

Abstract
The utilization of inverse docking methods for target identification has been driven by an increasing demand for efficient tools for detecting potential drug side effects. Despite impressive achievements in the field of inverse docking, identifying true positives from a pool of potential targets still remains challenging. Notably, most of the developed techniques have low accuracies, limit the pool of possible targets that can be investigated or are not easy to use for non-experts due to a lack of available scripts or webservers.
Guided by our finding that the absolute docking score was a poor indication of a ligand’s protein target, we developed a novel “combined Z-score” method that used a weighted fraction of ligand and receptor-based Z-scores to identify the most likely binding target of a ligand. With our combined Z-score method, an additional 14%, 3.6%, and 6.3% of all ligand-protein pairs of the Astex, DUD, and DUD-E databases, respectively, were correctly predicted compared to a docking score-based selection. The combined Z-score had the highest area under the curve in a ROC curve analysis of all three datasets and the enrichment factor for the top 1% predictions using the combined Z-score analysis was the highest for the Astex and DUD-E datasets. Additionally, we developed a user-friendly python script (compatible with both Python2 and Python3) that enables users to employ the combined Z-score analysis for target identification using a user-defined list of ligands and targets. We are providing this python script and a user tutorial as part of the supplemental information.
Keywords: Inverse Docking, Virtual Screening, molecular docking, z-score
Graphical abstract
Guided by our finding that the absolute docking score was a poor indication of a ligand’s protein target, we developed a novel “combined Z-score” method that successfully improved the identification of the most likely binding target of a ligand. We are also providing a user-friendly python script that enables non-expert users to employ the combined Z-score analysis for target identification using a user-defined list of ligands and targets.
Introduction
While some drugs can be proteins or peptides, most pharmaceuticals are small molecules. These small molecules generally interact with a protein target, or receptor, and modulate its function. While in most drug discovery workflows the identity of the drug target is known and even required (1, 2), this is not always the case. Small molecule protein-target identification is important in a drug discovery process and for understanding molecular function. Being able to identify protein targets of small molecules has important implications for the detection of potential drug side effects (3–6) and in the repurposing of FDA approved drugs (7, 8). Additionally, protein target identification can be important to follow up on experimental cell-based screens or to confirm the binding target of a compound identified by either structure-based drug discovery or high-throughput screening in cases where the experimental assay contained multiple proteins. Protein target identification has certainly benefited from the dramatic increase in available high-resolution protein structures in the protein databank (9, 10). In combination with these experimentally determined high-resolution protein structures, computational methods have the potential to play an important role in the process of protein target identification.
Molecular protein docking methods are used widely in the field of drug discovery as part of structure-based drug discovery (11). The docking process involves the prediction of ligand conformation and orientation within a specific targeted protein binding site (12) by modeling the interaction between a small molecule and a protein at the atomic level using a docking score (13). Since the implementation of the first docking algorithm in the early 1980s (14), there have been countless docking algorithms developed since, including Glide (15), Fred (16), AutoDock Vina (17), GOLD (18), FlexX (19), and RosettaLigand (20). Application of these algorithms has played a significant role in obtaining FDA approval for several pharmaceutical drugs (21–23). Additionally, virtual screening, sometimes in combination with algorithms accounting for receptor flexibility (24, 25), has identified thousands of hit compounds for a variety of disease targets (26–32). Application of molecular docking methods in protein target identification (frequently also referred to as inverse virtual screening (33)) seems straightforward but is plagued by shortcomings. Most notably, molecular docking methods have been developed to identify a number of potential ligands for a given target by screening thousands to millions of ligands against a single protein (34, 35). It has become apparent, however, that molecular docking methods are not particularly well equipped to identify a small number of potential targets (from a large set of possible targets) for a given ligand. Due to the binding environment’s significant contribution toward the docking score, selecting targets based on the raw docking scores has been shown to negatively impact the selection accuracy of inverse docking methods (36–38). Numerous protocols have been developed over the last 10 to 15 years to address this challenge.
The challenge of identifying true positives from a pool of potential targets has encouraged the development of various analysis methods and web-servers, predominantly with a focus on drug side effects detection. Among those protocols, INVDOCK (39), TarFisDock (40), SePreSA (41), and idTarget (42) are widely known molecular docking target identification servers (34, 35), where each server selects potential interactive targets of the users’ query compound from its own protein library. INVDOCK is the earliest version of a target identification server, and currently, the database contains 9,000 proteins and nucleic acids for screening. The selection method of INVDOCK is based on the energy threshold of interactive proteins, by which it compares the scoring of the query compound with the absolute energy threshold of the overall interactive energy of known ligand-protein complexes, including the competitor compounds. The performance of INVDOCK was evaluated with two test cases, Vitamin E and 4H-tamoxifen, which successfully identified 50% of the experimentally verified targets. The TarFisDock server was developed in 2006, with a target library containing 698 proteins from the PDTD database (43). TarFisDock selects the targets by comparing the docking scores of the query compound within the proteins of the target library and selecting the top 2, 5 or 10 % ligand-target pairs as the potential interactive proteins. TarFisDock has also been benchmarked with Vitamin E and 4H-tamoxifen and successfully identified 4 out of 12 experimentally verified binding proteins of Vitamin E, and 3 out of 10 known binding proteins of 4H-tamoxifen from the top 2% candidates. The SePreSA server was developed in 2009, and currently, the server allows users to screen nearly all the well-known SADR (serious adverse drug reactions) targets (44, 45). Unlike the aforementioned servers, which compared the interactive energy and the docking scores of the query compound-protein complexes, SePreSA introduced a new algorithm, the 2-directional Z-transformation (2DIZ). The authors demonstrated that SePreSA’s Z-transformation matrix (defined as the Z-score matrix normalized to a mean and a standard deviation of 0 and 1 respectively) enhanced the selection of true positives compared to the matrix of docking score and the Z-score matrix. The SePreSA algorithm was evaluated using a ROC curve and indeed resulted in the highest area under the curve (AUC), 0.82, among the three investigated matrices. Finally, the idTarget server is a more recent target identification tool, which covers 2,091 proteins in its library. The server virtually screens the target library with the query compound using AutoDock4 (46) and ranks the target by the predicted binding affinity, which then filters out targets with positive Z-scores. The idTarget server also demonstrated its performance with three different test cases, and one of them was tested on an experimentally verified kinase inhibitor 6-bromo-indirubin-3’ oxime (6BIO). It was reported that after screening 5,821 PDB entries of the protein kinases, the server successfully identified protein kinase targets that were known to interact with inhibitor 6BIO, resulting in an enrichment factor of 6.54 for the top 1% of compounds (42, 47, 48).
