

Abstract
Parkinson's disease (PD) is associated with olfactory defects in addition to dopaminergic degeneration. Dopaminergic signalling is necessary for subventricular zone (SVZ) proliferation and olfactory bulb (OB) neurogenesis. Alpha-synuclein (α-syn or Snca) modulates dopaminergic neurotransmission, and SNCA mutations cause familial PD, but how α-syn and its mutations affect adult neurogenesis is unclear. To address this, we studied a bacterial artificial chromosome transgenic mouse expressing the A30P SNCA familial PD point mutation on an Snca−/− background. We confirmed that the SNCA-A30P transgene recapitulates endogenous α-syn expression patterns and levels by immunohistochemical detection of endogenous α-syn in a wild-type mouse and transgenic SNCA-A30P α-syn protein in the forebrain. The number of SVZ stem cells (BrdU+GFAP+) was decreased in SNCA-A30P mice, whereas proliferating (phospho-histone 3+) cells were decreased in Snca−/− and even more so in SNCA-A30P mice. Similarly, SNCA-A30P mice had fewer Mash1+ transit-amplifying SVZ progenitor cells but Snca−/− mice did not. These data suggest the A30P mutation aggravates the effect of Snca loss in the SVZ. Interestingly, calbindin+ and calretinin (CalR)+ periglomerular neurons were decreased in both Snca−/−, and SNCA-A30P mice but tyrosine hydroxylase+ periglomerular OB neurons were only decreased in Snca−/− mice. Cell death decreased in the OB granule layer of Snca−/− and SNCA-A30P mice. In the same region, CalR+ numbers increased in Snca−/− and SNCA-A30P mice. Thus, α-syn loss and human A30P SNCA decrease SVZ proliferation, cell death in the OB and differentially alter interneuron numbers. Similar disruptions in human neurogenesis may contribute to the olfactory deficits, which are observed in PD.
Introduction
Parkinson's disease (PD) is associated with severe disruptions in olfactory function that precede the overt motor disturbances (1,2). The aetiology of PD olfactory defects is unclear, although one possibility is that abnormal olfactory bulb (OB) neurogenesis contributes to it. The subventricular zone (SVZ) is a mitotic layer lining the lateral ventricles; it harbours stem cells that generate Mash1+ transit-amplifying progenitor cells, which make neuroblasts (3). The newborn neuroblasts migrate from the SVZ along the rostral migratory stream (RMS), differentiate into periglomerular and granular interneurons and integrate into the OB circuitry. The adult SVZ-derived interneurons have been ascribed a variety of olfactory roles including odorant discrimination (4,5). One type of SVZ-derived periglomerular interneuron expresses tyrosine hydroxylase (TH) and secretes dopamine (DA) but does not appear to be vulnerable to the factors that kill substantia nigra pars compacta dopaminergic neurons in PD (6–8). Many intrinsic and environmental factors regulate SVZ neurogenesis, amongst them DA and DA receptors (9–11). For example, DA D3 receptor antagonism decreases postnatal neurogenesis (12).
Alpha-synuclein (α-syn) encoded by the Snca gene is a neuronal protein predominantly localized to presynaptic axon terminals that has been linked to a variety of functions (13,14). α-syn is a major constituent of Lewy bodies, one of the pathological hallmarks of PD. Five point mutations in SNCA (A30P, A53T, E46K, H50Q and G51D) have been identified in rare Mendelian forms of familial PD (15–19). Thus, the A30P mutation in α-syn that causes familial PD and thereby reduces DA concentrations may also regulate neurogenesis. A variety of animal models carrying SNCA mutations found in PD patients have been generated (20). For example, a mouse model carrying A53T exhibited decreased SVZ neurogenesis and increased apoptosis (21). However, the mutant A53T α-syn was driven by the platelet-derived growth factor (PDGF) promoter and was co-expressed at slightly higher levels than endogenous mouse α-syn (21). Thus, it is likely that the PDGF promoter resulted in α-syn expression that did not match normal patterns. In addition, the role of wild type (WT) or mutated human α-syn may have been masked by endogenous mouse α-syn. An additional issue is that the A53T human mutation is not a `mutation’ in mouse; threonine (T) is the normal amino acid. In an elegant set of studies, another mutation, A30P, reduced SVZ neurogenesis and was then rendered inactive via a tetracycline-off conditional transgenic approach (22). Indeed, the loss of newborn OB neurons was reversed by turning off A30P (23). However, this transgene was driven off the calcium/calmodulin-dependent protein kinase II promoter and, thus, as in the case of the PDGF promoter-driven transgene, may not reflect endogenous patterns of Snca expression. In addition, overexpression of human WT α-syn decreased survival of SVZ cells, but there was no measurable effect on rates of proliferation (24). Overexpression of α-syn also caused defects in olfactory modalities similar to PD (25).
To address these issues, and to further probe the role of α-syn in SVZ neurogenesis, we used a bacterial artificial chromosome (BAC) transgenic mouse that contains all putative cis regulatory elements of Snca, the endogenous α-syn gene, to drive expression of human A30P α-syn (26). We backcrossed A30P mice to an Snca−/− background to remove potential effects of endogenous mouse α-syn on A30P function (27). SNCA-A30P mice have diminished DA release in the dorsal but not the ventral striatum, whereas norepinephrine release and spontaneous behaviour are normal (26). We compared SNCA-A30P mice to α-syn null mice and WT C57BL/6 mice and found reduced proliferation in the SVZ and altered OB neurogenesis.
Results
α-syn expression in the forebrain
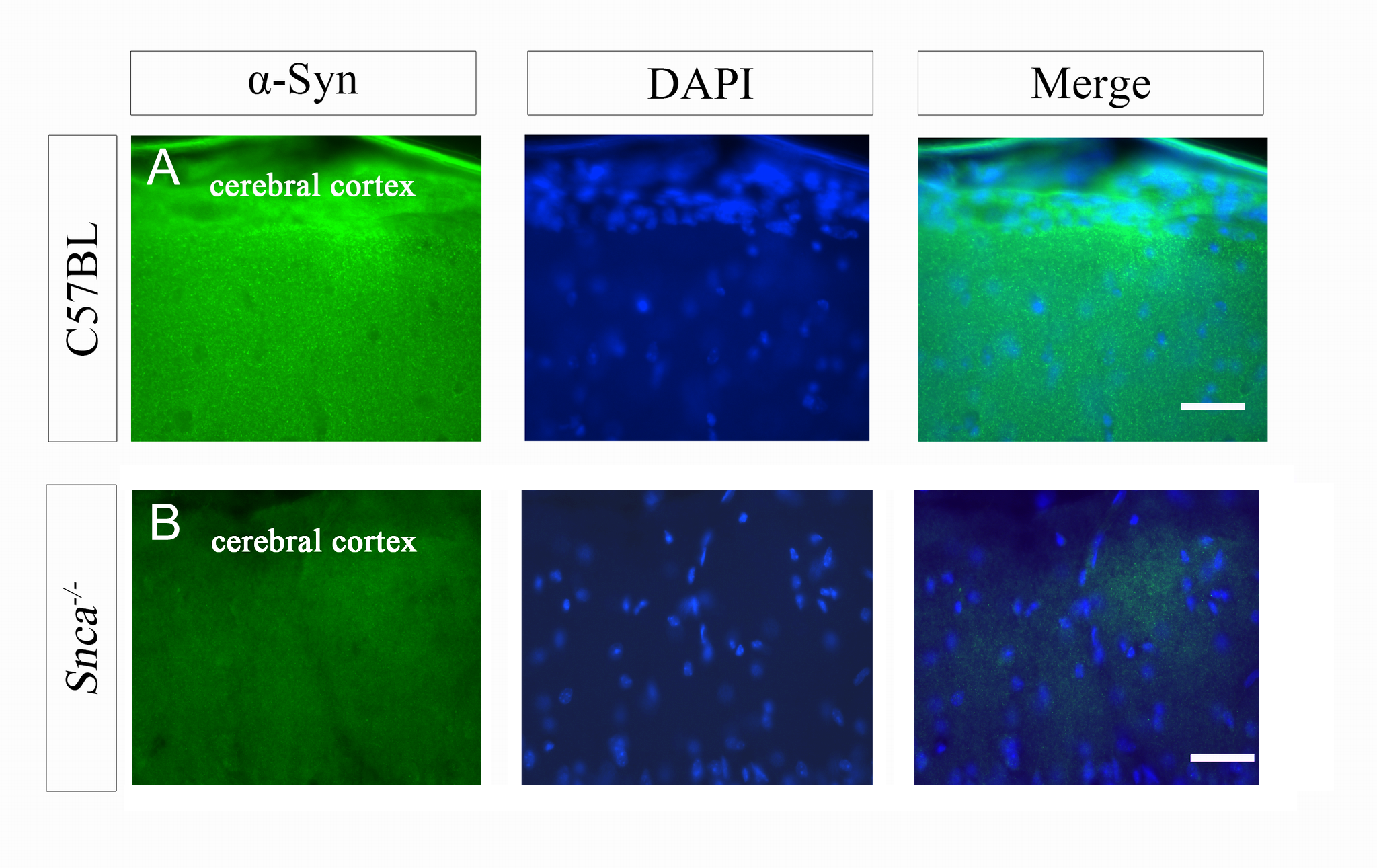
The monoclonal anti-human α-syn antibody resulted in weak positive immunoreactivity in WT but none in Snca−/− mice, as expected (Supplementary Material, Fig. S1). However, in the SNCA-A30P mouse forebrain, human α-syn+ neurons were consistently observed in the cerebral cortex (Fig. 1A) as previously described in this PD model (26). We did not observe any α-syn+ cells in the SVZ or RMS (Fig. 1B and C). As expected though, we found punctate to fibrous human α-syn immunoreactivity adjacent to the SVZ in the striatum (Fig. 1B and C). Numerous human α-syn+ cells were found in the OB granular and mitral cell layers (mcls; Fig. 1D) and a few in the periglomerular layer, recapitulating endogenous Snca expression (Fig. 1E).
