

ABSTRACT
The herbivore rumen ecosystem constitutes an extremely efficient degradation machinery for the intricate chemical structure of fiber biomass, thus, enabling the hosting animal to digest its feed. The challenging task of deconstructing and metabolizing fiber is performed by microorganisms inhabiting the rumen. Since most of the ingested feed is comprised of plant fiber, these fiber-degrading microorganisms are of cardinal importance to the ecology of the rumen microbial community and to the hosting animal, and have a great impact on our environment and food sustainability. We summarize herein the enzymological fundamentals of fiber degradation, how the genes encoding these enzymes are spread across fiber-degrading microbes, and these microbes' interactions with other members of the rumen microbial community and potential effect on community structure. An understanding of these concepts has applied value for agriculture and our environment, and will also contribute to a better understanding of microbial ecology and evolution in anaerobic ecosystems.
Keywords: glycoside hydrolase, microbial interaction, microbial ecosystem, plant biomass, fiber colonization, microbiome
Here we piece together a fiber degradation centric view of the rumen ecosystem by describing genomic, enzymatic, ecological and communal aspects of this key process. These aspects provide an overview and understanding of this intriguing nature's phenomenon, the obligatory dependence of ruminants on their microbes for the degradation and digestion of plants they ingest.
INTRODUCTION
Fiber digestion by ruminants results in a remarkable symbiosis between the host animal and the multitude of microorganisms inhabiting the rumen (Fig. 1). The host animal provides the plant material and enhances microbial degradation by repeatedly grinding it via rumination to increase the surface area to be used by the microbes. Moreover, the host animal provides a supportive environment for microbial fiber hydrolysis (constant mixing, stable pH and temperature conditions).
Figure 1.

The cow rumen contains a multitude of microorganisms that conjointly adhere to and degrade plant fiber. Here, SEM micrographs present several areas of a single fiber particle removed from a cannulated cow and submitted to intensive washes. The observed microorganisms are tightly attached to the fiber particle. The sample was coated with 5 nm chrome and imaged using a Verios 460 L FEI HRSEM. Scale bars are indicated.
Subsequent fiber deconstruction and fermentation are accomplished by the rumen microorganisms that release the energy stored in the complex plant carbohydrates, otherwise inaccessible to the animal, and convert them to short-chain fatty acids that are absorbed by the animal for its energy needs through the rumen wall (Mizrahi). These microorganisms also serve as an important protein source for the animal upon later digestion in the alimentary tract (Flint 1997; Mizrahi 2013). Hence, the fiber-degradation process is the basis for most of the essential metabolism performed by the rumen ecosystem. Decades of microbiological work on rumen microorganisms seem to have highlighted most of the key fiber-degrading microbes. Particular attention has been devoted to cellulose degraders, because cellulose is the most degradation-resistant polysaccharide in plant fiber. Hemicellulose and other polysaccharide degraders have also been thoroughly characterized. Multiple glycoside hydrolases (GHs) are required to deconstruct the intricate chemical structure of plant biomass, and since the enzymes that take part in this process are encoded in the microbial genomes of the rumen inhabitants, these 'enzymatic' interactions are in fact translated into 'microbial' interactions within the rumen ecosystem.
Although microbial interactions within such a complex and dynamic microbial community are difficult to predict, fiber-degrading isolates have been well studied in vitro, and co cultures or small microbial consortia have been used as tools to decipher the positive or negative interactions between them and other microbes that fail to degrade fiber. The fiber-degrading microorganisms (and the general rules governing the interactions among the fiber degraders themselves and the overall community within the unique rumen ecosystem) are highly important for understanding anaerobic microbial ecosystems in general, and the rumen ecosystem in particular. Indeed, the fundamental patterns of assembly and succession of rumen microbiomes have direct implications in microbial ecology, as analogous rules are observed in microbial communities from other gut ecosystems and anaerobic digesters (i.e. the human gut) (Flint et al. 2008), and may even apply to more distant ecosystems, such as soil. Moreover, a better understanding of these principles can help modulate this and other systems for applied purposes.
The overall richness of the bovine rumen microbiome is composed mostly of bacteria, at about 95% of the microorganisms, and a minority of archaea (2–5%), while eukaryotic protozoa and fungi are also present at very low percentages (0.1–1% of the total microbial richness) (Flint 1997; Mizrahi 2013). Organisms that are capable of fiber degradation have been described in each of these domains of life, thus emphasizing the strong environmental filtering toward the fiber-degrading function. It has been shown that key fiber-degrading bacteria appear in the rumen only a few days after birth, with the rumen ecosystem being functional with respect to fiber degradation at that early age (Fonty et al. 1987). Recent reports have even confirmed that they are present just after birth (Jami et al. 2013; Guzman et al. 2015). Such an early predisposition to the fiber-degrading function emphasizes the importance of the fiber-degrading microorganisms as the founders of the rumen microbial community in a functioning ecosystem. Furthermore, in a large-scale study, across diverse animals and countries, a conserved ruminant core microbiome containing the main cellulose- and hemicellulose-degrading families was observed in all ruminants (Henderson et al. 2015a). In addition, host genetics has been recently shown to be linked to one of the cardinal cellulose degraders, as well as to several hemicellulose degraders in the rumen microbiome, thereby suggesting host control of these potential keystone species (Sasson et al. 2017). This indicates selection toward a general, gut-specific and specialized core microbiome in ruminants (Jami and Mizrahi 2012; Weimer 2015; Henderson et al. 2015a). Here we will review the key functionalities that enable fiber degradation in the rumen ecosystem. We will then explain how these functions are distributed across the different rumen fiber-colonizing and degrading microbes and discuss the strategies employed by the different microbes for fiber degradation. Importantly, as these strategies and functions represent different ecological niches, we will try to deduce and understand the potential interactions between the different fiber-degrading microbes and their interactions with other rumen microbes, to infer the effect of the key fiber-degrading microbes on overall rumen microbiome structure.
In the plant cell walls, cellulose fibrils are embedded in a matrix of hemicellulose (mostly xylan, but also mannan, xyloglucan and β-glucan), lignin and pectin. The cellulose component is crystalline and insoluble. It is even more recalcitrant to degradation in its natural state, where it is physically embedded into a colloidal hemicellulose and lignin matrix (Bayer et al. 1998). Therefore, to achieve efficient plant fiber degradation, the concerted action of multiple GHs acting on different parts of the complex biopolymer is required.
Fiber-degrading functions are represented by a multiplicity of known enzyme activities (e.g. cellulases and hemicellulases), which are clustered into about 150 GH families, classified according to their sequence, function and structural properties (Lombard et al. 2014). In the following, we describe the most important enzymatic functions for plant biomass breakdown in the rumen.
Cellulose degradation
Although cellulose is a polysaccharide made up entirely of glucose units, it is a very tough substrate for enzymes to degrade, since the cellulose chains are tightly packed together with extensive internal and interchain hydrogen bonding, resulting in an insoluble, crystalline chemical structure. The kinetics of cellulose degradation is relatively slow, and the extent of degradation is incomplete. In nature, microbial cellulose degraders contain a variety of potent cellulases from several main GH families and in some cases, have many representatives of the same GH family, suggesting the importance of diverse substrate specificities for the cellulases to maximize enzymatic potential.
During cellulose degradation, the cellulose fibrils are attacked at the ends of the chain by exoglucanases that are generally from GH family 48 in bacteria, GH family 6 or 7 in aerobic fungi and GH family 6 and 48 in anaerobic fungi. These specific enzymes are highly important for cellulose breakdown. In cellulolytic bacteria, the GH48 enzymes are highly expressed and represent a major portion of the secreted proteome when grown in pure culture (Artzi, Bayer and Moraïs 2017). A second group of cellulases that act in concert with the exoglucanases are the endoglucanases, which cleave the cellulose chain internally. The main reported endoglucanases are from GH families 5, 8 and 9 in bacteria and GH families 6, 7 and 45 in fungi (Lombard et al. 2014). A subgroup of endoglucanases, known as ‘processive’ endoglucanases, act first as endoglucanases by cleaving the chain internally and then as exoglucanases by continuing chain cleavage.
Hemicellulose degradation
The second major component of the plant cell wall, hemicellulose, is comprised of various polysaccharides, with xylan being the most abundant. Xylan is organized in chains composed of xylose (xylobiose units), which are branched with different side-chain sugars. Xylanases are generally from GH families 10, 11 and 30 for the endoxylanases (cleaving the main chain internally) and 43 for exoxylanases (cleaving at the chain ends) and side-chain cleavage enzymes. Despite its complex chemical composition, xylan is generally less challenging for the microorganisms and their enzymes to deconstruct, and the kinetics of xylan degradation is relatively rapid. Mannan, xyloglucan and β-glucan present also in hemicellulose are hydrolyzed by additional GH families (mainly GH5 and GH26 for mannanases; GH5, GH12, GH44 or GH74 for xyloglucanases and GH5, GH16 and GH17 for β-glucanases).
Degradation of additional plant components
Aside from the structural polysaccharides cellulose and hemicellulose, another important source of carbon in plant biomass is the storage polysaccharide starch, a biopolymer of glucose. Storage polysaccharides are easier to degrade, since they have to be readily available to the plant for metabolic processes. Amylases can hydrolyze starch and are mainly from GH family 13. α-Amylases act randomly on starch substrates, whereas β-amylases act only from the non-reducing end of the chain. Complete degradation of starch requires the action of debranching enzymes, and the main product of starch degradation is maltose, a disaccharide composed of two α-linked molecules of glucose, as opposed to the cellobiose unit of cellulose, which is a β-linked glucose. Additional GH families can hydrolyze pectin and other polysaccharides present in lower amounts in the plant biomass. Furthermore, the action of GHs is complemented by other accessory enzymes, such as carbohydrate esterases, polysaccharide lyases and lytic polysaccharide mono-oxygenases (Lombard et al. 2014). Finally, the aromatic lignin polymer is degraded by different classes of oxidative enzymes, such as peroxidases and laccases, mainly produced by fungi (Pollegioni, Tonin and Rosini 2015). Additional details on the enzymology of fiber degradation are available in specific reviews (Bourne and Henrissat 2001; Bayer, Shoham and Lamed 2013).
Enzymatic potential across the rumen microbiome
Metagenomic studies, accompanied by biochemical verification and analyses of the cow rumen, have revealed a highly abundant set of enzymes produced by the resident microbial community, numbering 27 755 putative carbohydrate-active genes (Hess et al. 2011). Among them, 3442 putative amylases (GH13) were identified. Subsequent metatranscriptomics studies of the cow rumen demonstrated that enzymes of selected GH families are more largely expressed (Dai et al. 2015). GH families 5, 9, 45 and 48 represented 98% of the total expressed cellulases, of which GH48 was the most highly expressed. For hemicellulases, GH10, GH11 and GH26 (mannan- or xylan-degrading enzymes) were the most highly represented. The authors also identified that both cellulases and hemicellulases were mostly of bacterial origin, with most of the cellulase sequences expressed by the genera Ruminococcus and Fibrobacter and the hemicellulase sequences by Ruminococcus, Prevotella and Fibrobacter (Dai et al. 2015). When comparing the distribution of functions discovered in those reports (Hess et al. 2011; Dai et al. 2015), to the genomic potential of the Hungate1000 genomes (Seshadri et al. 2018), interesting similarities are revealed, among them the percentage of GH13 genes is similar in the genomes of microbial isolates and the metagenomes, which suggests that most of the enzymatic functionalities are represented in the isolated genomes.
DNA chips were also developed to measure the expression levels of the different GH families in rumen samples (Abot et al. 2016; Comtet-Marre et al. 2018). The FibroChip (Comtet-Marre et al. 2018) was designed with targeted GH families selected among the 20 most highly expressed bacterial GH families detected by the CAZyChip (Abot et al. 2016). Sequences originating from protozoan and fungal genomes were included in the FibroChip. Transcripts from GH families 5, 10 and 43 were the most strongly represented in the rumen, with Bacteroides, Fibrobacter and Ruminococcus genera being the major contributors. Fungal and protozoan transcripts represented only 8.1 and 6.7% of the total activity, respectively (Comtet-Marre et al. 2018), but the results may have been biased by the design of the chip, which only includes currently known sequences.
