

Abstract
Heterozygous missense mutations in several genes in the WNT5A signaling pathway cause autosomal dominant Robinow syndrome 1 (DRS1). Our objective was to clarify the functional impact of a missense mutation in WNT5A on the skeleton, one of the main affected tissues in RS. We delivered avian replication competent retroviruses (RCAS) containing human wild-type WNT5A (wtWNT5A), WNT5AC83S variant or GFP/AlkPO4 control genes to the chicken embryo limb. Strikingly, WNT5AC83S consistently caused a delay in ossification and bones were more than 50% shorter and 200% wider than controls. In contrast, bone dimensions in wtWNT5A limbs were slightly affected (20% shorter, 25% wider) but ossification occurred on schedule. The dysmorphology of bones was established during cartilage differentiation. Instead of stereotypical stacking of chondrocytes, the WNT5AC83S-infected cartilage was composed of randomly oriented chondrocytes and that had diffuse, rather than concentrated Prickle staining, both signs of disrupted planar cell polarity (PCP) mechanisms. Biochemical assays revealed that C83S variant was able to activate the Jun N-terminal kinase-PCP pathway similar to wtWNT5A; however, the activity of the variant ligand was influenced by receptor availability. Unexpectedly, the C83S change caused a reduction in the amount of protein being synthesized and secreted, compared to wtWNT5A. Thus, in the chicken and human, RS phenotypes are produced from the C83S mutation, even though the variant protein is less abundant than wtWNT5A. We conclude the variant protein has dominant-negative effects on chondrogenesis leading to limb abnormalities.
Introduction
In this study we explore the connection between WNT (Wingless-related) signaling and a human disease, Robinow syndrome. The syndrome was originally described in 1969 in a family that had short stature (1). Eventually, the genetic mutations were identified starting with the receptor ROR2 in recessive Robinow syndrome (OMIM#268310) (2–4). Subsequently the dominant forms of Robinow syndrome (DRS) were diagnosed and these patients either had missense or non-frameshift mutations in WNT5A ligand (dominant Robinow syndrome 1, OMIM#180700) (5–7), missense and truncating variants in the Frizzled 2 receptor (FZD2) (7) or nonsense frameshift mutations in intracellular signal transduction protein Dishevelled (DVL; DRS2, DVL1 OMIM#616331; DRS3, DVL3 OMIM#616894) (7–10). Recently a canine version of Robinow-like syndrome was reported to be caused by similar frameshift mutations in DVL2 (11).
All forms of RS are characterized by skeletal dysplasias affecting primarily the face and limbs (5–9,12). Patients typically present with short stature and mesomelic (middle bones) limb shortening. There are many possible steps in development that could cause limb shortening. There may be early effects on limb bud outgrowth caused by decreased proliferation, later effects on chondrogenesis and finally, precocious maturation of the growth plate. We can rule out the last explanation since patients have shortened limbs from birth and yet they continue to grow postnatally. If the early outgrowth of the limb bud were inhibited, there would be more severe phenotypes including phocomelia (agenesis of bones). In DRS, there appears to be a defect in WNT signaling that targets the middle stages of skeletogenesis, after chondrogenesis has initiated.
WNT signaling is a conserved pathway in metazoan animals. This pathway is involved in many aspects of embryonic development, such as regulating cell fate, cell migration, cell polarity and organogenesis (13,14). WNTs are short-range, secreted glycoproteins that are post-translationally modified by receiving a lipid palmitoylation chain at cysteine 104 and are glycosylated (15). Upon WNT ligand binding to its receptors, a cascade of intracellular events are triggered, often involving recruitment of activated DVL adaptor protein to the receptors. Depending on the receptors involved signaling will be directed to the β-catenin-dependent pathway (canonical) or β-catenin-independent (non-canonical), planar cell polarity (PCP) or calcium pathways (16). Of the 19 different WNT ligands, it is not always possible to predict which pathway will be stimulated since there are 10 different Frizzled receptors and multiple co-receptors (16–18).
WNT5A is thought to signal primarily via the Jun N-terminal kinase-PCP (JNK-PCP) pathway, which mediates tissue polarity (19). It involves the participation of membrane proteins such as VANGL, as well as intracellular proteins such as PRICKLE and DVL (13,20). The activation of the PCP pathway leads to stimulation of Rho and Rac GTPases. The small GTPases phosphorylate JNK, which leads to transcription of activating transcription factor 2 (ATF2), leading to the rearrangement of actin cytoskeleton (21). The loss of PCP genes such as Vangl2 or Prickle causes chondrocytes to lose their orientation (22–26). We hypothesized that the missense mutations in WNT5A would affect PCP in the cartilage and that the biochemical defects would lie in the JNK-PCP non-canonical signaling pathway.
Detailed studies of the role of Wnt5a in skeletogenesis have been made in the mouse model, offering some insights into the tissue requirements for Wnt5a. Germline knockouts of Wnt5a do not survive beyond birth. Embryos have major shortening of the body axis, appendicular skeleton and jaws (19,26–28). There appears to be disruption of chondrocyte polarity in the mutant mice (23,29).
Conditional overexpression of Wnt5a also causes moderate shortening of the long bones (27) and a delay in ossification in both intramembranous and endochondral bones (30). Using retroviral misexpression of gallus WNT5A in chicken limbs, there was a general shortening of posterior skeletal elements in the autopod and zeugopod (31,32). Thus, gain- or loss-of-function of WNT5A has profound effects on skeletal development, some of which resemble those caused by DRS mutations.
The functional impact of missense mutations in human WNT5A has only been studied twice. The C83S and C182R variants were studied in Xenopus and zebrafish embryos by others (5) and in the mandible of chicken embryos by our group (33). The authors of the first report on the WNT5A missense variants ruled out the possibility of WNT5A variants acting as a dominant negative mutation and instead suggested that the mutations are having a hypomorphic affect based on functional expression experiments in zebrafish and Xenopus embryos (5). Mutant WNT5A phenotypes were less extreme compared to either wild-type- or dominant negative-WNT5A zebrafish embryos (5). However, their data from overexpression could also be the result of over-activation of the non-canonical pathway leading to disruption of convergent extension, rather than the effect of a less active form of WNT5A. Overactivation of the non-canonical pathway impacts convergent extension indirectly, as seen with data from others (34,35). In order to determine effects of the missense mutations in WNT5A it is important to carry out functional assays on the skeleton because this is the major tissue affected in RS.
We therefore carried out the present study to explore the effects on limb development. Limb mesenchyme is derived from lateral plate mesoderm (36) as opposed to the facial mesenchyme which is largely neural crest cell derived (37). Furthermore, in the limb, bone forms by endochondral ossification whereas in the mandible cartilages persist to adulthood (38). Here we use the chicken model to misexpress wild-type or C83S variant human WNT5A in the limb bud over top of the gallus genome. Using this strategy, we can distinguish mutations that cause a loss-of-function from those that result in a gain of activity. In addition, comparison to the wtWNT5A gene will allow us to distinguish general effects of raising the level of WNT5A expression on the skeleton from those of the mutation. The chicken is an appropriate model because the patterning molecules involved in limb outgrowth and skeletal formation are conserved with mammals (39–41). In our study, we find that there are skeletal phenotypes uniquely associated with the expression of mutant WNT5A. In addition, biochemical work revealed a gain in JNK-PCP under certain contexts, and an inability to antagonize canonical WNT signaling. Finally, unexpectedly we determined that mutant protein is synthesized and secreted at lower levels than wild-type protein.
Results
Human WNT5A mutation causes wider and shorter skeletal elements in the chicken limb
To investigate the effects of WNT5A mutations on limb development, we compared the effects of expressing wtWNT5A to one of the six pathogenic variants of WNT5A that cause RS (NCBI, ClinVar database). We selected the NM_003392.4 variant which converts G at bp248 to a C resulting in conversion of cysteine 83 to a serine. This variant was studied by us in the mandible and had the most penetrant phenotypes (33). Results of the experimental viruses were compared to the GFP or AlkPO4 control viruses which do not affect development (33). Injections in to the limb field were carried out at stage 15 in order to maximize infection of the chondrocyte progenitor cells (36). Virus RNA levels were lower for WNT5AC83S as compared to wtWNT5A initially, 48 h after injection but by 96 h the levels of expression were similar since the virus is replication competent. In quantitative polymerase chain reaction (qPCR) experiments carried out on bulk limb bud RNA, wtWNT5A RNA was 65-fold higher than WNT5AC83S after 48 h (ΔΔCT, n = 4, SEM = 28, P = 0.001). By 96 h, wtWNT5A was 6.4-fold higher than WNT5AC83S (N = 4, SEM = 3, P = 0.03). In order to determine the ultimate phenotype caused by the viruses, animals were grown until endochondral ossification had begun and the full pattern of the limb was present (stage 38, Fig. 1; stage 36, Figs 2–4). All injections were made into the right limb since chick embryos turn during development, leaving the right side uppermost.
Figure 1.
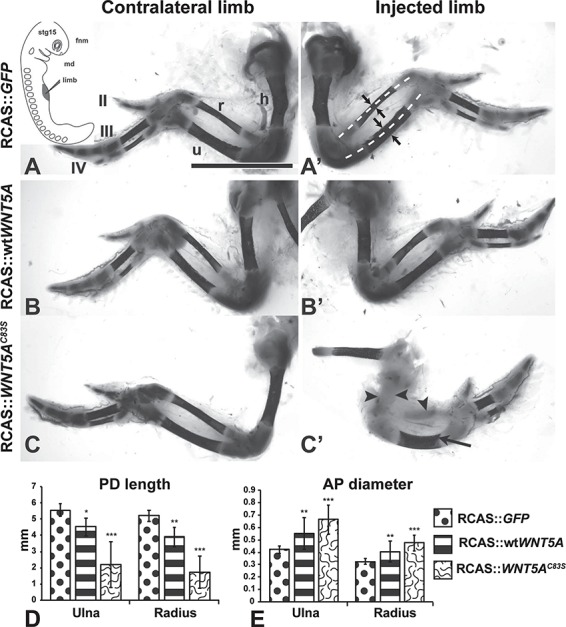
Skeletal phenotypes caused by injection of RCAS viruses expressing GFP, wtWNT5A or WNT5AC83S. The embryos were injected at stage 15 into the right forelimb field (inset, A) and fixed at stage 38. (A, A’) Wholemount staining with Alcian blue (cartilage) and Alizarin red (bone) shows the normal bone length (dashed white lines) and normal AP diameter (arrows). The expression of GFP does not affect patterning or size of the skeletal elements. (B, B’) The right limb is slightly shorter than the contralateral side and shorter than the GFP-injected limbs. (C, C’) The mutant virus has inhibited bone formation in the radius and humerus (arrowheads) while the ulna was still able to ossify (arrow). (D, E) Quantification of bone length and AP diameter from photographs. All bones are shorter and have increased AP diameter compared to GFP control limbs. Tukey’s post hoc, *P < 0.05, **P < 0.01, ***P < 0.001. Scale bar = 5 mm. For sample size refer to Table 1. Key: AP, anterior-posterior; fnm, frontal nasal mass; h, humerus; md, mandible; PD, proximo-distal; r, radius; u, ulna; II, III, IV, digit numbers. For colour version see Supplementary Material, Fig. S7.
Figure 2.