Besides the four target identification servers, other computational protocols that focus on drug side effect detection have been developed as well. Those protocols allow for a screening of a custom set of receptors. One study reported that applying a consensus scoring method (combining ICM (49) docking scores with the probability of the drug-protein interaction) resulted in the highest accuracy, 48.8%, after screening 252 human protein drug targets with 4,621 experimentally approved small molecules from the DrugBank (50). Another study proposed adding a custom score term to the Glide SP scoring function and improved the selection rate by 27% after cross docking (i.e. docking ligands into non-target proteins) a pre-filtered subset of 58 proteins from the Astex dataset (36).
Despite the listed achievements in the field of inverse docking, the challenge of reliable protein target identification is far from solved and many shortcomings remain. Notably, most of the developed techniques have low accuracies and are not easy to use for non-experts due to a lack of available scripts or webservers. Additionally, many of the available drug side effect detection servers (33, 40–42) have a preset list of protein targets that are screened, making them inadequate for screening specific assay proteins or if a custom list of protein is desired to be screened.
In this work, we aimed to address these above limitations. Guided by our finding that the absolute docking score was a poor indication of a ligand’s protein target, we developed a novel “combined Z-score” method that used a weighted fraction of ligand and receptor-based Z-scores to identify the correct target for each ligand. We benchmarked our protocol using the Astex, DUD, and DUD-E databases. With our combined Z-score method, an additional 14%, 3.6%, and 6.3% ligand-protein pairs of the Astex, DUD, and DUD-E datasets, respectively, were correctly predicted compared to a docking score-based selection as shown in Table 1. The combined Z-score had the highest areas under the curve (AUCs) in a ROC curve analysis among the score based, receptor-average Z-score, and ligand-average Z-score selection protocols for all three datasets: Astex (AUC=0.82), DUD (AUC=0.76), and DUD-E (AUC=0.74). Furthermore, the enrichment factor for the top 1% of compounds using the combined Z-score analysis was the highest in Astex (EF=36.5), and DUD-E (EF=18.0). Additionally, we developed a user-friendly python script (compatible with both Python2 and Python3) that enables users who are familiar with python to analyze docking results for target identification. Unlike other web-servers, our python script allows users to screen a custom list of query ligands to a custom list of proteins. We are providing this python script and a user tutorial as part of the supplemental information.
Table 1.
Prediction accuracy. The number of correctly selected ligand-protein pairs and the prediction accuracy of the four different selection methods are shown. The first column lists the results of the Astex dataset, in which 85 ligands were docked to every 85 proteins. The second column lists the results for the DUD dataset, in which 279 ligands were docked to 8 proteins, and the third lists the results for the DUD-E, in which 2,040 ligands were docked to 102 proteins.
| Number of hits |
85 × 85 | 279 × 8 | 2040 × 102 |
|---|---|---|---|
| Astex | DUD | DUD-E | |
| Score | 23 (27.1%) | 121 (43.4%) | 248 (12.2%) |
| Receptor-average Z-score | 34 (40.0%) | 111 (39.8%) | 368 (18.1%) |
| Ligand-average Z-score | 23 (27.1%) | 121 (43.4%) | 248 (12.2%) |
| Combined Z-score | 35 (41.2%) | 131 (47.0%) | 376 (18.5%) |
2. Materials and Methods
2.1. Datasets
Three datasets of protein targets with known binding ligands were used to investigate our combined Z-score method for the enhanced selection of true positives from inverse virtual screening. We used the Astex, DUD, and DUD-E databases for our study and detailed their properties below. Two of the three datasets (DUD and DUD-E) had multiple active ligands for each target protein, whereas the remaining dataset (Astex) only contained a single active ligand for each protein.
2.1.1. Astex
The Astex Diverse Set (51) contained a total of 85 proteins and a single unique corresponding active ligand for each target (totaling 85 active compounds), as shown in SI Figure 1. 99.9% of ligand pairs had Tanimoto indices below 0.6, suggesting that all 85 ligands have a significantly unique structure. Therefore, each target has a unique active ligand and 84 decoys. The Astex Diverse Set provided the 3D structure of the target protein in mol2 format and its 3D active ligand in mol format. No separate box file with binding site coordinates of the target protein was provided. Since the 3D ligands of the Astex Diverse Set were directly extracted from the original PDB file, the midpoint of the given active ligand coordinates was used as the center for the docking box.
2.1.2. DUD
The DUD (A Database of Useful Decoys) database assembled 40 different targets with 2,950 active compounds and over 100,000 decoys (52). DUD provided individual downloadable packages for each target, which contained a 3D structure of the target in PDB format with a box file that listed the binding site coordinates. Each target package also included two separate sets of 3D compounds in mol2 format, actives and decoys. To ensure computational tractability, 8 targets were randomly selected from the 40 targets, along with their respective active compound sets: ACE (49 actives and 230 decoys), ADA (23 actives and 256 decoys), ALR2 (26 actives and 253 decoys), AmpC (21 actives and 258 decoys), AR (74 actives and 205 decoys), CDK2 (50 actives and 229 decoys), COMT (11 actives and 268 decoys), and COX1 (25 actives and 254 decoys). We randomly selected at least 2 to 3 proteins from three different protein size categories: Protein size ranging from 100 to 300 residues (Group 1: small proteins), size ranging from 300 to 500 residues (Group 2: medium proteins), and size ranging from 500 to 700 residues (Group 3: large proteins). The targets AR, CDK2 and COMT were selected from Group 1, ADA, ALR2 and AmpC from Group 2, and ACE and COX1 from Group 3.
2.1.3. DUD-E
The DUD-E (A Database of Useful Decoys: Enhanced) database is an enhanced version of the DUD database, containing 102 diverse targets with 22,886 active compounds and over a million decoys (53). DUD-E provided different subsets of target proteins: subsets categorized by the proteins’ biological functions, a diverse set containing representative targets of the entire database, and a set containing the entirety of the proteins available in DUD-E. Similar to the DUD database, DUD-E also provided a 3D structure of the target protein (“receptor.pdb”) with a box file listing the binding site coordinates. Finally, for each target, DUD-E also provided a list of active compounds categorized by their biological functions: actives, marginal actives, marginal inactives, and inactives. Among those lists of compounds, “actives_combined.ism” was used for this study. From the combined actives list, the top 20 compounds with the strongest binding affinity to each target protein were extracted for our DUD-E ligand library. Therefore, each target has 20 active ligands and 2,020 decoys.