Figure 1.
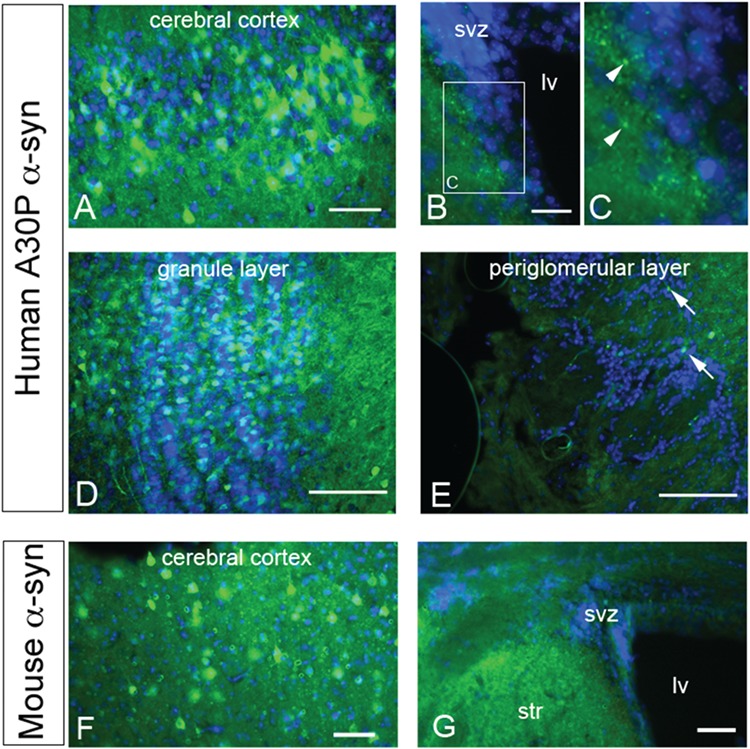
α-syn expression. (A) α-syn expression in a subset of cells in the cerebral cortex. DAPI nuclear counterstain shown in blue. Scale bar, 50 μm. (B and C) Low and high magnification showing lack of α-syn immunoreactivity in the SVZ. Note the punctate immunoreactivity (arrowheads) in the striatum next to the SVZ. Scale bar, 30 μm. Lv, lateral ventricle. (D) Many cells in the OB granule layer express human α-syn. Scale bar, 200 μm. (E) Few periglomerular cells (arrows) express human α-syn. (F) Endogenous murine α-syn expression in the cerebral cortex of a WT mouse. Scale bar, 50 μm. (G) α-syn was expressed in the striatum but not in the SVZ. Scale bar, 100 μm. str, striatum.
Similar to the human transgene, endogenous α-syn was detected in the cerebral cortex (Fig. 1F). Immunohistochemistry for mouse α-syn in Snca−/− mice was negative in the forebrain; the lack of signal was similar to the no primary antibody negative controls (data not shown). The SVZ of C57BL/6 mice was also devoid of α-syn immunofluorescence (Fig. 1G). However, diffuse α-syn+ immunofluorescence was seen adjacent to the SVZ in the striatum (Fig. 1G), RMS and OB of C57BL/6 mice in a pattern resembling human α-syn immunofluorescence in SNCA-A30P. Our results indicate that endogenous α-syn expression is below the level of detection in the SVZ, but is detectable in adjacent tissues such as the cerebral cortex, striatum and in the OB, the target of the SVZ. They also indicate that the A30P mutant SNCA human transgene reflects endogenous levels and patterns of expression in these forebrain regions.
No overt pathology was previously observed in SNCA-A30P mice (26), but to exclude any potential gross morphological malformations or obvious differences in cell density in the regions studied, we examined the distribution of DAPI+ nuclei in the SVZ, RMS and OB. There was no remarkable shape or size difference in the lateral ventricles, the RMS or OB in genetically modified mice compared with C57BL/6 (Supplementary Material, Fig. 2A–J).
Figure 2.
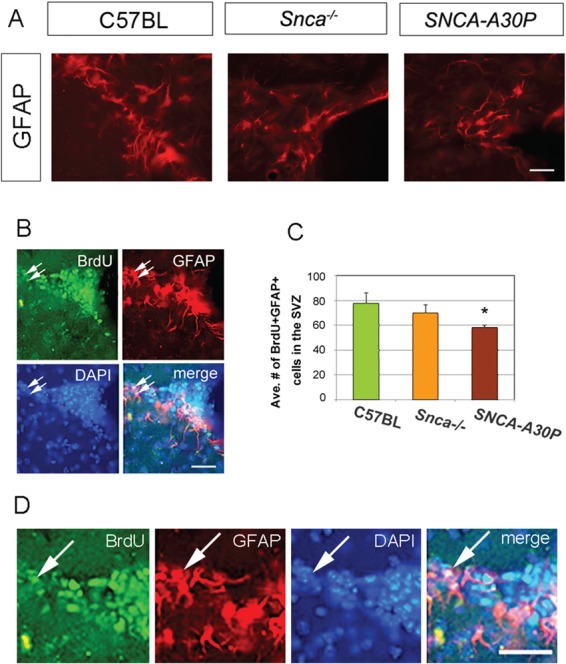
SNCA-A30P mice have fewer mitotic GFAP+ cells in the SVZ. (A) GFAP immunohistochemistry in the SVZ of the three cohorts. Scale bar, 30 μm. (B) Example of double immunofluorescence in the SVZ of a control (C57BL/6) mouse for BrdU and GFAP, with DAPI+ nuclei counterstain and merged image. Arrows show double labelled BrdU+GFAP+ cells. Scale bar, 30 μm. (C) The average number of BrdU+GFAP+ double labelled cells in the SVZ was significantly decreased in the SNCA-A30P group. DF = 39 and F = 3.098. (D) High-magnification view of B. Scale bar, 30 μm.