The recent global effort to isolate and sequence rumen microbial isolates (Seshadri et al. 2018) revealed that GH families are scattered across a multitude of microbial genomes. Examining the available genomes of the most abundant and prevalent bacterial species in the rumen (Henderson et al. 2015b), we observed that all of them code for at least one of the major GH families listed above, with the exception of the phylum Euryarchaeota (Fig. 2). Half of the sequenced genomes encoded multiple GHs, suggesting high fibrolytic activity in those microorganisms. This distribution of repetitive functions across most of the rumen bacterial population reflects both the importance of these GH enzymes for the fiber-degrading function and the subsequent ability of their host bacteria to grow in abundance and dominate this ecosystem. In addition, while most of the sequenced microorganisms in the rumen contain GH13 enzymes (Fig. 2), the distribution of hemicellulases and cellulases is more specific to certain phylogenies. Cellulases are mainly found in Ruminococcus, Butyrivibrio, Pseudobutyrivibrio and Lachnospiraceae genera. It should be noted that the members of the Ruminococcus genus that comprise the most known fiber degraders capable of crystalline cellulose deconstruction are distinguished by having at least one GH48 representative (exoglucanase). Conversely, cellulases are rare within the Bacilli and Negavicutes classes of the Firmicutes and in the phyla Proteobacteria and Actinobacteria, whereas some hemicellulases are present in those phylogenies. Furthermore, when cellulase genes are present in the genomes, the number of hemicellulases increases (Fig. 2).
Figure 2.

Distribution of the major GH families across rumen microbial genomes. Phylogeny of the 501 16S RNA sequences from the Hungate1000 collection (Seshadri et al. 2018). The sequences were aligned, and the tree was constructed using MEGA7.0 software. The tree displayed in the figure was condensed by collapsing branches with less than 50% statistical significance. Phyla and selected classes are indicated using either a color range that covers the full clades or branch symbols. Major cellulase GH families 5, 6, 8, 9, 45 and 48 are indicated with a green color gradient, major hemicellulase GH families 10, 11, 26, 30, 43 and 74 with a blue gradient, and GH13 enzymes (amylases) are indicated in yellow. The multiple bars indicate both the presence and quantity of each GH within each microbial genome.
Main enzymatic strategies for fiber degradation
In nature, GHs (as well as carbohydrate esterases and polysaccharide lyases) are multimodular enzymes, which may also include one or more carbohydrate-binding modules (CBMs), whose major role is to target the catalytic subunit to its substrate. The enzyme can also bear several catalytic GH subunits, and such multifunctional enzymes may thus exhibit several substrate specificities (Himmel et al. 2010). In addition, simple or multifunctional GHs can be either secreted as free enzymes in the external milieu or assembled in highly ordered enzymatic complexes termed cellulosomes (Artzi, Bayer and Moraïs 2017). Cellulosomes are self-assembling protein complexes produced by anaerobic microorganisms (bacteria and fungi) and composed of a non-enzymatic subunit (scaffoldin) containing multiple cohesin modules that integrate dockerin-containing enzymatic subunits via high-affinity and highly-specific cohesin–dockerin interactions. While details on fungal cellulosome architecture have yet to be revealed (Haitjema et al. 2017), bacterial cellulosome complexes have been extensively studied over the past several decades. Bacterial cellulosomes can be anchored to the microbial cell surface or released into the external milieu. They can contain varying numbers of enzymes (from 2 to 160) and scaffoldins (forming simple or highly structured cellulosomes), and a given bacterium can produce various modular architectures as a function of the available carbon source and additional types of substrate-independent regulatory signals (Artzi, Bayer and Moraïs 2017). Cellulosome producers are well known to exhibit high catalytic efficiencies toward fiber degradation, as the proximity between the catalytic subunits results in a well-ordered substrate-channeling effect (Artzi, Bayer and Moraïs 2017).
Enzymatic complementarity translates to microbial diversity
As already noted, due to the naturally elaborate complexity of the chemical and structural compositions of plant fibers, fiber deconstruction requires the combination and cooperation of multiple enzymatic functions. Synergy among the specific enzyme classes (cellulases, hemicellulases and amylases) is essential, generally among different GH families, but also within the same family and between cellulases and hemicellulases (Bayer, Shoham and Lamed 2013; Artzi, Bayer and Moraïs 2017). This prerequisite for enzymatic diversity is observed at the genome level in the enzymatic repertoire of each of the various microorganisms that efficiently degrade plant biomass (Tables 1 and 2). In the next section, we discuss how the main enzymatic functions and strategies for fiber degradation are scattered across the genomes of the various fiber degraders that inhabit the rumen, suggesting both niche partitioning and fine-tuned interactions among these microorganisms.
Table 1.
Number of enzymes from the major cellulase and hemicellulase GH families in selected key fibrolytic bacterial species inhabiting the rumen (Lombard et al. 2014; Seshadri et al. 2018).

Species in red are part of the 50 prevalent and abundant bacterial taxa in the rumen (Henderson et al. 2015a). Cellulases, in green, can be members of GH families 5, 8, 9, 45 and 48. Hemicellulases, in blue, are members of families 10, 11, 26, 30, 43 and 74 (but also families 5 and 8), and amylases (or pullulanases) (yellow/orange) are characteristic of family 13.
Table 2.
Number of enzymes from the major cellulase and hemicellulase GH families in selected sequenced fibrolytic fungal species inhabiting the rumen (Youssef et al. 2013; Kameshwar and Qin 2018).

Cellulases, highlighted in green, include members of GH families 5, 6, 8, 9, 45 and 48, and hemicellulases, highlighted in blue, are from families 10, 11, 30, 43 and 74 (but also from families 5 and 8).
KEY FIBROLYTIC MICROORGANISMS IN THE RUMEN
Fiber degraders are microorganisms that are able to deconstruct at least one component of the fiber biomass. Here, the most prominent fiber degraders will be discussed. Recent global efforts to isolate and characterize rumen bacteria and archaea (Hungate1000 project) have revealed ∼75% of the genus-level taxa from the core microbiome. These have been sequenced and extensively analyzed (Henderson et al. 2015b; Seshadri et al. 2018), thereby increasing our ability to understand microbial fiber degradation in the rumen.
Bacterial degraders
Bacteria are obligate inhabitants of the rumen; without them, the animal host would not survive. While most of the more prevalent bacteria across the rumen of various animals contain the major GH families (Fig. 2), some bacteria have been identified by decades of microbiological work on rumen isolates as essential fiber degraders. These bacteria have been thoroughly characterized and their various strategies for fiber-degrading specializations have been described.
Cellulose degradation
Only a few isolated bacteria that are present in relatively low abundance in the rumen have been reported to be capable of crystalline cellulose degradation. The main bacterial cellulose degraders are from three distinct species: Ruminococcus flavefaciens (which has been recently linked to host genetics; Sasson et al. 2017), Ruminococcus albus and Fibrobacter succinogenes (Mizrahi 2013). Interestingly, while R. flavefaciens assembles an incredibly elaborate enzymatic machinery on its bacterial cell wall to degrade cellulose (Lamed et al. 1983; Artzi, Bayer and Moraïs 2017; Israeli-Ruimy et al. 2017), R. albus and F. succinogenes adopt alternative enzymatic paradigms (Dassa et al. 2014; Arntzen et al. 2017).
The cellulosomal system of R. flavefaciens has been studied mostly by biochemical means, revealing that R. flavefaciens potentially assembles multiple architectures of complex cellulosomes via a multiplicity of protein assemblies (Jindou et al. 2008; Rincon et al. 2010; Dassa et al. 2014; Israeli-Ruimy et al. 2017). The cellulosomal proteins of R. flavefaciens vary from strain to strain (Dassa et al. 2014), and the cohesin–dockerin interactions are mostly (but not exclusively) strain-specific (Israeli-Ruimy et al. 2017). Moreover, the ability to digest cellulose has been shown to differ from strain to strain (Krause et al. 1999a). Cellulosomal interactions were examined in depth for strain FD-1 (Israeli-Ruimy et al. 2017). This strain could potentially assemble a cellulosome composed of four distinct scaffoldins interacting in a specific manner (color-coded interactions in Fig. 3A). The resultant cellulosomal complex can contain a maximum of 14 enzymes, whereas the total number of dockerin-containing proteins in strain FD-1 is 223. The disparity between dockerin-bearing proteins and dockerin-binding sites on the multi-scaffoldin platform attests to the variability in enzymatic content of the R. flavefaciens cellulosomes (Israeli-Ruimy et al. 2017). This extraordinary heterogeneity of cellulosomal architectures can account for the highly efficient fiber-degrading abilities of R. flavefaciens. In addition to the cellulosomal GHs, R. flavefaciens codes for a similar number of GHs that lack dockerin, suggesting that the bacterium combines free enzymatic and cellulosomal paradigms to achieve high levels of fiber degradation (Dassa et al. 2014). Cellulosomal complexes of R. flavefaciens still await dedicated in vivo analysis, to pinpoint the most expressed enzymatic subunits comprising the R. flavefaciens cellulosome.
Figure 3.

Schematic representations of cellulose-degrading machineries of rumen microorganisms. (A)R. flavefaciens produces a highly structured cellulosome complex. (B)R. albus produces cellulosomal enzymes along with CBM37-containing enzymes. (C)F. succinogenes contains extended cell-surface pili that target the bacterial cells to the cellulose surface. The bacterium produces surface-anchored enzymes and outer membrane vesicles that contain multiple GHs and a fibro-slime protein complex. (D) The polycentric anaerobic fungus, Orpinomyces sp., produces fungal cellulosomes. (E) The rumen ciliate Polyplastron both secretes GH and exhibits high enzymatic activity in its food vacuole. The ciliate protozoon is represented at 1:100 scale.
Surprisingly, R. albus enzymes also contain dockerin modules but the sequenced genome of the various isolated strains did not reveal suitable multiple cohesin-containing scaffoldin(s) (Dassa et al. 2014). Therefore, the ability of this clearly cellulolytic bacterial species to produce bona fide cellulosomes (Ohara et al. 2000) remains in doubt. Intriguingly, numerous R. albus enzymes contain a CBM from family 37 that seems to be unique to this bacterium (Fig. 3B) (Dassa et al. 2014). These CBMs display broad substrate-binding affinities (Xu et al. 2004), and may play a role in cell-surface attachment of the parent enzymes (Xu et al. 2004; Ezer et al. 2008). Aside from R. flavefaciens and R. albus, it has been revealed that many ruminal bacteria encode cellulosomal elements; this may indicate a broad role for the cellulosomal machinery in the rumen (Bensoussan et al. 2017).
In contrast to the cellulolytic ruminococci, the bacterium F. succinogenes neither produces cellulosomes nor secretes free enzymes (Suen et al. 2011), but employs an alternative strategy for fiber degradation. While growing on cellulose, it produces outer membrane vesicles (Fig. 3C) that are able to degrade cellulose and other polysaccharides (Groleau and Forsberg 1981; Gong and Forsberg 1993). In a recent study, these vesicles were isolated and their content examined by proteomics (Arntzen et al. 2017). That study revealed large outer membrane vesicles (average size of 49 nm) containing more than 300 different proteins, 21% of them with predicted fiber-degrading functions. In addition, multiprotein complexes in the outer membrane vesicles seem to play a role in cellulose-binding and degradation (Arntzen et al. 2017). The bacterium also produces fibro-slime and pili proteins that both display cellulases on the bacterial wall and adhere to cellulose (Burnet et al. 2015). Early studies on fiber degradation that compared the enzymatic potential of pure isolates of the cellulose degraders concluded that F. succinogenes is the most efficient bacterium for cellulosic fiber degradation in the rumen (Dehority 2003). Moreover, in most reports (Kobayashi, Shinkai and Koike 2008), it is claimed to be the most abundant of the three main cellulose-degrading strains.
