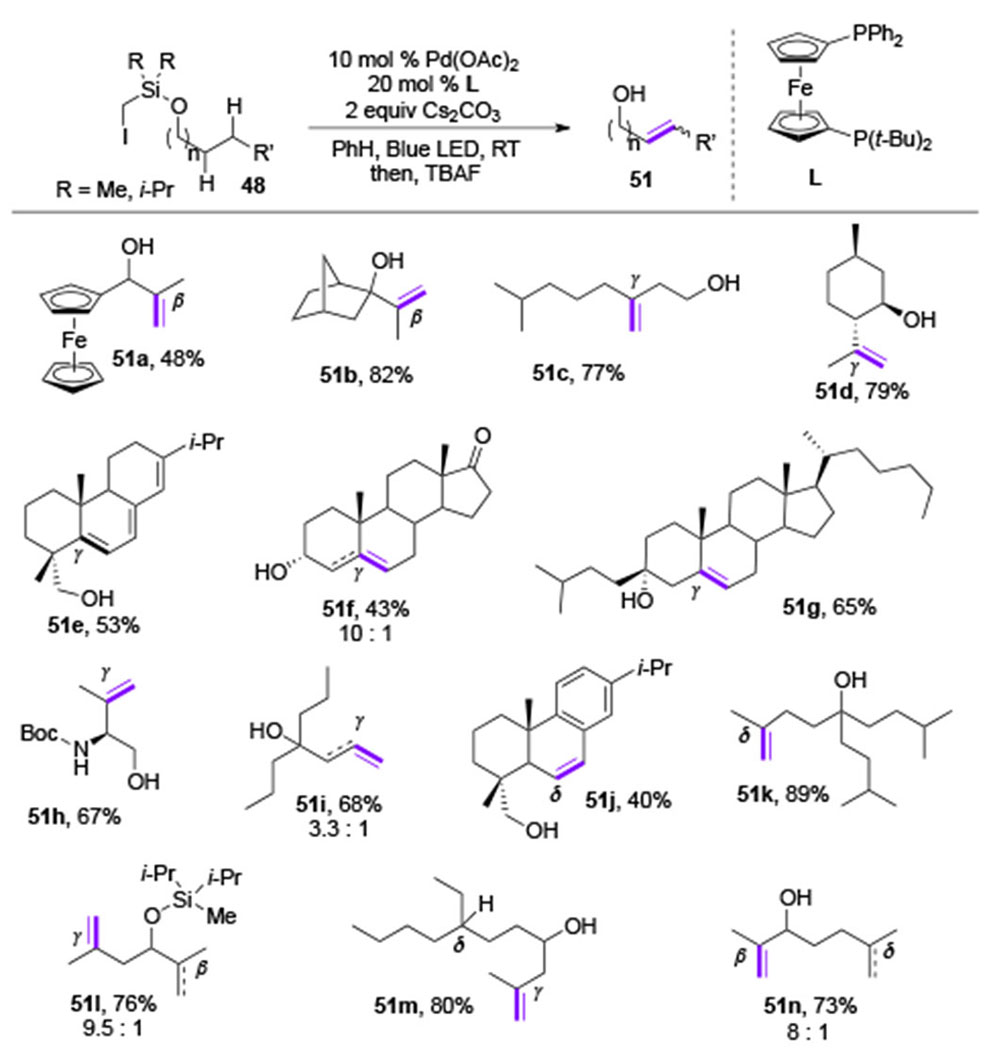 |
In 2017, Yu, independently from works by Gevorgyan, and Shang and Fu, reported the photoinduced Pd-catalyzed generation of alkyl radicals from alkyl halides, which was employed in the α-C–H alkylation of various heterocycles.[28] This reaction proceeded smoothly with a broad scope of alkyl halides, as unactivated primary, secondary, and tertiary alkyl bromides, and even benzyl chlorides, were efficiently coupled with N-aryltetrahydroisoquinolines 52 at the α-position (Table 8, 53a-53s).
Table 8.
Scope of Yu’s C(sp3)–C(sp3) cross-coupling reaction
 |
A radical-type mechanism was proposed for this C–H alkylation reaction (Scheme 12). Similarly to the mentioned methods involving alkyl halides, an SET process from photoexcited Pd-complex to an alkyl bromide was proposed at the initial step (37→42). Then, electron-rich tetrahydroisoquinoline 52 reduces the PdIX species to generate radical cation 54 and regenerates the Pd0 catalyst. Next, the base-assisted deprotonation of the α-hydrogen atom in 54 produces persistent benzylic radical 55, which undergoes a radical recombination with 42 to deliver the coupling product 53. The intermediacy of the alkyl radical 42 and the benzylic radical 55 were supported by radical trapping experiments. The performed Stern-Volmer studies confirmed the quenching of the excited Pd0-complex by an alkyl halide.
Scheme 12.