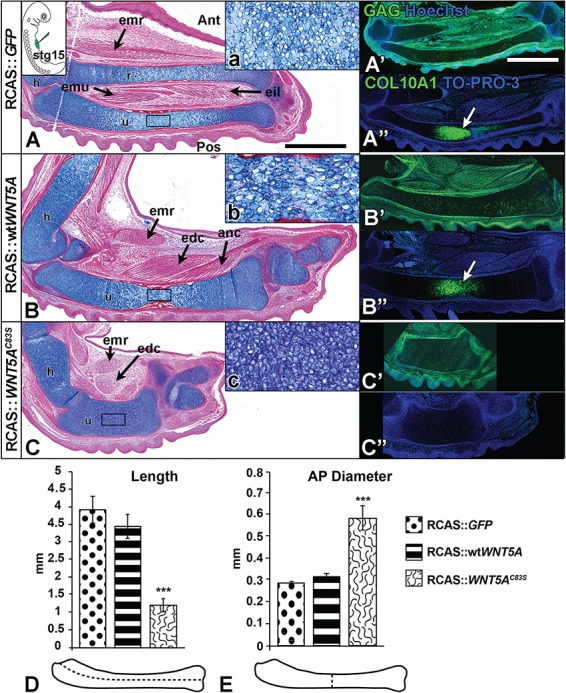
Mutant WNT5A shortens and widens dimensions of the developing long bone. Embryos were injected into the presumptive limb field at stage 15 (inset in A) and fixed at stage 36. Sagittal sections of injected forelimbs were stained with Alcian blue and Picrosirius red (A–C) and adjacent sections were stained with anti-GAG to locate the virus in the tissues (A’–C’), or with anti-COL10A1 to mark hypertrophic chondrocytes (A”–C”). (A) GFP virus has no effect on development. The onset of endochondral bone formation in normal limbs is seen first in the diaphysis (A, A”, white arrows). (B) Wild-type WNT5A virus has minimal effects on limb development or muscle development, and COL10A1 staining was normal (B”, white arrows). (C) The C83S variant virus causes significantly shorter and wider cartilages to develop and lacked COL10A1 staining. The limb musculature is present but displaced and shorter than normal. (D, E) The dimensions of the ulna were measured: (D) length and (E) diameter in AP plane. Mutant WNT5A variants showed significantly shorter length (D) and wider diameter (E) of the developing ulna as compared to the wtWNT5A virus. One-way analysis of variance (ANOVA), Tukey’s post hoc test, ***P < 0.001, n = 4. Scale bar = 1 mm for bright field views and fluorescent images. Key: Ant, anterior; anc, anconeus; AP, anterior-posterior; edc, extensor digitorum communis; eil, extensor indicis longus; emr, extensor metacarpi radialis; emu, extensor metacarpi ulnaris; h, humerus; Pos, posterior; r, radius; u, ulna.
Figure 4.
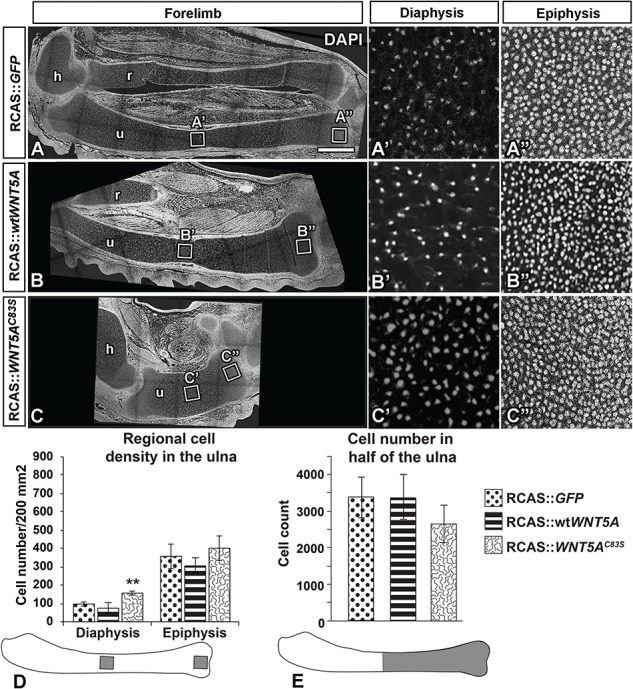
RCAS::WNT5AC83S virus showed increased cell density in the diaphysis of the ulna. (A–C) Sagittal sections of injected forelimbs fixed 10 days post-injection (stage 36) and stained with DAPI for nuclei. Cell density was quantified in the same area size in the diaphysis (A’–C’) and epiphysis (A”–C”) of the ulna. (D) WNT5AC83S virus increased cell density in the diaphysis, which was significantly different than GFP and wtWNT5A. In contrast, there was no significant difference in cell density in the epiphyses. One-way ANOVA post hoc Tukey, **P < 0.01, n = 4. (E) Cell count was quantified in half of the ulna bone closest to the digits. WNT5AC83S injected limbs showed no significant difference in cell count compared to GFP control or wtWNT5A injected limbs. Scale bar = 500 microns. Key: h, humerus; r, radius; u,ulna.
There were several possible outcomes for these experiments. If the missense mutations removed all function, then the phenotypes of the mutant WNT5A viruses should resemble GFP or AlkPO4 controls. If the phenotypes were similar to but not as strong as for wtWNT5A then the mutations may be hypomorphic (reduced function). If the phenotypes caused by mutant viruses resembled wtWNT5A but are more severe, then the mutations may cause a gain-of-function. Finally, if the mutant viruses induced unique phenotypes, not represented in the wtWNT5A-infected limbs, then this would suggest more complex functional alterations that need further elucidation. We confirmed that the human WNT5A genes were expressed using qPCR with human-specific primers and that gallus WNT5A expression levels were not changed as a result of the exogenous gene (data not shown).
Limbs injected with GFP virus were indistinguishable from the contralateral, uninjected side (Fig. 1A and A’). The wtWNT5A virus reduced the ulnar length by 20% and increased the diameter by 28% (Fig. 1A, B’, D and E; Table 1). In contrast, the C83S virus caused major shortening of the limb and delayed osteogenesis in the majority of limbs (ulna 60% shorter and 55% wider; Fig. 1C–E, Table 1). The absence of ossification in some bones (Fig. 1C’) was likely correlated with higher levels of virus expressed in certain cartilage primordia. In order to specifically correlate the areas with phenotypic changes and the presence of the retrovirus, the subsequent characterizations were done at the histological level. We allocated sections from each embryo for staining with anti-GAG antibody that recognizes the viral coat (Fig. 2A’, B’ and C’ and 3A–F insets; Supplementary Material, Fig. S2A, C and E). In the sagittal plane, limbs infected with wtWNT5A containing virus, had slightly disorganized cartilage in the diaphysis (Fig. 2Bb) but hypertrophy was taking place as shown by the COL10A1 antibody staining (Fig. 2B”). In contrast, the ulna of a WNT5AC83S-infected limb had a much wider diameter and significantly shorter length (Fig. 2C–E). Differentiation of cartilage was delayed (Fig. 2C”) and chondrocytes appeared more closely packed (Fig. 2C). At stage 36 the lengths of the bones in wtWNT5A-infected limbs was the same as in GFP controls (Fig. 2D). In contrast, the WNT5AC83S variant resulted in limbs that were approximately one-third the length (Fig. 1D) and double the anterior–posterior (AP) diameter in the diaphysis (Fig. 1E); similar to the data obtained from wholemount skeletal measurements.
Table 1.
Skeletal phenotypes observed at in stage 38 limbs stained in wholemount
| Injected virus (total n) | Normal | Stylopod and/or zeugopod abnormalitiesa | Delayed or absent ossification in any boneb |
|---|---|---|---|
| RCAS::GFP (n = 22) | 22 | 0 | 0 |
| RCAS::wtWNT5A (n = 12) | 2 | 10 | 0 |
| RCAS::WNT5AC83S (n = 17) | 0 | 17 | 16 |
a Abnormalities include shorter and bent bones.
b Ossification phenotypes occur concurrently with bone abnormalities.
Figure 3.
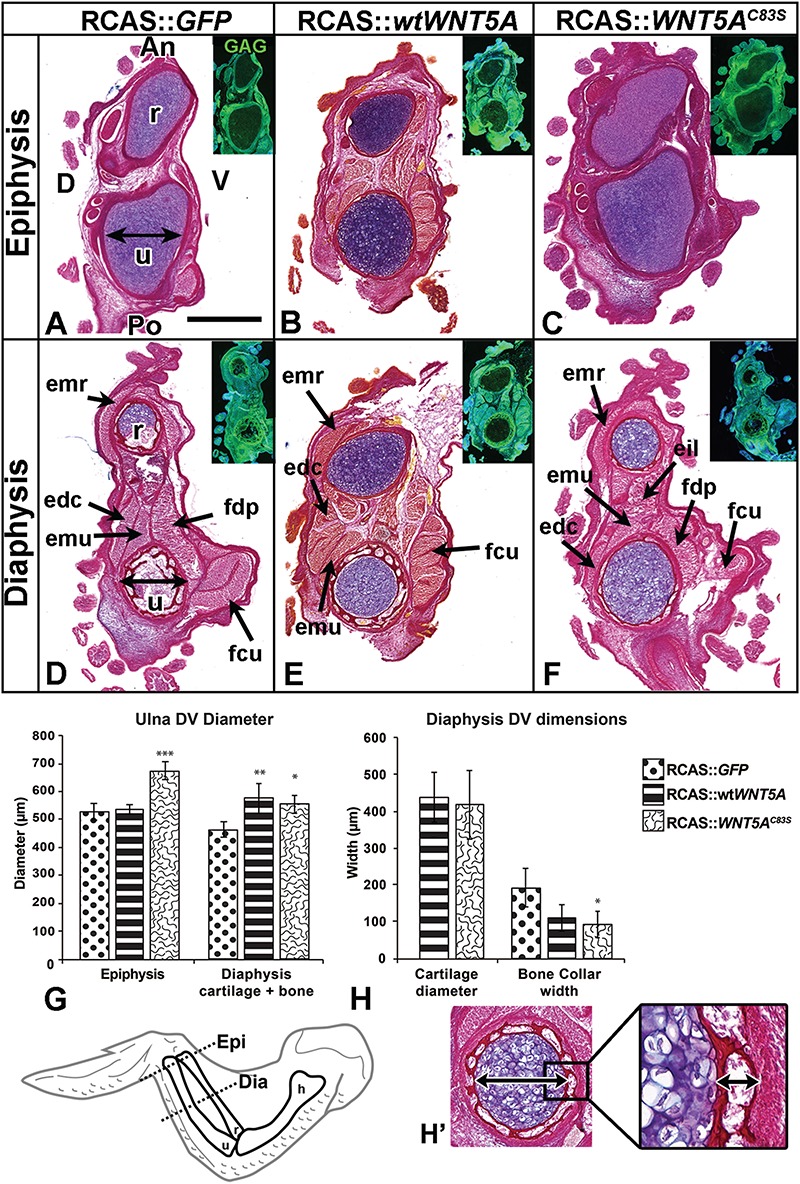
WNT5A viruses widen the ulna in chicken embryos. (A–H) Serial transverse sections of the injected forelimbs fixed 10 days post-injection (stage 36). Insets show viral (anti-GAG) expression. The dorso-ventral diameter of the ulna bone was measured in the epiphysis and diaphysis (see double arrows in A and D). WNT5AC83S injected forelimbs were significantly wider in their epiphyses (C, F, G) compared to both GFP control (A, D, G) and wtWNT5A viruses (B, E, G). Wild-type and mutant WNT5A injected forelimbs had wider cartilage compared to GFP controls (G). The WNT5AC83S injected specimens had a thinner bone collar relative to controls. (H, H’). One-way ANOVA followed by Tukey post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001; n = 3 for WNT5AC83S, GFP and n = 4 for wtWNT5A. Scale bar = 500 μm and applies to all brightfield images. Key: An, anterior; D, dorsal; dia,diaphysis; DV, dorsal-ventral; epi, epiphysis; edc, extensor digitorum communis; eil, extensor indicis longus; emr, extensor metacarpi; emu, extensor metacarpi ulnaris; fdp, flexor digitorum profundus; fcu, flexor carpi ulnaris; h, humerus; Po, Posterior; r, radius; u, ulna; V, ventral.
It appeared that the C83S variant had preferentially increased growth in the AP dimension but we wondered whether this trend was also true in the perpendicular, dorso-ventral axis. We sectioned other specimens in the transverse plane (Fig. 3). The epiphyses from the GFP and wtWNT5A-infected limbs were indistinguishable in terms of dorso-ventral measurements (Fig. 3A, B and G); however, the C83S variant significantly increased the diameter of the cartilage (Fig. 3C and G). In the diaphysis, the majority of GFP-infected limbs had replaced most of the cartilage with bone therefore we measured the distance between the outer edges of the bone collar (Fig. 3D). Both the wtWNT5A and C83S variants still had cartilage present and both were significantly wider than GFP controls (Fig. 3E–G). There was no significant difference in the thickness of the cartilage itself or the bone collar between the two WNT5A viruses (Fig. 3H). The skeletal muscles were present in their normal positions but were proportionately shorter and displaced, especially in the C83S-infected limbs (Figs 2C and 3F). This lack of muscle patterning phenotype fits with negative effects of wtWNT5A on muscle fiber type in chicken embryos (42). The subset of changes caused by the mutant virus include major shortening of the bones and preferential widening of the diaphysis in the AP plane. There was also a delay in hypertrophy of the diaphysis. These phenotypes are distinct from the wtWNT5A viruses.
