

The South Pacific Gyre, due to its vast size and remoteness, is one of the least-studied oceanic regions on earth. However, both remote sensing and in situ measurements indicated that the activity of its microbial community contributes significantly to global biogeochemical cycles. Presented here is an unparalleled investigation of the microbial community of the SPG from 20- to 5,000-m depths covering a geographic distance of ∼7,000 km. This insight was achieved through the development of a novel onboard analysis pipeline, which combines next-generation sequencing with fluorescence in situ hybridization and automated cell enumeration. The pipeline is well comparable to onshore systems based on the Illumina platforms and yields microbial community data in less than 35 h after sampling. Going forward, the ability to gain on-site knowledge of a remote microbial community will permit hypothesis-driven research, through the generation of novel scientific questions and subsequent additional targeted sampling efforts.
KEYWORDS: AEGEAN-169, biogeography, microbial community structure, Prochlorococcus, South Pacific Subtropical Gyre
ABSTRACT
The South Pacific Gyre (SPG) covers 10% of the ocean’s surface and is often regarded as a marine biological desert. To gain an on-site overview of the remote, ultraoligotrophic microbial community of the SPG, we developed a novel onboard analysis pipeline, which combines next-generation sequencing with fluorescence in situ hybridization and automated cell enumeration. We tested the pipeline during the SO-245 “UltraPac” cruise from Chile to New Zealand and found that the overall microbial community of the SPG was highly similar to those of other oceanic gyres. The SPG was dominated by 20 major bacterial clades, including SAR11, SAR116, the AEGEAN-169 marine group, SAR86, Prochlorococcus, SAR324, SAR406, and SAR202. Most of the bacterial clades showed a strong vertical (20 m to 5,000 m), but only a weak longitudinal (80°W to 160°W), distribution pattern. Surprisingly, in the central gyre, Prochlorococcus, the dominant photosynthetic organism, had only low cellular abundances in the upper waters (20 to 80 m) and was more frequent around the 1% irradiance zone (100 to 150 m). Instead, the surface waters of the central gyre were dominated by the SAR11, SAR86, and SAR116 clades known to harbor light-driven proton pumps. The alphaproteobacterial AEGEAN-169 marine group was particularly abundant in the surface waters of the central gyre, indicating a potentially interesting adaptation to ultraoligotrophic waters and high solar irradiance. In the future, the newly developed community analysis pipeline will allow for on-site insights into a microbial community within 35 h of sampling, which will permit more targeted sampling efforts and hypothesis-driven research.
IMPORTANCE The South Pacific Gyre, due to its vast size and remoteness, is one of the least-studied oceanic regions on earth. However, both remote sensing and in situ measurements indicated that the activity of its microbial community contributes significantly to global biogeochemical cycles. Presented here is an unparalleled investigation of the microbial community of the SPG from 20- to 5,000-m depths covering a geographic distance of ∼7,000 km. This insight was achieved through the development of a novel onboard analysis pipeline, which combines next-generation sequencing with fluorescence in situ hybridization and automated cell enumeration. The pipeline is well comparable to onshore systems based on the Illumina platforms and yields microbial community data in less than 35 h after sampling. Going forward, the ability to gain on-site knowledge of a remote microbial community will permit hypothesis-driven research, through the generation of novel scientific questions and subsequent additional targeted sampling efforts.
INTRODUCTION
Oligotrophic gyres are vast ocean biomes which represent 60% of the oceans and 40% of the Earth’s surface. The largest of these gyres is the South Pacific Gyre (SPG), which has a total area of 37 million km2 and represents ∼10% of the ocean’s total area (328 × 106 km2) (1). The SPG is a unique, ultraoligotrophic habitat, which has some of the clearest waters ever reported, near-undetectable surface nitrate concentrations, and the lowest sea surface chlorophyll a concentrations (0.023 nmol liter−1) (2, 3). Although often defined as ultraoligotrophic and a “biological desert,” estimates of the SPG’s contribution to global biogeochemical cycles show that it plays a significant role in global carbon and nitrogen cycling (4–7). This contribution is calculated primarily from remote sensing data obtained via satellites because, due to its large size, the SPG has received only limited direct scientific attention (8). The two major SPG expeditions (BIOSCOPE and Ocean Drilling Program [IODP] Expedition 329) have shown that while the waters are ultraoligotrophic, there is still a considerable amount of microbial activity, specifically carbon and nutrient cycling (4–6).