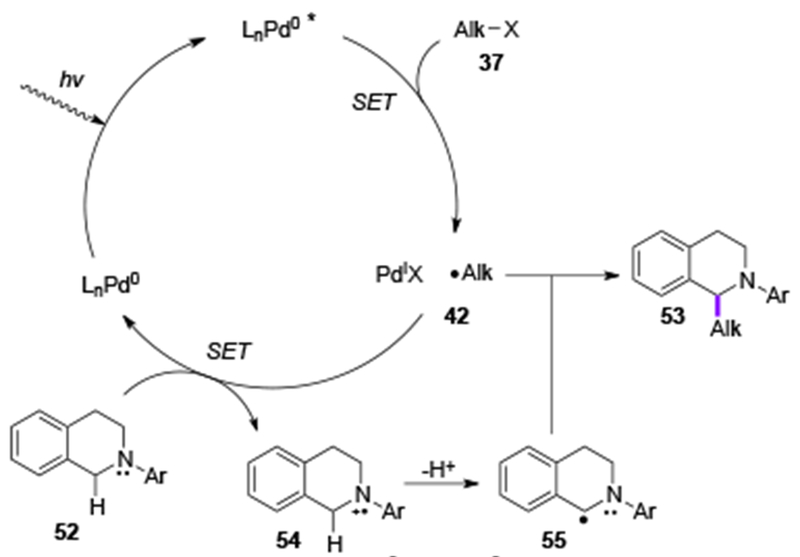
Mechanism of Yu’s C(sp3)–C(sp3) cross-coupling reaction.
In the same report, the authors showed that alkyl radicals can also undergo intramolecular radical addition to indoles under photoinduced conditions at room temperature (eq 3). Moreover, this method can be applied for intermolecular radical addition to heteroarenes using 1-adamantyl bromide (Table 9A, 59a-59e).
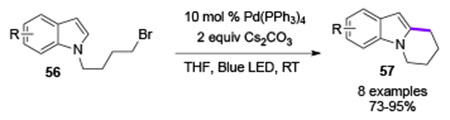 |
(eq 3) |
Table 9.
Radical addition of alkyl halides to arenes and heteroarenes (THF = tetrahydrofurane) .
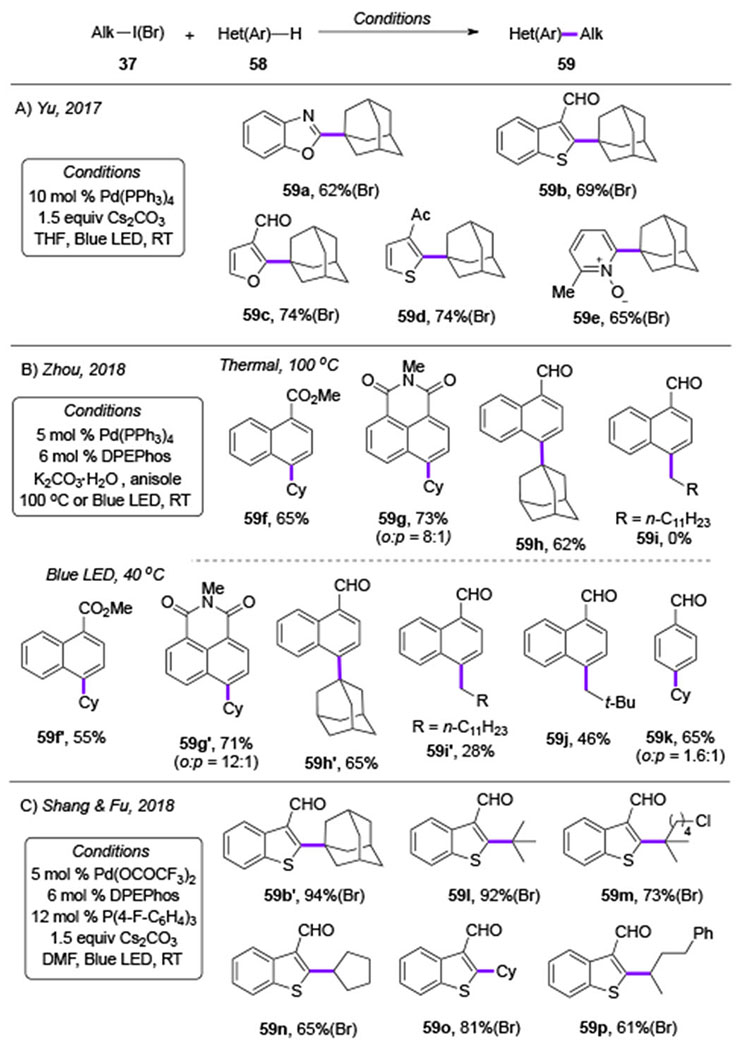 |
Later, Zhou group further explored this intermolecular alkyl radical addition,[29] where the the scope of this reaction was considerably expanded (Table 9B). In this work, two sets of reaction conditions were reported, thermal and visible light-induced conditions. Under thermal conditions, the C(sp3)–C(sp2) coupling reaction proceeded well for secondary and tertiary alkyl iodides with electron-poor naphthalenes (59f-59h). However, the primary alkyl iodide did not yield the desired product, as a premature HI-elimination byproduct was observed predominantly (59i). Visible light-induced conditions have shown enhancement in the reaction efficiency. The same coupling product that was obtained previously at 100 °C can now be obtained with the same efficiency under substantially lower temperature (59f-59h vs 59f’-59h’). Furthermore, primary alkyl iodides, which failed to react under the thermal conditions, were found to be capable reaction partners under the photoinduced conditions (59i’, 59j). Besides, the radical addition to benzaldehyde also proceeded smoothly, although with low regioselectivity between ortho- and para- positions (59k).