These three major cellulose degraders in the rumen are present in multiple representative strains of each bacterial species with variations in their gene content and physiology. F. succinogenes can be divided into four phylogenetic groups with significant physiological differences, three of which have been detected in the rumen with different dynamic populations (Koike et al. 2004). Moreover, the gene encoding the very conserved scaffoldin ScaC from R. flavefaciens and that encoding the major cellulase GH48 of R. albus were used to analyze the diversity of those species' phenotypes in steer. Large intraspecies diversity was observed for R. flavefaciens (Jindou et al. 2008), whereas less variation was observed for R. albus strains (Brulc et al. 2011; Grinberg et al. 2015).
It should be noted that bacterial cellulose degraders in the rumen are not solely limited to these three enzymatic paradigms, and some bacteria can employ a polysaccharide-utilization locus (PUL) mechanism for cellulose degradation, as has been suggested for various species of the phylum Bacteroidetes (Naas et al. 2014). These sets of genes were shown to be affiliated mostly to Prevotella genus in the bovine rumen (Rosewarne et al. 2014).
Hemicellulose degradation
The degradation of hemicellulose is performed by a more varied array of bacteria, which may reflect the large variation and complexity of hemicellulose types in the cell wall. This could imply that the different hemicelluloytic bacterial species are more specialized for given types of hemicellulose. Two of the most abundant bacterial genera in the rumen, Prevotella and Butyrivibrio (and Pseudobutyrivibrio), are highly efficient hemicellulose degraders. Individual host animals typically contain multiple species and strains (dozens to hundreds) of these two genera (Pitta et al. 2010; Jami and Mizrahi 2012). Isolates have been characterized, and they display an extremely broad enzymatic repertoire, with different enzymatic abilities (Avgustin, Flint and Whitehead 1992). The abundance of a hemicellulose degrader does not necessarily reflect its enzymatic efficiency toward its substrate. For example, Prevotella strains, which are considered to be less hemicellulolytic than Butyrivibrio, Ruminococcus or Fibrobacter, have been recently shown to dominate consortia in the hemicellulose-enriched rumen (Emerson and Weimer 2017).
Besides their strong ability to hydrolyze starch and hemicellulose, some bacteria can also hydrolyze proteins, and they can ferment both sugars and amino acids (Flint 1997; Hobson and Stewart 2012). The genomes of many Prevotella, Butyrivibrio and others have been sequenced. Their genomes contain multiple GHs from various families (Table 1) (Seshadri et al. 2018), and they produce free enzymatic subunits (no cellulosome formation). Since hemicellulose degraders are associated with both the planktonic and fiber-adherent fractions in the rumen (Klevenhusen et al. 2017), the production of free enzymes seems to be the most suitable strategy for substrate degradation, independent of the location of the bacterium relative to its substrate. Even if produced by a variety of bacterial strains, the sugar end products of many of the secreted enzymes are similar. This should increase the probability of promoting cheaters that will only use such products without expressing the enzymes. Such a phenomenon might be prevented or reduced by increasing the local concentration of the sugar products near the enzyme producers, potentially giving them a competitive advantage (Celiker and Gore 2012). It should be also noted that hemicellulases organized in polysaccharide-utilization loci are abundant in Bacteroidetes (and Prevotella) in the rumen (Pope et al. 2012; Rosewarne et al. 2014).
Another long-standing question is how do these species coexist when each carries many overlapping degradation abilities, which should result in harsh competition among them? Perhaps the large abundance of fiber biomass in the rumen decreases the potential competition and is the reason for their ability to coexist. It has indeed been observed that rumen fiber degradation is incomplete, with 50% of the fiber biomass present in the animal's feces (Weimer 1996; Russell, Muck and Weimer 2009; Shabat et al. 2016). Alternatively, these species might express different repertoires of enzymes depending on the microbial composition, potentially creating a niche-partitioning situation via changes in their gene-expression patterns. It is intriguing that some cellulose degraders (such as R. flavefaciens and F. succinogenes) also produce hemicellulases (see Table 1), even if they do not necessarily have the ability to process pentoses, which comprise the monomeric units of the hemicellulose polymers (such as xylose) (Matte, Forsberg and Gibbins 1992). In this case, it is hypothesized that since the cellulose can be physically embedded in hemicelluloses, these enzymes will only serve to give the cellulases access to the cellulose fibers. The fact that the cellulose degraders produce enzymes that release sugar monomers that they will not consume would also imply interspecies interactions.
Starch degradation
A large number of bacterial starch degraders inhabit the rumen. Among them, Ruminobacter amylophilus, Prevotella ruminicola, Streptococcus bovis, Succinimonas amylolytica, Selenomonas ruminantium and Butyrivibrio fibrisolvens (Hobson and Stewart 2012) produce free enzymatic subunits (Seshadri et al. 2018). Resistant starch is specifically degraded by the specialist Ruminococcus bromiithat produces amylosome complexes, analogous to cellulosomes (Ze et al. 2015; Mukhopadhya et al. 2018). Protozoa and fungi also exhibit amylolytic activities but are not essential for starch degradation (Hobson and Stewart 2012).
Fungal degraders
Rumen fungi are not obligatory inhabitants of the rumen and in some animals, they are not detected. Nevertheless, they have very high potential for fiber degradation, as they encode a variety of plant fiber-degrading enzymes. Indeed, it has been reported that the plant cell wall is weaker after incubation with fungi, compared to incubation with rumen prokaryotes (bacteria and archaea). This is attributed to the fungi's ability to better penetrate the plant cell wall and solubilize the lignin component, where the resultant phenolic compounds are not metabolized by these organisms (Akin and Borneman 1990). Nevertheless, it should be noted that under anaerobic conditions, lignin solubilization is quite limited (Susmel and Stefanon 1993).
Since lignin is covalently linked to hemicellulose via ferulate bridges, it interferes with microbial degradation of fiber polysaccharides by acting as a physical barrier. Therefore, rumen fungi are considered to play an important role in the initial degradation of large particles, thus increasing the rumen bacteria's access to the plant cell wall polysaccharides (Akin et al. 1989). Anaerobic fungi also produce a wide range of polysaccharide-degrading enzymes (Dehority 2003), and some studies suggest that their contribution to fermentation is greater than that of bacteria (Lee, Ha and Cheng 2000). Rumen fungi are active on both cellulose and hemicellulose, and six genera have been described: Neocallimastix, Caecomyces, Piromyces, Anaeromyces, Orpinomyces and Cyllamyces, with highest efficiencies observed in Neocallimastix and Piromyces (Fliegerova et al. 2015). Genome analysis of some anaerobic fungal species has revealed a gene repertoire that is extremely rich in hemicellulases and cellulases (Table 2) (Youssef et al. 2013; Kameshwar and Qin 2018). It has been speculated that GHs were acquired by anaerobic fungi via horizontal gene transfer from bacteria (Haitjema et al. 2017). Nevertheless, as can be seen from Table 2, it seems that in parallel, they have broadened their enzymatic repertoire considerably beyond these events.
Fungal ‘cellulosomes’ have been described for species of Neocallimastix, Piromyces and Orpinomyces (Fig. 3D). However, their dockerin modules lack definitive sequence homology to the bacterial dockerins, and the putative scaffoldin and cohesin modules described in ‘cellulosomal’ fungi are also significantly different, thus suggesting an independent origin from bacterial cellulosomes (Haitjema et al. 2017). Unlike the latter, the fungal scaffoldin system is broadly conserved across the anaerobic fungal phylum, allowing for interspecies interactions that might occur in the rumen environment (Fanutti et al. 1995).
Rumen protozoa
Ciliate protozoa in the rumen environment usually account for about 50% of the rumen biomass, and are believed to impact the ecosystem's metabolism (Newbold et al. 2015). However, defaunation procedures have demonstrated that the rumen ecosystem functions without the protozoa, suggesting that they are not essential to the ecosystem. Therefore, like the fungi, the protozoa are not obligatory inhabitants of the rumen. In a vast study examining 742 samples spanning 32 animal species and 35 countries, rumen protozoa were assigned to 12 species and were found to be highly variable across individual animals. The most dominant and prevalent genera were Entodinium and Epidinium, occupying 90% of the animals and 54% of the sequenced data (Henderson et al. 2016).
Some protozoa are able to consume substrate particles along with other microorganisms, and have apparent cellulolytic activity that could stem from the ingested fibrolytic microorganisms or from their own fiber-degrading enzymes (Fig. 3E). Cellulolytic activity in rumen protozoa has been measured for several species, and the most efficient cellulose degraders were Eudiplodinium maggii, Epidinium ecaudatum and Ostracodinium dilobum (Dehority 2003). None of these species have yet been sequenced. Nevertheless, it has been shown that the cellulolytic protozoan Polyplastron multivesiculatum encodes its own xylanases (Fig. 3E) (Devillard, Newbold and Scott 1999; Devillard et al. 2003). Later, horizontal gene transfer of GHs from bacteria to protozoa was suggested (Ricard et al. 2006). Twelve putative xylanase genes were detected in the entodiniomorphids Polyplastron multivesiculatum, Epidinium ecaudatum, Eudiplodinium maggii, Diploplastron affine and Metadinium medium, with high homology to Clostridium acetobutylicum genes. Nine putative cellulases were also detected in the entodiniomorphids E. ecaudatum and P. multivesiculatum from the GH5 and GH9 families. Methanogenic screening coupled with biochemical characterization served to retrieve and characterize hemicellulases from E. ecaudatum (Findley et al. 2011). Another study that examined the metagenome of the muskoxen rumen recovered protozoal sequences from GH5, GH9, GH10, GH11 and GH13, indicating cellulase, hemicellulase and amylase activities encoded by these rumen microbes (Qi et al. 2011). In contrast to bacterial and fungal fiber degraders, the number of GH genes seems to be low in protozoa, suggesting either a technical bias or that their contribution to fiber degradation does not rely on their enzymatic potential.
PRODUCTS OF FIBER DEGRADATION DRIVE MICROBIAL INTERACTION NETWORKS
Hydrolysis of fiber carbohydrates releases pentoses and hexoses as degradation byproducts. Subsequent diffusion of sugars into the extracellular matrix allows the growth of secondary consumers, thereby propagating the rumen food web by enabling subsequent construction of ecological niches to the trophic level. Furthermore, the fiber-degradation process and successive fermentation of the sugar monomers under anaerobic conditions produce a high quantity of hydrogen that accumulates in the rumen environment. In the following, we describe how these byproducts of fiber degradation are used by the rumen microbial community, creating microbial networks, and how the metabolites of these microbial interactions lay the foundation for rumen ecosystem function.
The role of methanogens and hydrogen-utilizing bacteria
Cellulose hydrolysis has been shown many times in the past to be increased by the presence of methanogens that play the role of hydrogen sink, thus releasing its partial pressure and inhibitory effect (Cazier et al. 2015). In the rumen, anaerobic cellulolytic fungi and bacteria, as well as protozoa, have shown beneficial interactions with methanogens. For example, the rumen fungus Neocallimastix sp. was shown to have positive interactions with methanogens (Mountfort, Asher and Bauchop 1982). Similarly, a clear beneficial effect was demonstrated when coculturing F. succinogenes with Methanobrevibacter smithii (Rychlik and May 2000), while a moderate effect was observed in cocultures of M. smithii and Rumonococcus flavefaciens or R. albus. A correlation was also observed between the number of methanogens and that of cellulolytic microorganisms in samples from various animals (Morvan et al. 1996a). Protozoa are also believed to play a part in methanogenesis by providing hydrogen to methanogens (Newbold et al. 2015), as defaunation procedures were shown to decrease methanogenesis (Hobson and Stewart 2012). By relieving the hydrogen partial pressure, these positive relationships with cellulolytic bacteria are not limited to methanogens, as also observed between the hydrogen-consuming bacterium S. ruminantium and the anaerobic fungus Neocallimastix sp. (Marvin-Sikkema et al. 1990). Moreover, hydrogen-utilizing acetogenic bacteria were also demonstrated to increase cellulose breakdown when cocultured with cellulolytic microorganisms (Morvan et al. 1996b). In contrast, no strong associations were detected between archaea and ciliate protozoa in two independent studies examining microbial networks in the rumen (Henderson et al. 2016; Tapio et al. 2017). Furthermore, methanogen concentration is higher in defaunated animals, suggesting predation of the methanogens by protozoa (Morgavi et al. 2012; Levy and Jami 2018).