Although microorganisms appear to be major players in the SPG, our understanding of their abundance and distribution patterns is very limited. The few available studies have begun to highlight the abundance of specific clades (4, 9, 10) or the community composition at individual depths (6, 11), but due to the diverse array of methodologies applied in these studies, it is difficult to obtain a comprehensive picture. This shortcoming hinders our ability to draw conclusions about the potential metabolic contributions of individual microorganisms to biogeochemical cycles in the SPG.
The primary reason for the lack of microbial data on the SPG is due to its vast size and remoteness; few scientific expeditions have traversed it due to high expedition costs. Moreover, in-depth analyses of microbial communities of remote sampling sites, such as the SPG, are hindered by onboard methodological limitations. Specifically, unlike direct measurements (temperature and salinity), samples for microbial ecology cannot readily be analyzed on-site and need to be preserved for later analysis in a laboratory. Realistically, the analysis of these samples occurs only weeks to months after sampling. Furthermore, in-depth follow-up studies must wait for future sampling campaigns, which may take years due to site remoteness and limitations in project funding. The consequences of this discrepancy between sampling and obtaining results are costly “shot-in-the-dark” sampling efforts and prevent targeted on-site experimentation.
The goal of our study was 2-fold. First, we wanted to address the lack of on-site microbial diversity and abundance analyses by developing a mobile, high-throughput sequencing and data analysis pipeline and combining this with fluorescence in situ hybridization (FISH) and onboard automated cell counting (12). The pipeline should have the capacity to quickly and inexpensively give comprehensive insight into a microbial community on-site. Second, we tested and operated the newly developed pipeline on board the R/V Sonne during the SO-245 “UltraPac” cruise in the SPG from the Chilean upwelling waters (84°W), crossing the center of the oligotrophic gyre, to the coast of New Zealand (159°W; 7,000 km).
RESULTS
Onboard next-generation sequencing (NGS).
During the SO-245 cruise, we investigated the bacterial diversity, composition, and abundance of the SPG using a newly designed field-based analysis pipeline, which functions even under challenging conditions, such as shipboard pitch and roll movements (e.g., at station 12, the maximum wave height was 3.1 m, the maximum heave was 4.7°, the maximum pitch was 5.2°, and the maximum roll angle was 8.5°).
A total of 147 samples were taken from multiple depths at 11 stations, during the SO-245 cruise, to validate the pipeline’s capacity for on-site diversity profiling (see Table S1a in the supplemental material). The samples were examined directly on board the R/V Sonne to test the individual steps of the pipeline and to ensure that a high number of high-quality reads could be obtained within the shortest possible time. We advanced previous efforts of onboard sequencing by optimizing each step from DNA extraction to data processing (Table S2) and by addressing the previously encountered issues of unexpected equipment failure due to transportation and computational limitations (13).
The pipeline had a minimum sampling requirement of 107 cells liter−1, and DNA could be extracted from all 147 samples, with an average concentration of 4.2 ng μl−1. The DNA concentration was proportional to the total cell count (TCC), with lower DNA concentrations obtained from deeper waters (3,000 to 5,000 m) (2.1 × 104 cells ml−1; 0.5 ng μl−1) and the highest DNA concentrations acquired from above the deep chlorophyll maximum (DCM) (75 to 100 m) (6 × 105 cells ml−1; 7.5 ng μl−1) (Table 1).
TABLE 1.
Onboard sequencing pipeline results for DNA extraction and total cell countsa
| Depth (m) | DNA concn (ng μl−1) |
TCC (cells ml−1) | No. of samples | |
|---|---|---|---|---|
| Avg | SD | |||
| 20 | 4.8 | 1.95 | 5.25 × 105 | 8 |
| 40–60 | 5.8 | 2.84 | 5.93 × 105 | 11 |
| 75–100 | 7.4 | 2.85 | 5.95 × 105 | 12 |
| 125–150 | 7.7 | 3.61 | 4.57 × 105 | 16 |
| 160–175 | 6.9 | 4.71 | 3.41 × 105 | 11 |
| 200–250 | 4.9 | 3.11 | 1.91 × 105 | 22 |
| 300–500 | 2.2 | 1.3 | 1.17 × 105 | 21 |
| 750–1,500 | 0.9 | 0.72 | 4.14 × 104 | 10 |
| 2,000–3,000 | 0.4 | 0.15 | 2.22 × 105 | 7 |
| 3,500–5,000 | 0.5 | 0.21 | 2.14 × 104 | 7 |
| Avg | 4.2 | 2.90 × 105 | 13 | |
Shown are averaged DNA concentrations (nanograms per microliter) with standard deviations and average total cell counts (TCC) (DAPI-stained cells per milliliter) over different depth ranges of the SPG.
