

Abstract
Myotonic dystrophy type 1 (DM1) is caused by an expanded (CTG)n tract in the 3′UTR of the DM protein kinase (DMPK) gene. The RNA transcripts produced from the expanded allele sequester or alter the function of RNA-binding proteins (MBNL1, CUGBP1, etc.). The sequestration of MBNL1 results in RNA-splicing defects that contribute to disease. Overexpression of MBNL1 in skeletal muscle has been shown to rescue some of the DM1 features in a mouse model and has been proposed as a therapeutic strategy for DM1. Here, we sought to confirm if overexpression of MBNL1 rescues the phenotypes in a different mouse model of RNA toxicity. Using an inducible mouse model of RNA toxicity in which expression of the mutant DMPK 3′UTR results in RNA foci formation, MBNL1 sequestration, splicing defects, myotonia and cardiac conduction defects, we find that MBNL1 overexpression did not rescue skeletal muscle function nor beneficially affect cardiac conduction. Surprisingly, MBNL1 overexpression also did not rescue myotonia, though variable rescue of Clcn1 splicing and other splicing defects was seen. Additionally, contrary to the previous study, we found evidence for increased muscle histopathology with MBNL1 overexpression. Overall, we did not find evidence for beneficial effects from overexpression of MBNL1 as a means to correct RNA toxicity mediated by mRNAs containing an expanded DMPK 3′UTR.
Introduction
Myotonic dystrophy type 1 (DM1) is the most common form of adult muscular dystrophy and is a multisystemic disorder (1). DM1 is an autosomal dominant disorder with a CTG repeat tract in the 3′ untranslated region (3′UTR) of the dystrophia myotonica protein kinase (DMPK) gene (2). The mutant DMPK mRNA forms aggregates that are retained in the nucleus as RNA foci (3). The repeat RNA sequesters muscleblind-like 1 (MBNL1) protein, and it is proposed that the functional loss of MBNL1 results in RNA splicing defects that contribute to DM1 phenotypes (1). Consistent with this hypothesis, mice in which the Mbnl1 gene is functionally deleted develop myotonia and skeletal muscle pathology similar to human DM1 (4,5). In addition, increasing MBNL1 expression either by localized expression of MBNL1 in tibialis anterior muscle using adeno-associated virus (AAV)-mediated overexpression of MBNL1 in the human-skeletal actin-long repeat (HSA-LR) mice or transgenic overexpression of MBNL1 in HSA-LR mice has been shown to rescue some of the DM1 features (6,7). These data suggest that up-regulation of MBNL1 might be good strategy to treat DM1.
We have developed an inducible mouse model of RNA toxicity in which expression of the toxic RNAs results in the RNA foci formation, MBNL1 sequestration, splicing defects, myotonia and cardiac conduction defects. In this study, we tested if overexpression of MBNL1 rescues the phenotypes in this mouse model of RNA toxicity. Our results indicate that MBNL1 overexpression rescues splicing defects variably and that it worsens myopathy in the mouse model of RNA toxicity.
Results
Characterization of mice overexpressing MBNL1
We obtained transgenic mice that overexpress MBNL1 (40 kDa isoform) from Dr Laura Ranum (6). These mice have been used in a prior study to demonstrate that they rescue key DM1 phenotypes such as myotonia, central nuclei and RNA-splicing defects in the HSA-LR mouse model of RNA toxicity (6). The two mouse lines, MBNL1-OE14685+/WT and MBNL1-OE14686+/WT, were first characterized by electromyography (EMG), electrocardiography (ECG), treadmill running and forelimb grip strength. We found no significant differences between the MBNL1-OE14685+/WT mice and their wild-type littermates. In contrast, MBNL1-OE14686+/WT mice show significantly reduced body weight, grip strength and run distance as compared to wild-type littermates (Supplementary Material, Table S1). These results were comparable to previously reported results for these mice, notably that the MBNL1-OE14686+/WT mice were smaller than the wild-type mice (6). The mice were then given doxycycline (D) (0.2%) for 6 weeks and reanalyzed by the various phenotypic analyses. We found that MBNL1-OE14685+/WT mice did not develop any muscle phenotypes at the end of 6 weeks. This ruled out any effect of D related to the measured phenotypes. Similarly, the phenotypes for the MBNL1-OE14686+/WT mice were not affected by D administration (Supplementary Material, Table S1). The relative levels of transgene expression in the MBNL1-OE14685+/WT and MBNL1-OE14686+/WT mice was confirmed by western blotting using an anti-MBNL1 monoclonal antibody (Supplementary Material, Fig. S1). These results were comparable to those previously reported for these mice. Immunofluorescence using an anti-MBNL1 monoclonal antibody (Supplementary Material, Fig. S2A) and an anti-FLAG antibody (Supplementary Material, Fig. S2B) was also performed to determine the relative levels as compared to wild-type littermates. This showed increased expression of MBNL1 and expression of the FLAG-tagged MBNL1 transgenic protein in the MBNL1-OE14685+/WT mice.