SNCA-A30P decreases the number of SVZ stem cells, proliferating and Mash1+ cells
Niche astrocytes in the SVZ can become reactive, such as in a stroke model (28,29). However, GFAP expression and the morphology of GFAP+ cells in the SVZ were not altered in the Snca−/− and SNCA-A30P mice (Fig. 2A). Thus, we do not believe that BrdU was incorporated into reactive niche astrocytes.
To detect proliferating SVZ stem cells, we administered BrdU via drinking water for 7 days and harvested brains 10 days after BrdU was discontinued. The regimen of BrdU combined with GFAP expression (Fig. 2B and D) labels SVZ stem cells. The number of BrdU+GFAP+ cells was not significantly decreased in Snca−/− mice compared to controls (Fig. 2C). In contrast, SNCA-A30P mice had significantly fewer BrdU+GFAP+ cells in the SVZ compared to controls (Fig. 2C). High-magnification images of BrdU+GFAP+ cells in a WT mouse are shown in Figure 2D. This was the first result showing that addition of A30P exacerbates effects caused by Snca loss.
Phospho-histone 3 (PHi3) immunohistochemistry was carried out to mark acutely proliferating transit-amplifying and neuroblast progenitor cells in the SVZ (Fig. 3A). Snca−/− and SNCA-A30P mice both exhibited a significant reduction in PHi3+ cells in the SVZ compared to C57BL/6 (Fig. 3B). Interestingly, SNCA-A30P mice also exhibited a significant reduction in PHi3+ cells compared to Snca−/− mice in the SVZ, suggesting that SNCA-A30P aggravates effects caused by Snca loss. This reduction in proliferating cells could be due to either fewer transit-amplifying progenitors or fewer neuroblasts, which are the two most frequently dividing SVZ cell types.
Figure 3.
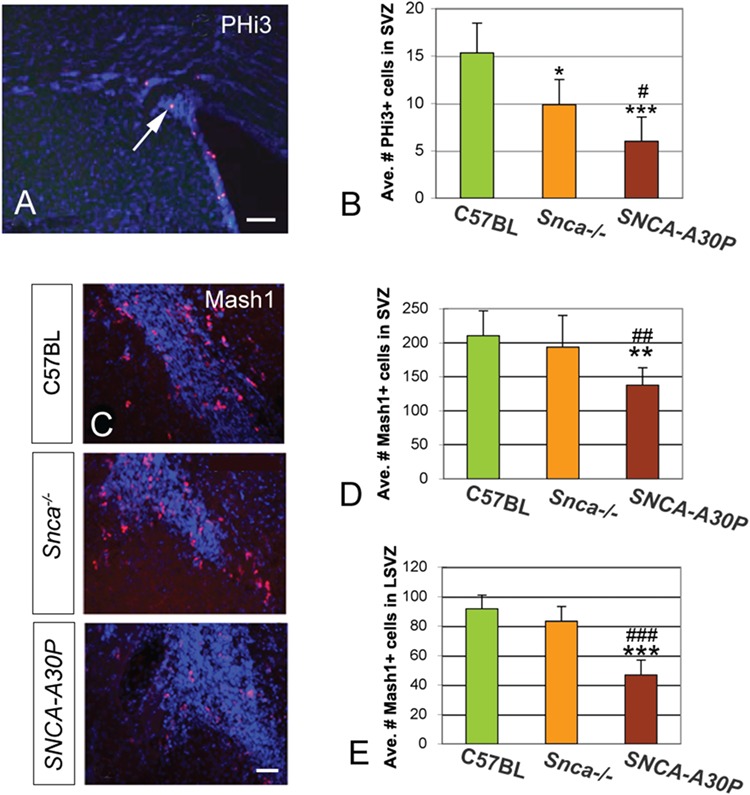
Proliferation deceases in the SVZ of Snca−/− and SNCA-A30P mice. (A) PHi3 immunohistochemistry in the SVZ of C57BL/6 mice. Scale bar, 100 μm. (B) The average number of PHi3+ cells per SVZ decreased in Snca−/− compared to C57BL/6 mice. The average number of PHi3+ cells also decreased in the SNCA-A30P SVZ compared to both C57BL/6 and Snca−/− (DF = 59, F = 8.07). We included the dorsal, lateral, medial and anterior SVZ in this analysis. (C) Mash1 immunohistochemistry in the anterior SVZ in C57BL/6 and mutant mice. Scale bar, 50 μm. (D) The average number of Mash1+ cells decreased in the total SVZ of SNCA-A30P mice compared to C57BL/6 and Snca−/− (DF = 73, F = 5.69). (E) The average number of Mash1+ cells decreased in the LSVZ of SNCA-A30P mice compared to C57BL/6 and Snca−/− (DF = 51, F = 25.05).
To assess if the observed reduction in proliferating cells was a result of decreased transit-amplifying cells, we examined Mash1 expression, which is specific to this cell type (Fig. 3C). Loss of Snca alone did not affect the numbers of Mash1+ cells in different subdivisions of the SVZ (Fig. 3D and E). In contrast, the number of Mash1+ cells in the SVZ decreased significantly in the SNCA-A30P mice compared to both C57BL/6 and Snca−/− mice (Fig. 3D and E). This was apparent in multiple subdivisions of the SVZ including the lateral SVZ (LSVZ; Fig. 3E). These results suggest that overall, decreased SVZ proliferation is more pronounced in Snca−/− mice harbouring the human A30P SNCA mutation.
OB neurogenesis is disrupted in Snca−/− and SNCA-A30P mice
To examine the effects of reduced SVZ proliferation and its downstream effects on OB interneurons, we studied doublecortin (Dcx) expression. Dcx is expressed by SVZ-derived neuroblasts for a few weeks after the cells are born allowing newborn neurons to be tracked in the RMS and OB (30,31). However, Dcx+ cells in the RMS are densely packed making quantification of cell numbers difficult. We, therefore, qualitatively examined Dcx+ neuroblasts in the RMS and the core of the OB and found that, compared to controls (Fig. 4A), the population appeared to be reduced in Snca−/− mice (Fig. 4B) and even more reduced in SNCA-A30P mice (Fig. 4C). Dcx+ neuroblasts disperse in the outer layers of the OB and are thus easier to quantify therein (Fig. 4D–F). The numbers of glomerular layer (gl) Dcx+ cells in Snca−/− and SNCA-A30P mice were significantly lower than in controls (Fig. 4G). The decreases in PHi3+ and Mash1+ SVZ cell numbers in mutant mice correlated with, and likely caused, the reduction of newborn Dcx+ neuroblasts in the OB.
Figure 4.