WNT5AC83S virus increased cell density specifically in the diaphysis
The chondrocytes appeared to be smaller and more numerous in the WNT5AC83S-infected cartilage (Fig. 2Cc). This could be explained by recruiting more cells to the condensation, by increased proliferation or by a change in the packing of the chondrocytes. We quantified DAPI stained nuclei in defined areas in stage 36 limbs. The cell density was significantly increased in the diaphysis of the WNTAC83S (Fig. 4C’ and D) as compared to the control GFP (Fig. 4A’ and D), wtWNT5A (Fig. 4B’ and D; n = 4 per virus type). The higher cell density suggested a less mature limb which is consistent with the lack of COL10A1 expression (Fig. 2C”). The density in the epiphyses was unchanged by the viruses (Fig. 4A”–C” and D). Interestingly, the total number of chondrocytes over half of the cartilage element was the same in mutant (C83S), wtWNT5A and GFP controls (Fig. 4E). Thus, the original number of cells recruited to the condensation was likely the same in both wtWNT5A and the C83S variant infected limbs.
Correlation between morphological change in cartilage and lack of differential cell proliferation across the cartilage
We asked when the cartilage phenotypes first appeared in order to understand the mechanism behind the dysmorphic changes induced by the DRS mutations. We measured the ulnae at 4 and 5 days post-injection (stage HH29, 30, Fig. 4 and Supplementary Material, Fig. S1). Initially, the condensations were equivalent in length for the GFP, wtWNT5A and C83S variant (stage 29; n = 4; Supplementary Material, Fig. S1A); however, the diameter of the cartilage was wider for the wtWNT5A compared to the other conditions (Supplementary Material, Fig. S1B, P < 0.02). There were no significant differences in overall proliferation index at stage 29 within SOX9-positive chondrocytes (Supplementary Material, Fig. S2A–G). By stage 30, the wtWNT5A limbs were shorter than the GFP controls (Supplementary Material, Fig. S1A) which was carried through until full ossification (Fig. 1D, Table 1). Signs of the RS phenotype were present in the C83S variant at stage 30, including significant shortening and increased AP diameter compared to GFP (Supplementary Material, Fig. S1A and B). By stage 30, proliferation differences were seen in both the wtWNT5A and C83S variant-infected limbs (Fig. 5A–G). The GFP controls had a significantly lower level of proliferation in the diaphysis compared to the epiphyses (Fig. 5B and G; P < 0.001). In contrast, the wtWNT5A and C83S variant had no differential proliferation between the epiphysis and diaphysis (Fig. 5G). Proliferation in the diaphyses was significantly increased by both WNT5A constructs compared to controls (Fig. 5G). Apoptosis was unchanged (TUNEL at HH29 or HH30, data not shown). Thus, the mutation did not impair the ability of WNT5A to stimulate proliferation in the diaphyseal cartilage.
Figure 5.
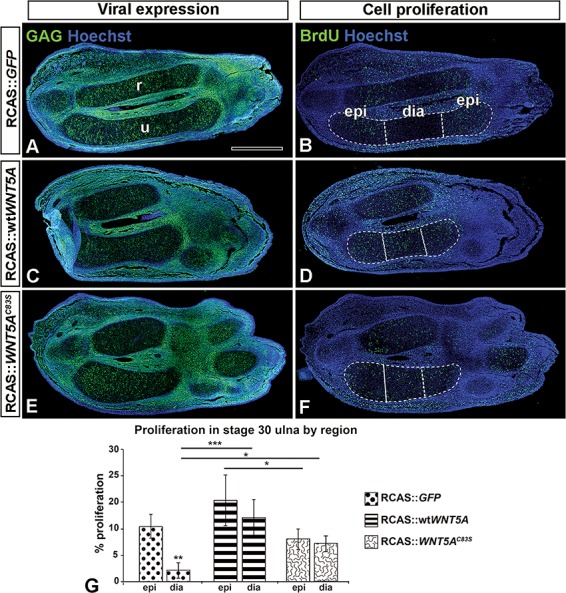
wtWNT5A- or WNT5AC83S-injected forelimbs have increased cell proliferation in the cartilage. (A, C, E) Sagittal sections of injected forelimbs fixed 5 days post-injection (stage 30) show viral (anti-GAG) expression (green) and (B, D, F) neighboring sections probed for anti-BrdU labeling. Note that the normal limbs diaphysis is devoid of proliferating cells (B). In contrast there are labeled cells in the diaphysis of wtWNT5A- or WNT5AC83S-injected ulnae (D, F). (G) Percentage BrdU-positive cells were similar in the epiphysis regardless of treatment. In the diaphysis, wtWNT5A showed significantly increased cell proliferation compared to GFP and C83S, whereas C83S was only significantly different than wtWNT5A. One-way ANOVA and Fisher’s least significant difference (LSD) post hoc test, *P < 0.05, ***P < 0.001; n = 4 GFP; n=5 wtWNT5A and WNT5AC83S. Scale bar = 500 μm. Key: dia, diaphysis; epi, epiphysis; r, radius; u, ulna.
Chondrocyte polarity and elongation is lost in the embryos infected with RCAS::WNT5AC83S
One reason for the failure of cartilage to narrow and elongate in the embryos treated with variant forms of the human WNT5A gene is that chondrocytes are not stacking properly. The movement of chondrocytes results in daughter cells that were initially lined up beside each other, slipping underneath one another to become stacked in a column (41,43). The adhesion of chondrocytes to each other helps to stabilize the rearrangements (44). It has previously been shown that non-canonical WNT signaling in the JNK-PCP pathway is required for organized orientation of chondrocytes in the chicken (41) and mouse limb (22,23). Since Golgi complexes in chondrocytes are located on one side of the nucleus (29) we used the angle between the Golgi nucleus axis and the long axis of the bones in the zeugopod cartilage as a measure of cell orientation. Angles were measured at stage 29 just prior to the onset of BrdU or cartilage shape phenotypes. Here, we found that in control embryos, Golgi were oriented perpendicular to the long axis of the developing cartilage (65.28° ± 3.2°; Fig. 6A and D). Overexpression of wtWNT5A did not significantly affect cell orientation (Fig. 6B and E); however, WNT5AC83S mutation caused randomization of chondrocytes compared to controls (47.7° ± 0.7°, P < 0.005) (Fig. 6C and F). We also measured chondrocyte shape since in control limbs it is typical for chondrocytes to become flattened and elongated in the AP axis (41). We found that chondrocytes in controls were flattened (Fig. 6A). In contrast, chondrocytes were significantly rounder in both RCAS::wtWNT5A- and RCAS::WNT5AC83S-infected limbs compared to controls (P < 0.01; Fig. 6B, C and J). Therefore, loss of proper orientation in chondrocytes is the main distinction between mutant and wild-type WNT5A.
Figure 6.
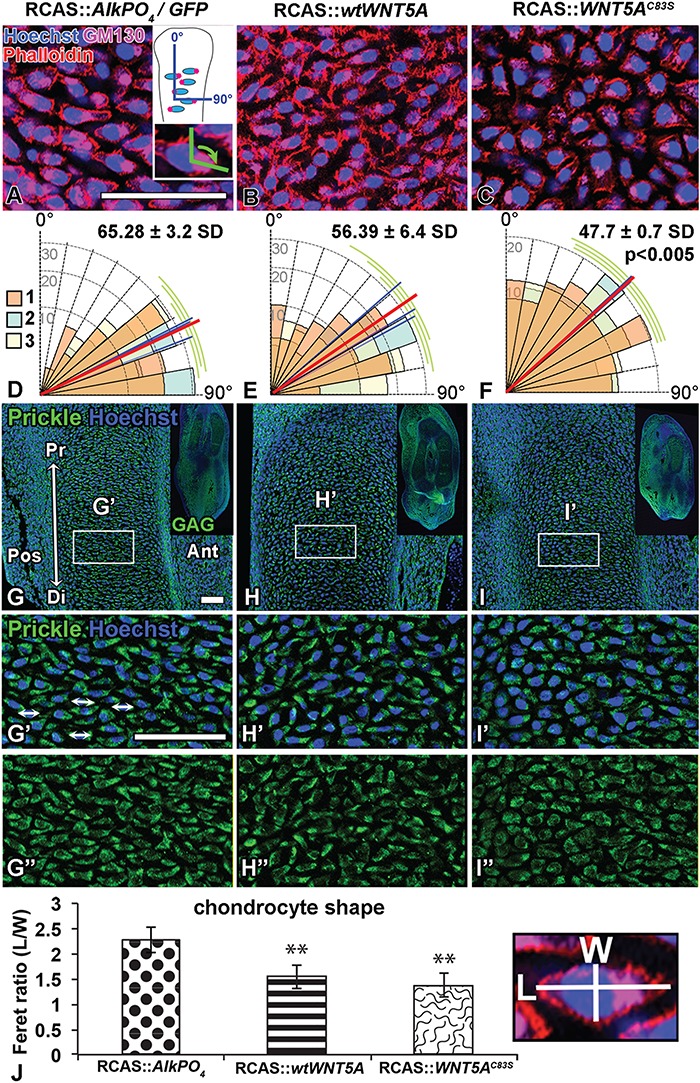
WNT5AC83S virus randomizes chondrocyte polarity. (A–C) Sagittal slices (200 μm) of injected forelimbs fixed 4 days post-injection (stage 29) and stained in wholemount with anti-Golgi (GM130, pink) and actin filaments with Phalloidin-568 (red). Expression in chondrocytes (SOX9-positive) was identified in adjacent sections (not shown). The angle between the Golgi-nucleus axis and the long axis of the bones in the ulna was measured (inset in A). (D–F) Graphical representation of angular data for each virus type (70 cells measured/specimen). The blue line is the average of each specimen and the red line is the overall mean for the group. The cells were more randomly oriented with WNT5AC83S compared to AlkPO4 control virus (One-way ANOVA and Tukey’s post hoc test, P < 0.005; n = 3). (G–I) Sagittal sections of injected forelimbs fixed 4 days post-injection (stage 29) show staining for pan-Prickle and counterstained with Hoechst for nuclei; viral expression (GAG) shown in the inset. (G’–I”) Increased magnification of box region in the zeugopod cartilage of the forelimb showing combined signal for nuclei and prickle as well as the prickle stain alone. (G’, G”) Double arrows indicate Prickle distribution at the ends of the elongated chondrocytes in the GFP control forelimbs. (H’, H”) The wtWNT5A has not affected Prickle distribution; however, (I’, I”) WNT5AC83S caused a general reduction in staining and a more diffuse pattern. (J) Chondrocyte shape was analyzed using Phalloidin-stained cell membranes (from A–C). Ferret diameter was measured in perpendicular axes in 50 cells/specimen (length/width). Significantly rounder cells were observed in both wtWNT5A and WNT5AC83S viruses (closer to 1) compared to AlkPO4. One-way ANOVA post hoc Tukey **P < 0.01, n = 4. Scale bar = 50 microns. Key: Ant, anterior; Di, distal; L, length; Pos, posterior; Pr, proximal; W, width.