Phenotypic effects of MBNL1 overexpression in RNA toxicity mice
We have an inducible/reversible mouse model of RNA toxicity (DM200+). Despite some leakiness of the transgene, in the absence of D, the DM200+ mice are phenotypically normal (Supplementary Material, Table S2, D− mice). Upon D administration, the expression of a (CUG)n-expanded DMPK 3’UTR mRNA results in RNA foci formation (Supplementary Material, Fig. S3), MBNL1 sequestration (Fig. 1C), splicing defects (Fig. 2), myotonia and cardiac conduction defects (Supplementary Material, Table S2) (8). Notably, cessation of toxic RNA expression by withdrawing D induction results in correction of DM1 related phenotypes (Supplementary Material, Table S2), thus demonstrating the potential utility of this mouse model in assessing the effects of genetic modifiers or potential therapeutics (9).
Figure 1.
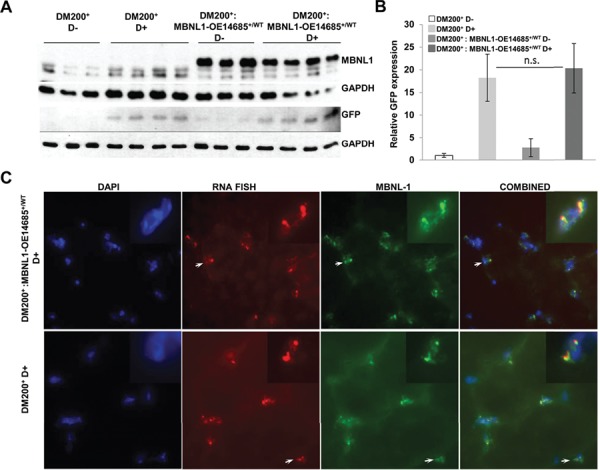
Characterization of DM200 mice overexpressing MBNL1. (A) Western blot analysis showing expression of MBNL1 in skeletal muscle (gastrocnemius/soleus) from DM200+ and DM200+: MBNL1-OE14685+/WT uninduced and induced mice (anti MBNL1-A2764 antibody). Western blot for GFP confirmed transgene induction. GAPDH was used as a loading control. (B) Quantification of the GFP expression from western blot in (A) shows no significant difference between DM200+- and DM200+: MBNL1-OE14685+/WT-induced mice. N = 3–4 mice per group. Errors bars are mean ± S.D. Student’s t-test used. n.s. indicates significance P-value > 0.05. (C) Immunofluorescence for MBNL1 (green) was performed using the M02-clone 3E7 (anti-MBNL1 monoclonal antibody, Abnova) in skeletal muscle (quadriceps femoris) from DM200+ and DM200+: MBNL1-OE14685+/WT mice induced for toxic RNA. RNA-FISH (red) was performed to study MBNL1 co-localization with RNA foci, with insets showing higher magnification focusing on indicated RNA foci. DAPI (blue) was used to stain nuclei. Representative images are also presented in Supplementary Material, Figure S4C.
Figure 2.
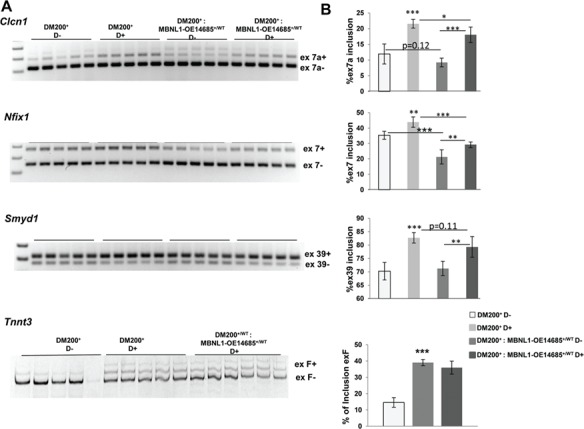
Analysis of RNA-splicing defects. (A) RT-PCR analysis of Clcn1 splicing in skeletal muscle (gastrocnemius/soleus) from DM200+ mice showing increased inclusion of ex7a. RT-PCR analysis of other RNA-splicing targets, Nfix, Smyd1 and Tnnt3, shows that RNA toxicity leads to splicing defects in the DM200+ mice for all the splicing targets. (B) Quantification of the gels in (A) shows significant splicing defects in the target tested. MBNL1 overexpression has significant effect on splicing of Clcn 1(ex7a) and Nfix (ex7) and trends towards significance for Smyd1 (ex39) but no effect on Tnnt3 (exF). In uninduced mice, MBNL1 overexpression has significant effect of splicing on Nfix (ex7) and trends towards significance for Clcn1 (ex7a) but no effect on other targets. N = 5/groups; (*P = 0.05, **P = 0.01, ***P = 0.001, Student’s t-test); error bars are mean ± S.D.