Next, Shang and Fu[30] groups reported the visible light-induced coupling reaction of secondary and tertiary alkyl bromides with benzothiophene in high yields using a combination of phosphine ligands at the Pd-catalyst (Table 9C, 59b’, 59l-59p).
A hybrid radical-Pd0/PdI mechanism was proposed for these C(sp3)–C(sp2) cross-coupling reactions (Scheme 13). After the generation of nucleophilic alkyl radical 42 from the excited Pd0-complex and an alkyl halide, it adds to an electron-poor arene or heteroarene 58 producing radical intermediate 60, which can be detected by an EPR measurement.[30] The subsequent oxidation by PdI, followed by the base-assisted deprotonation produces the coupling product 59 and regenerates the Pd0 catalyst. Also, the proposed by Zhou[29] radical-Pd0/PdI/PdII mechanism cannot be ruled out at this stage.
Scheme 13.
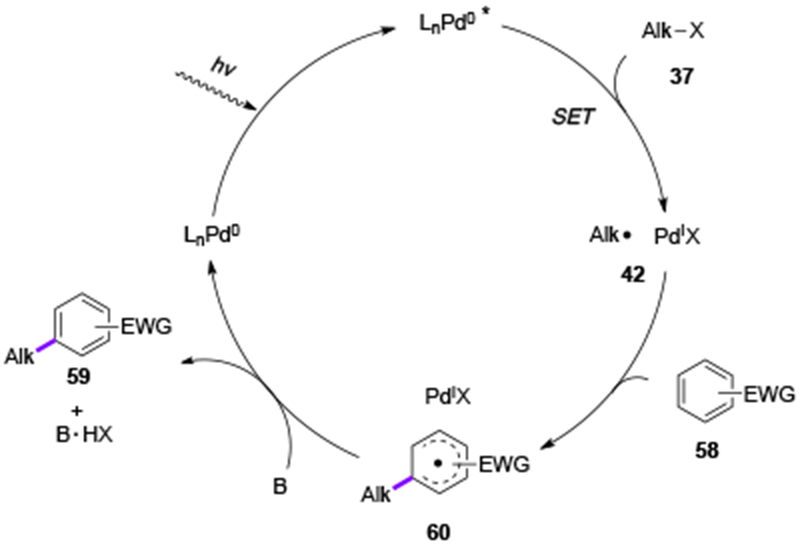
Mechanism of photoinduced C(sp3)–C(sp2) cross-coupling reactions of alkyl halides with electron-poor arenes and heteroarenes.
In addition to alkyl halides, redox active esters have also proved capable substrates for the photoinduced Pd-catalysis. In 2018, groups of Shang and Fu[31] and Glorius[32] independently discovered that the photoexcited Pd0-complex can be engaged in an SET event with aliphatic N-(acyloxy)phthalimides 61, a redox-active ester,[33] that has a similar redox potential to unactivated alkyl bromides (Scheme 14). A subsequent decarboxylative fragmentation of the latter would produce an alkyl Pd-radical hybrid species 42, which, similarly to the aforementioned Heck reactions of alkyl halides, would undergo radical addition to styrene derivatives, ultimately generating the corresponding Heck reaction products 62. Mechanistic studies of this transformation supported radical nature of the reaction. Furthermore, photophysical studies revealed that the Pd-complex is the only light-absorbing species, while N-(acyloxy)phthalimides 61 showed no absorption in the visible light region.
Scheme 14.
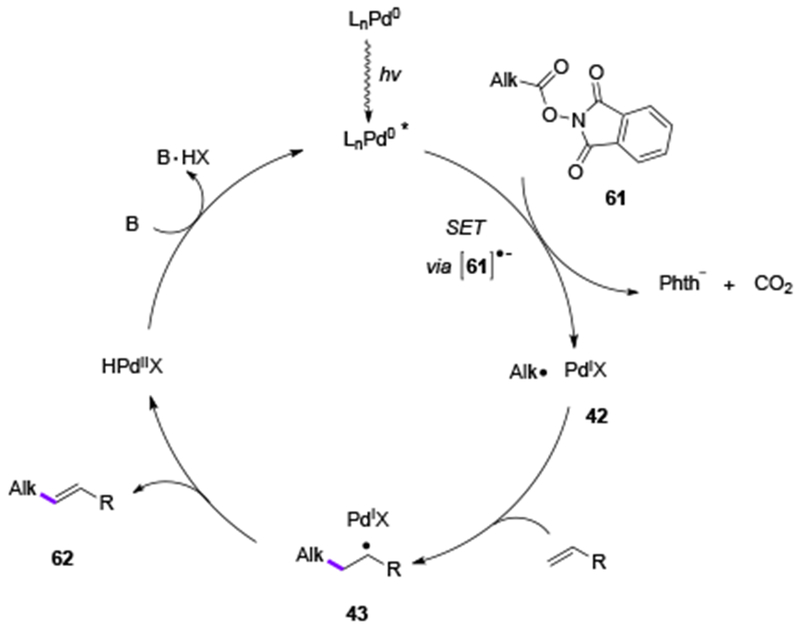
Proposed mechanism for Heck reaction of redox-active esters.