Since we observed several signs that polarity was disrupted, we examined the distribution of a core PCP molecule, Prickle. Prickle is concentrated on opposite ends of the flattened chondrocytes in the mouse (29) and when knocked out in mice an RS phenotype in the skeleton is produced (24). We found that in the developing ulna at stage HH29, GFP-infected control limbs showed Prickle molecule concentrated on opposite ends of the developing chondrocytes (Fig. 6G–G”). A similar result was observed with wtWNT5A (Fig. 6H–H”). However, WNT5AC83S-injected limbs displayed qualitatively more diffuse expression of Prickle (Fig. 6I–I”). Taken together, these cell polarity and Prickle changes suggest that the C83S variant is disrupting the JNK-PCP pathway.
wtWNT5A and WNT5AC83S plasmids activate JNK-PCP signaling
In order to measure the activity of the JNK-PCP pathway we used the ATF2 luciferase reporter in transfected limb mesenchyme cells or HEK293 cells which are commonly used in WNT luciferase assays (33,45,46). We used plasmids containing the wtWNT5A or C83S variants rather than RCAS viruses, since with plasmids, DNA concentration at the start of the transfection can be controlled. We found that both wild-type and WNT5AC83S mutant plasmids are taken up by similar numbers of cells; thus, luciferase results are comparable (Supplementary Material, Fig. S3A–C). Both forms of WNT5A significantly activated ATF2 reporter activity in primary cultures of limb cells (Fig. 7Aa). A similar result was obtained in HEK293 cells (Fig. 7Bb) and therefore the remainder of the luciferase assays were carried out in HEK293 cells.
Figure 7.
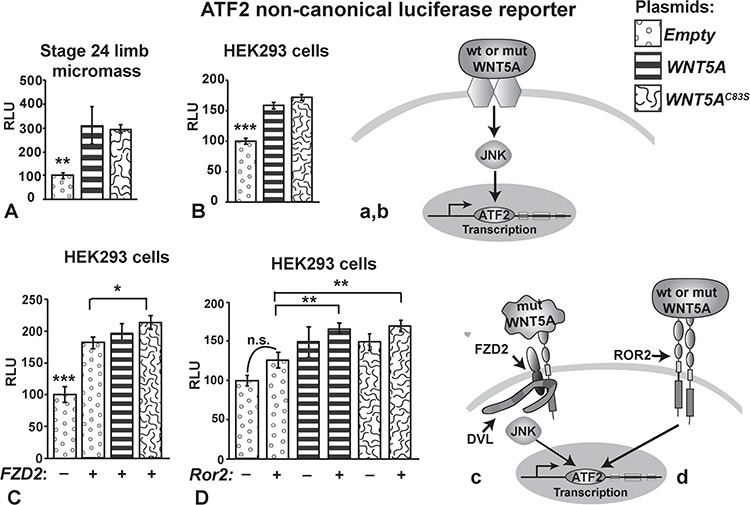
Pathway activity measured with luciferase reporters for JNK-PCP signaling (ATF2). ATF2-luciferase assays were conducted on primary limb mesenchyme (A) or HEK293 cells (B–D). Plasmids expressing wtWNT5A or WNT5AC83S were transiently transfected 48 h prior to lysing the cells. (A, B) Wild-type and mutant WNT5A plasmids significantly activate non-canonical JNK-PCP signaling. The likely pathway involves WNT5A binding to receptor dimers (unknown) and activating ATF2 transcription (a, b). (C) FZD2 receptor enhanced activity of the ATF2 reporter compared to empty plasmid. Thus, endogenous WNTs present in the HEK293 cells were able to utilize the transfected receptor. The addition of plasmids expressing WNT5A further increased activity of ATF2. The C83S variant is significantly more active than empty plasmid (c). (D) No significant change in activity occurred with Ror2 on its own suggesting there are insufficient endogenous ligands to bind to this receptor. Adding WNT5A or C83S variant combined with Ror2 significantly increased activity when compared to Ror2 alone (d). One-way ANOVA, Tukey's post hoc, *P < 0.05, **P < 0.01, ***P < 0.001. ATF2, activating transcription factor 2; DVL, Dishevelled; FZD2, Frizzled2; JNK, Jun N-terminal kinase; n.s., not significant; mut, mutant; RLU, relative light units; ROR2, Receptor Tyrosine Kinase-like orphan Receptor 2; STF, SuperTopflash; wt, wild-type.
We wondered whether the activation of the ATF2 reporter was facilitated by either the FZD2 or ROR2 receptors, both of which are mutated in DRS or recessive RS, respectively. When we added FZD2 to the WNT5A plasmids, activity was significantly increased only for the C83S variant (Fig. 7Cc). Thus, the mutant WNT5A protein may be more effective at utilizing the FZD2 receptor. ROR2 is thought to be one of the main receptors for WNT5A (19,23,47,48). Therefore we were surprised to see that the plasmid containing the mouse Ror2 receptor did not provide an advantage for ATF2-mediated signaling (Fig. 7Dd). The lack of difference in reporter activity between wt and mutant WNT5A when Ror2 was provided suggests that the mutation does not alter binding to the Ror2 receptor. Therefore, the WNT5AC83S protein could behave differently in different locations in the embryo, depending on the availability of certain receptors.
Mutant WNT5A is less effective at antagonizing canonical WNT signaling in the presence of WNT3A ligand
WNT5A is known to antagonize the canonical WNT pathway that is mediated by β-Catenin (49,50). WNT5A strongly inhibits the effects of WNT3A protein or a GSK3β antagonist, lithium chloride (LiCl) (51,52). The SuperTopFlash (STF) luciferase reporter measures canonical WNT activity through the TCF binding sites in the promoter for luciferase (53). T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factor is a major downstream target of the canonical pathway and can also activate transcription of other genes with TCF/LEF binding sites. The plasmid-derived wtWNT5A was able to significantly block endogenous canonical activity (Fig. 8Aa). The mutant WNT5A seemed to be less effective at antagonizing canonical activity but further experiments were needed, where cells are challenged with an activator of STF such as LiCl or WNT3A protein. Both wild-type and mutant WNT5A were able to bring down activation caused by LiCl to basal levels (Fig. 8Bb). This result indicates that WNT5A antagonism acts downstream of the GSK3β complex. When exogenous WNT3A was added, 50% of the STF activity was lost in the presence of wtWNT5A (Fig. 8C). The addition of Ror2 further inhibited signaling. In the presence of WNT3A ligand, the Ror2 receptor in combination with wtWNT5A significantly reduced STF activity to only 14% of the values achieved by the wtWNT5A plasmid alone. These data suggest that WNT5A antagonism of STF is facilitated by the Ror2 receptor (Fig. 8Cc).
Figure 8.
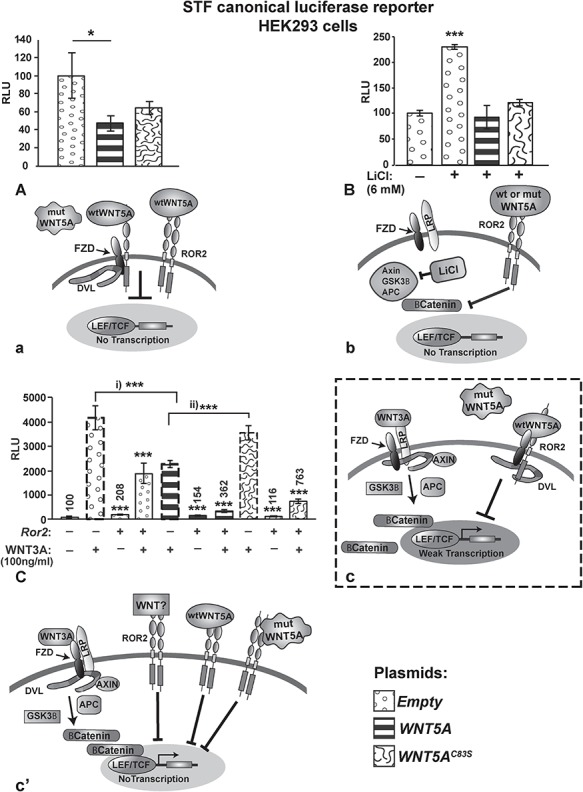
Pathway activity measured with luciferase reporters for canonical signaling (STF). (A) Wild-type WNT5A plasmid significantly inhibits endogenous canonical WNT signaling; however, WNT5AC83S is a less effective antagonist (a). (B) The wtWNT5A and variant plasmid were able to equally antagonize STF activity induced by LiCl (b). (C) WNT3A protein is a strong activator of canonical signaling via Frizzled-LRP dimers and this activity is significantly reduced by wtWNT5A but not by WNT5AC83S (bars with dashed outline; c). The addition of Ror2-expressing plasmid alone did change STF activity (bar 3). However, when Ror2 was added with WNT3A, the activation was greatly reduced, presumably facilitated by binding of endogenous WNT ligands (bar 4, c’). The addition of mutant DNA in combination with Ror2 and WNT3A significantly inhibited activity (bar 10, c’). Thus, WNT5AC83S may become a more potent inhibitor of canonical signaling when there is an excess of Ror2 receptors (c’). One-way ANOVA post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001. APC, Adenomatous Polyposis Coli; DVL, Dishevelled; FZD, Frizzled receptor; GSK3β, glycogen synthase kinase 3 β; LEF/TCF, Lymphoid Enhancer Factor/T-cell Factor; LiCl, lithium chloride; LRP, Low-density Lipoprotein receptor Related Protein; mut, mutant; RLU, relative light units; ROR2, Receptor Tyrosine kinase-like Orphan Receptor 2; wt, wild-type.
In contrast to the wtWNT5A DNA, the C83S variant DNA was unable to block signaling (Values not significantly different than adding WNT3A on its own; Fig. 8Cc). Adding Ror2 in combination with WNT5AC83S plasmid restored completely the ability to block STF activity (levels indistinguishable from basal levels of activity). In the presence of the strong stimulus, WNT3A, mutant WNT5A was still able to block most of the STF activity (Fig. 8Cc’). The excess of Ror2 receptor, appears to overcome a slight decrease in receptor affinity caused by the loss of a cysteine residue; however, the exact region of WNT5A that binds to ROR2 or FZD is not known.
To complete our signaling studies, we measured the ability of WNT5A variants to activate the calcium pathway using the NFAT luciferase reporter (measurement of calcineurin-dependent signaling) (7,54,55). HEK293 cells transiently transfected with the positive control plasmid expressing constitutively active NFAT (caNFAT) strongly activated the reporter (Supplementary Material, Fig. S4). In contrast, neither wild-type nor mutant WNT5A plasmids activated the calcium reporter (Supplementary Material, Fig. S4). Our results are in line with previous studies that found Wnt5a only weakly activates the NFAT reporter (55). Therefore, we found no evidence for an involvement of the calcium pathway in the DRS phenotypes.
Missense mutations decrease the amount of secreted mutant WNT5AC83S protein
The transient transfections used for luciferase assays demonstrated that the mutant protein is able to be synthesized from a plasmid, bind to cell surface receptors, transduce signals and activate reporter constructs. However, these transfection experiments do not address whether the protein is efficiently synthesized and secreted in a sustained manner. The RCAS viruses incorporate into the chicken fibroblast genome and thus stable expression is achieved without the need for antibiotic selection. The western blots of concentrated conditioned media from infected fibroblasts showed strong signal for wtWNT5A, indicating that the protein was secreted efficiently in our culture conditions. In contrast, there was significantly lower WNT5AC83S in the media (Fig. 9A and F). We excluded the possibility that the difference in staining was due to a problem with the antibody recognizing mutant WNT5A by reprobing the same blots with anti-Flag antibodies (Supplementary Material, Fig. S5A). It was possible that less secretion would mean there would be increased levels of protein hung up in the secretory pathway and these should show up in the cell lysate. Unexpectedly, decreased levels of mutant WNT5A protein were also observed in cell lysates (60% of wtWNT5A levels, Fig. 9B and F). Again, the Flag antibody confirmed this result (Supplementary Material, Fig. S5B). Viral protein levels were the same in GFP, wtWTN5A and C83S variant as shown by GAG antibody staining (Fig. 9B and F). It was possible that the loss of a disulfide bond could have slightly altered protein folding, perhaps facilitating degradation. We therefore ran proteins from the cell lysate on combination reducing and non-reducing gels (Fig. 9B and C). The wtWNT5A and WNT5AC83S bands were generally shifted to a similar, smaller molecular weight (Fig. 8C). However, the intensity of the band for WNT5AC83S was lower compared to wtWNT5A (n = 2). In addition the migration of the mutant protein was uneven across the lane such that there was more intense signal at the edges of the lane than in the center. There is a consistently lower level of WNT5AC83S protein out of the total protein loaded (40 μg/lane) in all blots which also contributes to the lower intensity band. It is unlikely that the WNT5A antibody binds with less affinity to the epitope in the non-reducing conditions. We showed that the antibody recognizes wtWNT5A and WNT5AC83S with equal affinity in immunocytochemistry staining (Supplementary Material, Fig. S3A–C). Thus, it seems unlikely that a partially unfolded protein would have less exposure of the relevant epitope than the native protein.