To test whether overexpression of MBNL1 ameliorates the adverse phenotypes in the RNA toxicity mice, we crossed DM200+ mice with both the MBNL1transgenic lines. Despite repeated attempts over a period of a year (more than 14 litters), we were never able to generate any adult DM200+ mice that also carried the MBNL1-OE14686+/WT transgene. A total of 22 DM200+: MBNL1-OE14686+/WT mice were found dead on the first post-natal day. There were another 24 mice that were DM200+: MBNL1-OE14686+/WT mice that survived to weaning (about 4–6 weeks at most). We were able to do analyses on 19 uninduced (i.e. D−) mice (11 of which were DM200+: MBNL1-OE14686+/WT and 8 were DM200+). Every one of the DM200+: MBNL1-OE14686+/WT mice was much smaller as compared to the DM200+ littermates (average total weight 8.8 g versus 17.8 g, P = 3.1 × 10−8). This was in sharp contrast to the MBNL1-OE14686+/WT line by itself, which on average was only about 20% smaller at 2 months of age as compared to wild-type mice (Supplementary Material, Table S1). When we did phenotypic analyses on these mice, none had myotonia. All the DM200+ mice had normal heart rhythms. However, all the DM200+: MBNL1-OE14686+/WT mice had significant degrees of heart block (2nd- and 3rd-degree heart blocks with marked bradycardia). Furthermore, when we compared the ratio of heart weight to total body weight (HW/BW), we found that the HW/BW in MBNL1-OE14686+/WT and the DM200+ mice were comparable (5.47% versus 5.86%, P = 0.96), whereas the DM200+: MBNL1-OE14686+/WT mice had a HW/BW ratio of 8.71% (P = 0.0005), demonstrating relative cardiomegaly as compared to the individual lines. The MBNL1-OE14686+/WT mice overexpress MBNL1 at a much greater level than the MBNL1-OE14685+/WT mice (6) (Supplementary Material, Fig. S1). Our results suggest that the combination of baseline DM200+ transgene leakiness and the level of MBNL1 overexpression in the MBNL1-OE14686+/WT line were highly detrimental to their viability.
However, we were readily able to breed the MBNL1-OE14685+/WT mice with the DM200+ mice to generate litters of DM200+: MBNL1-OE14685+/WT as well as a littermate control group of DM200+ only. Baseline analyses were done on mice at 2 months of age prior to D administration. Treadmill running showed that the DM200+: MBNL1-OE14685+/WT mice ran for far shorter distances (about 40% less than DM200+, P = 0.01; Fig. 3A). Neither group exhibited myotonia (Fig. 3B). Interestingly, some of the DM200+: MBNL1-OE14685+/WT mice were already exhibiting prolongation of the PR interval and 2nd-degree heart block in contrast to the normal sinus rhythm found in the DM200+ mice (Fig. 3C). Though these changes were not as drastic as those seen in the DM200+: MBNL1-OE14686+/WT mice, they showed a similar deleterious effect of MBNL1 overexpression on the cardiac rhythms of DM200+ mice.
Figure 3.
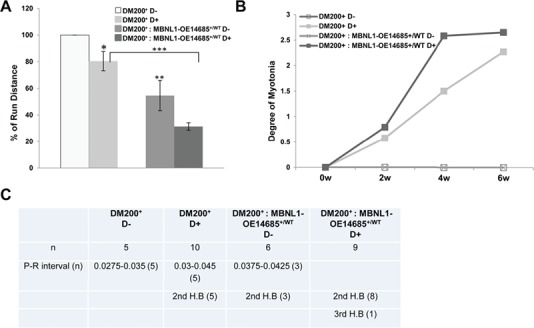
Phenotypic analysis of MBNL1 overexpression. (A) Graph showing percent run distance (relative to pre-induction DM200+ D−) at the end of 6 weeks induction of toxic RNA. Run distance is significantly different between uninduced and induced DM200+ mice (P = 0.019), between DM200+: MBNL1-OE14685+/WT-uninduced and DM200+-uninduced mice (P = 0.01) and between DM200+ D+ and DM200: MBNL1-OE14685+/WT D+ mice (*P < 0.05, **P = 0.01, ***P = 0.000048); (n = 5 to 10/group). (B) Graph showing that there was no significant difference in myotonia between DM200:MBNL1-OE14685+/WT-induced and DM200+-induced mice at 6 weeks post-induction. For all graphs (in A and B), errors bars are mean ± standard error of the mean (SEM). Student’s t-test applied to all comparisons. (C) ECG studies in DM200+ and DM200+: MBNL1-OE14685+/WT mice induced for toxic RNA (n = 5 to 10 mice per group used in the study).
Induction of toxic RNA expression by D administration for 6 weeks made the DM200+: MBNL1-OE14685+/WT and the DM200+ mice weaker. The percentage drop in run distance in induced DM200+: MBNL1-OE14685+/WT mice was similar to that of induced DM200+ mice, suggesting that MBNL1 overexpression had no beneficial effect on this phenotype (Fig. 3A). We also examined myotonia using EMG and cardiac conduction by ECG. Uninduced DM200+ and DM200+: MBNL1-OE14685+/WT mice did not exhibit any myotonia. However, the DM200+: MBNL1-OE14685+/WT and DM200+ mice developed similar degree of myotonia at the end of 6 weeks of induction (Fig. 3B). The DM200+: MBNL1-OE14685+/WT- and DM200+-induced mice developed conduction abnormalities. Notably, more of the DM200+: MBNL1-OE14685+/WT D+ mice showed severe conduction abnormalities (Fig. 3C). These data suggest that myotonia and cardiac function are not beneficially affected by overexpression of MBNL1 in the RNA toxicity mice.
Skeletal muscle and cardiac tissues were collected from mice in both groups. Induction of the GFP-DMPK 3′UTR transgene was confirmed by western blotting for GFP (Fig. 1A and B) and by RNA-FISH to detect RNA foci (Fig. 1C, Supplementary Material, Fig. S3) and quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Supplementary Material, Fig. S4A). In the DM200+: MBNL1-OE14685+/WT mice, overexpression of MBNL1 was confirmed by western blot and immunofluorescence (Fig. 1A, Supplementary Material, Fig. S4B and C). In the DM200+: MBNL1-OE14685+/WT-induced mice and the DM200+-induced mice, MBNL1 co-localization with the RNA foci was noted (Fig.1B, Supplementary Material, Fig. S4D). We did not see any obvious difference in either the size or number of foci and saw no evidence of cytoplasmic RNA foci.