2.2. Preparation for docking
For each database, all ligands were docked to each individual protein. For the Astex database, a total of 85 ligands were docked to each of the 85 proteins. For the DUD database subset, a total of 279 ligands were docked to each of the 8 proteins. And finally, for the DUD-E database, 2,040 ligands were docked to each of the 102 proteins. Unless the database provided 3D compounds, compounds were prepared using Schrödinger’s LigPrep package (54) prior to virtual screening. The energy minimization step was conducted using the OPLS_2005 force field, and compounds were ionized at a target pH of 7.0 ± 2.0. An additional LigPrep step was applied to compounds that failed to dock with any of the targets. For this additional LigPrep step, the OPLS_3 force field was used for the energy minimization step.
2.3. In silico Docking
Each of the active ligands was cross docked to the receptor proteins with Glide. For Schrodinger’s Glide (15, 55), the grid was centered at the target’s given binding coordinates with an inner box size of 20Åx20Åx20Å, and an outer box size of 40Åx40Åx40Å. In Glide, compounds were docked to the receptor center with the OPLS_2005 forcefield, the van der Walls radii of ligand atoms were scaled by 0.8, a charge cutoff for polarity was set at 0.15, and we used GlideScore version SP 5.0.
2.4. Analysis (Methods)
We evaluated a total of 4 different selection protocols for the identification of the small molecule protein targets: 1) score, 2) receptor-average Z-score, 3) ligand-average Z-score, and 4) the combined Z-score. To calculate these measures, we evaluated the docking scores of all the possible ligand-target pairs. The ligands were ranked by their docking score for each target individually. Subsequently, we calculated the average docking scores (Equation 1) and score standard deviations (Equation 2) for each receptor (receptor-average Z-score) and for each ligand (ligand-average Z-score).
| #(1) |
| #(2) |
For the receptor-average Z-score, the summation index i represents each query ligand, while j represents each target receptor, and N represents the total number of ligands of a dataset. For the ligand-average Z-score, the representation changes, where the summation index i now represents each target receptor, while j represents each query ligand, and N represents the total number of target proteins of a dataset. Based on those target-specific and ligand-specific averages, we calculated Z-scores for each ligand docked into each receptor (Equation 3 and 4).
| #(3) |
| #(4) |
In both equations 3 and 4, the index i represents each query ligand for both the receptor-average and ligand-average Z-score. However, the average and the standard deviation (SD) values are different from each other. As mentioned above, the receptor-average Z-score was calculated by taking the target-specific average score and standard deviation, whereas the ligand-specific average score and standard deviation were used for the calculation of the ligand-average Z-score.
In the score analysis, for each ligand, the receptor where that ligand had the lowest (i.e. most favorable) docking score was selected as the ligand’s potential binding partner. For the receptor-average Z-score and the ligand-average Z-score analysis, the receptor with the lowest Z-score, respectively, was selected as the potential target for each of the query ligands.
The combined Z-score was calculated as a linear combination of the receptor-average and ligand-average Z-scores:
| #(5) |
As part of the combined Z-score analysis, for each ligand, the receptor with the lowest combined Z-score was selected as the potential target. For the parameters of the combined Z-score, a total of 10 different pairs of parameters were tested. Both coefficients of the receptor-averaged Z-score and the ligand-averaged Z-score ranged from 0.0 to 1.0 with a step size of 0.1, and the summation of each coefficient pair was equal to 1.
2.5. Percent accuracies, ROC curves, AUC calculation, and enrichments
The 4 selection methods for the identification of the small molecule protein targets were evaluated by the percent accuracy and the AUC (area under the curve) value of the ROC (receiver operating characteristic) curves. The percent accuracy of the selection was calculated by counting the total number of correctly predicted ligand-receptor pairs and then dividing the number of correct hits by the total number of ligand-receptor pairs in the dataset. For the generation of the ROC curve, the ligand-receptor pairs were sorted by their scores and different versions of Z-scores, respectively. All the correct pairs were defined as the positives along the y-axis, and the rest of the pairs were considered as decoys along the x-axis for the ROC curve generation. A total of 6 different enrichment factors (for the top 1%, 2%, 5%, 10%, 20%, and 50% of the respective ligand-receptor lists) for each selection method were calculated. We also compared enrichments for each receptor by calculating the top 5% enrichment factor of the individual receptors. The receptor enrichment factor of the ligands ranked by the raw docking score was compared to that of when ligands were ranked by the combined Z-score.
2.6. Baseline calculations for AUC and percent accuracies
For the evaluation of the overall performance of the 4 different protocols, each selection protocol’s AUC and selection accuracies were compared with the respective baseline value expected for random predictions. For the baseline calculation, we generated matrices with random docking scores for each dataset. For the Astex model, we generated an 85×85 matrix with random docking scores of 85 ligands to 85 target proteins of the Astex dataset. Similarly, a 279×8 matrix was generated for the DUD, and a 2040×102 matrix was generated for the DUD-E dataset. From the random docking score matrix, we then assigned targets to ligands based on their respective scores and various versions of the Z-score. The above procedure was repeated 100,000 times, then the average percent accuracy and the AUC of the ROC curves of the 4 different selection methods were calculated and used as the baseline.
Results and Discussions
Historically, the primary goal of molecular docking methods was to effectively identify potential ligand binders to a single protein. Existing docking algorithm scoring functions have been optimized to accomplish its primary purpose: ranking the true positive ligands towards the top of the list of sorted docking scores within one target receptor. However, limitations of these molecular docking methods emerged when they were applied to inverse docking, i.e. when the same set of ligands was docked into multiple target receptors (36–38). To investigate optimal ways of selecting true positive ligand-protein pairs from the inverse docking results, we worked with three different databases: Astex, DUD, and DUD-E. Our Astex dataset was comprised of 85 unique ligand-protein pairs, from the DUD we collected 8 different proteins each paired with 11 to 74 active ligands, and from the DUD-E we selected 102 diverse proteins each paired with 20 unique active ligands. After docking all these ligands into each of the database protein targets, we then assigned targets to ligands based on their respective scores and various versions of the Z-score.