Figure 9.
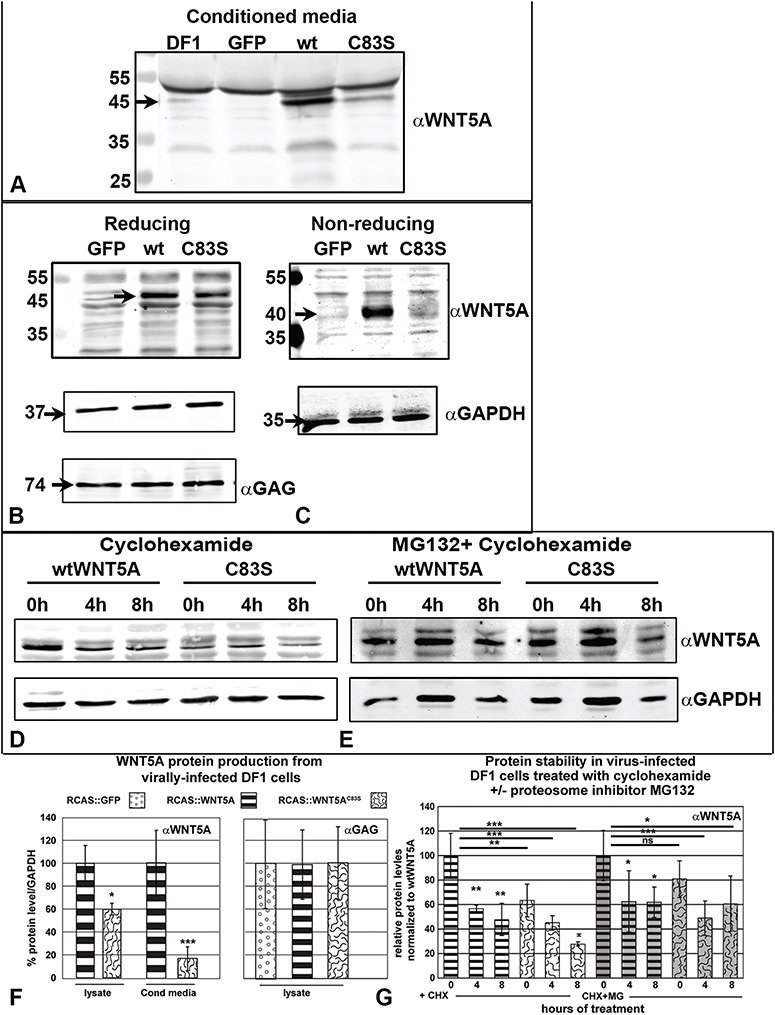
WNT5AC83S protein is translated and secreted at lower levels than wtWNT5A. Western blot analysis of WNT5A protein. (A) DF1 (chicken fibroblast) cells were infected with RCAS viruses (GFP, WNT5A or WNT5AC83S) or no virus and propagated for at least 1 month. Heparin (100 μg/ml) was added 24 h prior to collecting conditioned media. Blots were probed with anti-human WNT5A polyclonal antibody. The conditioned media from wtWNT5A-infected cells contains a strong band at 45 kDa, the predicted size of WNT5A protein (90 μg protein per lane). The lane with WNT5AC83S protein has considerably lighter staining. (B) In the lysate of DF1 cells, the 45 kDa band representing WNT5A was absent in the GFP controls and at lower intensity in the C83S protein sample (40 μg protein loaded per lane). The loading was equal as shown by the anti-GAPDH staining. There were also equal amounts of viral protein present (anti-GAG, F). (C) In non-reducing conditions, the bands for wild-type and mutant proteins have shifted equally suggesting there are no major changes in protein conformation. The band for mutant WNT5A is lighter than the control, in part due to lower expression of the protein. (D) The treatment of cultures with cyclohexamide blocked new synthesis of protein. Existing pools of WNT5A protein decreased over 8 h, suggesting they were degraded by the proteasome (40 μg per lane). (E) Pretreatment with MG132 for 1 h protected mutant WNT5A from being degraded to a similar extent to wtWNT5A (40 μg per lane). (F) Densitometry readings were made for three blots and normalized to GAPDH for the lysate or non-specific bands for the media. Significantly lower values for WNT5AC83S in the media suggests that protein is either synthesized at lower efficiency, fails to be secreted or is degraded prior to secretion. Lower levels in the lysate exclude retention inside the cell due to a problem with protein processing (Students t-test, *P < 0.05, ***P < 0.001). There was no quantitative difference in the levels of GAG viral protein. (G) Densitometry readings for three blots, normalized to GAPDH. Left side CHX: There were significant differences in the overall levels of wtWNT5A protein at 4 and 8 h of treatment with cyclohexamide (4 h, P = 0.004, 8 h, P = 0.001). The WNT5AC83S levels were unchanged at 4 h but by 8 h there was a significant decrease (P = 0.002). Right side MG132 + CHX: Pretreatment with MG132 protected the WNT5AC83S protein from being degraded at time zero. The pool of wtWNT5A was significantly reduced over 8 h, most likely due to secretion (P = 0.03). However, the C83S variant protein did not decrease over time, most likely due to lower efficiency secretion. ANOVA, Fisher’s LSD post hoc test for each treatment separately (CHX alone, CHX + MG132), *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
Next, we tested whether lower levels of the mutant protein were due to preferential degradation. DF1 fibroblasts were treated with cyclohexamide to block new protein synthesis and levels of WNT5A were measured over 8 h (Fig. 9D). Significant loss of protein occurred for the wtWNT5A over 8 h (decrease of 50%, P = 0.03; Fig. 9G). The WNT5AC83S protein similarly dropped to 50% by 8 h (P = 0.001; Fig. 9G). The levels of WNT5AC83S protein started off significantly lower at time 0 (P = 0.002; Fig. 9G), in accordance with previous data (Fig. 9F). There does not appear to be any difference in the rate of protein loss between wtWNT5A and mutant WNT5A.
Finally we blocked the ubiquitin proteasome pathway (56) with MG132 for 1 h prior to the addition of cyclohexamide. At time zero, or after 1 h of treatment with MG132, wtWNT5A and WNT5AC83S proteins were not significantly different. This indicates that blocking the proteasome for 1 h was sufficient to normalize the levels of the mutant protein. Further culture led to significantly lower levels of wtWNT5A (62% lower than time zero, P = 0.03; Fig. 9E and G). However, there was no significant difference in the three time points for WNT5AC83S. Since neither drug blocks secretion, it is likely that the decrease in wtWNT5A is due to protein leaving the cell through the secretory pathway. However, since the C83S variant diminishes secretion, the majority of protein remains in the cell after surviving proteasomal degradation. Levels of wtWNT5A were also significantly increased after 1 h of treatment with MG132 compared to no treatment (P = 0.01, data not shown). Since there is no significant difference between the levels of protein in mutant or wtWNT5A-infected cultures at time zero, there does not appear to be a selective decrease in protein stability caused by the mutation.
Finally, we tested whether in vivo there is a synthesis and secretion defect caused by the C83S variant. We therefore injected limbs with the GFP, wtWNT5A or C83S variant viruses and performed immunofluorescence staining, 5 days after virus was injected (HH30). This time point was chosen due to equivalent viral load between virus types. Antibodies to WNT5A recognize the human protein and may cross react with chicken based on sequence homology; however, in our western blots there was minimal signal in the GFP control lanes (Chicken, DF1 cell lysate, Fig. 9B). Similarly, limb sections stained for virus and WNT5A protein showed no expression of WNT5A in the GFP control limbs (Supplementary Material, Fig. S6A–A”’, n = 3). In contrast, wtWNT5A virus-infected limbs had strong signal for WNT5A protein that overlapped expression of GAG in the cytoplasm (Supplementary Material, Fig. S6B–B”’, n = 3). The C83S-expressing limbs also had cytoplasmic staining that overlapped with the GAG antibody (Supplementary Material, Fig. S6C–C”’, n = 3). Thus, WNT5A protein is being made in vivo from both forms of virus. The levels of expression were not quantifiable in the immunostaining assays.
Discussion
Skeletal malformations are a major finding in DRS including jaw hypoplasia, short stature and mesomelic limb shortening (5–7). DRS patients have mutations that were theoretically linked to the non-canonical WNT signaling pathway (7). Through local viral transgenesis in the chicken embryo, we provided an in vivo context for the human wild-type or mutant WNT5A gene to act over a period of time critical for skeletal development. Our experiment simulates the human situation where both wild-type and mutant protein are present since the retroviruses are expressed in the presence of gallus WNT5A. We have recapitulated the short limb phenotypes seen in RS. Even though there may be lower levels of mutant protein as compared to wtWNT5A present in RS patients and chicken embryos, a major impact on development of the skeleton is produced. Here we report that the main reason for pathogenesis of the C83S variant is the disruption of PCP and hypertrophy within the chondrocytes which has a dominant effect on elongation of the cartilage elements.
WNT5AC83S selectively and dominantly distorts early morphogenesis of the bones in the perpendicular axis
We reported a significant, consistent change in the morphology of cartilage rods in the C83S mutant viral infected limbs, where growth in the AP axis was exaggerated at the expense of the proximo-distal axis. These differences in direction of growth, while keeping the total cell number the same, imply that there are convergent extension defects (Fig. 10A–C). There are five neomorphic changes in the skeleton that are uniquely seen with WNT5AC83S misexpression (Table 2). These differences include (1) proximo-distal length that is only 30–50% of wtWNT5A, (2) a nearly 200% increase in AP diameter of the diaphysis compared to wtWNT5A, (3) a relative increase in cell density in the diaphysis followed by (4) a delay in chondrocyte hypertrophy and (5) decreased bone deposition. At the cellular level there were two additional changes only induced by the C83S variant that could explain the distorted morphogenesis (Table 2): (1) randomized orientation of the chondrocytes relative to the long axis, and (2) diffuse Prickle expression. All the aforementioned effects dominate over those of the endogenous gallus WNT5A gene and support the idea of interference by the mutant version of WNT5A with normal functions of the protein. The results are even more striking when we consider that expression levels are likely to be lower for the mutant protein. It is clear that even a small amount of the variant protein is sufficient to disrupt morphogenesis.
Figure 10.
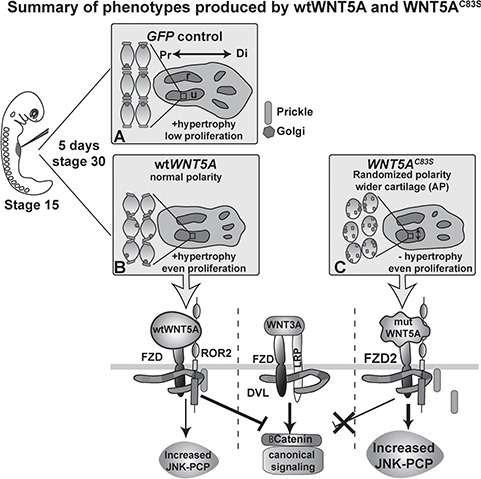
Summary of the dominant effects of WNT5A mutations on skeletal morphogenesis. The forelimb field is injected at stage 15 prior to budding. (A) Controls (empty plasmid, GFP or AlkPO4 virus) show flattened chondrocyte shape, hypertrophy and low proliferation in the diaphysis and elongation of the cartilage condensations in the proximo-distal axis. (B) wtWNT5A virus slightly altered chondrocyte flattening and increased proliferation in the diaphysis. Hypertrophy was present similar to controls. wtWNT5A increased JNK-PCP activity and blocked canonical WNT signaling. (C) In the presence of WNT5AC83S virus, chondrocytes were rounded and randomized which also correlated with a lack of elongation. The mutWNT5A was less effective at antagonizing the canonical pathway. Mutant WNT5A blocked hypertrophy leading to distortion of growth. Instead of elongating proximo-distally, cartilages grew in the perpendicular (AP and dorsi-ventral axes). AP, anterior-posterior; DVL, dishevelled; FZD, frizzled; LRP, low density lipoprotein receptor related protein; Pk, Prickle; Pr, proximal; Di, distal; r, radius; u, ulna.