Alternative splicing is partially corrected by MBNL1 overexpression in the RNA toxicity mice
To determine whether overexpression of MBNL1 would correct the mis-splicing events caused by the toxic RNA, we analyzed splicing of Clcn1 (ex7a), Nfix1 (ex7), Smyd1 (exons 39) and Tnnt3 (fetal exon, 5a), targets that are mis-spliced in skeletal muscles of individuals with DM1 (6,10). All these targets were found to be mis-spliced in the DM200+ induced mice (Fig 2A and B). In contrast to the previous report where MBNL1 overexpression rescued splicing defects in the HSA-LR mice (6), we found that MBNL1 overexpression results in variable rescue of a number of splicing targets in the DM200 mouse model (Fig. 2A and B, Supplementary Material, Fig. S5).
MBNL1 overexpression worsens myopathy in the RNA toxicity mice
To test whether overexpression of MBNL1 affects myopathy, we compared hematoxylin and eosin staining of quadriceps muscle sections from 3- to 4-month-old DM200+: MBNL1-OE14685+/WT and DM200+ mice that had been induced with D for 6 weeks. Surprisingly, the DM200+: MBNL1-OE14685+/WT-induced mice showed marked increased pathology including variation in fiber size and increased central nuclei (Fig. 4A). Analysis of central nuclei and fiber size (<30 μm) showed a significant increase in the percentage of centrally nuclei (40.6% versus 3.4%, P = 1.4E-05) and small fibers (25.4% versus 2.4%, P = 3.147E-10) in DM200+: MBNL1-OE14685+/WT-induced mice as compared to DM200+-induced mice (Fig. 4B and C). Thus, in contrast to previous results (6), we found that the skeletal muscle of DM200+: MBNL1-OE14685+/WT-induced mice had worse histopathology as compared to DM200+-induced mice.
Figure 4.
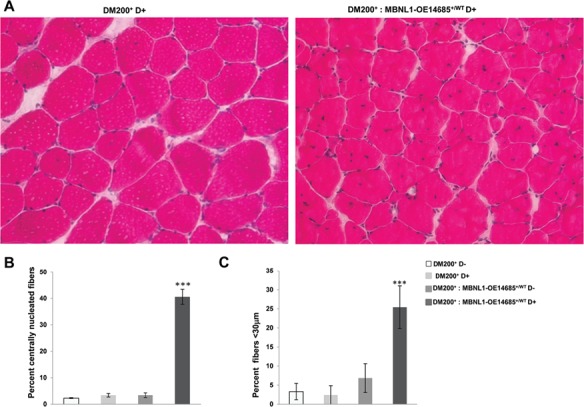
Overexpression of MBNL1 worsens the myopathy in DM200 mice. (A) H&E staining analysis of skeletal muscle (quadriceps femoris) shows increased central nuclei and smaller fiber size in DM200+: MBNL1-OE14685+/WT mice as compared to the DM200+ mice alone. (B) Quantification of centrally nucleated fibers in DM200+ D−, DM200+ D+, DM200+: MBNL1-OE14685+/WT D− and DM200+: MBNL1-OE14685+/WT D+ mice. (C) Percentage of small fibers (average fiber cross-sectional area < 30 μm) in skeletal muscle from DM200+ D−, DM200+ D+, DM200+: MBNL1-OE14685+/WT D− and DM200+: MBNL1-OE14685+/WT D+ mice. At least n = 4 mice analyzed each group; ***P < 0.001. Errors bars are mean ± SEM. (Student’s t-test).
The presence of central nuclei is considered by many to be an indicator of muscle regeneration (11). For example, the increased central nuclei in skeletal muscles of the mdx mouse (a mouse model of Duchenne muscular dystrophy) are attributed to the regenerative response to repeated episodes of muscle damage. This is associated with increased expression of embryonic and neonatal myosins such as Myh3 and Myh8. To evaluate if the increased central nuclei in induced DM200+: MBNL1-OE14685+/WT skeletal muscles is due to muscle regeneration, we analyzed expression of mRNAs encoding several myogenic markers of regeneration. We found that the expression of Pax7 (2-fold) and Myog (1.5-fold) were up-regulated in DM200+: MBNL1-OE14685+/WT as compared to DM200+ with RNA toxicity (Fig. 5A). We also found that Myh3 mRNA levels were increased 10-fold, and Mhy8 mRNA levels were increased 2-fold in quadriceps muscles (Fig. 5A). However, the expression of Myh3 was vastly lower as compared to MDX muscle where its expression is increased by 150-fold (Fig. 5B). Congruently, immunofluorescence analysis of embryonic MHC protein (MYH3) showed only rare expression in DM200+: MBNL1-OE14685+/WT muscle fibers as compared to MDX muscle (Fig. 5C). These data indicate that there is little evidence of muscle regeneration in DM200+: MBNL1-OE14685+/WT to account for the extensive central nucleated fibers observed in these mice.
Figure 5.