3.1. Variation in score ranges for different binding site environments make score-based target selection problematic
We first used the docking scores to assign the target-ligand pairs. In the score analysis, for each ligand, the receptor where that ligand had the lowest (i.e. most favorable) docking score was selected as the ligand’s potential binding partner. Figure 1 summarizes the docking results of the Astex, DUD, and DUD-E databases. The targets are listed on the X-axes and the scores of the ligands docked into each target are shown on the Y-axes. True positive ligands are colored orange. A major limitation of applying the inverse docking methodology became apparent if the potential targets were selected by comparing their respective docking scores. The scoring function of a docking program keeps track of the favorable and non-favorable interactions between the binding site and the query ligand, consequently resulting in a variation in score ranges for different binding site environments. Figure 1 clearly illustrates such score range variations of each protein. For example, Figure 1a shows the docking scores of 279 ligands docked into 8 protein targets (DUD dataset). All targets exhibited unique score ranges and score distribution widths. A similar trend was also found in the Astex (Figure 1b) and DUD-E datasets (Figure 1c), where docking score distributions were notably target-dependent. This effect renders the challenge of target identification by docking score since the targets exhibiting low score distributions would be predominantly favored. For example, in Figure 1a, the overall docking scores of AmpC and COMT are less favorable compared to the rest of the DUD proteins, which would most likely neglect those two proteins from the selection. Indeed, when each ligand’s potential binding partner was selected by the docking scores, none of the correct active ligands of AmpC and COMT were selected for these two proteins, resulting in 0% accuracy for the two receptors. However, the score-based selection accuracy for the DUD dataset was 43.4% (Table 1). This was not as low as it could have been based on the score distribution. The reason for this was that the DUD was the only dataset that consisted of unequal numbers of active ligands for each protein, which consequently led to an uneven distribution of true positive pairs. For example, the score distribution of AR ranged from 0.52 to −11.4 kcal/mol and had 74 active ligands, which is 27% of all 279 DUD subset ligands. Also, CDK2 had a wide range of score distribution, ranging from −1.05 to −10.2 kcal/mol, and contained 50 active ligands, covering 19% of the DUD ligands. This virtually diminished the negative effect of docking score-based selection for the DUD dataset. Even though the prediction accuracy for AmpC and COMT was 0% with the docking score-based selection, the total number of mispaired active ligands of the two proteins was only 11% of the entire DUD subset active ligands. Not surprisingly, however, when targets were selected based on the ligand docking scores for the other two datasets (Figure 1b and 1c), the selection prediction accuracy was significantly lower, compared to other selection methods. The Astex and DUD-E datasets exhibited accuracies of 27.1% and 12.2%, respectively, as shown in Table 1. In summary, due to the binding environment’s significant contribution toward the docking score, selecting targets based on the raw docking scores will generally negatively impact the selection accuracy of the inverse docking method. Hence, the idea for enhancing the selection accuracy by normalizing the docking scores prior to the selection step initiated this study.
Figure 1.
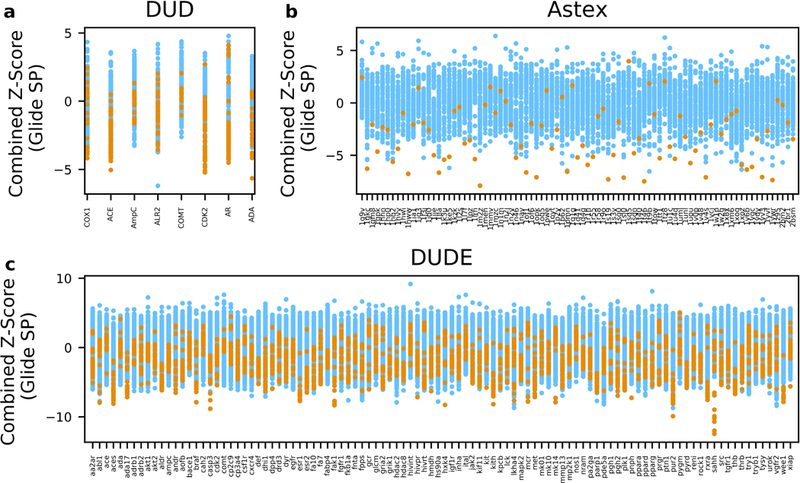
Glide SP docking results of the Astex, DUD, and DUD-E databases. The proteins of each dataset are shown along the x-axis and the Glide SP scores of the ligands docked into each target are shown on the y-axis. Correct ligands of each target are colored orange. (a) The 8 targets of the DUD dataset and the Glide SP scores of the 279 ligands docked into each target. (b) The 85 targets of the Astex dataset and the Glide SP scores of 85 ligands docked into each target. (c) The 102 targets of the DUD-E dataset and the Glide SP scores of 2,040 ligands docked into each target.
3.2. Receptor-average Z-score vs. Score
The variation in score ranges for different binding site environments prompted us to use a Z-score metric instead of the raw docking score to identify the target of a particular ligand. We used the receptor-average Z-score which normalized the raw docking score of a ligand by its deviation from the average ligand docking score of all ligands docked into that receptor. Thus, the receptor-average Z-score enabled a fairer comparison of different targets for a single ligand. As a result, when a receptor with the lowest receptor-average Z-score was selected as the potential binding target of a ligand, the prediction accuracy for successfully matching the true target for a ligand increased by 12.9 and 5.9 percentage points for the Astex and DUD-E dataset. However, the receptor-average Z-score did not improve the prediction accuracy of the DUD dataset, in which the raw docking score performed 3.6 percentage points better than the receptor-average Z-score. A ROC curve analysis evaluated how well the receptor-average Z-score distinguishes the true positives from the decoys. As shown in Figure 2, the AUCs for both Astex (AUC=0.82) and DUD-E (AUC=0.73) exceeded the respective docking score AUCs.
Figure 2.