Sequencing was performed on an Ion Torrent personal genome machine (PGM) platform, which was selected due to its physical robustness and compact dimensions and because it has previously been highlighted as a suitable platform for onboard sequencing (13). In total, 1,100 Mbp were sequenced on board the R/V Sonne, which equated to 3.9 million reads, with a median read length of 290 bases. Onboard sequencing was tested using multiple chip types (Ion v2 314, 316, and 318) and raw data processing methods (default and stringent) (Table 2). There was no difference in the read quality between chip types, but more stringent quality trimming decreased the total number of bases (by 30%) and the total number of reads (47%). Additionally, a more stringent processing method resulted in quicker processing times and increased the mean read length from 278 bp to 368 bp (Table 2).
TABLE 2.
Onboard sequencing pipeline results for sequencing performancea
| Chip type (Ion v2) | No. of runs | Setting | Total no. of bases (Mbp) | Total no. of reads (Q20) | Loading (%) | Mean length (bp) | Median length (bp) |
|---|---|---|---|---|---|---|---|
| Ion 314 | 5 | Default | 83 | 3.06 × 105 | 72 | 265 | 258 |
| Stringent | 56 | 1.48 × 105 | 379 | 439 | |||
| Difference | −33% | −52% | 114 | 181 | |||
| Ion 316 | 1 | Default | 465 | 1.49 × 106 | 52 | 312 | 364 |
| Stringent | 322 | 8.47 × 105 | 380 | 433 | |||
| Difference | −31% | −43% | 68 | 69 | |||
| Ion 318 | 2 | Default | 551 | 2.12 × 106 | 50 | 259 | 248 |
| Stringent | 404 | 1.17 × 106 | 345 | 362 | |||
| Difference | −27% | −45% | 86 | 114 | |||
Torrent Suite analysis of raw sequencing data was performed using three different sequencing chip types (Ion v2 314, 316, and 318) and two analysis methods (default and stringent). Loading refers to the percentage of wells of each sequencing chip that are filled with ISPs.
A major issue encountered in previous attempts at producing a remote sequencing pipeline was field-based computational issues. We specifically addressed this by developing a novel offline version of the SILVAngs pipeline (14) on a dedicated mobile server (Table S2). Before the cruise, the server was preinstalled with all necessary software and tested in controlled settings (see Materials and Methods). For validation, two mock community data sets were analyzed on both the SILVAngs online Web service and the newly developed offline version of SILVAngs using the SILVA 16S rRNA database (SSUref123) as a reference (14). For the offline server, the alignment of the rRNA in the SSUref123 database was shortened to match the amplified 16S region to increase the classification speed (see Materials and Methods). Cluster analysis showed that samples analyzed by the normal and modified SILVAngs versions yielded highly similar results (Fig. S1). Mantel tests showed no significant difference between the community compositions of the two systems (R = 0.996; P = 0.001 [based on 1,000 permutations]).
All 147 sequencing samples were processed on the SILVAngs offline server on board the R/V Sonne, which equated to a total of 3.2 × 107 reads. For all stations, a minimum of 3,500 reads per sample were obtained. The median read abundance for main stations was ∼24,000, and that for intermediate stations was ∼8,800 reads (Table 3). A higher sequencing depth was obtained for the main stations to test the ability of the SILVAngs offline server to classify rare bacterial populations.
TABLE 3.
Onboard sequencing pipeline results for read abundances for all stations and within different station types (main and intermediate)
| Station type | No. of sequence reads |
||||
|---|---|---|---|---|---|
| Avg | Median | Mode | Min | Max | |
| All | 18,630 | 13,051 | 9,321 | 3,454 | 61,928 |
| Main | 23,247 | 24,035 | 8,531 | 5,433 | 61,928 |
| Intermediate | 10,274 | 8,809 | 9,321 | 3,454 | 34,095 |
Overall, the field-based sequencing and data analysis pipeline yielded equivalent results, at similar costs, to previous laboratory-based investigations. Results could be obtained within 30 to 34 h of sampling (Table S3). The cost of DNA extraction, PCR, size selection, and sequencing using our field-based pipeline was approximately 450 euros for a single run, yielding 60 to 100 Mb (400-bp reads, not including machine or personnel costs).