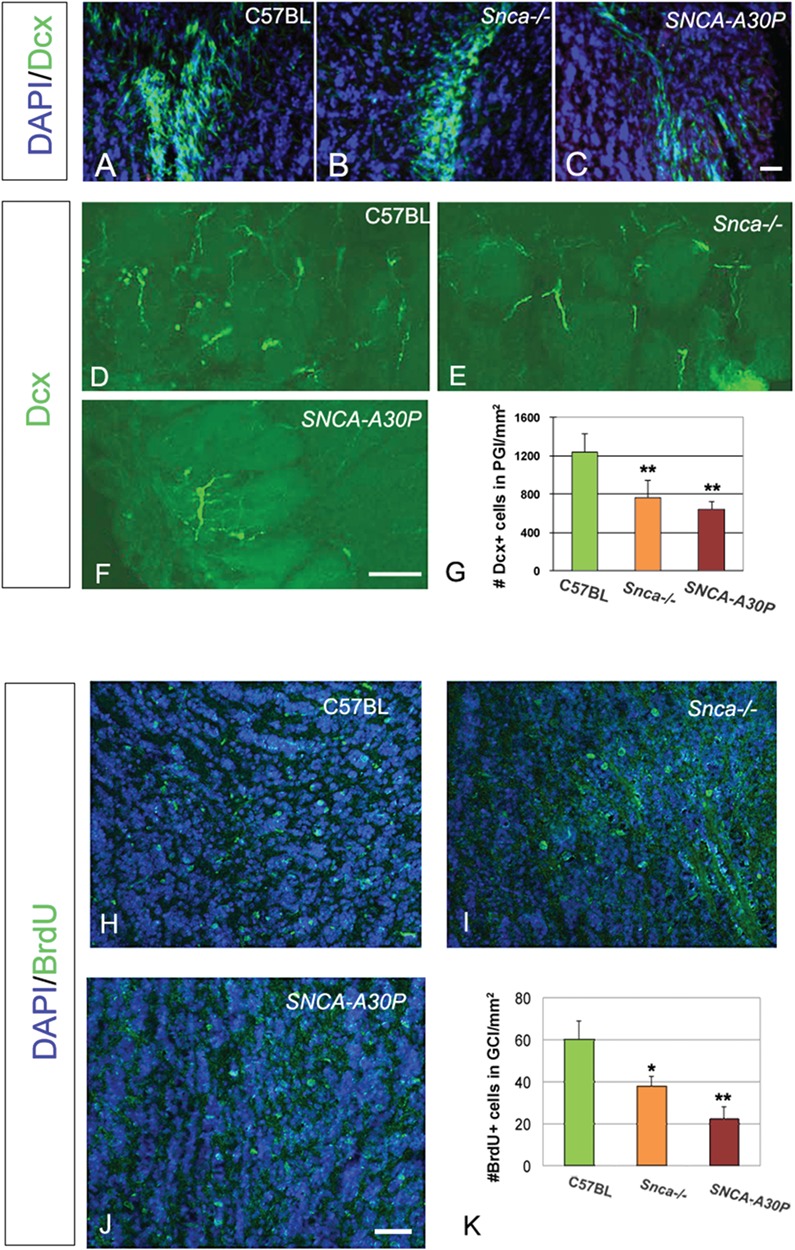
Snca −/− and SNCA-A30P mice have fewer Dcx+ and BrdU+ cells in the OB. (A–C) Dcx+ cells (green) in the core of the OB (DAPI blue counterstain of nuclei). Note that Dcx+ cells in Snca−/− and SNCA-A30P mice are sparser than in C57BL/6 mice. Scale bar, 30 μm. (D–F) Dcx+ cells in gl (glomeruli have slightly higher background and can be faintly seen). Scale bar, 100 μm. (G) The density per mm2 of Dcx+ cells decreased in the periglomerular layer of Snca−/− and SNCA-A30P mice (DF = 35, F = 6.67). (H–J) BrdU+ cells (green) in the core of the OB (DAPI blue counterstain of nuclei). Scale bar, 100 μm. (K) The density per mm2 of BrdU+ cells decreased in the OB of Snca−/− and SNCA-A30P mice (DF = 21, F = 4.273).
The regimen of 1 week BrdU in drinking water followed by a 10 day wash out labels mitotic SVZ neuroblasts that have migrated into the OB. We found that the number of BrdU+ cells in the granule cell layer (GCL) of the OB was significantly decreased in the Snca−/− mice and even more so in the SNCA-A30P mice (Fig. 4H–K). These results correspond with the reduced number of OB Dcx+ cells shown above.
The three types of periglomerular interneurons in the OB express distinct markers: TH, calbindin (CalB) and calretinin (CalR). Due to the reduction in proliferating SVZ cells and Dcx+ neuroblasts, immunostaining of these markers was carried out to examine if the different subpopulations of interneurons were affected in Snca−/− and SNCA-A30P mice (Fig. 5A–C, E–G and I–K). Snca−/− mice had significantly fewer interneurons of all three subtypes compared to WT mice (Fig. 5D, H and L). SNCA-A30P mice had fewer CalB+ and CalR+ interneurons (Fig. 5D and H). Remarkably, the number of TH+ cells in the periglomerular layer was similar in C57BL/6 and SNCA-A30P mice (Fig. 5L). The morphology, size and juxtaglomerular positioning of CalB+, CalR+ and TH+ interneurons were not different in Snca−/− and SNCA-A30P mice from controls. These results suggest that loss of Snca is sufficient to decrease numbers of all periglomerular newborn interneuron subtypes but that the SNCA-A30P affects CalR+ and CalB+ interneurons differently from TH+ neurons.
Figure 5.
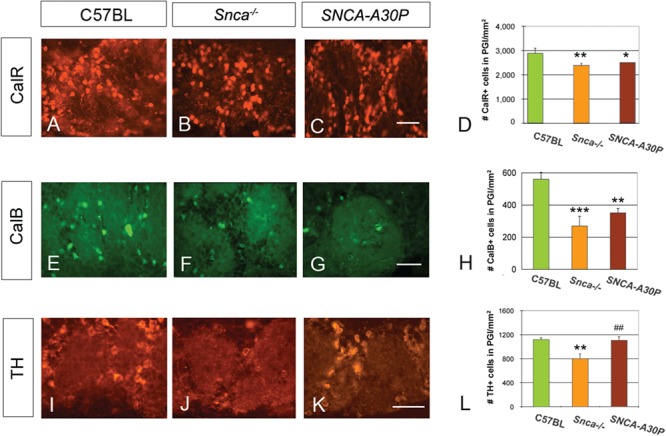
Snca −/− and SNCA-A30P mice have fewer periglomerular interneurons. (A–D) CalR+ immunoreactivity and graph showing loss of cells in mutants (DF = 65, F = 4.26). The reported cell numbers in D, H and L are densities of labelled cells per surface area. Scale bar, 100 μm. (E–H) CalB+ immunoreactivity and graph showing loss of cells in mutants (DF = 35, F = 9.73). Scale bar, 100 μm. (I–L) TH immunoreactivity and graph showing loss of cells in Snca−/− mice (DF = 65, F = 5.54). PGl, periglomerular layer. Scale bar, 100 μm.
We next quantified the number of CalR+ cells in the GCL. The majority of adult-born granule neurons reside in the deep layers; thus, we subdivided the GCL into evenly spaced deep to superficial bins (GCL-1 to GCL-5) for quantification (Fig. 6A). The number of CalR+ cells in C57BL/6 mice was greatest in intermediate layers (GCL-3; Fig. 6B). In contrast to the decreases in interneuron numbers in the periglomerular layer of Snca−/− mice, CalR+ neuron numbers remained unchanged in most granule layers but increased in the deepest granule layer (GCL-1; Fig. 6B). Similarly, the greatest increase in granule CalR+ cells in SNCA-A30P compared to the WT controls was in the deepest layer (GCL-1), and these mice also showed increases in layers 2 and 4 (GCL-2 and GCL-4; Fig. 6B). Again, the addition of the A30P mutation seemed to augment the effect of Snca loss.
Figure 6.
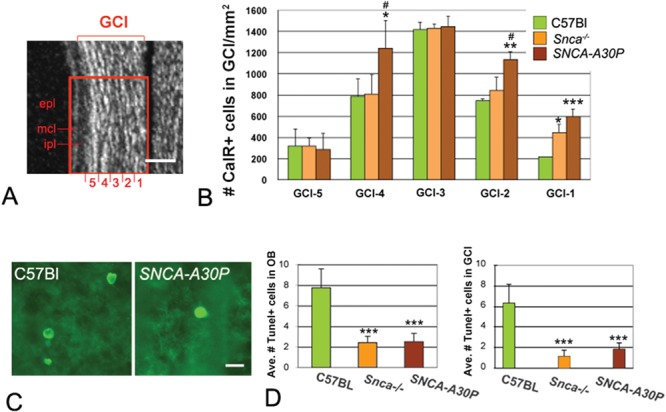
GCL CalR+ neurons and TUNEL+ cells in Snca−/− and SNCA-A30P mice. (A) DAPI stained section showing how the GCL was subdivided for quantification. ipl, internal plexiform layer; mcl, mitral cell layer; epl, external plexiform layer. Scale bar, 100 μm. (B) Quantification of CalR+ cells in the GCL (layer 1: DF = 35, F = 10.72; layer 2: DF = 35, F = 6.08; layer 3: DF = 35, F = 0.04; layer 4: DF = 35, F = 5.09; layer 5: DF = 35, F = 0.05). The reported cell numbers in B are densities of labelled cells per surface area. (C) TUNEL+ cells in the granule layer of the OB. Scale bar, 10 μm. (D) The average number of TUNEL+ cells was significantly decreased throughout the OB of Snca−/− and SNCA-A30P mice compared to C57BL/6 mice (DF = 35, F = 19.24). The average number of TUNEL+ cells was also significantly decreased in the GCL of Snca−/− and SNCA-A30P mice (DF = 30, F = 12.09).