The scope of these two transformations were comparable with that of the Heck reaction of alkyl halides. Primary, secondary and tertiary radicals produced from the corresponding redox-active esters all readily coupled with the styrene derivatives (Table 10, 62a-62p). Overall, this method represents an alternative approach towards Heck reaction products employing easily available carboxylic acid-derived substrates under mild photoinduced conditions. Importantly, these reports uncovered a novel feature of photoexcited Pd-complexes for an activation of N-(acyloxy)phthalimides (61), which notably expanded the scope of visible light-induced Pd-catalysis.
Table 10.
Scope for the Heck reaction of redox-active esters.
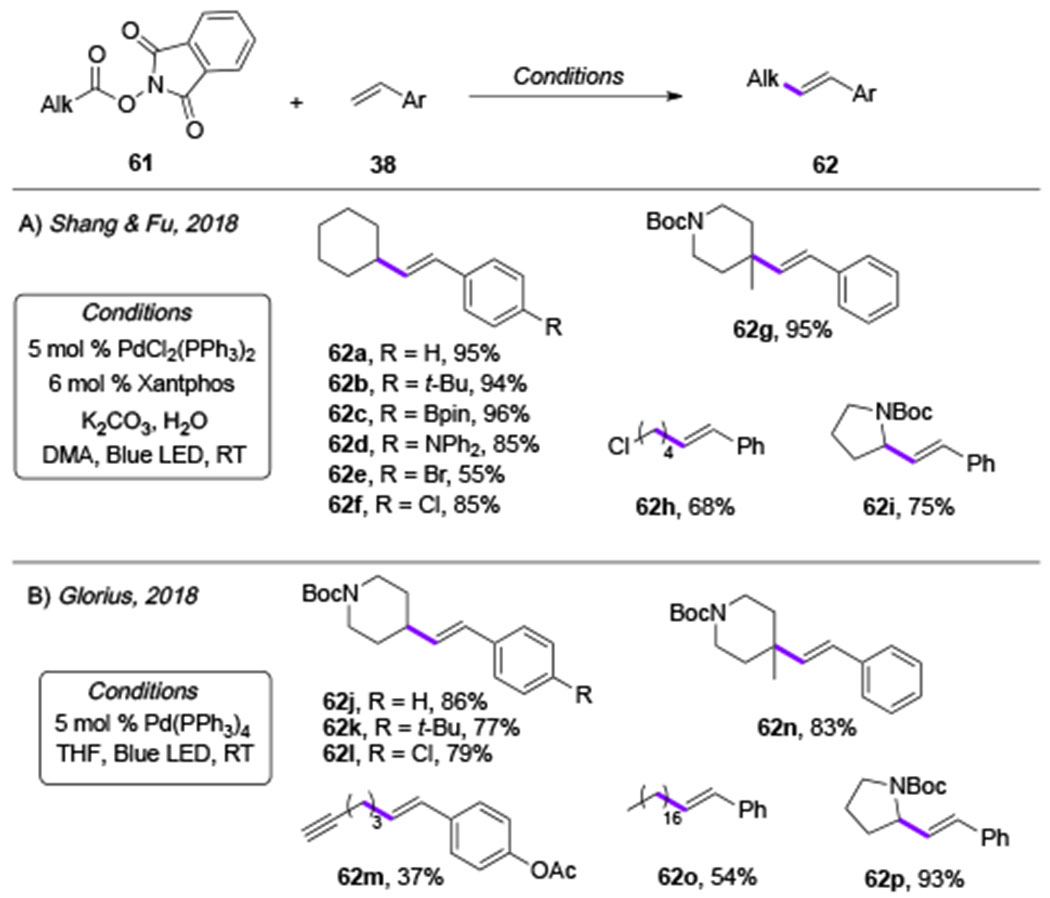 |
Later in 2018, Shang and Fu disclosed a Pd-catalyzed photoinduced decarboxylative desaturation reaction of aliphatic redox active esters in the presence of a dual ligand system (Table 11).[34] A variety of alkenes, including enol ethers, enamides, as well as natural products Chondriamide A and C were shown to be synthesized in high yields and E/Z ratios (63a-63j). Likewise to the prior work on the Heck reaction, an SET from excited Pd0 complex to N-(acyloxy)phthalimides, followed by decarboxylative fragmentation is believed to produce alkyl Pd-radical hybrid species 42, which, upon ligand dissosiation, undergoes recombination towards alkyl PdII intermediate 64 (Scheme 15). A subsequent β-H-elimination and a base-assisted deprotonation produce the alkene product 63 together with the active Pd0 catalyst. The system of mono- and biphosphine ligands was hypothesized to serve an important role in stabilization of PdI intermediates via tricoordinate binding, thus enabling a more efficient irradiation-induced palladium catalysis.