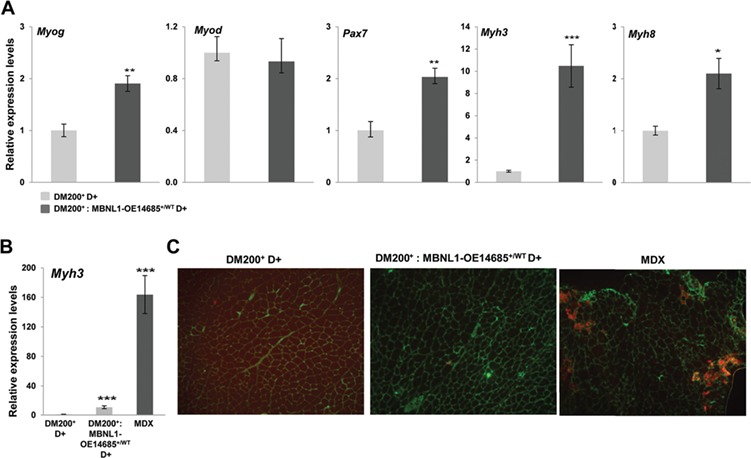
Effects of MBNL1 overexpression on target genes implicated in muscle regeneration. (A) Quantitative RT-PCR shows increased expression of markers of regeneration (Myog, Pax7, Myh3 and Myh8 mRNA levels) in skeletal muscle (gastrocnemius/soleus) from DM200+: MBNL1-OE14685+/WT-induced mice as compared to DM200+ induced (*P < 0.05, **P = 0.01). (B) Quantitative RT-PCR shows increased Myh3 mRNA levels in MDX mice as compared to induced DM200+ or DM200+: MBNL1-OE14685+/WT mice (***P = 0.0009). At least 5 mice per group were analyzed. For all graphs, errors bars are mean ± SEM. Student’s t-test applied to all comparisons. (C) Immunofluoresence for MYH3 shows little evidence for MYH3 expression in the skeletal muscles (quadriceps femoris) of DM200+ or DM200+: MBNL1-OE14685+/WT mice as compared to MDX mice (MYH3 (red); Laminin (green); nuclei (blue); genotypes of mice as indicated.
Discussion
In DM, MBNL1 sequestration has been shown to play key role in the pathogenesis of DM1 (4,12,13). Therapeutic strategies to either free up MBNL1 or increase the expression of MBNL1 to rescue DM1 pathogenesis are getting more attention (6,7,14). Here, we wanted to test whether increasing MBNL1 expression in a mouse model of RNA toxicity (DM200) can rescue the DM1 phenotypes. Our data show that MBNL1 overexpression was not effective in reversing DM1 phenotypes such as myotonia and cardiac conduction abnormalities. Also, the mice did not show improvement in function assays such as grip strength or treadmill running. We found that MBNL1 overexpression notably increased muscle histopathology and resulted in variable rescue of a number of splicing targets.
This is in contrast to the previous studies where overexpression of MBNL1 in the HSA-LR mice has been shown to rescue myotonia, myopathy and alternative splicing abnormalities (6,7). Because MBNL sequestration is thought to play an important role in DM pathogenesis through aberrant alternative splicing, we also analyzed splicing of MBNL1 targets in the DM200 mice overexpressing MBNL1. We found that there was very little rescue of splicing abnormalities associated with DM1, including splicing of Clcn1 (Fig. 2). This suggests that either free MBNL1 is not enough to rescue splicing of Clcn1or other factors are required in combination with MBNL1 to regulate Clcn1 splicing and myotonia. In addition to its role in splicing, MBNL1 has been implicated in a wide range of tissue-specific functions including RNA localization and translation, mRNA decay, alternative polyadenylation and regulation of miRNA processing (15–18). For example, in a recent study (19), the authors found that MBNL1 bound to over 2400 targets, and based on their bioinformatics analyses, they concluded that MBNL1 has a higher chance of binding to spliced RNAs rather than being a splice effector. Furthermore, they found that overexpression of MBNL1 in mice led to fibroblast differentiation and increased tissue fibrosis in a variety of tissues (19). Consistent with this, we found that expression of various markers of fibrosis was increased in the hearts of DM200+: MBNL1-OE14685+/WT mice (Supplementary Material, Fig. S6). All these studies only serve to highlight the complexity of MBNL1’s role in RNA metabolism. However, the functional consequences of these roles of MBNL1 in the pathology associated with RNA toxicity are yet to be well defined.