Coculture with hydrogenotrophs also revealed drastic changes in the fermentation products obtained from cellulose. For example, the interaction between R. flavefaciens and Methanobacterium ruminantium diverted final metabolite production from succinic acid to acetate (Latham and Wolin 1977). These changes in fermentation products could potentially impact overall microbial structure by rewiring the rumen food web to alternative routes.
In an in situ study that examined microbial succession on switchgrass, fiber degradation was observed to occur in three phases: (1) rapid degradation of 13% of the fiber in 30 min, (2) a lag phase during which no fiber degradation occurred and the abundance of methanogens rose considerably, and (3) continuation of fiber degradation after 4 h (Piao et al. 2014). That report suggested a key role for methanogens in relieving hydrogen pressure and therefore, strengthened the notion of an intimate cooperation between fiber degraders and methanogens for fiber degradation. These hydrogen-dependent interactions also translate to physical interactions, as was recently revealed in symbiotic interactions between the methanogen M. ruminantium, and hydrogen producers. In this interaction, the methanogen produced an adhesin-like protein that could adhere to the cell surface of the hydrogen producers. Indeed, binding was observed between a large number of rumen protozoa and the bacterium Butyrivibrio proteoclasticus (Ng et al. 2016).
While focus has centered on the improvement of cellulose hydrolysis by coculturing methanogens with cellulose degraders, enhancement of xylan degradation has also been reported upon cocultivation of R. flavefaciens and M. smithii (Williams, Withers and Joblin 1994). In addition, hemicellulose scavengers that are also hydrogen producers exhibited syntrophic interactions with methanogens (Leahy et al. 2010).
Nutritional interactions among rumen microorganisms during fiber degradation
Nutritional interactions have been observed between fiber-degrading microorganisms with distinct phylogenies and enzymatic functionalities. A possible mechanism for these symbiotic relationships could simply be that some fibrolytic bacteria depend on other members of the gut microbial community for certain vitamins and precursors for amino acid synthesis (Dehority 2003), as is the case for cellulolytic species. In turn, other microorganisms take advantage of the specialist microbes (e.g. cellulolytic species) that carry the burden of producing specialized enzymes (such as the generally high-molecular-weight modular cellulases) but do not utilize the products of fiber degradation to their fullest (Berlemont and Martiny 2013). It has indeed been shown that cellodextrins produced in the first step of cellulose degradation are utilized by several non-cellulolytic species, such as S. ruminantium and P. ruminicola (Russell 1985). The cellulose degraders thus allow the loss of hardly obtained cellodextrins to the benefit of other microbes in order to access valuable goods produced by these populations. For example, F. succinogenes cultured with group U2 bacterium R-25 and/or S. ruminantium exhibited enhanced succinate production and fiber hydrolysis (Sawanon, Koike and Kobayashi 2011; Fukuma, Koike and Kobayashi 2015). Similarly, Treponema bryantii (which also has the genomic potential to produce GHs) interacts with the cellulolytic bacterium F. succinogenes to enhance fiber degradation (Stanton et al. 1980). In other cases, there was no noticeable enhancement of fiber degradation upon microorganism coculturing, but clear cross-feeding has been established, as for P. ruminicola, which uses its proteolytic capabilities to supply ammonia ions to R. albus, which in turn degrades cellulose and provides the proteolytic bacterium with hydrolyzed soluble sugars (Bryant and Wolin 1975). Another study from Miura, Horiguchi and Matsumoto (1980) also introduced the concept of nutritional interdependence between distinct fiber degraders in the rumen: R. albus, Megasphaera elsdenii and Bacteroides amylophilus. These three bacteria could not be grown as single cultures in the basal media, but M. elsdenii could be co-cultured with either B. amylophilus or R. albus in the same medium containing starch and soluble sugars. By determining the concentration of free branched amino acids and volatile fatty acids in single and cocultures, the authors revealed that in the triculture, the production of branched amino acids by B. amylophilus is used directly by M. elsdenii to produce branched fatty acids necessary for the growth of R. albus. Hence, by providing soluble sugars to the community, R. albus gains essential branched fatty acids for its own growth. Since these nutritional interchanges are undoubtedly impossible by molecule diffusion alone in the rumen environment, one could assume that the three populations are physically close to one another.
MICROBIAL INTERACTIONS IN LIGHT OF FIBER DEGRADATION
Competition among microbes that occupy the same ecological niche
It is hypothesized that fiber biomass, although an abundant resource in the rumen ecosystem, can also be a source of competition among microbes sharing the same ecological niche. For example, mostly negative competitive interactions were reported between the cellulose degraders F. succinogenes, R. flavefaciens and R. albus when grown in co-culture in vitro on cellulosic substrates. While it was reported that there was no difference in the attachment rates of these three species in vivo (Rogar et al. 1990), Mosoni, Fonty and Gouet (1997) used radiolabeling to observe competition for adhesion to the cellulosic substrate between these three main bacterial cellulose degraders. Different inhibitory interactions were observed between R. flavefaciens, R. albus and F. succinogenes, varied according to their mode of inoculation, i.e. simultaneous or sequential. Aside from competition for adhesion, another study examined the specific growth of each of these three bacteria in coculture (Shi, Odt and Weimer 1997). Cell numbers of individual species were approximately equal upon coculture (in the excess of cellulose substrate) of R. albus with R. flavefaciens, R. albus with F. succinogenes, or R. flavefaciens with F. succinogenes. When cellulose was limiting, however, F. succinogenes outcompeted R. albus and R. flavefaciens outcompeted both bacteria (Shi, Odt and Weimer 1997). In the case of these species, additional mechanisms of competition, and not only resource utilization, were shown to play a part in their interaction. A mechanistic cause for the observed inhibition was indeed reported for R. albus, whereby this bacterium produced a bacteriocin-like protein that prevented growth of the competitor (R. flavefaciens), the specific action of this protein being strain dependent (Chan and Dehority 1999). This mechanism was also reported in R. flavefaciens competing against F. succinogenes (Chen and Weimer 2001). Similarly, multiple strains of Butyrivibrio fibrisolvens produce bacteriocin-like proteins as a mechanism to compete between strains (Kalmokoff and Teather 1997). These interfering competitive mechanisms were also observed between kingdoms: the ruminococci R. albus and R. flavefaciens both inhibited fungal activity while competing for the cellulose resource (Stewart et al. 1992; Bernalier et al. 1993). Furthermore, numerous additional reports have indicated that fibrolytic bacteria do not appear to interact synergistically with fungi (Hobson and Stewart 2012). Similarly, ciliate protozoa and fungi grown in coculture demonstrated mainly inhibitory interactions (Hobson and Stewart 2012).
Fiber, species interactions and rumen microbial richness
The above-observed competitions show that while functional redundancy generally occurs in vivo in the rumen, a complex environment where countless bacterial interactions are in place, the functional redundancy is less possible in vitro, where the rumen ecosystem is minimalized to comprise only a few fiber-degraders that share the same functions. This suggests that rock–paper–scissor-type interactions may be in play in complex ecosystems like the rumen. In this model, despite competitive exclusion principles between pairs of competitors (when one microorganism competes for a mutual resource and excludes the other), the outcome of competition and species composition is embedded in competitive networks with multiple factors allowing species coexistence and an increase in richness (Allesina and Levine 2011). Therefore, we should ask whether the competitive interactions in vitro really reflect the interactions that occur in vivo in the presence of a large species diversity, where metabolite production can lead to an interfeeding mechanism. With the design of a more complex bacterial consortium, Chen and Weimer (2001) examined competition between R. flavefaciens, R. albus and F. succinogenes in the presence or absence of the non-cellulolytic bacteria S. ruminantium and Streptococcus bovis. In continuous culture (substrate-limiting conditions) on cellulose, the introduction of non-cellulolytic bacteria did not change the outcome of the culture in which R. albus dominated. However, in batch culture (substrate non-limiting conditions), introduction of the non-competitor changed the proportions of the three cellulolytic bacteria, in both cases (S. ruminantium or Streptococcus bovis) to allow more growth of R. flavefaciens at the expense of F. succinogenes. This suggests that the non-cellulolytic bacteria affected the competitive interaction between the cellulolytic species, thereby reinforcing the notion that complex interactions increase species diversity in this ecosystem.
Additional studies have contradicted the competition observed in in vitro studies. Defaunation experiments demonstrated a symbiotic relationship among protozoa, anaerobic fungi and cellulolytic bacteria. The absence of protozoa resulted in a decrease in cellulose hydrolysis in several studies (Dehority 2003), as well as decreases in the concentrations of anaerobic fungi and some cellulolytic bacteria (R. albus and R. flavefaciens, but not F. succinogenes) (Newbold et al. 2015). While it is well known that rumen protozoa prey on bacteria and that they cannot grow in axenic culture, as the bacteria provide them with essential nutrients, the benefits to bacteria have yet to be determined (Hobson and Stewart 2012). Other studies have revealed that a decrease in redox potential by yeast in the rumen environment results in more favorable conditions for the growth of strict anaerobic cellulolytic bacteria, which could stimulate their attachment to fiber particles (Roger, Fonty and Komisarczuk-Bony 1990; Jouany et al. 1999) and thus increase the initial rate of cellulolysis.
Niche partitioning could also explain both the reduced competition and the observed functional redundancy in vivo. This mechanism was observed using fluorescent in situ hybridization (FISH), both intraspecies among the F. succinogenes groups and interspecies between F. succinogenes and R. flavefaciens. In both cases, the bacteria were distributed in different parts of the plant, using orchard grass hay as a native cellulosic target substrate (Shinkai and Kobayashi 2007).
Other groups also studied the outcome of fiber succession, after pre-colonization of the fiber by selected species. A successional experiment in which F. succinogenes was first inoculated in hay stem before other members of the sheep rumen microbiome served to determine the microorganisms with which the cellulose degrader has beneficial/symbiotic and stable interactions (Shinkai, Ueki and Kobayashi 2010). An unexpected outcome of that study was the different microbial consortia obtained when different groups of F. succinogenes (1, 2 and 3) were used. Group 1 developed a stable consortium with Butyrivibrio fibrisolvens, Pseudobutyrivibrio ruminis, Clostridium sp., members of the group 2 F. succinogenes, P. ruminicola and unclassified Bacteroides. In group 2, Treponema bryantii, B. fibrisolvens, Acinetobacter sp. and Wolinella succinogenes were more abundant. In the group 3 consortium, the F. succinogenes strains from group 3 disappeared and were replaced by R. albus and F. succinogenes strains from group 1, which emerged from the rumen fluid. These results reflect the extraordinary diversity of interactions that take place in the rumen, where physiological changes in one population (e.g. F. succinogenes) give rise to extremely diverse microbial interactions.
TEMPORAL DYNAMICS OF FIBER DEGRADATION
Experiments in which the fiber biomass was placed in closed nylon bags directly in the rumen of cannulated animals and removed at different time points (in sacco) were used to study the colonization of different types of fiber biomass including perennial ryegrass (Huws et al. 2016), switchgrass (Piao et al. 2014), rice straw (Liu et al. 2016), alfalfa (Liu et al. 2016) and wheat straw (Jin et al. 2018). In all of these studies, the fiber in the bags was washed and only the fiber-adherent microbial population was studied (via DNA extraction and 16S rRNA sequencing). The time frame for those experiments was generally between 0.5 h and 72 h, corresponding to the time feed can remain in the rumen. In each of those studies, following three phases were observed: (i) sharp degradation of about 10% of the fiber within 0.5–1 h of incubation, (ii) a latent phase with no noticeable changes in fiber mass loss between 1 h and 4–6 h and (iii) continuous degradation of the fiber between 4–6 h and 72 h (Fig. 4).