Table 11.
Scope for Shang & Fu’s decarboxylative desaturation of redox-active esters.
 |
Scheme 15.
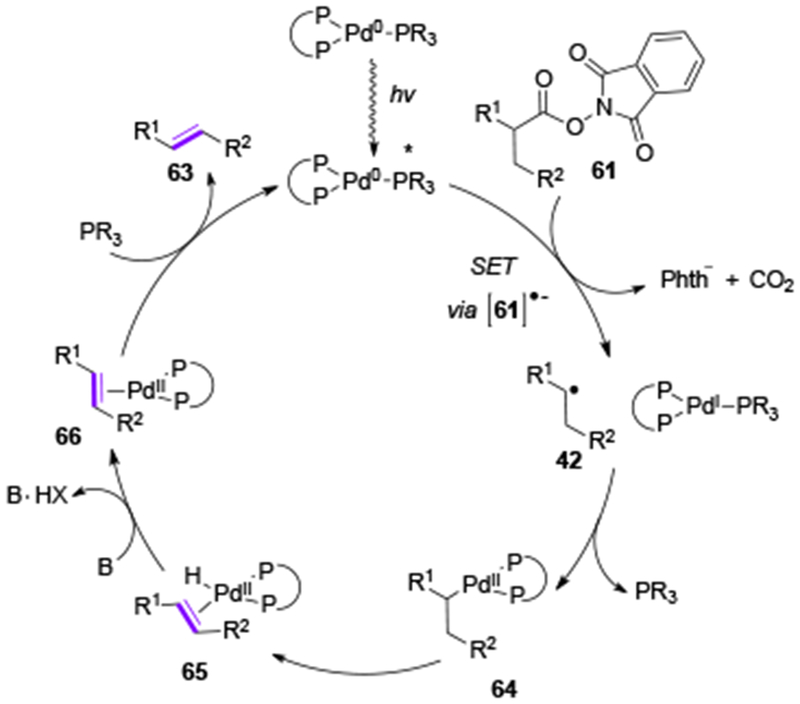
Proposed mechanism for Shang & Fu’s decarboxylative desaturation of redox-active esters
Recently, Yu and co-workers applied the concept of visible light-induced Heck reactions of alkyl halides towards the development of a novel transformation, an oxy-alkylation of allylamines.[35] This reaction proceeded smoothly under visible light irradiation to deliver oxazolidinones 68 via coupling of alkyl halides 37 with allylamines 67 under 1 atm of CO2 (Table 12, 68a-68j). Radical 42, formed via an SET process with photoecited Pd0 species, added to an allylcarbamate 69, generated in situ from an electron-rich allyamine 67 and CO2 gas producing a stable benzylic radical intermediate 70 (Scheme 16). The latter, via a radical-polar crossover mechanism with PdI species, was oxidized into a carbocation 71, which upon cyclization formed the oxazolidinone product 68. This general and mild reaction represents a convenient synthetic route towards substituted oxazolidinones.
Table 12.
Scope of Yu’s oxy-alkylation of allylamines (Bn = benzyl).
 |
Scheme 16.
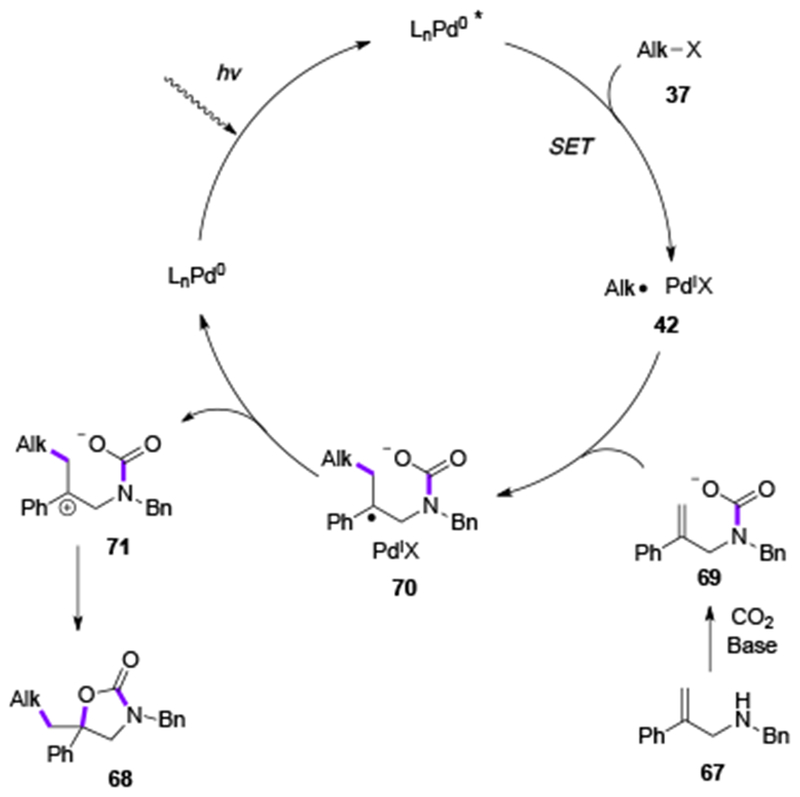
Proposed mechanism for Yu’s oxy-alkylation of allylamines.