Cardiac abnormalities are common in DM1 (1). In humans and mice, DMPK is highly expressed in the heart (20,21), at comparable levels to that seen in skeletal muscle. RNA foci and MBNL co-localization have been reported in cardiac tissues of individuals with DM1 (22) as well as in mouse models of RNA toxicity (23) including the DM200+ mice (Supplementary Material, Fig. S7). Previous studies have shown that loss of MBNL1 and concomitant depletion of MBNL2 result in cardiac functional abnormalities in mice (5,24). However, cardiac phenotypes have not been studied in mice overexpressing MBNL1. We examined the effect of MBNL1 overexpression on cardiac conduction in the MBNL1 transgenic lines. In both lines (i.e. MBNL1-OE14685+/WT and MBNL1-OE14686+/WT), neither the overexpression of MBNL1 by itself nor the administration of D to these mice resulted in observable cardiac conduction defects (Supplementary Material, Table S1). In our DM200 mice, even in the absence of D induction of the GFP transgene, the leakiness from the transgene is enough to generate RNA foci (Supplementary Material, Fig. S3 and S7) as well as splicing defects for Cacna1s (Cav1.1) and Myom (Myomesin) in the heart (Supplementary Material, Fig. S8). However, these were not associated with conduction abnormalities (Supplementary Material, Table S2). But, when the DM200+ mice were bred with either the MBNL1-OE14685+/WT or the MBNL1-OE14686+/WT line, we saw worsening conduction abnormalities, even in the absence of D (Fig. 3C). This worsened further in the DM200+: MBNL1-OE14685+/WT mice once RNA toxicity was induced (Fig. 3C). In fact, in the DM200+: MBNL1-OE14686+/WT mice, we observed severe cardiac conduction abnormalities that were associated with heart failure and cardiomegaly, resulting in death by 6 weeks of age, in the absence of any D induction. Furthermore, the splicing assays show minimal to no correction of Cacna1s and Myom splicing defects in the hearts of DM200+: MBNL1-OE14685+/WT mice (Supplementary Material, Fig. S8). Our results suggest that overexpression of MBNL1 did not beneficially correct the splicing defects or cardiac conduction defects in the RNA toxicity mice.
Previous studies suggested that overexpression of MBNL1 led to less skeletal muscle myopathy and reduced central nuclei in the HSA-LR mice (4,6). Surprisingly, and in contrast, we found increased myopathy and central nuclei after overexpression of MBNL1 in the DM200+ RNA toxicity mice (Fig. 4). This was associated with slight but significant increases in Pax7 and Myog mRNA levels but little evidence for active muscle regeneration (Fig. 5). Interestingly, MBNL1-OE14685+/WT mice by themselves showed increased Pax7 mRNA levels and increased PAX7 positive cells in skeletal muscles (Supplementary Material, Fig. S9). Similar results were seen with the MBNL1-OE14686+/WT line (data not shown). The significance of the increase in PAX7 positive cells is unclear. Though MBNL3 has been reported to play a role in muscle regeneration in older mice (25), the role of MBNL1 in muscle regeneration and skeletal muscle satellite cell function is undefined and will require further investigation.
The differences in the results between our study and the previous study (6) could be attributed to the mouse models used in the studies. In the HSA-LR mice, the CUG repeats are expressed outside the context of the DMPK gene, as they are incorporated in the 3′UTR of the human skeletal actin gene. Unlike the HSA-LR mice, the DM200 mice express an expanded repeat as a part of the DMPK 3′UTR. Interestingly, in an independent mouse model overexpressing a (CUG)5DMPK 3′UTR, the DM5 mouse model of RNA toxicity (8), we again found that the MBNL1-OE14685 transgenic line did not rescue myotonia (Supplementary Material, Fig. S10) and adversely affected skeletal muscle histopathology with a striking increase in central nuclei and fiber size variation (Supplementary Material, Fig. S11). These mice do not form discernable RNA foci, but we have previously demonstrated that MBNL1 interacts with the transgene RNA (26). These differences between the HSA-LR mice and mice that express the DMPK 3′UTR could be relevant as it has been previously shown that the presence of the DMPK 3’UTR makes a significant difference with respect to deleterious effects of the toxic RNA on C2C12 mouse myoblast differentiation (27). Also, as the DM200 mouse model uses the human DMPK promoter (28) to drive transgene expression, the expression of the toxic RNA results in multi-systemic expression including skeletal, cardiac and smooth muscle (8). This difference in promoters could also lead to temporal as well as fiber-specific differences in transgene expression relative to the HSA-LR mouse. Other possibilities include that the DM200 mice produce more toxic RNA (i.e. increased CUG load) as compared to the HSA-LR mice or that more of the mutant transcripts may be processed fully. However, using an assay designed to measure CUG RNA load, we found that the DM200 mice produced about five times less CUG RNA than the HSA-LR mice in skeletal muscle (Supplementary Material, Fig. S12).
In conclusion, a number of strategies have been used to correct toxic RNA effects in mouse models either by degrading toxic RNAs or displacing MBNLs from toxic RNAs (29–32). And, although a pervious study has reported that MBNL1 overexpression can rescue some of the DM1 associated phenotypes, our current results strike a more cautionary note with regard to this strategy for treatment of RNA toxicity. Our results only emphasize the point that MBNL1 is a multifunctional protein with many as yet uncharacterized functions and that targeting it may require a more nuanced approach to modulating its levels and functions in order to develop a safe and effective treatment.
Materials and Methods
Transgenic mice
The DM200 mouse model expresses a GFP-DMPK 3′UTR (CTG) 200 transgene under the control of the human DMPK promoter upon induction with D (8). MBNL1-OE lines (MBNL1-OE14685+/WT and MBNL1-OE14686+/WT) were obtained from Dr Ranum (6). DM200+ mice were bred with MBNL1-OE14685+/WT to assess effects on phenotypes. The MBNL1-OE lines were in an FVB background, and the DM200+ lines were in a mixed background (FVB/Bl6). In all studies comparing the pure MBNL1-OE lines to wild-type mice, we used FVB control mice. In studies utilizing DM200+ crossed with MBNL1-OE14685+/WT, littermate controls were used. The breeding was such that parent of origin for either transgene made no difference. Also male and female were used throughout the studies. The transgenic mice were induced with 0.2% D in drinking water at the age of 2 months for 6 weeks. All animals were used in accordance with protocols approved by the Animal Care and Use Committee at the University of Virginia.