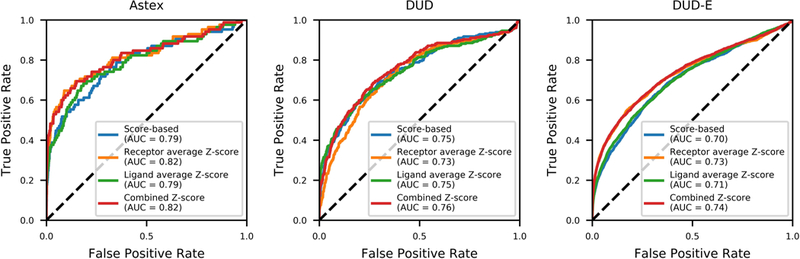
ROC curves. The ROC curves of 4 different selection methods for inverse docking into three datasets (Astex, DUD, and DUD-E) are shown: Score-based (blue); Receptor average Z-score (orange); Ligand average Z-score (green); Combined Z-score (red). The AUC value for each selection methods is shown in the legend.
3.3. Ligand-average Z-score vs. Score
Even though applying the receptor-average Z-score, instead of the raw docking score, for the selection of ligand-protein targets successfully enhanced the prediction accuracy for the Astex and DUD-E datasets, it was not the ultimate solution. This led us to investigate a different type of Z-score analysis, the ligand-average Z-score, which was a method introduced in SePreSA (41). For the ligand-average Z-score, each ligand’s raw docking score was normalized by its deviation from the average ligand docking score of that particular ligand docked into each receptor. Subsequently, the receptor with the lowest ligand-average Z-score was selected as the potential target for that ligand. As such, the ligand-average Z-score was closely related to the raw docking score, however, it normalized the values by their deviation from the respective average values. As a direct consequence, the prediction accuracy of the ligand-average Z-score was identical to the score-based selection for all three datasets. Despite this, the AUCs for the ligand-average Z-score were different from the score-based selection. As shown in Figure 2, the AUCs for both Astex (AUC=0.79) and DUD-E (AUC=0.71) performed slightly better than the respective docking score AUCs, but not as well as the receptor-average Z-score.
3.4. Combined Z-score vs. Score
The above results of the receptor-average and ligand-average Z-scores inspired the generation of a combined Z-score. The combined Z-score was defined as a linear combination of the receptor-average and ligand-average Z-scores as defined in Equation 5. Subsequently, we selected the receptor with the lowest combined Z-score as the potential target for that ligand. One of the advantages of the combined Z-score was that it did not rely on a single Z-score term, but rather it merged the strengths of the two individual Z-scores. As a result, the combined Z-score selection had the highest accuracy compared to the three alternative selection methods for all three datasets. With the combined Z-score method, an additional 14% of the Astex ligand-protein pairs, an additional 3.6% of the ligand-protein pairs for DUD, and an additional 6.3% of ligand-protein pairs for DUD-E were correctly identified (see Table 1 and SI Figure 2). As shown in Table 2, the prediction accuracy enhancement of the combined Z-score compared to a random selection was the highest for all three cases. The Astex dataset had a prediction accuracy enhancement of 34.9, DUD had a prediction accuracy enhancement of 3.8, and DUD-E had a prediction accuracy enhancement of 18.9. Additionally, the combined Z-score had the highest AUCs for all three datasets. Astex’s AUC was 0.82, DUD’s AUC was 0.76, and DUD-E’s AUC was 0.74. As shown in Table 3, the enrichment factor within the top 1% scored ligand-protein pairs for the combined Z-score was the highest in Astex (EF=36.5), and DUD-E (EF=18). Even though the top 1% enrichment factor of the combined Z-score for DUD (EF=7.2) was not the best among the other selection methods, it is important to note that it is seven times more likely to find correct ligand-protein pairs in the top 1% with the combined Z-score compared to a random selection.
Table 2.
Prediction accuracy enhancement of 4 different selection methods. The prediction accuracy of each selection methods was divided by the expected prediction accuracy of a random target selection to calculate the prediction accuracy enhancement, which is shown in the third column.
| Astex | Prediction Accuracy (%) | Random Prediction Accuracy (%) |
Prediction Accuracy Enhancement |
|---|---|---|---|
| Score | 27.1 | 1.2 | 23.0 |
| Receptor-average Z-score | 40.0 | 1.2 | 33.9 |
| Ligand-average Z-score | 27.1 | 1.2 | 23.0 |
| Combined Z-score | 41.2 | 1.2 | 34.9 |
| DUD | Prediction Accuracy (%) | Random Prediction Accuracy (%) |
Prediction Accuracy Enhancement |
|---|---|---|---|
| Score | 43.4 | 12.5 | 3.5 |
| Receptor-average Z-score | 39.8 | 12.5 | 3.2 |
| Ligand-average Z-score | 43.4 | 12.5 | 3.5 |
| Combined Z-score | 47.0 | 12.5 | 3.8 |
| DUD-E | Prediction Accuracy (%) | Random Prediction Accuracy (%) |
Prediction Accuracy Enhancement |
|---|---|---|---|
| Score | 12.2 | 1.0 | 12.4 |
| Receptor-average Z-score | 18.1 | 1.0 | 18.5 |
| Ligand-average Z-score | 12.2 | 1.0 | 12.4 |
| Combined Z-score | 18.5 | 1.0 | 18.9 |
Table 3.
ROC curve enrichment. A total of 6 different enrichment factors (for the top 1%, 2%, 5%, 10%, 20%, and 50% of the respective ligand-receptor lists) for each selection method were calculated.