Decreased number of TUNEL+ cells in Snca−/− and SNCA-A30P mice
Since the changes in OB interneuron numbers may have been due to altered rates of cell death, we performed terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) staining (Fig. 6C). The number of TUNEL+ cells across the entire OB, including the periglomerular and granule layers, was significantly decreased in both Snca−/− and SNCA-A30P mice (Fig. 6D). These two groups also had significant decreases in the number of TUNEL+ cells in the entire OB and in the GCL in particular (Fig. 6D), suggesting that the decreased cell death may have contributed to the increased numbers of CalR+ neurons in the granule layer. Snca−/− mice showed no changes in periglomerular cell death, whereas SNCA-A30P mice had reduced numbers of TUNEL+ cells in the gl (P < 0.05), suggesting that cell death did not contribute to decreasing periglomerular interneuron numbers [least significant difference (LSD) post hoc tests were employed, DF = 35, F = 2.646; P = 0.029 between SNCA-A30P mice and C57BL/6 mice]. The number of TUNEL+ cells in the external plexiform layer was unchanged in the different groups of mice.
Discussion
We examined the effects of the human SNCA-A30P mutation expressed from a 135 kb human genomic DNA transgene on an Snca−/− background compared to Snca−/− littermate controls and WT mice. Importantly, we showed that the human transgene expression was very similar to endogenous gene expression. We then demonstrated that the human A30P SNCA mutation driven by endogenous promoter enhancer sequences causes significant decreases in SVZ proliferation and transit-amplifying cell numbers in addition to altering OB neurogenesis. Overall, the A30P effects exacerbated the loss of Snca, which also resulted in decreased neurogenesis. Decreased SVZ neurogenesis has been reported in PD (9), and our results support previous evidence in animal models of PD. We hypothesize these results may be due to decreased DA release in the striatum adjacent to the SVZ.
Snca regulates SVZ proliferation
These BAC transgenic SNCA-A30P mice accurately reflect endogenous patterns of Snca expression in forebrain regions surrounding the SVZ including the striatum and cerebral cortex. We did not find evidence for expression of endogenous Snca or mutated A30P human transgene in the SVZ itself. However, it was expressed in the striatum adjacent to the SVZ, suggesting that loss of Snca or the A30P mutation could affect DA release in that region. The A30P BAC transgene is expressed in TH+ substantia nigra pars compacta neurons as well as in the ventral tegmental area in the same cell subcompartments as endogenous α-syn (26). As well, these SNCA-A30P mice exhibit reduced stimulus-evoked DA release in the dorsal striatum (26). Dopaminergic signalling in the SVZ is thought to positively regulate proliferation (9) through D2 and D3 receptors (12). We previously examined DA receptor expression on SVZ cells and reported that transit-amplifying progenitor cells express the D3 DA receptor and that neuroblasts express D1, D2 and D5 receptors (12). Although the normal cellular functions of α-syn are unclear, considerable evidence now suggests that it may serve to regulate DA release and presynaptic DA concentrations (32–34). The decreases in SVZ proliferation in the Snca knockout and the human BAC transgenic A30P mutant resulted in reduced proliferation overall and a decrease in the number of Mash1+ transit-amplifying progenitors. This reduction in cell number paired with our previous work on dopaminergic signalling in the SVZ, suggest that our results may be due to decreased DA release and thus receptor signalling. These mice did not exhibit any synucleinopathy, loss of substantia nigra DA neurons or gliosis (26) suggesting that the effects were not due to DA-ergic neurodegeneration. Thus, our data were not likely influenced by inflammation resulting from synucleinopathy (35) or associated with death of nigrostriatal neurons (36). The SNCA-A30P mice were analysed for a wide variety of motor and sensory behaviours associated with PD and were not different from controls except for a mild increase in night-time running behaviour (26). If anything, this may be expected to increase proliferation in the SVZ (37); however, we observed decreases.
Snca regulates OB neurogenesis
Transit-amplifying progenitor cells in the SVZ give rise to Dcx+ neuroblasts that migrate in the RMS to the OB. Dcx expression is lost in these neuroblasts a few weeks after neuronal birth (30), allowing use of Dcx expression as a marker for neurogenesis. We observed qualitatively decreased numbers of Dcx+ cells in the RMS and quantitatively decreased Dcx + cells in the OB suggesting that neurogenesis was decreased in Snca−/− and SNCA-A30P mice. The diminished number of Dcx + cells in the OB correlates with the decreased SVZ proliferation. In turn, the reduced number of TUNEL+ cells in the OB periglomerular layer may be a reflection of decreased SVZ neurogenesis. A human A30P P1 artificial chromosome (PAC) transgenic mouse, similar to ours, showed gastrointestinal defects typical of PD but did not exhibit olfactory defects (38). It would therefore be interesting to assess if indeed that lack of phenotype in the A30P PAC mice is due to unchanged OB neurogenesis. However, this was not carried out in their study.
Quantification of BrdU+ cells in the OB revealed that they were reduced similar to the reduction in Dcx+ cells. We believe that the large majority of BrdU+ cells in the OB 10 days after the BrdU washout period were neuroblasts born in the SVZ that had migrated into the OB. We showed that Snca−/− mice had significantly fewer OB BrdU+ cells and this effect was amplified in SNCA-A30P mice. These results match and confirm our findings that the consequence of SNCA-A30P in the SVZ was greater than that of endogenous Snca loss.
Loss of Snca function differentially regulates periglomerular and granule CalR+ interneuron numbers
The increased numbers of CalR+ cells in the granule layer compared to the decrease in the periglomerular layer in SNCA-A30P mice may theoretically have been due to decreased cell death in the granule layer. However, the small number of TUNEL+ cells makes this problematic to absolutely prove. There is evidence that both periglomerular and granular CalR+ interneurons derive from either anterior or medial (39) and dorsal subregions of the SVZ (40) and are mostly born postnatally (41). Although there is disagreement as to the exact source of these cells (39,40), both studies suggest that granular and periglomerular CalR+ cells are in the same lineage. Thus, it is not likely that mechanisms, such as decreased DA release induced by Snca loss and the A30P mutation, would work differently on granule versus periglomerular CalR neurogenesis. DA has been shown to affect interneuron progenitor migration during embryonic development (42) and similarly could also modulate adult SVZ neuroblast migration. One scenario compatible with the discrepancy is reduced DA release in the OB decreases neuroblast migration from the granule to periglomerular layer.
Differences between Snca−/− and SNCA-A30P mice
Some significant differences between the Snca−/− and SNCA-A30P mice were uncovered. Notably, the number of PHi3+ SVZ cells was significantly lower in these two groups, whilst the number of periglomerular TH+ cells was only reduced in the Snca−/− mice. A previous study demonstrated that SNCA-A30P mice had ~ 30% less DA release after single or burst stimulation compared to Snca−/− mice (26). This loss of dopaminergic tone throughout the animal's life may have significantly reduced SVZ proliferation in the SNCA-A30P mice compared to the Snca−/− mice. Snca−/− mice on the other hand do not have decreased electrically evoked DA release in the striatum compared to WT C57BL/6 (34). Importantly, only SNCA-A30P, but not Snca−/− mice, exhibited decreased numbers of Mash1+ cells in the SVZ. We believe that this distinction is important and may explain some of our findings. Mash1 is commonly used as a marker to label transit-amplifying progenitors (3). It also has important functions such as activating stem cells (43) and maintaining neurogenic proliferation (44). Thus, loss of Mash1 function in the SNCA-A30P mice may have exacerbated the effect of loss of Snca.