So far, the photoexcited Pd-catalysis has been used for alkyl-Heck reactions and for C–H activations. Very recently, Gevorgyan group reported a radical relay Heck reaction of aliphatic alcohols selectively at remote unactivated β-, γ- and δ-C(sp3)–H sites, which synergistically combines C(sp3)–H activation via an HAT process and an alkyl Heck reaction (Table 13).[36] The scope of this reaction was found to be quite general, as γ-, β-, and δ-Heck coupling at unactivated tertiary and secondary C–H sites proceeded smoothly (72a-72q). Acrylonitrile, acrylate and styrene derivatives were all capable coupling partners for this transformation. The reaction proceeded through the formation of Pd-radical hybrid species 49 formed via an SET process of alkyl iodide 48 with the photoexcited Pd-complex (Scheme 17). Then, a subsequent 1,n-HAT process produces translocated Pd-radical hybrid species 50. Interestingly, an iodine-atom transfer intermediate 74 was observed during the reaction, presumably generated by a reversible I-atom transfer from the PdII species. Next, these active intermediates 50 or 74 can couple with an alkene producing the remote Heck product 72. Remarkably, the formed Pd-radical intermediate 49 bypassed the potential side-processes, such as premature Heck coupling reaction (49→73) or desaturation reaction (49→51), thus allowing for the remote Heck reaction at the aliphatic C(sp3)–H sites.
Table 13.
Scope of Gevorgyan’s radical relay Heck reaction (Ac = acetyl).
 |
Scheme 17.
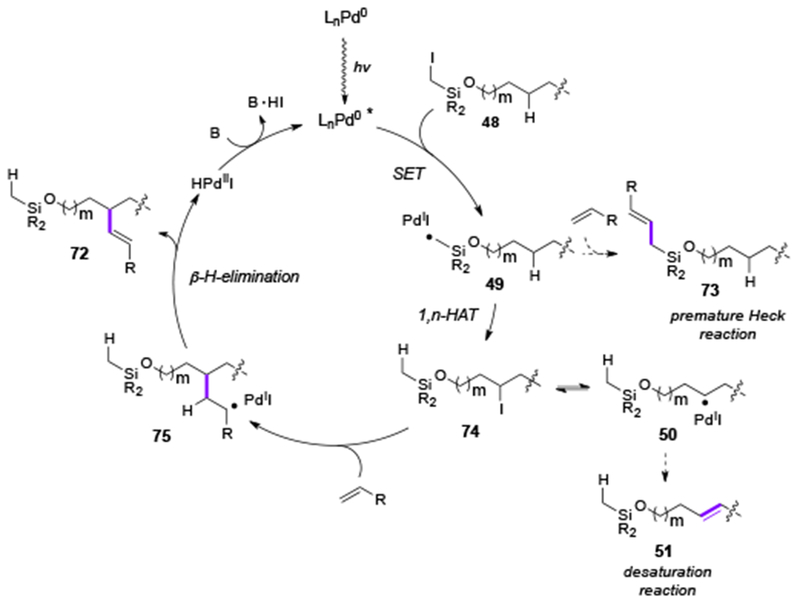
Proposed mechanism for Gevorgyan’s radical relay Heck reaction.
5. Conclusion
Photoexcited Pd-catalysis has enabled the new reactivity mode for palladium-catalyzed reactions, such as general alkyl Heck reaction, oxidative remote C(sp3)–H functionalization reactions via a hydrogen atom transfer (HAT) process, and coupling reactions of usually unreactive aryl chlorides. Furthermore, these reactions proceed under mild conditions at room temperature without an employment of often-expensive photosensitizers. With remarkable reactivity of the radical species and versatility of a Pd-catalysis, it is expected that this approach opens a new avenue for development of novel mild and efficient transformations. One of the future directions in this area may be the development of stereoselective transfromations. Obviously, the more detail mechanistic studies are highly desired for better understanding the mechanism of these new reactions, which could guide the development of novel useful transformations.
Acknowledgements
We thank the National Institutes of Health (GM120281) and National Science Foundation (CHE-1663779) for the financial support of this work.
Biography
Padon Chuentragool obtained his BS and MS degrees from Chulalongkorn University, Bangkok, Thailand. He received his PhD in 2018 under guidance of Prof. Gevorgyan at the University of Illinois at Chicago, where he was focusing on the development of selective methods for C(sp3)-H functionalizations via photoexcited Pd-catalysis.

Daria Kurandina received her BS from St. Petersburg State University, Russia. In 2014, she joined Gevorgyan’s group at the University of Illinois at Chicago as a PhD student. Her work focuses on the development of novel transition metal-catalyzed synthetic methodologies.

Vladimir Gevorgyan received his PhD in 1984 from the Latvian Institute of Organic Synthesis, where then he worked as a group leader. After postdoctoral research at Tohoku University with Prof. Y. Yamamoto as the JSPS- and then Ciba Geigy International Postdoctoral Fellow, in 1996 he joined the faculty position there. In 1999, he moved to UIC as an Associate Professor, and was promoted to the Full Professor in 2003. From 2012, he is a Distinguished Professor of LAS. His group is interested in the development of novel synthetic methodologies.