Figure 4.
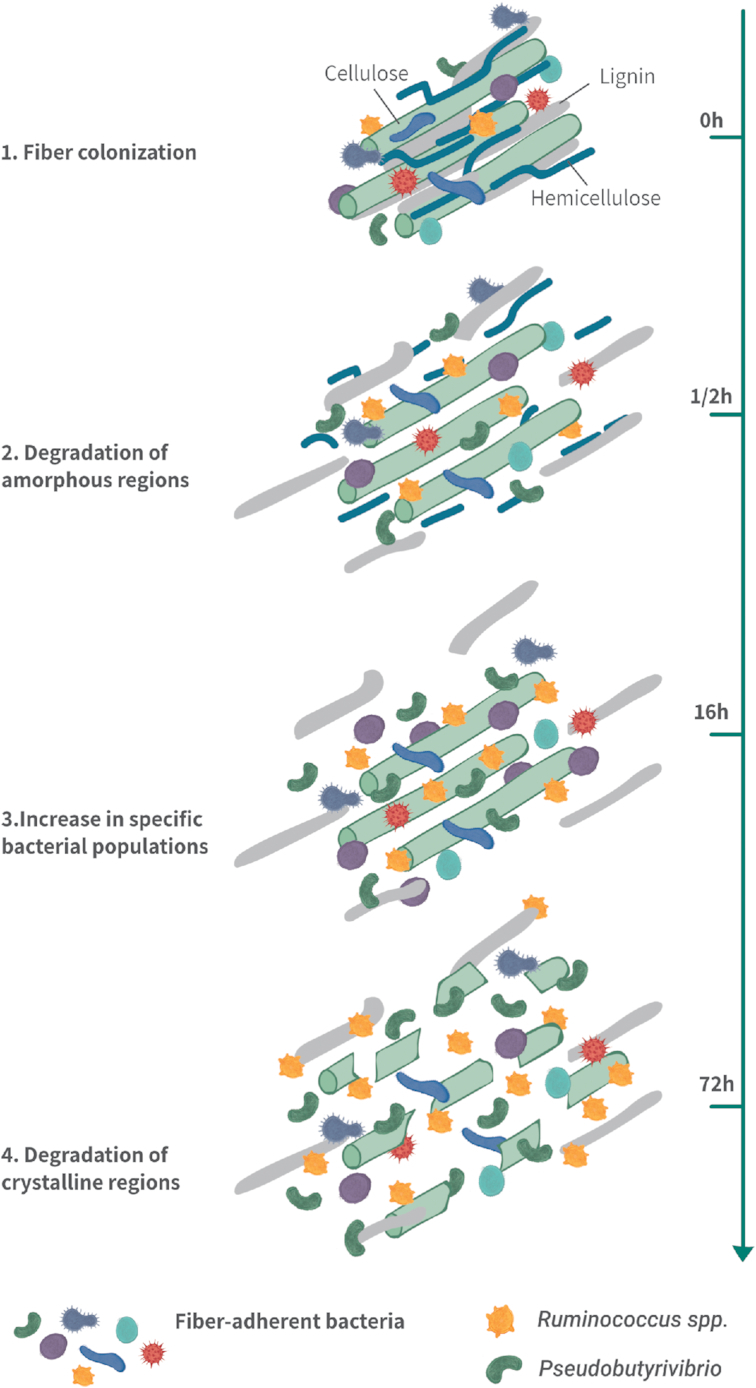
Four phases in fiber colonization and degradation as revealed by in sacco experiments in cannulated cows. (1.)Fiber colonization. (2.)Degradation of amorphous regions of the fiber. (3.)Increase in specific bacterial population (latent phase). (4.)Degradation of crystalline regions.
The two degradation phases might reflect, respectively, the first microbial degradation of accessible amorphous regions of the fiber, which is performed rapidly, and the slower degradation of intricate crystalline regions of the fiber. It could be hypothesized that the latent phase reflects a microbial shift between degradation of the amorphous and complex regions of the fiber, which may be performed by distinct microorganisms. However, it seems that the overall microbial structure is stable within a short period of incubation (0.5–1 h) and changes occur mostly in abundance among the different bacterial taxa. Indeed, it has been suggested that the two phases of degradation correspond to two phases of colonization, where changes in bacterial abundance (during the latent phase) lead to the second phase of degradation. Huws et al. (2016) observed that Pseudobutyrivibrio, Roseburia and Ruminococcus spp. are less abundant during primary colonization events than in the second phase of degradation. Similarly, Piao et al. (2014) also saw increases in Pseudobutyrivibrio and Ruminococcus spp. during the second degradation phase. These studies suggest that fiber degradation is subject to community dynamics processes that might be driven by niche partitioning and microbial interactions, such as the hydrogen transfer mentioned earlier.
Support for this premise was apparent from studies that also revealed that the fiber-adherent bacteria that colonized the fiber were not all fiber degraders. It was observed that, on switchgrass (Piao et al. 2014), the presumed fiber degraders from the genera Prevotella, Fibrobacter, Ruminococcus, Butyrivibrio, Bacteroides, Ruminobacter, Treponema, Selenomonas and Clostridium colonized the fiber very rapidly after 30 min of exposure and reached maximal growth at around 16 h. Conjointly, additional genera (with less or without fiber-degradation potential) increased during the same timeframe. These include Anaerostipes, Coprococcus, Oscillospira, Succiniclasticum, Desulfovibrio and members of the families Anaerolinaceae and Anaeroplasmataceae (Piao et al. 2014). These results would suggest that these taxa benefit from, or interact with the fiber degraders and have thus developed and maintained the potential to tightly adhere to the fiber. This is in accordance with early studies that demonstrated that a large fraction (around 70%) of the bacterial cells in the rumen are attached to plant fiber (Forsberg and Lam 1977).
Nonetheless, fiber degraders represented the dominant population in the bacterial community attached to fiber. On wheat straw, Prevotella, Succiniclasticum, Butyrivibrio and Ruminococcus were dominant (Jin et al. 2018). On rice straw and alfafa, Prevotella, Butyrivibrio, Ruminococcus, Fibrobacter and Treponema spp. dominated the microbial community attached to the fiber (Liu et al. 2016). On switchgrass, high abundances of Prevotella, Butyrivibrio, Ruminococcus and Fibrobacter were reported (Piao et al. 2014). Abundant Butyrivibrio, Fibrobacter, Olsenella and Prevotella were observed in the bacterial community colonizing fresh perennial ryegrass. Independent of fiber type, Prevotella, Butyrivibrio and Ruminococcus were consistently dominant, indicating that these taxa have the potential to express a broader enzymatic repertoire and adapt to the available fiber.
In addition, it should be noted that by using only the 16S rRNA gene for taxon identification, these studies ignored the potential of fungal and protozoal colonizers and how they could impact both the degradation profile of the biomass and interact with the adherent bacterial populations.
FIBER DEGRADERS AT EARLY LIFE OF THE HOST ANIMAL
It was demonstrated in lambs that the establishment of ciliate protozoa takes place by animal contact at a very early age of the animal, between days 9 and 21 (Margaret Eadie 1962). The colonization of anaerobic fungi appeared to be linked to the introduction of solid food and therefore, fiber. However, it was demonstrated that the appearance of the fibrolytic bacterial population occurs at a very early age in the young rumen. Therefore, the establishment of fiber degraders is not dependent on the feeding of the animals, since at such an early age, the rumen has no functional activity (the animals commence eating fiber biomass at around 8 weeks of age).
In an early study, a group of 24 lambs were studied from birth for microbial colonization. The animals' rumens were sampled by stomach tubes daily and microbial isolation was performed on selective media. It appeared that cellulolytic bacteria were established at day 4 of age in most of the animals, and until day 7 in all animal groups (Fonty et al. 1987). Microscopic analyses revealed that the Ruminococcus genus was the most represented genus. Methanogenic bacteria were established conjointly from day 2 to the end of the first week in all animals (also reported by Morvan et al. 1994). Anaerobic fungi appeared later on, from day 8 to day 20 in all animals and microscopic analyses revealed a morphological resemblance to Neocallimastix frontalis. In this study, animals were also divided into several groups to test the effects of the feed, maternal contact and isolation of the animal host on microbial colonization. It appeared that cellulolytic bacteria could colonize the rumen of animals fed only with commercial milk replacement (no solid feed) and that total isolation of the animal did not prevent the colonization of cellulolytic bacteria. It could be argued, though, that isolation of the animal occurred 24 h after birth, which is long enough for microbial transmission to proceed from the mother to her newborn.
In a later study, four calves were examined from 1 day of age, after separation from their mothers, and the rumen contents were sampled via stomach tubes until 10 weeks age (Minato et al. 1992). Likewise, microbial populations were plated and enumerated on selective media. Amylolytic bacteria were present from day 1 at high number and increased to day 3, at which time they reached a stable number. Similarly, xylan and pectin fermenters were detected as soon as 1 day of life and their numbers stabilized at 3 days. Cellulolytic bacteria were isolated from day 3 and increased until the third week of life, while methanogens appeared at 1 week and increased gradually until 8 weeks.
The microbial composition of the bovine rumen was explored using more recent microbial ecology techniques, by examining the overall composition of the rumen microbiome using 16S rRNA amplicon sequencing, thus allowing phylogenetic identification by Jami et coworkers (Jami et al. 2013). In this study, different animals of four age groups were studied: 1–3-days old, 2-months old, 6-months old and 2-years old. Samples were taken via stomach tubes, DNA was extracted and the 16S rRNA was amplified and sequenced. Firmicutes and Bacteroidetes (i.e. phyla containing fiber degraders) were present in all four age groups in varying proportions. The genera Prevotella and Ruminococcus were detected very early in 3-day-old calves. To further strengthen the analysis, real-time PCR with specific primers was performed. This revealed the appearance of R. flavefaciens at 1 day of life, followed by R. albus in 3-day-old calves. F. succinogenes could only be detected in samples from animals of the 2-months-old group. These results were in accordance with those obtained in lambs by Fonty and colleagues (Fonty et al. 1987). Concerning hemicellulose degraders, P. ruminicola was present in increasing concentrations from day 1, while P. bryantii and P. brevis were detected only in the 2-month-old group. The starch degrader S. bovis, was found in relatively high abundance in samples from 1- and 3-day-old calves. This study (Jami et al. 2013) also revealed that the microbiome structure (composition and abundance) stabilizes between 2 and 6 months of age, independent of animal diet.
Another study examined microbial colonization during the first hours of life, in 12 calves (Guzman et al. 2015). In that study, the animals were euthanized 20 min, 24 h, 48 h or 72 h after birth, in order to surgically collect the contents of the different parts of their digestive tracts. The 20-min-old calves were not exposed to food, and the others were separated from their mothers immediately after birth and fed with colostrum using feeders. After DNA extraction, real-time PCR was performed with specific primers for methanogenic and fibrolytic populations. The four methanogens tested were present in all age groups and in all samples tested from different anatomic locations (rumen fluid, tissues, abomasum fluid, cecum fluid, tissues and feces of calves) but with differences in abundance. Of the fibrolytic populations tested, R. flavefaciens, F. succinogenes and P. ruminicola were present at similar concentrations in all age groups and all anatomic locations, which would suggest that they were metabolically active.
An additional study revealed the presence of Ruminococcaceae and Prevotella in additional stomach compartments of goats (omasum, abomasum and reticulum) at as early as day 3 of life (Lei et al. 2018).
A timeline of rumen colonization is presented in Fig. 5. Although the rumen is considered to be functionally inactive in the early days after birth, these studies show that the animal host somehow ensures its inoculation by the microbes that carry the most essential functions for rumen functioning and its consequent ability to digest fiber. In particular are the bacterial fibrolytic populations and the methanogenic ones that might be transmitted at birth from the animal mother via skin, birth canal or saliva, as has been demonstrated in humans (Dominguez-Bello et al. 2010). These potential mechanisms ensure that a very conserved functionality of these populations is transmitted along the animal lineages.