We hypothesized that both Snca−/− and SNCA-A30P mice would have fewer TH+ neurons since Snca is expressed in periglomerular TH+ cells (45). However, only the Snca−/− mice had fewer TH+ neurons. This is interesting because in human PD, TH+ neurons in the OB may be resistant to degeneration (6). Decreasing dopaminergic input with 6OHDA lesions reduced SVZ-OB neurogenesis of all interneuron types except TH+, which were increased in number (46). This remarkable finding is similar to our findings in that A30P affected generation of all interneuron subtypes examined except for TH+ cells. Because PD may spare the TH+ OB population, further exogenous stimulation of TH+ neurons may be a therapeutic target. They may be induced to migrate into the striatum and replenish DA-ergic levels in the context of PD (47). Pax6, Meis2 and Id1 stimulate dopaminergic neurogenesis in the SVZ (48) and could be used to stimulate increased production of TH+ neurons. Interestingly, this concept has already been tested. Clustered Ephrin-A1 injected into the lateral ventricle not only restored neurogenesis that had been decreased in a rat PD model but also SVZ cells migrated into the striatum, differentiated into dopaminergic neurons and rescued behavioural abnormalities (49).
Comparison of results with other α-synuclein models
Previous studies using other models of PD with aberrant α-syn expression have also shown abnormal SVZ neurogenesis but with important distinctions from what we have detected. Overexpression of human α-syn decreased OB neurogenesis due to increased apoptosis (24). A53T and A30P human α-syn mutations driven off the TH promoter reduced SVZ granule neurogenesis in a transgenic rat model of PD (50); however, they increased periglomerular neurogenesis (50). This is opposite to the general pattern of decreased periglomerular and increased granule interneuron neurogenesis we observed. Finally, a Thy1 promoter-driven A30P α-syn model did not show differences in DA neuron integration into the periglomerular layer (51). Many of these differences may be explained by differing patterns of transgene expression of α-syn or mutant α-syn in the different models.
Conclusion
We examined adult SVZ neurogenesis in Snca−/− and SNCA-A30P mice in which the mutated gene expression pattern reflected endogenous α-syn expression. We found defects in proliferation, Mash1 expression and numbers of OB interneurons in both Snca−/− and SNCA-A30P mice, with the latter having an exaggerated phenotype (Fig. 7). Our study lends support to the notion that DA-ergic signalling regulates adult neurogenesis and that α-syn plays a central role in this process (Fig. 7).
Figure 7.
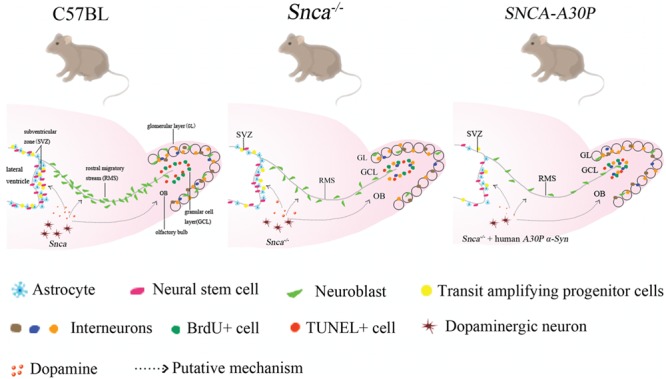
The A30P α-syn mutation decreases SVZ proliferation and OB neurogenesis. Endogenous Snca (α-syn) regulates adult neurogenesis. Snca−/− mice exhibit decreased SVZ proliferation and OB neurogenesis, and the exogenous human SNCA-A30P mutation exaggerates this phenotype, which is putatively regulated by α-syn-modulated dopaminergic signalling.
Materials and Methods
Animals
Experiments were performed on adult C57BL/6 mice with (1) endogenous Snca knocked out (Snca−/−), (2) endogenous Snca knocked out, but expressing human SNCA with an A30P mutation (SNCA-A30P) (26), and (3) C57BL/6 control mice (Ctl). SNCA-A30P mice were created with a high-capacity BAC containing the complete 111 kb SNCA DNA locus plus a ~ 20 kb upstream promoter region microinjected into C57BL/6 embryos to create lines carrying the complete human A30P SNCA locus backcrossed onto an Snca−/− background (26). Throughout the study, all groups had an N of 4, except for the separate BrdU study that had an N of 3–4 (see below). Animals were kept in rooms with a lighting schedule of 12 h light/darkness and with standard diet and water ad libitum. The experiments were performed in accordance with the UK Animals (Scientific Procedures) 1986 Act, UK Home Office. All animal work was approved by the UK Home Office, License #30/2496, and the University of Oxford Department of Physiology, Anatomy and Genetics Departmental Ethical Review Committee.
BrdU administration
Mice (N = 4, WT; N = 3, Snca−/−; N = 3, SNCA-A30P) were provided with 1 mg/ml of BrdU in normal drinking water for 1 week followed by a 10 day chase period without BrdU. BrdU in water was replaced twice a week. For the first 3 days, 1% glucose was added to BrdU/water in order to avoid taste aversion.
Immunohistochemistry
Mice were perfused under deep anaesthesia with PBS (phosphate buffer saline) buffered 4% paraformaldehyde. A total of 30 μm coronal sections were collected on a sliding microtome (Leica, Wetzlar, Germany) and transferred to cryoprotectant. Free-floating sections were treated with PBS 3 times for 10 min each, 50 mm glycine for 15 min, PBS 3 times for 10 min each and PBS+ (10% Donkey Serum/0.1% Triton X-100 in PBS) for 1 h and incubated in primary antibodies [mouse anti-human α-synuclein monoclonal (1:500; Abcam, Cambridge, UK 1904), rabbit anti-α-synuclein (for human, mouse and rat) (1: 1000; ab5038, Chemicon, Billerica, MA, USA), rabbit anti-PHi3 (1:500; Millipore, Burlington, MA, USA), goat anti-Dcx (1:100; Santa Cruz, Santa Cruz, CA, USA), mouse anti-Mash1 (1:200; BDscience, New York, NY, USA), mouse anti-CalB (1:2000; Sigma-Aldrich, St Louis, MO, USA), rabbit anti-CalR (1:2000; Chemicon, Billerica, MA, USA) or rabbit anti-TH (1:2000; Chemicon, Billerica, MA, USA) in PBS+] at 4°C overnight. Then, slices were washed with PBS 3 times for 10 min each, incubated with Cy3 donkey anti-mouse, donkey anti-rabbit or donkey anti-mouse (all 1:500; Jackson Immunoresearch, West Grove, PA, USA) or Alexa Fluro 488 donkey anti-goat (1:500; Jackson Immunoresearch, West Grove, PA, USA) in PBS+ for 1 h, PBS 3 times 10 min each, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), 40 mg/ml DAPI stock solution (1:1000 in PBS) for 10 min, phosphate buffer 3 times 10 min each, air dried and coverslipped with Fluorsave (Calbiochem, Merck KGaA, Darmstadt, Germany).
TUNEL
The TUNEL assay was performed using the In Situ Cell Death Detection Kit Fluorescein (Roche, Penzberg, Germany) in a modified procedure for free-floating sections as previously described (52). Briefly, the sections were mounted on slides and air dried at least for 1 h. After rinsing in Tris-buffered saline (TBS) 2 times for 5 min each, the sections were incubated in 70 and 90% isopropanol for 2 min each, followed by incubation in 100% isopropanol for 10 min and 90 and 70% isopropanol for 2 min each. After 3 rinses in TBS for 5 min each, positive control sections were incubated with DNase buffer for 20 min, DNase buffer, +2 mg/ml DNase for 15 min at 37°C; TBS, 3 times for 5 min each. The areas around samples were dried, and the samples were incubated with 50 μl TUNEL staining mixture (45 μl labelling solution + 5 μl enzyme for positive control and experimental sections, 50 μl labelling solution for negative control) for 1 h at 37°C in a dark box. After rinsing in TBS 3 times each for 5 min, all the sections were air dried in the dark and coverslipped with Fluorsave (Calbiochem) for microscopy.