Onboard microbial abundance profiling.
Although 16S rRNA tag sequencing provides in-depth insight into the composition of a microbial community, it is semiquantitative and, consequently, does not provide a comprehensive interpretation of a microbial community (15). Knowing on-site if a target organism is present or absent and, even more, its absolute cellular abundance and vertical distribution improves sampling and experimental efforts, particularly for cultivation, metagenomics, or single-cell analyses (16, 17). We therefore combined our newly developed onboard sequencing pipeline with the high-throughput image acquisition and cell enumeration system described previously by C. M. Bennke et al. (12). We applied the cell enumeration pipeline, in parallel to the sequencing pipeline, during the SO-245 cruise to the total and relative microbial abundances in 257 samples from 15 stations at various depths (see Table S1b in the supplemental material).
Absolute abundances of particular bacterial clades were determined using specific FISH probes, which were selected based on previously acquired sequencing results (10). By combining the two methods, the specificity and coverage of each FISH probe could be tested before FISH, preventing unnecessary, labor-intensive FISH procedures. One limitation of the combined approach is that onboard FISH analysis can be done only using previously described probes, which are selected based on previous studies. For the “unknown” clades, such as AEGEAN-169 in this study, there were no available probes, and new specific probes needed to be designed. The counts for such unknown clades cannot be performed directly on board.
Physicochemical properties of the SPG.
The SO-245 UltraPac cruise crossed through the oligotrophic “eye” of the SPG (Fig. 1a and b). The most pronounced changes in physicochemical conditions occurred in the top 500 m of the SPG (Fig. S2a to c). The central gyre region (stations 4 to 9; 100°W to 120°W) had characteristically high surface water temperatures of between 20°C and 25°C, and there was virtually no chlorophyll fluorescence measurable in the surface waters down to 70 m. At station 6 (110°W), marking the very center of the gyre, the temperature peaked at 24.9°C at the surface and was 19.9°C at a 200-m depth (Fig. 1b). There, the DCM descended down to a maximum depth of 190 to 200 m, with 0.5 μg liter−1 fluorescence. Along the transect, chlorophyll fluorescence was highest in the surface waters at station 14 (160°W; 1.9 μg liter−1), indicating an increase in primary productivity toward New Zealand (Fig. S2e and f). The depth of the euphotic layer, representing the depth where downward photosynthetic available radiation (PAR) (as defined from 400 to 700 nm) irradiance is reduced to 1% of its surface value, varied between 162 m in the SPG (stations 4 to 9), 110 m for station 1, and 69 m for station 14 (Fig. 1c). The DCM depths are below the euphotic layer for all SPG stations but within the 1% irradiance layer if only blue light (430 to 490 nm) is considered (down to 210 m for the SPG) (data not shown). Below 500 m, the physicochemical parameters stayed relatively consistent across the SPG, except for the oxygen profile, which showed the extent of the well-documented oxygen-minimum zone (OMZ) within the water column (Fig. S2) (18, 19).
FIG 1.
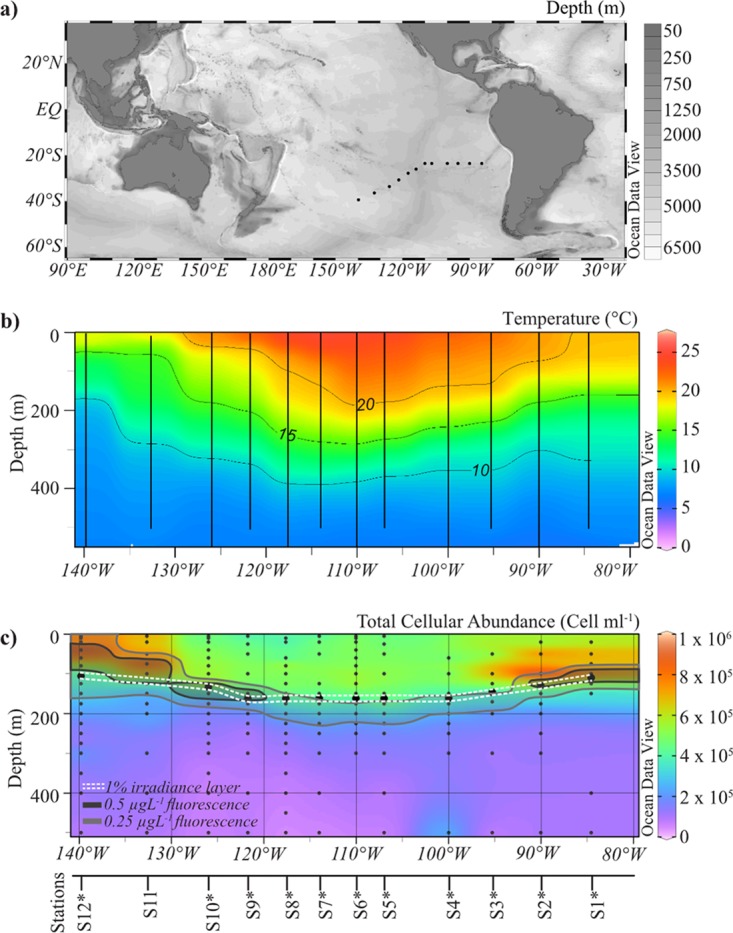