| Astex EF | 1% | 2% | 5% | 10% | 20% | 50% |
|---|---|---|---|---|---|---|
| Score | 23.5 | 17.1 | 8.7 | 5.2 | 3.1 | 1.69 |
| Receptor-average Z-score | 32.9 | 19.4 | 11.1 | 6.5 | 3.6 | 1.65 |
| Ligand-average Z-score | 25.9 | 17.1 | 8.0 | 5.2 | 3.4 | 1.65 |
| Combined Z-score | 36.5 | 21.8 | 10.6 | 6.1 | 3.5 | 1.69 |
| DUD EF | 1% | 2% | 5% | 10% | 20% | 50% |
|---|---|---|---|---|---|---|
| Score | 7.2 | 6.1 | 4.8 | 3.8 | 2.6 | 1.56 |
| Receptor-average Z-score | 6.1 | 4.5 | 3.7 | 3.1 | 2.4 | 1.60 |
| Ligand-average Z-score | 7.5 | 7.2 | 5.8 | 3.9 | 2.6 | 1.54 |
| Combined Z-score | 7.2 | 6.8 | 4.7 | 3.7 | 2.7 | 1.62 |
| DUD-E EF | 1% | 2% | 5% | 10% | 20% | 50% |
|---|---|---|---|---|---|---|
| Score | 12.7 | 8.5 | 5.0 | 3.5 | 2.4 | 1.52 |
| Receptor-average Z-score | 16.7 | 12.1 | 6.9 | 4.3 | 2.8 | 1.56 |
| Ligand-average Z -score | 12.8 | 9.1 | 5.4 | 3.7 | 2.5 | 1.52 |
| Combined Z-score | 18.0 | 11.9 | 6.8 | 4.4 | 2.8 | 1.57 |
As shown in the above results, when the combined Z-score was used as a tool for target identification instead of the raw docking score, we achieved the highest prediction accuracy among the other methods for all three datasets. We next compared the individual receptors’ enrichment of active ligands in the top 5% for a score and Z-score based selection. Figure 3 illustrates the difference between the combined Z-score’s enrichment factor and the raw docking score’s enrichment factor for each receptor. As shown in Figure 3, the y-axis is the enrichment factor difference (ΔEF) between the two methods. A positive ΔEF represents a higher enrichment factor when using the combined Z-score based ranking, whereas a negative ΔEF represents a higher enrichment factor when docking-score based ranking is employed. If, for a particular receptor, both the combined Z-score and the raw docking score had an identical enrichment factor, ΔEF = 0 and no bar is shown. According to Figure 3, when ligands were ranked by the combined Z-score, 8 proteins of the Astex, 3 proteins of the DUD, and 47 proteins of the DUD-E dataset showed improvement in the enrichment factor, while only 2, 3, and 12 proteins, respectively, showed improvement when the ranking was performed based on the docking score. The range of improvement was significantly different between the two methods. For the DUD dataset, the improvement of the 3 proteins from the combined Z-score ranged from 1.6 to 6.1, whereas the docking score improvement ranged from 0.4 to 1. Similarly, for the DUD-E dataset, the combined Z-score enrichment factor improvement ranged from 1 to 14 for the 47 proteins, whereas the docking score enrichment factor improvement ranged from 1 to 6 for the 12 proteins.
Figure 3.
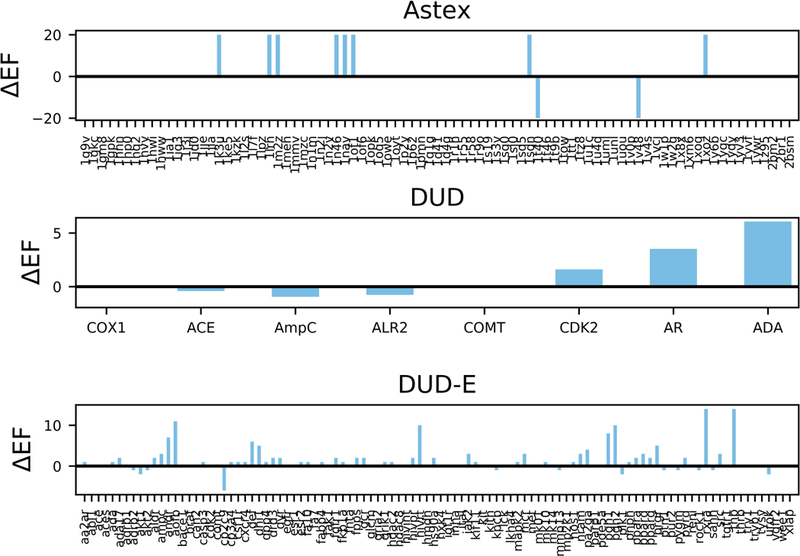
Top 5% enrichment factor enhancement of individual receptors. This figure illustrates the difference between the combined Z-score’s top 5% enrichment factor and the raw docking score’s top 5% enrichment factor for each receptor. The y-axis is the enrichment factor difference (ΔEF) between the two methods, where the length of the bar is a measurement of relative improvement. A positive ΔEF represents a larger improvement of the enrichment factor by the combined Z-score based ranking. If both the combined Z-score and the docking score had an identical enrichment factor, no bar is shown in the figure.
3.5. Development of a user-friendly python script
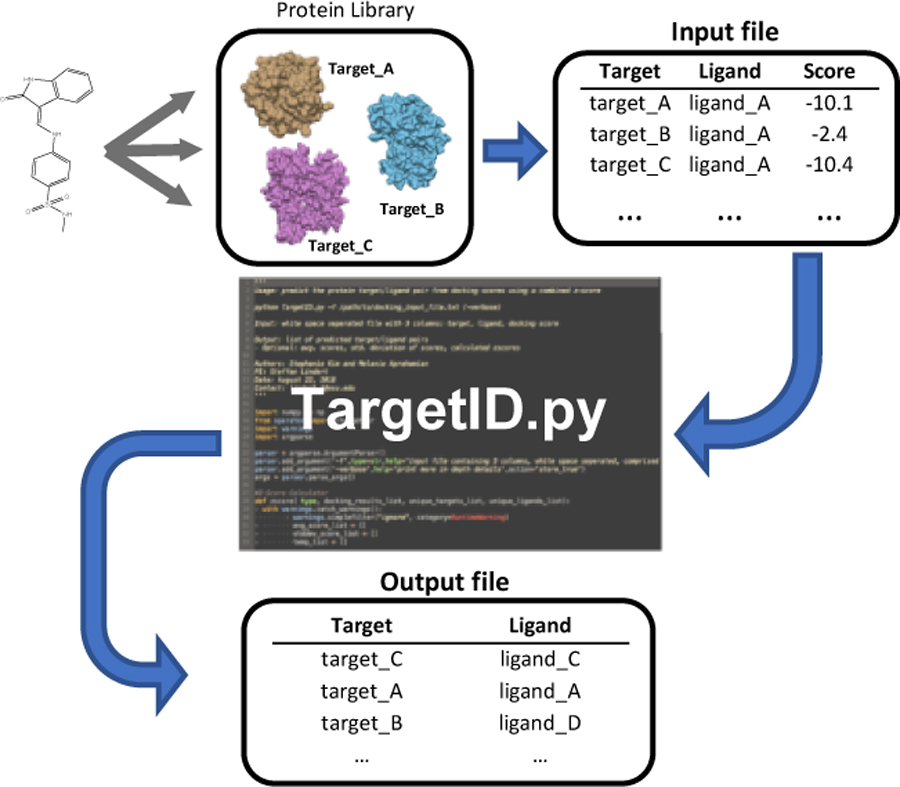
A Python script, TargetID.py, was created that reads in a user-generated input file comprised of a list of the user’s docking results (regardless of the software used to perform the docking) and outputs the predicted target-ligand combination. The script was written with a focus on user-friendliness and only requires a single user input. The input docking results need to be formatted in a three column, whitespace separated list containing the protein receptor name, ligand name, and docking score (one set per line). This script, along with a tutorial, has been made freely available to whomever wishes to use it and is accessible through the supporting information.