Quantification and statistical analysis
The sections used were cut in the coronal plane, and BrdU+GFAP+, Phi3+ and Mash1+ cells in the SVZ were quantified directly through a Leica DM IRB inverted epifluorescent microscope with a 40× PL Fluotar objective lens. We selected the area analysed based on experience and the Mouse Brain Atlas (53). To cover the entire SVZ of each mouse, we selected 3 equivalent sections including anterior, middle and posterior SVZ, Bregma 0–+1.2 mm. For each section, we imaged, counted and analysed the dorsal, medial, lateral and ventral regions of the right and left SVZ, separately. Finally, we averaged the numbers of labelled cells. For periglomerular cell counts (Dcx+, CalR+, Calb+ and TH+ cells), multiple focal planes were photographed (3–6, depending on the number of cells) and counted in 3 evenly spaced coronal OB sections per animal (Bregma +3.9–+4.2 mm). Cells were photomicrographed with a 40× PL Fluotar objective lens quantified from images and the numbers expressed as density per surface area. BrdU+ cells in the GCL of 3 evenly spaced coronal OB sections (Bregma +3.9 − +4.2 mm) throughout the GCL were imaged and quantified. BrdU+ cells with labelling above background were included and the numbers expressed as density per surface area. For counting CalR+ cells in the GCL, images taken with a 20× PL Fluotar lens were divided evenly into 5 fields ranging from the intersection of the GCL and RMS, medially and the GCL and internal plexiform layer, laterally. CalR+ cells were then counted in fields 1–5 from medial to lateral, as shown in Figure 6A. TUNEL+ cells are comparatively rare in the gl of the OB. Therefore, photomicrographs were not used in this case but the entire OB was analysed in three evenly spaced sections directly through the microscope.
All statistical analysis was performed using ANOVA. We used the LSD and Tukey post hoc tests to determine significance within ANOVAS. * = P < 0.05, ** = P < 0.01 and *** = P < 0.001 comparing Snca−/− or SNCA-A30P with the C57BL/6 mice. # = P < 0.05, ## = P < 0.01 and ### = P < 0.001 comparing Snca−/− mice with the SNCA-A30P group.
Supplementary Material
{kind=link}
{kind=link}
Present address: College of Medicine, Penn State University, Hershey, PA 17036, USA.
Acknowledgements
The authors thank Christopher Young, Rachel E. James and Zheng-Zhu Wang for their technical help. We are also grateful to Professor Ziyi Li for financial support in the BrdU experiments.
Conflict of Interest statement. None declared.
Funding
China Scholarship Council (X.Z.); National Key Research and Development Program of China Stem Cell and Translational Research (2017YFA0105101 to X.Z.); National Institutes of Health Grant (NS-42253 to F.G.S.); Monument Trust Discovery Award from Parkinson's UK (R.W.-M.).
References
- 1. Kovács T. (2004) Mechanisms of olfactory dysfunction in aging and neurodegenerative disorders. Ageing Res. Rev., 3, 215–232. [DOI] [PubMed] [Google Scholar]
- 2. Braak H., Del Tredici K., Rüb U., Vos R.A., Jansen Steur E.N. and Braak E. (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging, 24, 197–211. [DOI] [PubMed] [Google Scholar]
- 3. Giachino C., Basak O., Lugert S., Knuckles P., Obernier K., Fiorelli R., Frank S., Raineteau O., Alvarez-Buylla A. and Taylor V. (2014) Molecular diversity subdivides the adult forebrain neural stem cell population. Stem Cells, 32, 70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gheusi G., Cremer H., McLean H., Chazal G., Vincent J.D. and Lledo P.M. (2000) Importance of newly generated neurons in the adult olfactory bulb for odor discrimination. Proc. Natl. Acad. Sci. U. S. A., 97, 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mouret A., Lepousez G., Gras J., Gabellec M.M. and Lledo P.M. (2009) Turnover of newborn olfactory bulb neurons optimizes olfaction. J. Neurosci., 29, 12302–12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cave J.W. and Baker H. (2009) Dopamine systems in the forebrain. Adv. Exp. Med. Biol., 651, 15–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hökfelt T., Halasz N., Ljungdahl A., Johansson O., Goldstein M. and Park D. (1975) Histochemical support for a dopaminergic mechanism in the dendrites of certain periglomerular cells in the rat olfactory bulb. Neurosci. Lett., 1, 85–90. [DOI] [PubMed] [Google Scholar]
- 8. Maher B.J. and Westbrook G.L. (2008) Co-transmission of dopamine and GABA in periglomerular cells. J. Neurophysiol., 99, 1559–1564. [DOI] [PubMed] [Google Scholar]
- 9. Höglinger G.U., Rizk P., Muriel M.P., Duyckaerts C., Oertel W.H., Caille I. and Hirsch E.C. (2004) Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci., 7, 726–735. [DOI] [PubMed] [Google Scholar]
- 10. Borta A. and Höglinger G.U. (2007) Dopamine and adult neurogenesis. J. Neurochem., 100, 587–595. [DOI] [PubMed] [Google Scholar]
- 11. Baker S.A., Baker K.A. and Hagg T. (2004) Dopaminergic nigrostriatal projections regulate neural precursor proliferation in the adult mouse subventricular zone. Eur. J. Neurosci., 20, 575–579. [DOI] [PubMed] [Google Scholar]
- 12. Kim Y., Wang W.Z., Comte I., Pastrana E., Tran P.B., Brown J., Miller R.J., Doetsch F., Molnár Z. and Szele F.G. (2010) Dopamine stimulation of postnatal murine subventricular zone neurogenesis via the D3 receptor. J. Neurochem., 114, 750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spinelli K.J., Taylor J.K., Osterberg V.R., Churchill M.J., Pollock E., Moore C., Meshul C.K. and Unni V.K. (2014) Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson's disease. J. Neurosci., 34, 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uchihara T. and Giasson B.I. (2016) Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol., 131, 49–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J.T., Schöls L. and Riess O. (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet., 18, 106–108. [DOI] [PubMed] [Google Scholar]
- 16. Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R. et al. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science, 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- 17. Zarranz J.J., Alegre J., Gómez-Esteban J.C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B. et al. (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol., 55, 164–173. [DOI] [PubMed] [Google Scholar]
- 18. Appel-Cresswell S., Vilarino-Guell C., Encarnacion M., Sherman H., Yu I., Shah B., Weir D., Thompson C., Szu-Tu C., Trinh J. et al. (2013) Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord., 28, 811–813. [DOI] [PubMed] [Google Scholar]
- 19. Lesage S., Anheim M., Letournel F., Bousset L., Honoré A., Rozas N., Pieri L., Madiona K., Dürr A., Melki R. et al. (2013) G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol., 73, 459–471. [DOI] [PubMed] [Google Scholar]
- 20. Chesselet M.F. (2008) In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson's disease. Exp. Neurol., 209, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Winner B., Rockenstein E., Lie D.C., Aigner R., Mante M., Bogdahn U., Couillard-Despres S., Masliah E. and Winkler J. (2008) Mutant alpha-synuclein exacerbates age-related decrease of neurogenesis. Neurobiol. Aging, 29, 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nuber S., Petrasch-Parwez E., Winner B., Winkler J., von Hörsten S., Schmidt T., Boy J., Kuhn M., Nguyen H.P., Teismann P. et al. (2008) Neurodegeneration and motor dysfunction in a conditional model of Parkinson's disease. J. Neurosci., 28, 2471–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marxreiter F., Nuber S., Kandasamy M., Klucken J., Aigner R., Burgmayer R., Couillard-Despres S., Riess O., Winkler J. and Winner B. (2009) Changes in adult olfactory bulb neurogenesis in mice expressing the A30P mutant form of alpha-synuclein. Eur. J. Neurosci., 29, 879–890. [DOI] [PubMed] [Google Scholar]