Table 2.
Summary of phenotypic changes in different biological functions
| Biological function | Experimental readout | Loss of function—C83S similar to GFP or lower levels than wtWNT5A | Gain of function—C83S similar to wtWNT5A | Neomorphic function—C83S different than wtWNT5A |
|---|---|---|---|---|
| Cell cycle | Proliferation |
|
||
| Signaling | Activation of the JNK pathway in ATF2 luciferase assays |
|
|
|
| Inhibition of the canonical WNT pathway |
|
|
||
| Translation | Protein synthesis and secretion |
|
|
|
| Morphogenesis | Skeletal morphology |
|
|
|
| Planar cell polarity | PCP phenotypes |
|
|
The main mechanism that contributes to bone shortening is likely a failure of chondrocytes to flatten and stack in the proximo-distal axis. The stacking mechanism involves a slipping of adjacent chondrocytes under each other, after cell division (41,43). Misexpression of RCAS viruses containing dominant-negative (dn) forms of FZD7 and ROCK2 or overexpression of VANGL2 (41) disrupt the formation of organized columns due to interference with the non-canonical WNT pathway. In this previous study, the susceptible chondrocytes were located in the proliferative zone of the epiphysis. In a study by the same authors using live imaging, either increased or decreased PCP signaling resulted in misoriented chondrocytes that fail to form columns (43). The role of cell adhesiveness was also studied (43,44) and was found to be important for pivoting of daughter cells after medio-lateral (or dorso-ventral) proliferation. In our static imaging, the rounder chondrocytes do not appear to be stacking into columns in the presence of WNT5AC83S and further analysis of intercalation is a subject for a future study.
The increase in chondrocyte size is timed with the most rapid period of bone elongation in the appendicular skeleton (57). In normal GFP- or wtWNT5A-infected limbs, the progression to hypertrophy occurs in the diaphysis on schedule which correlates with normal length bones. Others have consistently reported that either an increase (27,31) or decrease in the levels of WNT5A (27) shortens skeletal elements and delays expression of COL10A1 RNA, secreted by hypertrophic chondrocytes. It is interesting that in our hands in contrast to Hartmann and Tabin (2000) the overexpression of wtWNT5A allowed the normal expression of COL10A1, similar to GFP virus controls. We examined limbs at slightly later stages than those used for the chicken (31) or mouse (27) which may have allowed sufficient time for the COL10A1 protein to be expressed. In contrast, the C83S variant protein derived from the virus was expressed at lower levels than wtWNT5A based on western blot experiments. One might have expected the expression of COL10A1 to be similar to GFP; however, the mutant protein prevented COL10A1 expression in all embryos. The C83S mutation also dominantly interferes with the expansion of chondrocytes as shown by the lack of lacunae in Alcian blue stained sections. This data suggests that hypertrophy is not occurring in a timely manner. Our wholemount skeletal preparations show that bone can form if embryos are allowed to develop to stage 38 (Table 1). All chicken embryo experiments end prior to hatching so the ultimate effect on adult bone morphology cannot be determined. Nevertheless, it is these early effects on cartilage that will have long lasting effects on bone length and stature.
The slight difference in levels of WNT5A protein produced by the mutated gene in patients with RS are likely not the reason for the specific changes in extension of cartilage. Our data suggests that the local, disruption of cell polarity and hypertrophy are more important for the manifestation of the syndrome and that the secretion problems are not the main mechanism explaining the RS phenotype. We would like to draw attention to a difference in the findings of the present study and those of our previous study on the protein derived from the WNT5AC83S variant. Previously we found that WNT5AC83S present in media failed to activate non-canonical WNT signaling via ATF2 luciferase reporter (33). In the previous study we performed luciferase assays using non-concentrated conditioned media collected from RCAS-infected cells which now we know to contain minimal levels of WNT5AC83S protein, far too low to activate the reporter.
RS phenotypes are highly unlikely to be due to a gain-of-WNT5A function
The use of the retroviral system helps to clarify the effects of a gain-of-function mutation where we compare the mutant to wild-type genes. If the mutation caused a gain of function we would expect to see even higher proliferation and higher JNK-PCP activity than wtWNT5A levels, but this was not the case. There should also have been significant disruption of chondrocyte orientation and polarity caused by the wtWNT5A but only the mutant form of WNT5A caused a phenotype in the chondrocyte morphology (Table 2). There is no evidence of a more exaggerated phenotype of the mutant compared to wtWNT5A. Based on these data, we can exclude a gain-of-function as the reason for the craniofacial and limb phenotypes of RS.
The partial loss of WNT5AC83S function in antagonizing canonical signaling may have low biological significance
Some of the data with the C83S variant of WNT5A was similar to GFP results or was quantitatively lower than that obtained with wtWNT5A (Fig. 10A–C; Table 2). The C83S variant was completely unable to antagonize canonical signaling in the presence of WNT3A (Fig. 10C). While in our biochemical experiments there appears to be a net gain in canonical WNT activity since the mutant WNT5A is a poor antagonist of canonical signaling, the same may not be true in chicken embryos or patients. We showed that the mutated protein can interact with Ror2 to antagonize canonical signaling. Thus, in the limb cartilage where Ror2 is expressed in cartilage condensations (58) there should be adequate antagonism of canonical signaling from both the normal and mutant allele of WNT5A.
WNT5A missense mutations may affect protein conformation
The direct effect of point mutations on protein structure of WNT5A is challenging to determine since WNT proteins are notoriously hard to express, purify and crystallize (59). Nevertheless, using the chicken retroviral system we were able to partially characterize the mutant protein. The missense mutation leading to the C83S substitution consistently leads to lower protein levels than wtWNT5A in lysate and media. We found no evidence for increased degradation of the mutant protein in the presence of MG132, nor were RNA levels lower for the mutant transcripts in the cultured cells (data not shown). Instead it is possible that the loss of a disulfide bond might trigger an unfolded protein response (UPR) which leads to lower protein levels (60,61). The UPR consists of a set of interlinking intracellular pathways that maintain the protein-folding capacity of the endoplasmic reticulum (ER). When a UPR has been induced, the cell increases the amount of ER available and reduces the translation of the protein to restore homeostasis. Since the RCAS system resulted in stable integration of the WNT5AC83S transgene, the ‘new’ homeostasis for these fibroblasts was to have lower levels of protein expression, as shown in western blots with cell lysate. An unregulated UPR will cause cell stress. In our in vivo studies, there was no evidence for increased cell death (negative TUNEL data) and long term cell cultures containing the RCAS:WNT5AC83S grew at a similar rate to the wtWNT5A virus or GFP virus. Thus, it appears that by lowering the levels of WNT5AC83S, cells in vivo and in vitro are able to avoid cell stress. Taken together, the patients with RS will have a deficiency in WNT5A but we do not think this is related to the phenotypes. Instead, the mutated protein interferes with normal functions of WNT5A at the signaling level as well as during chondrocyte differentiation.
In summary, the chicken provides a unique opportunity to isolate the dominant effects of mutation. It is well accepted that variability in expressivity of a phenotype is due to the interaction of the gene of interest with the rest of the genome (modifier genes) and environmental factors (55–57). Our results suggest that if all genetic and epigenetic factors are held equal as in the chicken embryo system, that C83S variant has major pathogenic effects on morphogenesis. Unfortunately, due to the rarity of the syndrome and lack of detailed phenotypic data from radiographs it is not possible to determine whether subjects with different variants of WNT5A have greater or lesser severity in the skeletal phenotypes. The avian embryo is a valuable model system in which we can gain insights into autosomal dominant human genetic disorders such as RS.
Materials and Methods
Cloning of virus and plasmid constructs
The ultimate ORF containing human WNT5A (Genbank Ref seq: NM_003392.4) was purchased from Invitrogen (#IOH39817). We used restriction-free cloning (62) to knock in a mutation that causes autosomal dominant RS2 (OMIM: 180700): the 248G-C mutation in exon 3 of the WNT5A gene, resulting in cysteine 83 converting to serine (C83S). A C-terminal Flag tag was cloned after the coding sequence and a stop codon was added to the 3′ end of the tag. Gateway cloning (Invitrogen, Burlington, ON, Canada) was used to move the mutant or wild-type sequences from pENTRY into pcDNA3.2 for plasmid transfection and RCASBPY for retroviral expression as described (33). The human FZD2 coding sequence in a shuttle vector was purchased from GeneCopoeia (Rockville, MD, clone #GC-S0193-B) and recombined into pcDNA3.2 using Gateway recombinase (LR Clonase II, Invitrogen). Other viruses containing GFP (A. Gaunt) or Alkaline Phosphatase (AlkPO4) (L. Niswander) were generously provided by other investigators.
Virus preparation, injection and measurement of expression levels
White leghorn eggs were incubated until Hamburger Hamilton stage 15 (E2.5) (63) and concentrated RCAS retrovirus viral particles were injected into the forelimb region (between somites 15 and 16) using a Picospritzer microinjector (General Valve Corp.). We chose this stage in order to give the virus sufficient time to replicate prior to lineage specification in the limb field (36). The key events in limb morphogenesis are limb budding (stage 18, E3.0, 24 h post-injection) and start of cartilage differentiation (stage 28, E5.5; 72 h post-injection). Embryos were euthanized at various stages (stage 29 or E6.0; stage 30 or E6.5; stage 36, E10 or stage 38, E12).
To quantify viral expression, primers specific to human WNT5A were used to amplify the viral insert as described (33). Total RNA was isolated from batches of limb buds using a Qiagen RNAeasy kit (#75144, Toronto, Canada) (3–4 limb buds were pooled to give one biological replicate). There were four biological replicates collected at 48 h and 96 h post-injection. Sybr green-based qRT-PCR was carried out using an ABI Step One plus instrument (95°C for 5 s, 60°C for 20 s, 40 repeats). Levels of expression were normalized to 18S RNA. The ΔΔCt method was used to calculate relative fold-change expression between wtWNT5A- and WNT5AC83S-infected limbs and T-tests were used to determine significance.
Histology, immunofluorescence and immunocytochemistry
Forelimbs were fixed in 4% paraformaldehyde for 2–3 days. Limbs at stage HH36 were decalcified in 12% EDTA for 4–5 days prior to processing into wax. Selected sections (sagittal or transverse) were stained with Alcian blue and Picrosirius red to see the differentiated cartilage and bone (64). For immunofluorescence, antigen retrieval was carried out either with sodium citrate (10 mm) or Diva Decloaker (WNT5A, Biocare Medical, Inter Medico, Markham, Ontario, Canada, #DV2004MX). For WNT5A antibody, sections were treated with 0.3% Triton in PBS for 10 min and then put into block containing 1% donkey serum, 0.3% Triton in PBS for 30 min. For collagen X antibody staining, sections were pretreated with 0.5% hyaluronidase in Hank’s Balanced Salt Solution for 30 min prior to antigen retrieval. Sections were blocked with 5% or 10% goat serum plus 0.1% tween/PBS at room temperature for 1 h and incubated overnight at 4°C with primary antibodies: anti-3C2/GAG (Developmental Studies Hybridoma bank, Iowa City, Iowa, 1:4 #AMV-3C2); anti-collagen X (COL10A1, Developmental Studies Hybridoma bank, 1:250 #X-AC9-c); anti-pan-Prickle (Abcam Toronto, ON, Canada, 1:50, #15577) and anti-WNT5A (R&D systems, Minneapolis, MN, 1:500, #AF645). Secondary antibodies included Alexa 488, 647 and Cy5 (Invitrogen, 1:250) and these were applied for 1 h at room temperature followed by staining with 10 μg/ml Hoechst for 10 min (#33568, Sigma Aldrich, Oakville, ON, Canada). Slides were coverslipped with Prolong Gold (Invitrogen).