Conclusions
The development of inverse docking methods and web-servers for target identification has been driven by an increasing demand for efficient tools for identifying off-target interactions to predict potential drug side effects. By virtually docking a single compound to multiple proteins, inverse docking methods allow facile screening of large protein libraries. However, limitations of applying such molecular docking methods for target identification became apparent when compound docking scores were used as the main criterion for target selection. Thus, in this study, we investigated optimal ways of correctly selecting ligand-protein pairs from inverse docking results by working with three different datasets: Astex, DUD, and DUD-E.
The variation in score ranges for different binding site environments prompted the use of a Z-score metric instead of the raw docking score to identify the target of a particular ligand. We introduced a novel “Combined Z-score” method for target identification of a ligand, which significantly enhanced the selection of correct ligand-protein pairs. With our combined Z-score method, an additional 14%, 3.6%, and 6.3% of ligand-protein pairs of the Astex, DUD, and DUD-E, respectively, were correctly predicted compared to a docking score-based selection. Additionally, the combined Z-score had the highest AUCs for all three datasets, and the enrichment factor at the top 1% for the combined Z-score was the highest in Astex and DUD-E. We also developed a user-friendly python script that will allow the non-expert users to readily analyze their inverse docking results for target identification. Unlike other web-servers, our python script allows users to screen their query ligands to a custom protein library.
As mentioned earlier, being able to identify protein targets of small molecules has become an important tool for detecting potential side effects of novel and commercialized drugs. Among other known detectors of drug side effects, inverse docking is an efficient tool for screening large protein libraries. By applying our combined Z-score method to the inverse docking, it will assist in further increasing the accuracy of target identification with molecular docking methods.
Supplementary Material
Acknowledgments
We thank the members of the Lindert lab for many useful discussions. We would like to extend a special thanks to Dr. Bill Coldren for sharing his knowledge of ligand structure similarity measurement. We would like to thank the Ohio Supercomputer Center for valuable computational resources (56). This work was supported by NIH (R03 AG054904) to S.L. Additionally, work in the Lindert laboratory is supported through NIH (R01 HL137015, P41 GM128577, R01 AI140541), NSF (CHE 1750666) and a Falk Medical Research Trust Catalyst Award.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Schenone M, Dancik V, Wagner BK, Clemons PA (2013) Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol;9: 232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Comess KM, McLoughlin SM, Oyer JA, Richardson PL, Stöckmann H, Vasudevan A, et al. (2018) Emerging Approaches for the Identification of Protein Targets of Small Molecules - A Practitioners’ Perspective. J Med Chem; [DOI] [PubMed]
- 3.Ivanov SM, Lagunin AA, Rudik AV, Filimonov DA, Poroikov VV (2018) ADVERPred-Web Service for Prediction of Adverse Effects of Drugs. J Chem Inf Model;58: 8–11. [DOI] [PubMed] [Google Scholar]
- 4.Chen YZ, Ung CY (2001) Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand-protein inverse docking approach. J Mol Graph Model;20: 199–218. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Huang T, Zhang J, Zheng MY, Feng KY, Cai YD, et al. (2013) Predicting drugs side effects based on chemical-chemical interactions and protein-chemical interactions. Biomed Res Int;2013: 485034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizutani S, Pauwels E, Stoven V, Goto S, Yamanishi Y (2012) Relating drug-protein interaction network with drug side effects. Bioinformatics;28: i522–i8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng W, Sun W, Simeonov A (2018) Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br J Pharmacol;175: 181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez JJ, Pryszlak M, Smith L, Yanchus C, Kurji N, Shahani VM, et al. (2017) Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Frontiers in oncology;7: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. (2002) The Protein Data Bank. Acta Crystallogr D Biol Crystallogr;58: 899–907. [DOI] [PubMed] [Google Scholar]
- 10.Berman HM, Coimbatore Narayanan B, Di Costanzo L, Dutta S, Ghosh S, Hudson BP, et al. (2013) Trendspotting in the Protein Data Bank. FEBS Lett;587: 1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leelananda SP, Lindert S (2016) Computational methods in drug discovery. Beilstein journal of organic chemistry;12: 2694–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov;3: 935–49. [DOI] [PubMed] [Google Scholar]
- 13.Meng XY, Zhang HX, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Current computer-aided drug design;7: 146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE (1982) A geometric approach to macromolecule-ligand interactions. J Mol Biol;161: 269–88. [DOI] [PubMed] [Google Scholar]
- 15.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. (2004) Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. Journal of Medicinal Chemistry;47: 1739–49. [DOI] [PubMed] [Google Scholar]
- 16.McGann M (2011) FRED Pose Prediction and Virtual Screening Accuracy. Journal of Chemical Information and Modeling;51: 578–96. [DOI] [PubMed] [Google Scholar]
- 17.Trott O, Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry;31: 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD (2003) Improved protein-”ligand docking using GOLD. Proteins: Structure, Function, and Bioinformatics;52: 609–23. [DOI] [PubMed] [Google Scholar]
- 19.Kramer B, Rarey M, Lengauer T (1999) Evaluation of the FLEXX incremental construction algorithm for protein- ”ligand docking. Proteins: Structure, Function, and Bioinformatics;37: 228–41. [DOI] [PubMed] [Google Scholar]
- 20.Meiler J, Baker D (2006) ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins;65: 538–48. [DOI] [PubMed] [Google Scholar]
- 21.Talele TT, Khedkar SA, Rigby AC (2010) Successful Applications of Computer Aided Drug Discovery: Moving Drugs from Concept to the Clinic. Current Topics in Medicinal Chemistry;10: 127–41. [DOI] [PubMed] [Google Scholar]
- 22.Clark DE (2006) What has computer-aided molecular design ever done for drug discovery? Expert Opinion on Drug Discovery;1: 103–10. [DOI] [PubMed] [Google Scholar]