Footnotes
These authors made equal contribution to this paper.
References
- [1].(a) Johansson Seechurn CC, Kitching MO, Colacot TJ, Snieckus V, Angew. Chem. Int. Ed 2012, 51, 5062–5085 [DOI] [PubMed] [Google Scholar]; (b) de Meijere A, Bräse S, Metal-Catalyzed Cross-Coupling Reactions and More, Oestreich M, Eds., Wiley-VCH, Weinheim, Germany, 2014 [Google Scholar]; (c) de Meijere A, Handbook of Organopalladium Chemistry for Organic Synthesis, Negishi E, Eds., Wiley, New York, 2002 [Google Scholar]; (d) Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].For examples of ground-state Pd-catalysis involving single electron processes, see: (a) Powers DC, Ritter T, Nat. Chem 2009, 1, 302–309 [DOI] [PubMed] [Google Scholar]; (b) Powers DC, Ritter T, Acc. Chem. Res 2012, 45, 840–850 [DOI] [PubMed] [Google Scholar]; (c) Lanci MP, Remy MS, Kaminsky W, Mayer JM, Sanford MS, J. Am. Chem. Soc 2009, 131, 15618–15620 [DOI] [PubMed] [Google Scholar]; (d) Mazzotti AR, Campbell MG, Tang P, Murphy JM, Ritter T, J. Am. Chem. Soc 2013, 135, 14012–14015. For C-H/C-H coupling, that potentially involves Pd(I) species, see [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Curio JM, Kozlowski MC J. Am. Chem. Soc 2015, 137, 18–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].(a) Narayanam JMR, Stephenson CRJ, Chem. Soc. Rev 2011, 40, 102–113 [DOI] [PubMed] [Google Scholar]; (b) Tucker JW, Stephenson CRJ, J. Org. Chem, 2012, 77, 1617–1622 [DOI] [PubMed] [Google Scholar]; (c) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schultz DM, Yoon TP, Science, 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Meggers E, Chem. Commun 2015, 51, 3290–3301 [DOI] [PubMed] [Google Scholar]; (f) Romero NA, Nicewicz DA, Chem. Rev, 2016, 116, 10075–10166 [DOI] [PubMed] [Google Scholar]; (g) Shaw MH, Twilton J, MacMillan DWC, J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA, Acc. Chem. Res, 2016, 49, 1429–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skubi KL, Blum TR, Yoon TP, Chem. Rev, 2016, 116, 10035–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lang X, Zhao J and Chen X, Chem. Soc. Rev, 2016, 45, 3026–3038 [DOI] [PubMed] [Google Scholar]; (d) Twilton J, Le C, Zhang P, Shaw MH, Evans RW, MacMillan DWC, Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]
- [5].For a review for photoexcited transition metal catalysis beyond conventional photosensitizers, see: Parasram M, Gevorgyan V, Chem. Soc. Rev, 2017, 46, 6227–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu Q, Dong X, Li J, Xiao J, Dong Y, Liu H, ACS Catal H. 2015, 5, 6111–6137. [Google Scholar]
- [7].For selected examples on visible light-induced Pd-catalyzed reactions with photosensitizers, see: (a) Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS, J. Am. Chem. Soc 2011, 133, 18566–18569 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zoller J, Fabry DC, Ronge MA, Rueping M, Angew. Chem. Int. Ed 2014, 53, 13264–13268 [DOI] [PubMed] [Google Scholar]; (c) Xuan J, Zeng T-T, Feng Z-J, Deng Q-H, Chen J-R, Lu L-Q, Xiao W-J, Alper H, Angew. Chem. Int. Ed 2015, 54, 1625–1628 [DOI] [PubMed] [Google Scholar]; (d) Choi S, Chatterjee T, Choi WJ, You Y, Cho EJ, ACS Catal. 2015, 5, 4796–4802 [Google Scholar]; (e) Cheng W-M, Shang R, Yu H-Z, Fu Y, Chem. Eur. J 2015, 21, 13191–13195 [DOI] [PubMed] [Google Scholar]; (f) Xu N, Li P, Xie Z, Wang L, Chem. Eur. J 2016, 22, 2236–2242 [DOI] [PubMed] [Google Scholar]; (g) Zheng C, Cheng W-M, Li H-L, Na R-S, Shang R, Org. Lett 2018, 20, 2559–2563. [DOI] [PubMed] [Google Scholar]
- [8].Sumino S, Fusano A, Fukuyama T, Ryu I, Acc. Chem. Res, 2014, 47, 1563–1574. [DOI] [PubMed] [Google Scholar]
- [9].Parasram M, Chuentragool P, Sarkar D, Gevorgyan V, J. Am. Chem. Soc 2016, 138, 6340–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lenges CP, White PS, Brookhart M, J. Am. Chem. Soc, 1999, 121, 4385–4396. [Google Scholar]