- 24. Winner B., Lie D.C., Rockenstein E., Aigner R., Aigner L., Masliah E., Kuhn H.G. and Winkler J. (2004) Human wild-type alpha-synuclein impairs neurogenesis. J. Neuropathol. Exp. Neurol., 63, 1155–1166. [DOI] [PubMed] [Google Scholar]
- 25. Fleming S.M., Tetreault N.A., Mulligan C.K., Hutson C.B., Masliah E. and Chesselet M.F. (2008) Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur. J. Neurosci., 28, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taylor T.N., Potgieter D., Anwar S., Senior S.L., Janezic S., Threlfell S., Ryan B., Parkkinen L., Deltheil T., Cioroch M. et al. (2014) Region-specific deficits in dopamine, but not norepinephrine, signaling in a novel A30P α-synuclein BAC transgenic mouse. Neurobiol. Dis., 62, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cabin D.E., Gispert-Sanchez S., Murphy D., Auburger G., Myers R.R. and Nussbaum R.L. (2005) Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiol. Aging, 26, 25–35. [DOI] [PubMed] [Google Scholar]
- 28. Young C.C., Al-Dalahmah O., Lewis N.J., Brooks K.J., Jenkins M.M., Poirier F., Buchan A.M. and Szele F.G. (2014) Blocked angiogenesis in Galectin-3 null mice does not alter cellular and behavioral recovery after middle cerebral artery occlusion stroke. Neurobiol. Dis., 63, 155–164. [DOI] [PubMed] [Google Scholar]
- 29. Young C.C., van der Harg J.M., Lewis N.J., Brooks K.J., Buchan A.M. and Szele F.G. (2013) Ependymal ciliary dysfunction and reactive astrocytosis in a reorganized subventricular zone after stroke. Cereb. Cortex, 23, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brown J.P., Couillard-Després S., Cooper-Kuhn C.M., Winkler J., Aigner L. and Kuhn H.G. (2003) Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol., 467, 1–10. [DOI] [PubMed] [Google Scholar]
- 31. Gleeson J.G., Lin P.T., Flanagan L.A. and Walsh C.A. (1999) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron, 23, 257–271. [DOI] [PubMed] [Google Scholar]
- 32. Anwar S., Peters O., Millership S., Ninkina N., Doig N., Connor-Robson N., Threlfell S., Kooner G., Deacon R.M., Bannerman D.M. et al. (2011) Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci., 31, 7264–7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Janezic S., Threlfell S., Dodson P.D., Dowie M.J., Taylor T.N., Potgieter D., Parkkinen L., Senior S.L., Anwar S., Ryan B. et al. (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. U. S. A., 110, E4016–E4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Senior S.L., Ninkina N., Deacon R., Bannerman D., Buchman V.L., Cragg S.J. and Wade-Martins R. (2008) Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur. J. Neurosci., 27, 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mori K., Kaneko Y.S., Nakashima A., Nagasaki H., Nagatsu T., Nagatsu I. and Ota A. (2012) Subventricular zone under the neuroinflammatory stress and Parkinson's disease. Cell. Mol. Neurobiol., 32, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. L'Episcopo F., Tirolo C., Testa N., Caniglia S., Morale M.C., Deleidi M., Serapide M.F., Pluchino S. and Marchetti B. (2012) Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson's disease involves cross talk between inflammatory and Wnt/β-catenin signaling pathways: functional consequences for neuroprotection and repair. J. Neurosci., 32, 2062–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Praag H., Kempermann G. and Gage F.H. (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci., 2, 266–270. [DOI] [PubMed] [Google Scholar]
- 38. Kuo Y.M., Li Z., Jiao Y., Gaborit N., Pani A.K., Orrison B.M., Bruneau B.G., Giasson B.I., Smeyne R.J., Gershon M.D. et al. (2010) Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet., 19, 1633–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merkle F.T., Mirzadeh Z. and Alvarez-Buylla A. (2007) Mosaic organization of neural stem cells in the adult brain. Science, 317, 381–384. [DOI] [PubMed] [Google Scholar]
- 40. Young K.M., Fogarty M., Kessaris N. and Richardson W.D. (2007) Subventricular zone stem cells are heterogeneous with respect to their embryonic origins and neurogenic fates in the adult olfactory bulb. J. Neurosci., 27, 8286–8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Batista-Brito R., Close J., Machold R. and Fishell G. (2008) The distinct temporal origins of olfactory bulb interneuron subtypes. J. Neurosci., 28, 3966–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crandall J.E., McCarthy D.M., Araki K.Y., Sims J.R., Ren J.Q. and Bhide P.G. (2007) Dopamine receptor activation modulates GABA neuron migration from the basal forebrain to the cerebral cortex. J. Neurosci., 27, 3813–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersen J., Urbán N., Achimastou A., Ito A., Simic M., Ullom K., Martynoga B., Lebel M., Göritz C., Frisen J. et al. (2014) A transcriptional mechanism integrating inputs from extracellular signals to activate hippocampal stem cells. Neuron, 83, 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Castro D.S., Martynoga B., Parras C., Ramesh V., Pacary E., Johnston C., Drechsel D., Lebel-Potter M., Garcia L.G., Hunt C. et al. (2011) A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes. Dev., 25, 930–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alerte T.N., Akinfolarin A.A., Friedrich E.E., Mader S.A., Hong C.S. and Perez R.G. (2008) Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: lessons from viral transduction of knockout mice. Neurosci. Lett., 435, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winner B., Geyer M., Couillard-Despres S., Aigner R., Bogdahn U., Aigner L., Kuhn G. and Winkler J. (2006) Striatal deafferentation increases dopaminergic neurogenesis in the adult olfactory bulb. Exp. Neurol., 197, 113–121. [DOI] [PubMed] [Google Scholar]
- 47. van den Berge S.A., van Strien M.E. and Hol E.M. (2013) Resident adult neural stem cells in Parkinson's disease—the brain's own repair system. Eur. J. Pharmacol., 719, 117–127. [DOI] [PubMed] [Google Scholar]
- 48. Agoston Z., Heine P., Brill M.S., Grebbin B.M., Hau A.C., Kallenborn-Gerhardt W., Schramm J., Götz M. and Schulte D. (2014) Meis2 is a Pax6 co-factor in neurogenesis and dopaminergic periglomerular fate specification in the adult olfactory bulb. Development, 141, 28–38. [DOI] [PubMed] [Google Scholar]
- 49. Jing X., Miwa H., Sawada T., Nakanishi I., Kondo T., Miyajima M. and Sakaguchi K. (2012) Ephrin-A1-mediated dopaminergic neurogenesis and angiogenesis in a rat model of Parkinson's disease. PLoS One, 7, e32019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lelan F., Boyer C., Thinard R., Rémy S., Usal C., Tesson L., Anegon I., Neveu I., Damier P., Naveilhan P. et al. (2011) Effects of human alpha-synuclein A53T-A30P mutations on SVZ and local olfactory bulb cell proliferation in a transgenic rat model of Parkinson disease. Parkinsons Dis., 2011, 987084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Neuner J., Filser S., Michalakis S., Biel M. and Herms J. (2014) A30P α-synuclein interferes with the stable integration of adult-born neurons into the olfactory network. Sci. Rep., 4, 3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cooper-Kuhn C.M. and Kuhn H.G. (2002) Is it all DNA repair? Methodological considerations for detecting neurogenesis in the adult brain. Brain Res. Dev. Brain Res., 134, 13–21. [DOI] [PubMed] [Google Scholar]
- 53. Paxinos G. and Franklin K.B.J. (2001) The Mouse Brain in Stereotaxic Coordinates. Academic Press, San Diego, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.