- 23.Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov;3: [DOI] [PubMed] [Google Scholar]
- 24.Feixas F, Lindert S, Sinko W, McCammon JA (2014) Exploring the role of receptor flexibility in structure-based drug discovery. Biophysical chemistry;186: 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinko W, Lindert S, McCammon JA (2013) Accounting for Receptor Flexibility and Enhanced Sampling Methods in Computer-Aided Drug Design. Chem Biol Drug Des;81: 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aprahamian ML, Tikunova SB, Price MV, Cuesta AF, Davis JP, Lindert S (2017) Successful Identification of Cardiac Troponin Calcium Sensitizers Using a Combination of Virtual Screening and ROC Analysis of Known Troponin C Binders. J Chem Inf Model;57: 3056–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindert S, Zhu W, Liu YL, Pang R, Oldfield E, McCammon JA (2013) Farnesyl diphosphate synthase inhibitors from in silico screening. Chem Biol Drug Des;81: 742–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu YL, Lindert S, Zhu W, Wang K, McCammon JA, Oldfield E (2014) Taxodione and arenarone inhibit farnesyl diphosphate synthase by binding to the isopentenyl diphosphate site. Proc Natl Acad Sci U S A;111: E2530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alberts IL, Todorov NP, Dean PM (2005) Receptor flexibility in de novo ligand design and docking. J Med Chem;48: 6585–96. [DOI] [PubMed] [Google Scholar]
- 30.Chan AH, Wereszczynski J, Amer BR, Yi SW, Jung ME, McCammon JA, et al. (2013) Discovery of Staphylococcus aureus sortase A inhibitors using virtual screening and the relaxed complex scheme. Chem Biol Drug Des;82: 418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durrant JD, Cao R, Gorfe AA, Zhu W, Li J, Sankovsky A, et al. (2011) Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chem Biol Drug Des;78: 323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim MO, Feng X, Feixas F, Zhu W, Lindert S, Bogue S, et al. (2015) A Molecular Dynamics Investigation of Mycobacterium tuberculosis Prenyl Synthases: Conformational Flexibility and Implications for Computer-aided Drug Discovery. Chem Biol Drug Des;85: 756–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen YZ, Zhi DG. (2001) Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule United States: 2001 Wiley-Liss, Inc: p. 217–26. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Zhang G, Zhou Y, Lin C, Chen S, Lin Y, et al. (2018) Reverse Screening Methods to Search for the Protein Targets of Chemopreventive Compounds. Front Chem;6: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu X, Huang M, Zou X (2018) Docking-based inverse virtual screening: methods, applications, and challenges. Biophys Rep;4: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Zhou X, He W, Fan Y, Chen Y, Chen X (2012) The interprotein scoring noises in glide docking scores. Proteins;80: 169–83. [DOI] [PubMed] [Google Scholar]
- 37.Schomburg KT, Bietz S, Briem H, Henzler AM, Urbaczek S, Rarey M (2014) Facing the challenges of structure-based target prediction by inverse virtual screening. J Chem Inf Model;54: 1676–86. [DOI] [PubMed] [Google Scholar]
- 38.Luo Q, Zhao L, Hu J, Jin H, Liu Z, Zhang L (2017) The scoring bias in reverse docking and the score normalization strategy to improve success rate of target fishing. PLoS One;12: e0171433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Gao Z, Kang L, Zhang H, Yang K, Yu K, et al. (2006) TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res;34: W219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L, Luo H, Chen J, Xing Q, He L (2009) SePreSA: a server for the prediction of populations susceptible to serious adverse drug reactions implementing the methodology of a chemical-protein interactome. Nucleic Acids Res;37: W406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang JC, Chu PY, Chen CM, Lin JH (2012) idTarget: a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Nucleic Acids Res;40: W393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao Z, Li H, Zhang H, Liu X, Kang L, Luo X, et al. (2008) PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics;9: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang JX, Huang WJ, Zeng JH, Huang WH, Wang Y, Zhao R, et al. (2007) DITOP: drug-induced toxicity related protein database. Bioinformatics;23: 1710–2. [DOI] [PubMed] [Google Scholar]
- 44.Ji ZL, Han LY, Yap CW, Sun LZ, Chen X, Chen YZ (2003) Drug Adverse Reaction Target Database (DART) : proteins related to adverse drug reactions. Drug Saf;26: 685–90. [DOI] [PubMed] [Google Scholar]
- 45.Wang JC, Lin JH, Chen CM, Perryman AL, Olson AJ (2011) Robust scoring functions for protein-ligand interactions with quantum chemical charge models. J Chem Inf Model;51: 2528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zahler S, Tietze S, Totzke F, Kubbutat M, Meijer L, Vollmar AM, et al. (2007) Inverse in silico screening for identification of kinase inhibitor targets. Chem Biol;14: 1207–14. [DOI] [PubMed] [Google Scholar]
- 47.Kosmopoulou MN, Leonidas DD, Chrysina ED, Bischler N, Eisenbrand G, Sakarellos CE, et al. (2004) Binding of the potential antitumour agent indirubin-5-sulphonate at the inhibitor site of rabbit muscle glycogen phosphorylase b. Comparison with ligand binding to pCDK2-cyclin A complex. Eur J Biochem;271: 2280–90. [DOI] [PubMed] [Google Scholar]
- 48.Abagyan R, Totrov M. and Kuznetsov D. (1994) ICM-A new method for protein modeling and design: Application to docking and structure prediction from the distorted native conformation. ICM-A new method for protein modeling and design: Application to docking and structure prediction from the distorted native conformation.: J.Comput.Chem; p. 488–506.
- 49.Li YY, An J, Jones SJ (2011) A computational approach to finding novel targets for existing drugs. PLoS Comput Biol;7: e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WT, Mortenson PN, et al. (2007) Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem;50: 726–41. [DOI] [PubMed] [Google Scholar]
- 51.Huang N, Shoichet BK, Irwin JJ (2006) Benchmarking sets for molecular docking. J Med Chem;49: 6789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK (2012) Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem;55: 6582–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrödinger. (2018) Schrödinger Release 2018–2 : LigPrep. Schrödinger Release 2018–2 : LigPrep LLC, New York, NY. [Google Scholar]
- 54.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem;47: 1750–9. [DOI] [PubMed] [Google Scholar]
- 55.(1987) Ohio Supercomputer Center. Ohio Supercomputer Center
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.