RNA-FISH, immunofluorescence and western blot analysis
For RNA fluorescence in situ hybridization (RNA-FISH), tissue was fixed in 4% paraformaldehyde in 1× phosphate-buffered saline and a Cy3-CAG10 probe used for hybridization. Details about the RNA-FISH protocol are described elsewhere (33). Immunofluorescence was performed as described previously (33). Primary antibodies were anti-Flag (1:1000), anti-MBNL1 monoclonal antibody (Abnova, Taipei city, Taiwan) and anti-MBNL1 (A2764; 1:10 000). Secondary antibodies were from Molecular Probes, Eugene, OR (1; 1000 dilution). Total protein extracts were made using standard protocols in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1%NP40, 0.5% Na-deoxycholate and 0.1% sodium dodecyl sulfate) and protease inhibitor (Roche Inc., Mannheim, Germany, cat. #1873580). Proteins were detected with the following antibodies: GAPDH (Ambion, Foster City, CA #4300), MBNL1 (M02, 3E7, Abnova, Taipei city, Taiwan), MBNL1 (A2764, gift from Dr Charles A. Thornton) and GFP (Invitrogen, Carlsbad, CA).
Phenotypic analysis
All the details about protocols for treadmill running, EMG and ECG are described elsewhere (8,9,34). All results are reported as retained function with reference to baseline for each mouse.
RNA isolation, slot blot, qRT-PCR assays and splicing analysis
We extracted total RNA from skeletal muscle tissues using protocols as described in the previous study (35). All RNAs are DNAse treated and confirmed to be free of DNA contamination. Expression of (CUG)n RNA levels in mouse models was compared using slot blot analysis according to standard procedures. Briefly, three sets of total RNA from skeletal muscle (gluteus maximus) of induced DM200+ and HSALR mice, each consisting of four dilutions (2, 1, 0.5 and 0.25 μg), were loaded onto a membrane (Zeta-Probe GT membrane, Bio-Rad) with a slot blot apparatus (Bio-Rad). After the RNA was cross-linked with the membrane by exposure to UV light for 3 min, the membrane was incubated with a (CAG) 10 2′-O-methyl Digoxygenin (Dig)-labeled RNA probe (IDT) into Ultrahyb (#8669, Ambion, Foster City, CA) at 45°C for overnight. The Dig-labeled probe was detected by using anti-Digoxigenin-AP, Fab fragments (Roche).
QuantiTech Reverse Transcription Kit (Qiagen, Germantown, MD) was used for making cDNA from 1 μg of total RNA. qRT-PCR was done using the BioRad iCycler (Biorad, Hercules, CA) and detected with SYBER Green dye. Data were normalized using an endogenous control (Gapdh), and normalized values were subjected to a 2-ΔΔCt formula to calculate the fold changes between uninduced and induced groups. Primer sequences for qRT-PCR are given in Supplementary Material, Table S3. All the splicing assays were done in at least five mice or more per group. PCR primers and conditions have been described in Supplementary Material, Table S4.
Muscle histology and fiber size determination
Histopathology was assessed by H&E staining of quadriceps femoris (6 μm) cryosections. H&E staining was done according to standard procedures and examined under a light microscope. At least 3–5 mice per group were analyzed, and for each mouse, 3–5 images were analyzed.
Statistical analysis
Statistical significance was determined using a two-tailed Student’s t-test with equal or unequal variance as appropriate. All data are expressed as mean ± standard deviation (S.D.). *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t-test). P < 0.05 was considered statistically significant unless otherwise specified.
Conflict of Interest statement. None declared.
Supplementary Material
Funding
National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01-AR062189, R01-AR45992, R01-AR071170); Stone Circle of Friends.
Author contributions
M.S.M. did microscopy, phenotyping, experimental design and wrote the manuscript. R.S.Y co-wrote the manuscript and did experimental design and experiments on splicing and expression analyses. Y.K.K designed the experiments and did phenotyping in concert with M.M. J.T.G. and Q.Y. did expression analyses and statistical analyses. K.M. did fiber sizing and nuclei count analyses.