- [11].Chuentragool P, Parasram M, Shi Y, Gevorgyan V, J. Am. Chem. Soc 2018, 140, 2465–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fredricks MA, Drees M and Kohler K, ChemCatChem, 2010, 2, 1467–1476. [Google Scholar]
- [13].Abdiaj I, Huck L, Mateo JM, Hoz A, Gomez MV, Diaz-Ortiz A, Alcazar J, Angew. Chem. Int. Ed 2018, 57, 13231–13236. [DOI] [PubMed] [Google Scholar]
- [14].Hsu Y-C, Wang VC-C, Au-Yeung K-C, Tsai C-Y, Chang C-C, Lin B-C, Chan Y-T, Hsu C-P, Yap GPA, Jurca T, Ong T-G, Angew T-G. Chem. Int. Ed 2018, 57, 4622–4626. [DOI] [PubMed] [Google Scholar]
- [15].Ratushnyy M, Parasram M, Wang Y, Gevorgyan V, Angew. Chem., Int. Ed, 2018, 57, 2712–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Curran DP, Shen W, J. Am. Chem. Soc 1993, 115, 6051–6059. [Google Scholar]
- [17].Studer A, Curran DP, Angew. Chem. Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- [18].Firmansjah L, Fu GC, J. Am. Chem. Soc 2007, 129, 11340–11341. [DOI] [PubMed] [Google Scholar]
- [19].(a) Bloome KS, McMahen RL, Alexanian EJ, J. Am. Chem. Soc 2011, 133, 20146–20148 [DOI] [PubMed] [Google Scholar]; (b) McMahon CM ; Alexanian EJ Angew. Chem. Int. Ed 2014, 53, 5974–5977. [DOI] [PubMed] [Google Scholar]
- [20].Zou Y ; Zhou JS Chem. Commun 2014, 50, 3725–3728. [DOI] [PubMed] [Google Scholar]
- [21].(a) Netherton MR, Fu GC, Palladium-Catalyzed Cross-Coupling Reactions of Unactivated Alkyl Electrophiles with Organometallic Compounds. In Topics in Organometallic Chemistry: Palladium in Organic Synthesis, Tsuji J , Ed., Springer; New York, 2005 [Google Scholar]; (b) Frisch AC, Beller M, Angew. Chem. Int. Ed 2005, 44, 674–688 [DOI] [PubMed] [Google Scholar]; (c) Rudolph A, Lautens M, Angew. Chem. Int. Ed 2009, 48, 2656–2670 [DOI] [PubMed] [Google Scholar]; (d) Peacock DM, Roos CB, Hartwig JF, ACS Cent JF. Sci 2016, 2, 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kurandina D, Parasram M, Gevorgyan V, Angew. Chem., Int. Ed, 2017, 56, 14212–14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kurandina D, Rivas M, Radzhabov M, Gevorgyan V, Org. Lett 2018, 20, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang G-Z, Shang R, Cheng W-M, Fu Y, J. Am. Chem. Soc 2017, 139, 18307–18312. [DOI] [PubMed] [Google Scholar]
- [25].Rueping M, Angew. Chem. Int. Ed in press, DOI: 10.1002/anie.201811439 [DOI] [Google Scholar]
- [26].Parasram M, Chuentragool P, Wang Y, Shi Y, Gevorgyan V, J. Am. Chem. Soc 2017, 139, 14857–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Voica A-F, Mendoza A, Gutekunst WR, Fraga JO and Baran PS, Nat. Chem 2012, 4, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou W-J, Cao G-M, Shen G, Zhu X-Y, Gui Y-Y, Ye J-H, Sun L, Liao L-L, Li J, Yu D-G, Angew. Chem. Int. Ed 2017, 56, 15683–15687. [DOI] [PubMed] [Google Scholar]
- [29].Jiao Z, Lim LH, Hirao H, Zhou JS, Angew. Chem. Int. Ed 2018, 57, 6294–6298. [DOI] [PubMed] [Google Scholar]
- [30].Wang G-Z, Shang R, Fu Y, Synthesis 2018, 50, 2908–2914. [Google Scholar]
- [31].Wang G-Z, Shang R, Fu Y, Org. Lett 2018, 20, 888–891. [DOI] [PubMed] [Google Scholar]
- [32].Koy M, Sandfort F, Tlahuext-Aca A, Quach L, Daniliuc CG, Glorius F, Chem. Eur. J 2018, 24, 4552–4555. [DOI] [PubMed] [Google Scholar]
- [33].For a review on cross-coupling reactions using N-(acyloxy)phthalimide as a redox-active ester, see: Murarka S, Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]
- [34].Cheng W-M, Shang R, Fu Y, Nat. Commun 2018, 9, DOI: 10.1038/s41467-018-07694-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sun L, Ye J-H, Zhou W-J, Zeng X, Yu D-G, Org. Lett 2018, 20, 3049–3052. [DOI] [PubMed] [Google Scholar]
- [36].Chuentragool P, Yadagiri D, Morita T, Sarkar S, Parasram M, Wang Y, Gevorgyan V, Angew. Chem. Int. Ed in press, DOI: 10.1002/anie.201812398. [DOI] [PMC free article] [PubMed] [Google Scholar]