Figure 5.

Timescale of rumen colonization by fiber degraders and methanogens from birth to 2 months of age. Different publications are color-coded, animal types are represented by different line types (goats, normal; calves, dashed; lambs, bold) and the suggested mode of acquisition by F, C and T (see Figure captions).
MICROBIOME MANIPULATIONS FOR INCREASED FIBER DEGRADATION
Scientists have sought to improve rumen functioning in the last few decades by increasing fiber degradation in the rumen, but their attempts have been unsuccessful. Ruminal manipulations have been attempted by using feed additives such as chemical agents (Chalupa 1977) or enzymes (Beauchemin et al. 2003). The use of live bacteria as probiotics has been the focus of more recent ongoing research (Weimer 2015).
Initially, strains were introduced in a single dose and the outcome of the colonization was followed rapidly after inoculation (Flint, Bisset and Webb 1989). Since it was found that the foreign bacteria decrease very sharply during the days following their inoculation, a second strategy consisted of repeated dosing (Krause et al. 2001; Chiquette et al. 2007). In the latter case, concentrations of the fibrolytic bacteria (Ruminococcus sp. and/or F. succinogenes) also declined at a rapid rate post-dosing.
In all of these studies, the probiotic dosing effect on fiber digestibility was either absent or transitory; the dosing had a transient impact on microbiome structure with a rapid return to the initial state. In some of those studies, several fibrolytic populations (Ruminococcus, Fibrobacter, eukaryotes, anaerobic rumen fungi and protozoa), apart from the probiotic itself, increased in response to the dosing (Krause et al. 1999b). Additional protocols were also tested, such as increasing the fiber content of the feed-to-concentrate forage ratio (Chiquette et al. 2007), probiotic dosing after starvation (Præsteng et al. 2013), or replacing the entire content of the rumen with the rumen content from an animal with higher fiber-degrading abilities (Weimer et al. 2017; Zhou et al. 2018).
Probiotic dosing after a period of starvation was employed to enrich reindeer rumen microbiome with key fibrolytic species, such as R. flavefaciens, in an attempt to increase both plant fiber hydrolysis and animal welfare (Præsteng et al. 2013). As for Krause et al. (1999a), changes in microbiome structure were observed following the dosing, with an enrichment of Prevotella and a decline in Bacteroidetes populations; however, the probiotic itself was not maintained in the rumen environment. In many of those studies, increases in the eukaryotic population during dosing were noted (Krause et al. 1999b, 2001), suggesting predation of fiber-degrading bacteria by protozoa.
Whereas some studies reported establishment of the probiotic bacteria, it could be argued that the sampling after dosing was perfomed too early, and that subsequent sampling would have revealed a return to the initial microbiome state.
Anaerobic rumen fungi were also supplemented to ruminant animals with higher success for better utilization of fibrous feeds in terms of increased feed intake, but the concentration of the supplemented fungi after supplementation was not measured in that study (Puniya et al. 2015),
More extreme strategies to change microbiome composition have been employed in more recent attempts to modulate host energetic efficiency by transfusion of rumen microbiomes from efficient to inefficient animals (Weimer et al. 2017; Zhou et al. 2018). The replacement of 95% of the rumen content in four animals led to a transient effect on microbiome structure and function with a return to the initial condition after only 10 days (Weimer et al. 2017). These findings suggest that the composition of the rumen microbiome is highly resilient to changes and that its adaptation to its host is so strong that the initial microbial community in the rumen has the capacity to reestablish itself, even from a very low concentration. In a second study, in which intense washing of the rumen was applied before transfusion in an attempt to avoid persistence of the initial rumen composition, the authors reported high individual variability of microbiome composition 28 days post-transfusion (Zhou et al. 2018). Altogether, these reports highlight the resilience of the ruminal microbial community to perturbations and its host specificity, each host individual coevolving with its microbiome in a very specific manner.
Some encouraging results were nevertheless obtained when the probiotic bacteria were targeted to a free ecological niche. Indeed, a genetically modified Butyrivibrio fibrisolvens, able to break down fluoroacetate, could be established in sheep consuming fluoroacetate-containing plant material and protected the animal from poisoning (Gregg et al. 1998). In addition, resistance to the toxic amino acid mimosine could be conferred to sensitive goats by transferring rumen contents from resistant goats (Hammond 1995). Aside from a free ecological niche, another condition for easier establishment would be the microbial complexity of the rumen ecosystem, as it was shown that a diverse and high microbial diversity promote higher probiotic establishment (Fonty et al. 1983, 1988).
Modulation of the human gut microbiome has also proven very difficult, if not impossible (Walter, Maldonado-Gómez and Martínez 2018). A mechanism for engraftment has been proposed, in which the characteristics of the microbial invader and both microbiome- and host-related mechanisms are interlinked and would play a determinant role in invasion success. Applications of these principles to the rumen gut environment could favor superior outcomes of rumen manipulations.
As discussed earlier, the rumen microbial community is established and assembled very early in life (Jami et al. 2013; Guzman et al. 2015), and interventions to program and control the microbial composition and its function may be required just following birth (Yáñez-Ruiz et al. 2015). While it is possible to avoid establishments of protozoa (Yáñez-Ruiz et al. 2015) and anaerobic fungi (Puniya et al. 2015) in the young rumen, controlling the bacterial population is less controlled. Nevertheless, some effort focused on reducing methanogenesis by applying changes in early life resulted in positive outcomes (Abecia et al. 2013, 2014), and this could be a path toward increasing fiber-degrading efficiency.
CONCLUDING REMARKS
Rumen ecosystem functioning is centered around the degradation of plant fiber by a complex microbial community. This functionality enables ruminant animals to digest their feed, thereby underscoring its critical importance to humans needs, as it is the major contributor to the conversion of plant biomass into fundamental products (milk, meat and fiber products). A great majority of the microorganisms that inhabit the rumen have the genetic potential to participate in the fiber-degradation process, and they present an extremely broad diversity—both in their enzymatic repertoire and mechanisms for fiber deconstruction, which suggests complementarity at the functional level. The conversion of plant biomass to valuable products for the animal is also a result of the concerted action of the members of the fiber-degrading microbial community (via their enzymes), which interact intimately with other associated microbes that do not degrade fiber per se, but serve to support the fiber-degrading function. This interaction seems to be mainly based on the metabolic exchange that facilitates ecosystem functioning. Microbiome research is now focused on deconvoluting the mechanisms that promote rumen ecosystem function, and the mapping of these metabolic exchanges is essential for elucidating the role of each of the microorganisms that contributes in the fiber-degrading process. Furthermore, since deep knowledge of microbial interactions could be the key to successful microbial manipulations and perfected animal–microbial function, effort should focus on developing techniques to study microbial interactions in vivo and using appropriate tools to examine communities with high complexity in vitro.
ACKNOWLEDGEMENTS
The authors are particularly grateful to Prof. Edward A. Bayer (The Weizmann Institute of Science) for his critical review of the manuscript and to Dr Einat Nativ-Roth (Ben Gurion University of the Negev) for her expertise in SEM microscopy.
Contributor Information
Sarah Moraïs, Department of Life Sciences and the National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Sderot Ben Gurion 1, Beer-Sheva 8499000, Israel.
Itzhak Mizrahi, Department of Life Sciences and the National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Sderot Ben Gurion 1, Beer-Sheva 8499000, Israel.
FUNDING
The authors appreciate the support of the Israel Science Foundation (grant no. 1313/13), the Chief Scientist of the Israeli Ministry of Agriculture and Rural Development Fund (grant no. 362-0426), the Chief Scientist of the Israeli Ministry of Science Foundation (grant no.3-10880), the Israel Dairy Board Foundation Projects (grant nos. 362-0300 and 362-0524), the European Research Council under the European Union's Horizon 2020 research and innovation program (grant no. 640384) and the European Union Horizon 2020 contract: sustainable production of next generation biofuels from waste streams: Waste2Fuels.
Conflict of interest. None declared.
REFERENCES
- Abecia L, Martín-García AI, Martínez Get al.. Nutritional intervention in early life to manipulate rumen microbial colonization and methane output by kid goats postweaning. J Anim Sci. 2013;91:4832–40. [DOI] [PubMed] [Google Scholar]
- Abecia L, Waddams KE, Martínez-Fernandez Get al.. An antimethanogenic nutritional intervention in early life of ruminants modifies ruminal colonization by Archaea. Archaea. 2014;2014:841463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abot A, Arnal G, Auer Let al.. CAZyChip: dynamic assessment of exploration of glycoside hydrolases in microbial ecosystems. BMC Genomics. 2016;17:671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin DE, Borneman WS. Role of rumen fungi in fiber degradation. J Dairy Sci. 1990;73:3023–32. [DOI] [PubMed] [Google Scholar]
- Akin DE, Lyon CE, Windham WRet al.. Physical degradation of lignified stem tissues by ruminal fungi. Appl Environ Microbiol. 1989;55:611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allesina S, Levine JM.. A competitive network theory of species diversity. Proc Natl Acad Sci USA. 2011;108:5638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arntzen MØ, Várnai A, Mackie RIet al.. Outer membrane vesicles from Fibrobacter succinogenes S85 contain an array of carbohydrate-active enzymes with versatile polysaccharide-degrading capacity. Environ Microbiol. 2017;19:2701–14. [DOI] [PubMed] [Google Scholar]
- Artzi L, Bayer EA, Moraïs S. Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat Rev Microbiol. 2017;15:83–95. [DOI] [PubMed] [Google Scholar]
- Avgustin G, Flint HJ, Whitehead TR. Distribution of xylanase genes and enzymes among strains of Prevotella (Bacteroides) ruminicola from the rumen. FEMS Microbiol Lett. 1992;78:137–43. [DOI] [PubMed] [Google Scholar]
- Bayer EA, Chanzy H, Lamed Ret al.. Cellulose, cellulases and cellulosomes. Curr Opin Struct Biol. 1998;8:548–57. [DOI] [PubMed] [Google Scholar]
- Bayer EA, Shoham Y, Lamed R. Lignocellulose-decomposing bacteria and their enzyme systems. In: Rosenberg E, DeLong EF, Lory Set al. (eds). The Prokaryotes: Prokaryotic Physiology and Biochemistry. Berlin, Heidelberg:Springer Berlin Heidelberg, 2013, 215–66. [Google Scholar]
- Beauchemin KA, Colombatto D, Morgavi DPet al.. Use of exogenous fibrolytic enzymes to improve feed utilization by ruminants. J Anim Sci. 2003;81:E37–47. [Google Scholar]
- Bensoussan L, Moraïs S, Dassa Bet al.. Broad phylogeny and functionality of cellulosomal components in the bovine rumen microbiome. Environ Microbiol. 2017;19:185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlemont R, Martiny AC. Phylogenetic distribution of potential cellulases in bacteria. Appl Environ Microbiol. 2013;79:1545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernalier A, Fonty G, Bonnemoy Fet al.. Inhibition of the cellulolytic activity of Neocallimastix frontalis by Ruminococcus flavefaciens. J Gen Microbiol. 1993;139:873–80. [DOI] [PubMed] [Google Scholar]