Phalloidin staining to visualize cortical actin was carried out in wholemount on 200 μm thick slices of limbs since this reagent is incompatible with ethanol. Forelimb slices were fixed for 48 h at 4°C in 4% paraformaldehyde, permeabilized in 0.5% triton-X/PBS for 30 min, and blocked in 10% FBS, 0.5% triton-X/PBS for 30 min 2× at room temperature. Primary antibodies were incubated separately for 48–96 h at 4°C on a rocker in blocking solution; anti-Golgi antibody (GM130, 1:100, BD labs, Burlington, ON, Canada, #610822) and anti-SOX9 (Sigma-Aldrich, 1:200, #HPA001758). Slices were also incubated in Phalloidin (Molecular Probes, ThermoFisher, Mississauga, ON, Canada, Alexa Fluor-568, 1:50, #A12380). Secondary antibodies (Alexa-fluor-488, −647, Molecular Probes, Thermo Fisher, Mississauga, ON, Canada 1:200) were incubated 48–96 h at 4°C on a rocker in 0.5% triton-X/PBS. Signals were detected using Alexa Fluor conjugated secondary antibodies (Life Technologies 1:200), and nuclei were counter stained with 10 μg/ml Hoechst (#33568, Sigma), DAPI (Molecular Probes #P36935) or TO-PRO-3 iodide (Life technologies, #T3605). Prolong Gold antifade was applied to all slides (Life Technologies #P36930). Fluorescence images were collected with a Leica SP5 confocal microscope or with a 20X objective on a slide scanner (3DHISTECH Ltd, Budapest, Hungary).
For cell proliferation studies, chicken embryos 72 or 96 h post-virus injection (stage 29 or 30) were labeled with BrdU (10 mg/ml), injected into the heart with a Picospritzer, 3 h prior to euthanasia. Antigen retrieval was performed with 2N HCl for 30 min at room temperature and treated with 0.1% trypsin/PBS for 10 min at 37°C. Sections were blocked with 10% goat serum and 0.1% tween-20 for 1 h then incubated overnight at 4°C in blocking solution with anti-BrdU (Developmental Studies Iowa Hybridoma bank, 1:20, #G3G4). Slides were then probed for anti-SOX9 (1:100) in block overnight at 4°C. The proportion of BrdU-labeled cells in the SOX9-positive region was counted using ImageJ. Apoptosis was analyzed via TUNEL analysis using ApopTag Apoptosis Kit (Chemicon, S7101) and was detected using anti-digoxygenin tagged with fluorescein as described (46).
For immunocytochemistry experiments, HEK293 cells were seeded onto coated coverslips (18 mm2, Corning) and coated with Poly-L-Lysine (Sigma #RNBC8085) for 15 min. Cultures were grown to 40% confluency and transfected using Lipofectamine3000 (Invitrogen #L3000-008) following manufacturer’s instructions. A total of 2.5 μg DNA was transfected in each experiment (plasmids: WNT5A, WNT5AC83S or empty parent vector, pcDNA3.2). Cells were fixed in 4% PFA 48 h post-transfection for 20 min and then stored in PBS + 0.01% sodium azide at 4°C. Cultures were blocked in 10% normal horse serum (Sigma #H0146). Permeabilized with 0.2% triton-X for 1 h then incubated overnight at 4°C with primary antibody (WNT5A R&D, 2 μg/ml, #AF645). Secondary antibody was applied for 1 h at room temperature (anti-goat Sigma #11C1215) and counterstained with 10 μg/ml Hoechst. Cells were imaged using a Leica SP5 confocal microscope. Three random fields of view per treatment were analyzed; assay performed three times.
Quantification of cell polarity and cell density
We used ImageJ to measure cell polarity and shape as described (33). Measurements were conducted for 70 cells located in the diaphysis of the ulna. To measure cell density, images with nuclear stain from stage 36 embryos were imported into ImageJ software. Sagittal measurements included the distance from the epiphysis near the digits to epiphysis near the humerus, the width in the center of the diaphysis and area (around entire ulna). Transverse sections were perpendicular to the long axis of the cartilage, through the ulna spanning the epiphysis (closest to the digits) and the diaphysis. Regional cell density was quantified by counting nuclei located in a 200 μm2 region in the epiphysis and diaphysis of the ulna in sagittal sections. Cell density was also quantified in half of the developing ulna (closest to the digits).
Cell culture and luciferase reporter assays
Transient transfections for luciferase assays were performed on 30–40% confluent HEK293 cells (0.17–0.18 × 106 cells/ml) (33); or using non-treated HH24 chicken limb bud mesenchyme micromass cultures as described for facial mesenchyme (46,65). Cells were transfected using Lipofectamine3000 (Invitrogen #L3000-008; Nunc 24-well plates #142475). HEK293 cells were transfected 24 h after plating. Micromass cultures were allowed to attach for 30 min then transfection reagents were added to the culture spots for 30 min prior to flooding the dish with media. The following plasmids were used: empty (pcDNA3.2/V5-DEST), WNT5A, WNT5AC83S, mRor2 (Addgene, #22613), FZD2 or caNFAT (constitutively active nuclear factor of activated T-cells; Addgene, # 10959), or in combinations (totaling 0.3 μg or 0.6 μg for FZD2-ATF2 assay). Additionally, Renilla luciferase reporter plasmid was transfected for normalization (0.05 μg or 0.01 μg for FZD2-ATF2 assay), along with Firefly reporter plasmids: STF, ATF2 (45) or NFAT (Addgene #10959) luciferase reporters (0.2 μg). At 24 h post-transfection, a subset of cultures was treated with 100 ng/ml Wnt3a protein (R&D, #5036-WN-010). Luciferase assays were performed after 48 h of culture using Dual-luciferase reporter assay system (Promega #E1910) as described (65). A Tecan luminometer (Spark® multimode Tecan plate reader) was used to read luminescence activity at 1 s reading with OD1 filter. At least three technical and three biological replicates were carried out for each transfection mixture and the experiment was repeated on two different days.
Western blotting
To generate cells that stably express WNT5A and WNT5AC83S proteins, chicken DF1 cells (ATCC) were transfected with RCAS constructs as in the initial stages of preparing virus for embryo injections (66). After one month of passaging, viruses have been spread to all cells in the culture. At 24 h before collection, WNT5A-infected cells along with GFP-infected control DF1 cells were treated with 1% FBS and 100 μg/ml heparin. Conditioned media was collected and centrifuged 1000 xg at 4°C for 10 min. The supernatant was transferred to a centrifugal filter (Amicon Ultra Cell 10 K #UFC901024) and concentrated at 5000 xg at 4°C in a fixed rotor for 95 min. For cyclohexamide treatment a subset of cultures were grown in the following manner: cells cultured in 6-well plates until they reach 100% confluence. A concentration of 50 μg/ml cyclohexamide (Sigma Aldrich) was added and cultures incubated for 0, 4 and 8 h. Another group of DF1 cells were pretreated with 30 μM MG132 (Sigma Aldrich) for 1 h to block the proteasome and then incubated in cyclohexamide (50 μg/ml) for a further 4 and 8 h.
To prepare cell lysates, cells were washed 2× in cold PBS then cell lysate buffer was added (RIPA radioimmunoprecipitation assay buffer, buffer with SDS containing mini protease inhibitor cocktail, Roche #04693124001 Mississauga, ON, Canada) and phosphatase inhibitor cocktail (phosSTOP, Roche #04906845001). Lysed cells were removed with a cell scraper (Corning, 3010) then transferred to a 1.5 ml Eppendorf tube and held on ice for 15 min. Cell lysates were spun at 14 000 rpm at 4°C for 15 min and the supernatants were collected and stored at −20°C.
Cell lysate samples were mixed with sample buffer at 1.25 mg/ml with or without 0.3% β-Mercaptoethanol (Sigma Aldrich) to detect protein shifts. A total of 40 μg protein was added per lane for the lysate and 90 μg for conditioned media. Samples were resolved on SDS-10% acrylamide gels and wet-transferred (50V for 90 min) to 0.45 μm nitrocellulose membrane (Thermo Fisher #88018). Membranes were incubated for 1 h in blocking solution (according to R&D protocol) and primary antibodies incubated overnight at 4°C in 0.5% BSA in blotting buffer (R&D blotting buffer group 8); anti-Wnt5a (R&D, 2 μg/ml, #AF645); anti-Flag Sigma Aldrich (1:1000 #F7425); anti-GAG (Developmental Studies Hybridoma bank, AMV-3C2, 1:50); and anti-GAPDH (Thermo Fisher 1:1000 #AM4300). Licor (Cedarlane Laboratories, Burlington, ON, Canada) secondary antibodies were incubated at 1:10 000 for 1 h at room temperature. Membranes were scanned using near-infrared Licor scanner (Odyssey).
Statistical analysis
All statistical analyses for BrdU, luciferase, cell shape, cell density, Golgi orientation, limb morphometrics and luciferase assays were done using one-way ANOVA, followed by Tukey’s or Fisher’s LSD post hoc test for multiple comparisons. T-tests were used for paired densitometry readings. Statistica software v 6.0 was used. For densitometry, software from the Licor scanner was used and values were normalized to GAPDH for the cell lysate. The media values were normalized to two non-specific bands that were present in all samples.
Supplementary Material
Acknowledgements
The authors would like to thank members of the Richman lab and to M. Underhill and C. Roskelley for stimulating discussions. In addition we thank J. Cooper for her assistance with immuno-staining.
Conflict of Interest statement. None declared.
Funding
Canadian Institutes of Health Research (grant # MOP-123536 to J.M.R.); 4-year University of British Columbia doctoral fellowship (to S.G.).