References
- 1. Day J.W. and Ranum L.P. (2005) RNA pathogenesis of the myotonic dystrophies. Neuromuscul. Disord., 15, 5–16. [DOI] [PubMed] [Google Scholar]
- 2. Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barcelo J., O'Hoy K. et al. (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science, 255, 1253–1255. [DOI] [PubMed] [Google Scholar]
- 3. Taneja K.L., McCurrach M., Schalling M., Housman D. and Singer R.H. (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol., 128, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W. and Swanson M.S. (2003) A muscleblind knockout model for myotonic dystrophy. Science, 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- 5. Lee K.Y., Li M., Manchanda M., Batra R., Charizanis K., Mohan A., Warren S.A., Chamberlain C.M., Finn D., Hong H. et al. (2013) Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med., 5, 1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chamberlain C.M. and Ranum L.P. (2012) Mouse model of muscleblind-like 1 overexpression: skeletal muscle effects and therapeutic promise. Hum. Mol. Genet., 21, 4645–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A. and Swanson M.S. (2006) Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A., 103, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mahadevan M.S., Yadava R.S., Yu Q., Balijepalli S., Frenzel-McCardell C.D., Bourne T.D. and Phillips L.H. (2006) Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet., 38, 1066–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yadava R.S., Foff E.P., Yu Q., Gladman J.T., Kim Y.K., Bhatt K.S., Thornton C.A., Zheng T.S. and Mahadevan M.S. (2015) TWEAK/Fn14, a pathway and novel therapeutic target in myotonic dystrophy. Hum. Mol. Genet., 24, 2035–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Du H., Cline M.S., Osborne R.J., Tuttle D.L., Clark T.A., Donohue J.P., Hall M.P., Shiue L., Swanson M.S., Thornton C.A. et al. (2010) Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol., 17, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ciciliot S. and Schiaffino S. (2010) Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des., 16, 906–914. [DOI] [PubMed] [Google Scholar]
- 12. Ho T.H., Charlet B.N., Poulos M.G., Singh G., Swanson M.S. and Cooper T.A. (2004) Muscleblind proteins regulate alternative splicing. EMBO J., 23, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S. and Thornton C.A. (2006) Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet., 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- 14. Wheeler T.M., Sobczak K., Lueck J.D., Osborne R.J., Lin X., Dirksen R.T. and Thornton C.A. (2009) Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science, 325, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S. et al. (2012) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell, 150, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Masuda A., Andersen H.S., Doktor T.K., Okamoto T., Ito M., Andresen B.S. and Ohno K. (2012) CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep., 2, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Batra R., Charizanis K., Manchanda M., Mohan A., Li M., Finn D.J., Goodwin M., Zhang C., Sobczak K., Thornton C.A. et al. (2014) Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell, 56, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rau F., Freyermuth F., Fugier C., Villemin J.P., Fischer M.C., Jost B., Dembele D., Gourdon G., Nicole A., Duboc D. et al. (2011) Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol., 18, 840–845. [DOI] [PubMed] [Google Scholar]
- 19. Davis J., Salomonis N., Ghearing N., Lin S.C., Kwong J.Q., Mohan A., Swanson M.S. and Molkentin J.D. (2015) MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat. Commun., 6, 10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lam L.T., Pham Y.C., Nguyen T.M. and Morris G.E. (2000) Characterization of a monoclonal antibody panel shows that the myotonic dystrophy protein kinase, DMPK, is expressed almost exclusively in muscle and heart. Hum. Mol. Genet., 9, 2167–2173. [DOI] [PubMed] [Google Scholar]
- 21. Sarkar P.S., Han J. and Reddy S. (2004) In situ hybridization analysis of Dmpk mRNA in adult mouse tissues. Neuromuscul. Disord., 14, 497–506. [DOI] [PubMed] [Google Scholar]
- 22. Mankodi A., Lin X., Blaxall B.C., Swanson M.S. and Thornton C.A. (2005) Nuclear RNA foci in the heart in myotonic dystrophy. Circ. Res., 97, 1152–1155. [DOI] [PubMed] [Google Scholar]
- 23. Wang G.S., Kearney D.L., De Biasi M., Taffet G. and Cooper T.A. (2007) Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Invest., 117, 2802–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomas J.D., Sznajder L.J., Bardhi O., Aslam F.N., Anastasiadis Z.P., Scotti M.M., Nishino I., Nakamori M., Wang E.T. and Swanson M.S. (2017) Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev., 31, 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poulos M.G., Batra R., Li M., Yuan Y., Zhang C., Darnell R.B. and Swanson M.S. (2013) Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum. Mol. Genet., 22, 3547–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rehman S., Gladman J.T., Periasamy A., Sun Y. and Mahadevan M.S. (2014) Development of an AP-FRET based analysis for characterizing RNA-protein interactions in myotonic dystrophy (DM1). PLoS One, 9, e95957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Amack J.D., Reagan S.R. and Mahadevan M.S. (2002) Mutant DMPK 3'-UTR transcripts disrupt C2C12 myogenic differentiation by compromising MyoD. J. Cell Biol., 159, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Storbeck C.J., Sabourin L.A., Waring J.D. and Korneluk R.G. (1998) Definition of regulatory sequence elements in the promoter region and the first intron of the myotonic dystrophy protein kinase gene. J. Biol. Chem., 273, 9139–9147. [DOI] [PubMed] [Google Scholar]
- 29. Mulders S.A., van den Broek W.J., Wheeler T.M., Croes H.J., van Kuik-Romeijn P., de Kimpe S.J., Furling D., Platenburg G.J., Gourdon G., Thornton C.A. et al. (2009) Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A., 106, 13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee J.E., Bennett C.F. and Cooper T.A. (2012) RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U. S. A., 109, 4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F. and Thornton C.A. (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature, 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Warf M.B., Nakamori M., Matthys C.M., Thornton C.A. and Berglund J.A. (2009) Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A., 106, 18551–18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mankodi A., Teng-Umnuay P., Krym M., Henderson D., Swanson M. and Thornton C.A. (2003) Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann. Neurol., 54, 760–768. [DOI] [PubMed] [Google Scholar]
- 34. Kim Y.K., Mandal M., Yadava R.S., Paillard L. and Mahadevan M.S. (2014) Evaluating the effects of CELF1 deficiency in a mouse model of RNA toxicity. Hum. Mol. Genet., 23, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langlois M.A., Lee N.S., Rossi J.J. and Puymirat J. (2003) Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol. Ther., 7, 670–680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.