- Bourne Y, Henrissat B. Glycoside hydrolases and glycosyltransferases: families and functional modules. Curr Opin Struct Biol. 2001;11:593–600. [DOI] [PubMed] [Google Scholar]
- Brulc JM, Yeoman CJ, Wilson MKet al.. Cellulosomics, a gene-centric approach to investigating the intraspecific diversity and adaptation of Ruminococcus flavefaciens within the rumen. PLoS One. 2011;6:e25329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant MP, Wolin MJ. Rumen bacteria and their metabolic interactions. Proc Intersect Congr Int Assoc Microbiol Soc. 1975;2:297-306. [Google Scholar]
- Burnet MC, Dohnalkova AC, Neumann APet al.. Evaluating models of cellulose degradation by Fibrobacter succinogenes S85. PLoS One. 2015;10:e0143809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazier EA, Trably E, Steyer JPet al.. Biomass hydrolysis inhibition at high hydrogen partial pressure in solid-state anaerobic digestion. Bioresour Technol. 2015;190:106–13. [DOI] [PubMed] [Google Scholar]
- Celiker H, Gore J. Competition between species can stabilize public-goods cooperation within a species. Mol Syst Biol. 2012;8:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalupa W. Manipulating rumen fermentation. J Anim Sci. 1977;45:585–99. [Google Scholar]
- Chan WW, Dehority BA. Production of Ruminococcus flavefaciensgrowth inhibitor(s) by Ruminococcus albus. Anim Feed Sci Technol. 1999;77:61–71. [Google Scholar]
- Chen J, Weimer P. Competition among three predominant ruminal cellulolytic bacteria in the absence or presence of non-cellulolytic bacteria. Microbiology. 2001;147:21–30. [DOI] [PubMed] [Google Scholar]
- Chiquette J, Talbot G, Markwell Fet al.. Repeated ruminal dosing of Ruminococcus flavefaciensNJ along with a probiotic mixture in forage or concentrate-fed dairy cows: effect on ruminal fermentation, cellulolytic populations and in sacco digestibility. Can J Anim Sci. 2007;87:237–49. [Google Scholar]
- Comtet-Marre S, Chaucheyras-Durand F, Bouzid Oet al.. FibroChip, a functional DNA microarray to monitor cellulolytic and hemicellulolytic activities of rumen microbiota. Front Microbiol. 2018;9:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Tian Y, Li Jet al.. Metatranscriptomic analyses of plant cell wall polysaccharide degradation by microorganisms in the cow rumen. Appl Environ Microbiol. 2015;81:1375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassa B, Borovok I, Ruimy-Israeli Vet al.. Rumen cellulosomics: divergent fiber-degrading strategies revealed by comparative genome-wide analysis of six ruminococcal strains. PLoS One. 2014;9:e99221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehority BA. Rumen Microbiology. Nottingham University Press: Nottingham, 2003. [Google Scholar]
- Devillard E, Bera-Maillet C, Flint HJet al.. Characterization of XYN10B, a modular xylanase from the ruminal protozoan Polyplastron multivesiculatum, with a family 22 carbohydrate-binding module that binds to cellulose. Biochem J. 2003;373:495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devillard E, Newbold CJ, Scott KP. A xylanase produced by the rumen anaerobic protozoan Polyplastron multivesiculatumshows close sequence similarity to family 11 xylanases from Gram-positive. FEMS microbiology letters. 1999;181:145-52. [DOI] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras Met al.. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA. 2010;107:11971–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson EL, Weimer PJ. Fermentation of model hemicelluloses by Prevotella strains and Butyrivibrio fibrisolvensin pure culture and in ruminal enrichment cultures. Appl Microbiol Biotechnol. 2017;101:4269–78. [DOI] [PubMed] [Google Scholar]
- Ezer A, Matalon E, Jindou Set al.. Cell surface enzyme attachment is mediated by family 37 carbohydrate-binding modules, unique to Ruminococcus albus. J Bacteriol. 2008;190:8220–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanutti C, Ponyi T, Black GWet al.. The conserved noncatalytic 40-residue sequence in cellulases and hemicellulases from anaerobic fungi functions as a protein docking domain. J Biol Chem. 1995;270:29314–22. [DOI] [PubMed] [Google Scholar]
- Findley SD, Mormile MR, Sommer-Hurley Aet al.. Activity-based metagenomic screening and biochemical characterization of bovine ruminal protozoan glycoside hydrolases. Appl Environ Microbiol. 2011;77:8106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegerova K, Kaerger K, Kirk Pet al.. Rumen fungi. In: Puniya AK, Singh R, Kamra DN(eds). Rumen Microbiology: From Evolution to Revolution. New Delhi: Springer India, 2015, 97–112. [Google Scholar]
- Flint HJ, Bayer EA, Rincon MTet al.. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6:121–31. [DOI] [PubMed] [Google Scholar]
- Flint HJ, Bisset J, Webb J. Use of antibiotic resistance mutations to track strains of obligately anaerobic bacteria introduced into the rumen of sheep. J Appl Bacteriol. 1989;67:177–83. [DOI] [PubMed] [Google Scholar]
- Flint HJ. The rumen microbial ecosystem–some recent developments. Trends Microbiol. 1997;5:483–8. [DOI] [PubMed] [Google Scholar]
- Fonty G, Gouet P, Jouany J-Pet al.. Establishment of the Microflora and anaerobic fungi in the rumen of lambs. Microbiology. 1987;133:1835–43. [Google Scholar]
- Fonty G, Gouet P, Jouany JPet al.. Ecological factors determining establishment of cellulolytic bacteria and protozoa in the rumens of meroxenic lambs. J Gen Microbiol. 1983;129:213–23. [DOI] [PubMed] [Google Scholar]
- Fonty G, Gouet P, Ratefiarivelo Het al.. Establishment of bacteroides succinogenes and measurement of the main digestive parameters in the rumen of gnotoxenic lambs. Can J Microbiol. 1988;34:938–46. [DOI] [PubMed] [Google Scholar]
- Forsberg CW, Lam K. Use of adenosine 5′-triphosphate as an indicator of the microbiota biomass in rumen contents. Appl Environ Microbiol. 1977;33:528-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuma NM, Koike S, Kobayashi Y. Monitoring of gene expression in Fibrobacter succinogenes S85 under the co-culture with non-fibrolytic ruminal bacteria. Arch Microbiol. 2015;197:269–76. [DOI] [PubMed] [Google Scholar]
- Gong J, Forsberg CW. Separation of outer and cytoplasmic membranes of Fibrobacter succinogenes and membrane and glycogen granule locations of glycanases and cellobiase. J Bacteriol. 1993;175:6810–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg K, Hamdorf B, Henderson Ket al.. Genetically modified ruminal bacteria protect sheep from fluoroacetate poisoning. Appl Environ Microbiol. 1998;64:3496–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg IR, Yin G, Borovok Iet al.. Functional phylotyping approach for assessing intraspecific diversity ofRuminococcus albus within the rumen microbiome. FEMS Microbiol Lett. 2015;362:1–10. [DOI] [PubMed] [Google Scholar]
- Groleau D, Forsberg CW. Cellulolytic activity of the rumen bacterium Bacteroides succinogenes. Can J Microbiol. 1981;27:517–30. [DOI] [PubMed] [Google Scholar]
- Guzman CE, Bereza-Malcolm LT, De Groef Bet al.. Presence of selected methanogens, Fibrolytic bacteria, and proteobacteria in the gastrointestinal tract of neonatal dairy calves from birth to 72 hours. PLoS One. 2015;10:e0133048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haitjema CH, Gilmore SP, Henske JKet al.. A parts list for fungal cellulosomes revealed by comparative genomics. Nat Microbiol. 2017;2:17087. [DOI] [PubMed] [Google Scholar]
- Hammond AC. Leucaena toxicosis and its control in ruminants. J Anim Sci. 1995;73:1487–92. [DOI] [PubMed] [Google Scholar]
- Henderson G, Cox F, Ganesh Set al.. Erratum: rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2016;6:19175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson G, Cox F, Ganesh Set al.. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015a;5:14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson G, Cox F, Ganesh Set al.. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015b;5:14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess M, Sczyrba A, Egan Ret al.. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–7. [DOI] [PubMed] [Google Scholar]
- Himmel ME, Xu Q, Luo Yet al.. Microbial enzyme systems for biomass conversion: emerging paradigms. Biofuels. 2010;1:323–41. [Google Scholar]
- Hobson PN, Stewart CS. The Rumen Microbial Ecosystem. Netherlands: Springer Science & Business Media, 1997. [Google Scholar]
- Huws SA, Edwards JE, Creevey CJet al.. Temporal dynamics of the metabolically active rumen bacteria colonizing fresh perennial ryegrass. FEMS Microbiol Ecol. 2016;92, DOI: 10.1093/femsec/fiv137. [DOI] [PubMed] [Google Scholar]
- Israeli-Ruimy V, Bule P, Jindou Set al.. Complexity of the Ruminococcus flavefaciens FD-1 cellulosome reflects an expansion of family-related protein-protein interactions. Sci Rep. 2017;7:42355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jami E, Israel A, Kotser Aet al.. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013;7:1069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jami E, Mizrahi I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS One. 2012;7:e33306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindou S, Brulc JM, Levy-Assaraf Met al.. Cellulosome gene cluster analysis for gauging the diversity of the ruminal cellulolytic bacterium Ruminococcus flavefaciens. FEMS Microbiol Lett. 2008;285:188–94. [DOI] [PubMed] [Google Scholar]
- Jin W, Wang Y, Li Yet al.. Temporal changes of the bacterial community colonizing wheat straw in the cow rumen. Anaerobe. 2018;50:1–8. [DOI] [PubMed] [Google Scholar]
- Jouany J.P., Mathieu F., Senaud J.et al.. Effects of Saccharomyces cervisiae and Aspergillus orizae on the population of rumen microbes and their polysaccharidase activities. S Afr J Anim Sci. 1999;29:6–7. [Google Scholar]
- Kalmokoff ML, Teather RM. Isolation and characterization of a bacteriocin (Butyrivibriocin AR10) from the ruminal anaerobe Butyrivibrio fibrisolvens AR10: evidence in support of the widespread occurrence of bacteriocin-like activity among ruminal isolates of B. fibrisolvens. Appl Environ Microbiol. 1997;63:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameshwar AKS, Qin W. Genome wide analysis reveals the extrinsic cellulolytic and biohydrogen generating abilities of neocallimastigomycota fungi. J Genomics. 2018;6:74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevenhusen F, Petri RM, Kleefisch M-Tet al.. Changes in fibre-adherent and fluid-associated microbial communities and fermentation profiles in the rumen of cattle fed diets differing in hay quality and concentrate amount. FEMS Microbiol Ecol. 2017;93, DOI: 10.1093/femsec/fix100. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Shinkai T, Koike S. Ecological and physiological characterization shows that Fibrobacter succinogenes is important in rumen fiber digestion – review. Folia Microbiol. 2008;53:195–200. [DOI] [PubMed] [Google Scholar]
- Koike S, Pan J, Suzuki Tet al.. Ruminal distribution of the cellulolytic bacterium Fibrobacter succinogenes in relation to its phylogenetic grouping. Anim Sci J. 2004;75:417–22. [Google Scholar]
- Krause DO, Bunch RJ, Conlan LLet al.. Repeated ruminal dosing of Ruminococcus spp. does not result in persistence, but changes in other microbial populations occur that can be measured with quantitative 16S-rRNA-based probes. Microbiology. 2001;147:1719–29. [DOI] [PubMed] [Google Scholar]
- Krause DO, Bunch RJ, Smith WJMet al.. Diversity of Ruminococcus strains: a survey of genetic polymorphisms and plant digestibility. J Appl Microbiol. 1999a;86:487–95. [Google Scholar]
- Krause DO, Smith WJM, Ryan FMEet al.. Use of 16S-rRNA based techniques to investigate the ecological succession of microbial populations in the immature lamb rumen: tracking of a specific strain of inoculated Ruminococcus and interactions with other microbial populations in vivo. Microb Ecol. 1999b;38:365–76. [DOI] [PubMed] [Google Scholar]
- Lamed R, Setter E, Kenig Ret al.. The cellulosome—a discrete cell surface organelle of Clostridium thermocellum which exhibits separate antigenic, cellulose-binding and various cellulolytic. Biotechnol Bioeng Symp. 1983;13:163-81. [Google Scholar]