References
- 1. Robinow M., Silverman F.N. and Smith H.D. (1969) A newly recognized dwarfing syndrome. Am. J. Dis. Child., 117, 645–651. [DOI] [PubMed] [Google Scholar]
- 2. Afzal A.R., Rajab A., Fenske C.D., Oldridge M., Elanko N., Ternes-Pereira E., Tuysuz B., Murday V.A., Patton M.A., Wilkie A.O. et al. (2000) Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet., 25, 419–422. [DOI] [PubMed] [Google Scholar]
- 3. Schwabe G.C., Tinschert S., Buschow C., Meinecke P., Wolff G., Gillessen-Kaesbach G., Oldridge M., Wilkie A.O., Komec R. and Mundlos S. (2000) Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am. J. Hum. Genet., 67, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Bokhoven H., Celli J., Kayserili H., van Beusekom E., Balci S., Brussel W., Skovby F., Kerr B., Percin E.F., Akarsu N. et al. (2000) Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet., 25, 423–426. [DOI] [PubMed] [Google Scholar]
- 5. Person A.D., Beiraghi S., Sieben C.M., Hermanson S., Neumann A.N., Robu M.E., Schleiffarth J.R., Billington C.J. Jr., van Bokhoven H., Hoogeboom J.M. et al. (2010) WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev. Dyn., 239, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roifman M., Marcelis C.L., Paton T., Marshall C., Silver R., Lohr J.L., Yntema H.G., Venselaar H., Kayserili H., van Bon B. et al. (2015) De novo WNT5A-associated autosomal dominant Robinow syndrome suggests specificity of genotype and phenotype. Clin. Genet., 87, 34–41. [DOI] [PubMed] [Google Scholar]
- 7. White J.J., Mazzeu J.F., Coban-Akdemir Z., Bayram Y., Bahrambeigi V., Hoischen A., van Bon B.W.M., Gezdirici A., Gulec E.Y., Ramond F. et al. (2018) WNT Signaling Perturbations Underlie the Genetic Heterogeneity of Robinow Syndrome. Am. J. Hum. Genet., 102, 27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White J., Mazzeu J.F., Hoischen A., Jhangiani S.N., Gambin T., Alcino M.C., Penney S., Saraiva J.M., Hove H., Skovby F. et al. (2015) DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. Am. J. Hum. Genet., 96, 612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White J.J., Mazzeu J.F., Hoischen A., Bayram Y., Withers M., Gezdirici A., Kimonis V., Steehouwer M., Jhangiani S.N., Muzny D.M. et al. (2016) DVL3 Alleles Resulting in a −1 Frameshift of the Last Exon Mediate Autosomal-Dominant Robinow Syndrome. Am. J. Hum. Genet., 98, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bunn K.J., Daniel P., Rosken H.S., O'Neill A.C., Cameron-Christie S.R., Morgan T., Brunner H.G., Lai A., Kunst H.P., Markie D.M. et al. (2015) Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome. Am. J. Hum. Genet., 96, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mansour T.A., Lucot K., Konopelski S.E., Dickinson P.J., Sturges B.K., Vernau K.L., Choi S., Stern J.A., Thomasy S.M., Ho H.H. et al. (2018) Whole genome variant association across 100 dogs identifies a frame shift mutation in DISHEVELLED 2 which contributes to Robinow-like syndrome in Bulldogs and related screw tail dog breeds. PLoS Genet., 14, e1007850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mazzeu J.F., Pardono E., Vianna-Morgante A.M., Richieri-Costa A., Ae Kim C., Brunoni D., Martelli L., de Andrade C.E., Colin G. and Otto P.A. (2007) Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am. J. Hum. Genet. Part A, 143, 320–325. [DOI] [PubMed] [Google Scholar]
- 13. Yang Y. and Mlodzik M. (2015) Wnt-Frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Ann. Rev. Cell Dev. Biol., 31, 623–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wiese K.E., Nusse R. and Amerongen R. (2018) Wnt signalling: conquering complexity. Development, 145. [DOI] [PubMed] [Google Scholar]
- 15. Langton P.F., Kakugawa S. and Vincent J.P. (2016) Making, Exporting, and Modulating Wnts. Trends Cell Biol., 26, 756–765. [DOI] [PubMed] [Google Scholar]
- 16. Niehrs C. (2012) The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell. Biol., 13, 767–779. [DOI] [PubMed] [Google Scholar]
- 17. Stricker S., Rauschenberger V. and Schambony A. (2017) ROR-Family Receptor Tyrosine Kinases. Curr. Top. Dev. Biol., 123, 105–142. [DOI] [PubMed] [Google Scholar]
- 18. Nusse R. and Clevers H. (2017) Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell, 169, 985–999. [DOI] [PubMed] [Google Scholar]
- 19. Ho H.Y., Susman M.W., Bikoff J.B., Ryu Y.K., Jonas A.M., Hu L., Kuruvilla R. and Greenberg M.E. (2012) Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. U.S.A., 109, 4044–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Butler M.T. and Wallingford J.B. (2017) Planar cell polarity in development and disease. Nat Rev Mol Cell Biol, 18, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wallingford J.B. (2012) Planar cell polarity and the developmental control of cell behavior in vertebrate embryos. Ann. Rev. Cell Dev. Biol., 28, 627–653. [DOI] [PubMed] [Google Scholar]
- 22. Gao B., Ajima R., Yang W., Li C., Song H., Anderson M.J., Liu R.R., Lewandoski M.B., Yamaguchi T.P. and Yang Y. (2018) Coordinated directional outgrowth and pattern formation by integration of Wnt5a and Fgf signaling in planar cell polarity. Development, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao B., Song H., Bishop K., Elliot G., Garrett L., English M.A., Andre P., Robinson J., Sood R., Minami Y. et al. (2011) Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev. Cell, 20, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu C., Lin C., Gao C., May-Simera H., Swaroop A. and Li T. (2014) Null and hypomorph Prickle1 alleles in mice phenocopy human Robinow syndrome and disrupt signaling downstream of Wnt5a. Biol. Open, 3, 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang T., Bassuk A.G. and Fritzsch B. (2013) Prickle1 stunts limb growth through alteration of cell polarity and gene expression. Dev. Dyn., 242, 1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang B., Sinha T., Jiao K., Serra R. and Wang J.B. (2011) Disruption of PCP signaling causes limb morphogenesis and skeletal defects and may underlie Robinow syndrome and brachydactyly type B. Hum. Mol. Genet., 20, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y., Topol L., Lee H. and Wu J. (2003) Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development, 130, 1003–1015. [DOI] [PubMed] [Google Scholar]
- 28. Yamaguchi T.P., Bradley A., McMahon A.P. and Jones S. (1999) A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development, 126, 1211–1223. [DOI] [PubMed] [Google Scholar]
- 29. Kuss P., Kraft K., Stumm J., Ibrahim D., Vallecillo-Garcia P., Mundlos S. and Stricker S. (2014) Regulation of cell polarity in the cartilage growth plate and perichondrium of metacarpal elements by HOXD13 and WNT5A. Dev. Biol., 385, 83–93. [DOI] [PubMed] [Google Scholar]
- 30. van Amerongen R., Fuerer C., Mizutani M. and Nusse R. (2012) Wnt5a can both activate and repress Wnt/beta-catenin signaling during mouse embryonic development. Dev. Biol., 369, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hartmann C. and Tabin C.J. (2000) Dual roles of Wnt signaling during chondrogenesis in the chicken limb. Development, 127, 3141–3159. [DOI] [PubMed] [Google Scholar]
- 32. Kawakami Y., Wada N., Nishimatsu S.I., Ishikawa T., Noji S. and Nohno T. (1999) Involvement of Wnt-5a in chondrogenic pattern formation in the chick limb bud. Dev. Growth Differ., 41, 29–40. [DOI] [PubMed] [Google Scholar]
- 33. Hosseini-Farahabadi S., Gignac S.J., Danescu A., Fu K. and Richman J.M. (2017) Abnormal WNT5A Signaling Causes Mandibular Hypoplasia in Robinow Syndrome. J Dent. Res., 96, 1265–1272. [DOI] [PubMed] [Google Scholar]
- 34. Qi J., Lee H.J., Saquet A., Cheng X.N., Shao M., Zheng J.J. and Shi D.L. (2017) Autoinhibition of Dishevelled protein regulated by its extreme C terminus plays a distinct role in Wnt/beta-catenin and Wnt/planar cell polarity (PCP) signaling pathways. J. Biol. Chem., 292, 5898–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee H.J., Shi D.L. and Zheng J.J. (2015) Conformational change of Dishevelled plays a key regulatory role in the Wnt signaling pathways. eLife, 4, e08142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pearse R.V. II, Scherz P.J., Campbell J.K. and Tabin C.J. (2007) A cellular lineage analysis of the chick limb bud. Dev. Biol., 310, 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Creuzet S., Couly G. and Le Douarin N.M. (2005) Patterning the neural crest derivatives during development of the vertebrate head: insights from avian studies. J. Anat., 207, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eames B.F. and Helms J.A. (2004) Conserved molecular program regulating cranial and appendicular skeletogenesis. Dev. Dyn., 231, 4–13. [DOI] [PubMed] [Google Scholar]
- 39. Zeller R. (2010) The temporal dynamics of vertebrate limb development, teratogenesis and evolution. Curr. Opin. Genet. Dev., 20, 384–390. [DOI] [PubMed] [Google Scholar]
- 40. Towers M., Wolpert L. and Tickle C. (2012) Gradients of signalling in the developing limb. Curr. Opin. Cell. Biol., 24, 181–187. [DOI] [PubMed] [Google Scholar]
- 41. Li Y. and Dudley A.T. (2009) Noncanonical frizzled signaling regulates cell polarity of growth plate chondrocytes. Development, 136, 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anakwe K., Robson L., Hadley J., Buxton P., Church V., Allen S., Hartmann C., Harfe B., Nohno T., Brown A.M. et al. (2003) Wnt signalling regulates myogenic differentiation in the developing avian wing. Development, 130, 3503–3514. [DOI] [PubMed] [Google Scholar]
- 43. Li Y., Li A., Junge J. and Bronner M. (2017) Planar cell polarity signaling coordinates oriented cell division and cell rearrangement in clonally expanding growth plate cartilage. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Romereim S.M., Conoan N.H., Chen B. and Dudley A.T. (2014) A dynamic cell adhesion surface regulates tissue architecture in growth plate cartilage. Development, 141, 2085–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ohkawara B. and Niehrs C. (2011) An ATF2-Based Luciferase Reporter to Monitor Non-Canonical Wnt Signaling in Xenopus Embryos. Dev. Dyn., 240, 188–194. [DOI] [PubMed] [Google Scholar]
- 46. Hosseini-Farahabadi S., Geetha-Loganathan P., Fu K., Nimmagadda S., Yang H.J. and Richman J.M. (2013) Dual functions for WNT5A during cartilage development and in disease. Matrix Biol., 32, 252–264. [DOI] [PubMed] [Google Scholar]
- 47. Mikels A., Minami Y. and Nusse R. (2009) Ror2 Receptor Requires Tyrosine Kinase Activity to Mediate Wnt5A Signaling. J. Biol. Chem., 284, 30167–30176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nishita M., Itsukushima S., Nomachi A., Endo M., Wang Z.C., Inaba D., Qiao S., Takada S., Kikuchi A. and Minami Y. (2010) Ror2/Frizzled Complex Mediates Wnt5a-Induced AP-1 Activation by Regulating Dishevelled Polymerization. Mol. Cell. Biol., 30, 3610–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mikels A.J. and Nusse R. (2006) Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol., 4, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grumolato L., Liu G.Z., Mong P., Mudbhary R., Biswas R., Arroyave R., Vijayakumar S., Economides A.N. and Aaronson S.A. (2010) Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev., 24, 2517–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klein P.S. and Melton D.A. (1996) A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A., 93, 8455–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thorne C.A., Hanson A.J., Schneider J., Tahinci E., Orton D., Cselenyi C.S., Jernigan K.K., Meyers K.C., Hang B.I., Waterson A.G. et al. (2010) Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat. Chem. Biol., 6, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Veeman M.T., Slusarski D.C., Kaykas A., Louie S.H. and Moon R.T. (2003) Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol, 13, 680–685. [DOI] [PubMed] [Google Scholar]
- 54. Bradley E.W. and Drissi M.H. (2010) WNT5A regulates chondrocyte differentiation through differential use of the CaN/NFAT and IKK/NF-kappaB pathways. Mol. Endocrinol., 24, 1581–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Topol L., Jiang X., Choi H., Garrett-Beal L., Carolan P.J. and Yang Y. (2003) Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J. Cell Biol., 162, 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Amm I., Sommer T. and Wolf D.H. (2014) Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta, 1843, 182–196. [DOI] [PubMed] [Google Scholar]
- 57. Breur G.J., VanEnkevort B.A., Farnum C.E. and Wilsman N.J. (1991) Linear relationship between the volume of hypertrophic chondrocytes and the rate of longitudinal bone growth in growth plates. J. Orthop. Res., 9, 348–359. [DOI] [PubMed] [Google Scholar]
- 58. Stricker S., Verhey van Wijk N., Witte F., Brieske N., Seidel K. and Mundlos S. (2006) Cloning and expression pattern of chicken Ror2 and functional characterization of truncating mutations in Brachydactyly type B and Robinow syndrome. Dev. Dyn., 235, 3456–3465. [DOI] [PubMed] [Google Scholar]
- 59. Janda C.Y., Waghray D., Levin A.M., Thomas C. and Garcia K.C. (2012) Structural basis of Wnt recognition by Frizzled. Science, 337, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Korennykh A. and Walter P. (2012) Structural basis of the unfolded protein response. Ann. Rev. Cell Dev. Biol., 28, 251–277. [DOI] [PubMed] [Google Scholar]
- 61. Walter P. and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science, 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- 62. Bond S.R. and Naus C.C. (2012) RF-Cloning.org: an online tool for the design of restriction-free cloning projects. Nuc. Acids Res., 40, W209–W213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hamburger V. and Hamilton H. (1951) A series of normal stages in the development of the chick embryo. J. Morphol., 88, 49–92. [PubMed] [Google Scholar]
- 64. Ashique A.M., Fu K. and Richman J.M. (2002) Endogenous bone morphogenetic proteins regulate outgrowth and epithelial survival during avian lip fusion. Development, 129, 4647–4660. [DOI] [PubMed] [Google Scholar]
- 65. Geetha-Loganathan P., Nimmagadda S., Fu K. and Richman J.M. (2014) Avian Facial Morphogenesis Is Regulated by c-Jun N-terminal Kinase/Planar Cell Polarity (JNK/PCP) Wingless-related (WNT) Signaling. J. Biol. Chem., 289, 24153–24167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gordon C.T., Rodda F.A. and Farlie P.G. (2009) The RCAS retroviral expression system in the study of skeletal development. Dev. Dyn., 238, 797–811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.