- Latham MJ, Wolin MJ. Fermentation of cellulose by Ruminococcus flavefaciens in the presence and absence of Methanobacterium ruminantium. Appl Environ Microbiol. 1977;34:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy SC, Kelly WJ, Altermann Eet al.. The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One. 2010;5:e8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Ha JK, Cheng K. Relative contributions of bacteria, protozoa, and fungi to in vitro degradation of orchard grass cell walls and their interactions. Appl Environ Microbiol. 2000;66:3807–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Zhang K, Guo Met al.. Exploring the spatial-temporal microbiota of compound stomachs in a pre-weaned goat model. Front Microbiol. 2018;9:1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy B, Jami E. Exploring the prokaryotic community associated with the rumen ciliate protozoa population. Front Microbiol. 2018;9:2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang M, Xue Cet al.. Characterization and comparison of the temporal dynamics of ruminal bacterial microbiota colonizing rice straw and alfalfa hay within ruminants. J Dairy Sci. 2016;99:9668–81. [DOI] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula Eet al.. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margaret Eadie J. The development of rumen microbial populations in lambs and calves under various conditions of management. Microbiology. 1962;29:563–78. [Google Scholar]
- Marvin-Sikkema FD, Richardson AJ, Stewart CSet al.. Influence of hydrogen-consuming bacteria on cellulose degradation by anaerobic fungi. Appl Environ Microbiol. 1990;56:3793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matte A, Forsberg CW, Gibbins AMV. Enzymes associated with metabolism of xylose and other pentoses by Prevotella(Bacteroides)ruminicola strains, Selenomonas ruminantiumD, and Fibrobacter succinogenes S85. Can J Microbiol. 1992;38:370–6. [DOI] [PubMed] [Google Scholar]
- Minato H, Otsuka M, Shirasaka Set al.. Colonization of microorganisms in the rumen of young calves. J Gen Appl Microbiol. 1992;38:447–56. [Google Scholar]
- Miura H, Horiguchi M, Matsumoto T. Nutritional interdependence among rumen bacteria, Bacteroides amylophilus, Megasphaera elsdenii, and Ruminococcus albus. Appl Environ Microbiol. 1980;40:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi I. Rumen symbioses. In: Rosenberg E, DeLong EF, Lory Set al.(eds). The Prokaryotes: Prokaryotic Biology and Symbiotic Associations. Berlin, Heidelberg: Springer Berlin Heidelberg, 2013, 533–44. [Google Scholar]
- Morgavi DP, Martin C, Jouany J-Pet al.. Rumen protozoa and methanogenesis: not a simple cause–effect relationship. Br J Nutr. 2012;107:388–97. [DOI] [PubMed] [Google Scholar]
- Morvan B, Bonnemoy F, Fonty Get al.. Quantitative determination of H2-utilizing acetogenic and sulfate-reducing bacteria and methanogenic archaea from digestive tract of different mammals. Curr Microbiol. 1996a;32:129–33. [DOI] [PubMed] [Google Scholar]
- Morvan B, Dore J, Rieu-Lesme Fet al.. Establishment of hydrogen-utilizing bacteria in the rumen of the newborn lamb. FEMS Microbiol Lett. 1994;117:249–56. [DOI] [PubMed] [Google Scholar]
- Morvan B, Rieu-Lesme F, Fonty Get al.. In vitroInteractions between rumen H2-producing cellulolytic microorganisms and H2-utilizing acetogenic and sulfate-reducing bacteria. Anaerobe. 1996b;2:175–80. [Google Scholar]
- Mosoni P, Fonty G, Gouet P. Competition between ruminal cellulolytic bacteria for adhesion to cellulose. Curr Microbiol. 1997;35:44–7. [DOI] [PubMed] [Google Scholar]
- Mountfort DO, Asher RA, Bauchop T. Fermentation of cellulose to methane and carbon dioxide by a rumen anaerobic fungus in a triculture with Methanobrevibacter sp. strain RA1 and Methanosarcina. Appl Environ Microbiol. 1982;44:128-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhya I, Moraïs S, Laverde-Gomez Jet al.. Sporulation capability and amylosome conservation among diverse human colonic and rumen isolates of the keystone starch-degrader Ruminococcus bromii. Environ Microbiol. 2018;20:324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naas AE, Mackenzie AK, Mravec Jet al.. Do rumen Bacteroidetes utilize an alternative mechanism for cellulose degradation?. MBio. 2014;5:e01401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold CJ, de la Fuente G, Belanche Aet al.. The role of ciliate protozoa in the rumen. Front Microbiol. 2015;6:1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng F, Kittelmann S, Patchett MLet al.. An adhesin from hydrogen-utilizing rumen methanogen Methanobrevibacter ruminantium M 1 binds a broad range of hydrogen-producing microorganisms. Environ Microbiol. 2016;18:3010–21. [DOI] [PubMed] [Google Scholar]
- Ohara H, Karita S, Kimura Tet al.. Characterization of the cellulolytic complex (cellulosome) from Ruminococcus albus. Biosci Biotechnol Biochem. 2000;64:254–60. [DOI] [PubMed] [Google Scholar]
- Piao H, Lachman M, Malfatti Set al.. Temporal dynamics of fibrolytic and methanogenic rumen microorganisms during in situ incubation of switchgrass determined by 16S rRNA gene profiling. Front Microbiol. 2014;5:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitta DW, Pinchak E, Dowd SEet al.. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb Ecol. 2010;59:511–22. [DOI] [PubMed] [Google Scholar]
- Pollegioni L, Tonin F, Rosini E. Lignin-degrading enzymes. FEBS J. 2015;282:1190–213. [DOI] [PubMed] [Google Scholar]
- Pope PB, Mackenzie AK, Gregor Iet al.. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS One. 2012;7:e38571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Præsteng KE, Pope PB, Cann IKOet al.. Probiotic dosing of Ruminococcus flavefaciens affects rumen microbiome structure and function in reindeer. Microb Ecol. 2013;66:840–9. [DOI] [PubMed] [Google Scholar]
- Puniya AK, Salem AZM, Kumar Set al.. Role of live microbial feed supplements with reference to anaerobic fungi in ruminant productivity: a review. J Integr Agric. 2015;14:550–60. [Google Scholar]
- Qi M, Wang P, O'Toole Net al.. Snapshot of the eukaryotic gene expression in muskoxen rumen—a metatranscriptomic approach. PLoS One. 2011;6:e20521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard G, McEwan NR, Dutilh BEet al.. Horizontal gene transfer from Bacteria to rumen Ciliates indicates adaptation to their anaerobic, carbohydrates-rich environment. BMC Genomics. 2006;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon MT, Dassa B, Flint HJet al.. Abundance and diversity of dockerin-containing proteins in the fiber-degrading rumen bacterium, Ruminococcus flavefaciens FD-1. PLoS One. 2010;5:e12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger V, Fonty G, Komisarczuk-Bony S. Effects of physicochemical factors on the adhesion to cellulose avicel of the ruminal bacteria Ruminococcus flavefaciens and Fibrobacter succinogenes subsp. Applied and. 1990;56:3081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosewarne CP, Pope PB, Cheung JLet al.. Analysis of the bovine rumen microbiome reveals a diversity of sus-like polysaccharide utilization loci from the bacterial phylum Bacteroidetes. J Ind Microbiol Biotechnol. 2014;41:601–6. [DOI] [PubMed] [Google Scholar]
- Russell JB, Muck RE, Weimer PJ. Quantitative analysis of cellulose degradation and growth of cellulolytic bacteria in the rumen. FEMS Microbiol Ecol. 2009;67:183–97. [DOI] [PubMed] [Google Scholar]
- Russell JB. Fermentation of cellodextrins by cellulolytic and noncellulolytic rumen bacteria. Appl Environ Microbiol. 1985;49:572–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlik JL, May T. The effect of a methanogen, Methanobrevibacter smithii, on the growth rate, organic acid production, and specific ATP activity of three predominant ruminal cellulolytic bacteria. Curr Microbiol. 2000;40:176–80. [DOI] [PubMed] [Google Scholar]
- Sasson G, Kruger Ben-Shabat S, Seroussi Eet al.. Heritable bovine rumen bacteria are phylogenetically related and correlated with the Cow's capacity to harvest energy from its feed. MBio. 2017;8, DOI: 10.1128/mBio.00703-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawanon S, Koike S, Kobayashi Y. Evidence for the possible involvement of Selenomonas ruminantium in rumen fiber digestion. FEMS Microbiol Lett. 2011;325:170–9. [DOI] [PubMed] [Google Scholar]
- Seshadri R, Leahy SC, Attwood GTet al.. Cultivation and sequencing of rumen microbiome members from the Hungate1000 collection. Nat Biotechnol. 2018;36:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabat SKB, Sasson G, Doron-Faigenboim Aet al.. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016;10:2958–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai T, Kobayashi Y. Localization of ruminal cellulolytic bacteria on plant fibrous materials as determined by fluorescence in situ hybridization and real-time PCR. Appl Environ Microbiol. 2007;73:1646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai T, Ueki T, Kobayashi Y. Detection and identification of rumen bacteria constituting a fibrolytic consortium dominated by Fibrobacter succinogenes. Anim Sci J. 2010;81:72–9. [DOI] [PubMed] [Google Scholar]
- Shi Y, Odt CL, Weimer PJ. Competition for cellulose among three predominant ruminal cellulolytic bacteria under substrate-excess and substrate-limited conditions. Appl Environ Microbiol. 1997;63:734–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton TB, Canale-Parola E. Treponema bryantii sp. nov., a rumen spirochete that interacts with cellulolytic bacteria. Arch Microbiol. 1980;127:145–56. [DOI] [PubMed] [Google Scholar]
- Stewart CS, Duncan SH, Richardson AJet al.. The inhibition of fungal cellulolysis by cell-free preparations from ruminococci. FEMS Microbiol Lett. 1992;76:83–7. [DOI] [PubMed] [Google Scholar]
- Suen G, Weimer PJ, Stevenson DMet al.. The complete genome sequence of Fibrobacter succinogenes S85 reveals a cellulolytic and metabolic specialist. PLoS One. 2011;6:e18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susmel P, Stefanon B. Aspects of lignin degradation by rumen microorganisms. J Biotechnol. 1993;30:141–8. [Google Scholar]
- Tapio I, Fischer D, Blasco Let al.. Taxon abundance, diversity, co-occurrence and network analysis of the ruminal microbiota in response to dietary changes in dairy cows. PLoS One. 2017;12:e0180260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Maldonado-Gómez MX, Martínez I. To engraft or not to engraft: an ecological framework for gut microbiome modulation with live microbes. Curr Opin Biotechnol. 2018;49:129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer PJ, Cox MS, Vieira de Paula Tet al.. Transient changes in milk production efficiency and bacterial community composition resulting from near-total exchange of ruminal contents between high- and low-efficiency Holstein cows. J Dairy Sci. 2017, DOI: 10.3168/jds.2017-12746. [DOI] [PubMed] [Google Scholar]
- Weimer PJ. Redundancy, resilience, and host specificity of the ruminal microbiota: implications for engineering improved ruminal fermentations. Front Microbiol. 2015;6:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer PJ. Why don't ruminal bacteria digest cellulose faster?. J Dairy Sci. 1996;79:1496–502. [DOI] [PubMed] [Google Scholar]
- Williams AG, Withers SE, Joblin KN. The effect of cocultivation with hydrogen-consuming bacteria on xylanolysis by Ruminococcus flavefaciens. Curr Microbiol. 1994. [Google Scholar]
- Xu Q, Morrison M, Nelson KEet al.. A novel family of carbohydrate-binding modules identified with Ruminococcus albusproteins. FEBS Lett. 2004;566:11–6. [DOI] [PubMed] [Google Scholar]
- Youssef NH, Couger MB, Struchtemeyer CGet al.. The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Appl Environ Microbiol. 2013;79:4620–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Ruiz DR, Abecia L, Newbold CJet al.. Manipulating rumen microbiome and fermentation through interventions during early life: a review. Front Microbiol. 2015;6:1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ze X, Ben David Y, Laverde-Gomez JAet al.. Unique organization of extracellular amylases into Amylosomes in the resistant starch-utilizing human colonic firmicutes bacterium Ruminococcus bromii. MBio. 2015;6:e01058–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Peng Y-J, Chen Yet al.. Assessment of microbiome changes after rumen transfaunation: implications on improving feed efficiency in beef cattle. Microbiome. 2018;6:62. [DOI] [PMC free article] [PubMed] [Google Scholar]