(a) Map showing 12 sampling sites in the South Pacific Gyre, indicated by black dots. (b) Contour plots of temperature (degrees Celsius) data derived from CTD measurements at 12 stations during the SO-245 cruise with depths from 0 to 500 m. (c) Contour plot of total cellular abundance enumerated by DAPI staining (cells per milliliter) with depths from 0 to 500 m. The dashed white line represents the euphotic layer (meters). Also shown is the chlorophyll fluorescence, indicated by dark gray and light gray lines. The stations are indicated on the axis below the plots. All data are publicly available from Pangaea (51). All panels were created using Ocean Data View software (71).
Niche partitioning in the bacterial community of the SPG.
The bacterial community compositions of the SPG were highly similar across a geographic distance of ∼7,000 km (Fig. 2a and Table 4) but showed a significant change with depth, which could be directly correlated to the change in light availability (Fig. 2b and c and Table 4). Correspondingly, the total cellular abundance decreases with the decrease in available light (Fig. 1c). It was higher in the surface waters (top 200 m) (1 × 106 to 2 × 105 cells ml−1) and decreased to 7 × 104 cells ml−1 by a depth of 500 m, below which it stayed relatively constant (Fig. 1c and Fig. S3a and b). The highest total cell counts (TCC) of the SPG were found just above the 1% irradiance zone at 90°W (1.1 × 106 cells ml−1) and at a 40-m depth at 139°W (9.2 × 105 cells ml−1) (Fig. 1c). In the center of the gyre (100°W to 120°W), there were 3.9 × 105 cells ml−1 in the surface waters, and this increased to ∼5 × 105 cells ml−1 at a 100-m depth (Fig. 1c).
FIG 2.

NMDS plots showing Bray-Curtis dissimilarity in community composition across longitude (°E) (a), by depth (meters) (b), and by irradiance zone (c). Each dot represents an individual sample, and the communities are color-coded according to the keys.
TABLE 4.
Permutational multivariate analysis of variance, analysis of similarly, and Mantel tests of bacterial community composition based on Bray-Curtis dissimilarities of relative read abundancesa
| Factor | PERMANOVA |
ANOSIM R | Mantel test R | |||
|---|---|---|---|---|---|---|
| df | SS | Pseudo-F | R2 | |||
| Analysis by station (geographic distance) | ||||||
| Station (longitude) | 1 | 0.323 | 02.11 | 0.018 | 0.012 | 0.03 |
| Residuals | 116 | 17.92 | 0.15 | |||
| Total | 118 | 18.24 | 1 | |||
| Analysis by depth (m) | ||||||
| Depth | 1 | 3.32 | 26.05 | 0.18* | 0.69* | |
| Residuals | 117 | 14.92 | 0.13 | |||
| Total | 118 | 18.24 | 1 | |||
| Analysis by irradiance zone | ||||||
| Irradiance | 2 | 10.31 | 75.42 | 0.56* | 0.76* | |
| Residuals | 116 | 7.93 | 0.068 | |||
| Total | 118 | 18.24 | 1 | |||
Factors used for grouping observations were station (geographic distance [Mantel test]), irradiance, and depth (meters). * denotes significance at a P value of <0.001. PERMANOVA P values were obtained using sums of squares and 999 permutations. ANOSIM was performed with 999 permutations. The Mantel test was equated using a distance matrix of the geographic distance between stations. df, degree of freedom; SS, sum of squares.
There were 20 dominant bacterial clades within the SPG, with a relative read abundance of >0.5% in at least two stations (Fig. 3b). These clades showed a distinct distribution with depth, having higher read counts (determined by sequencing) and cellular abundances (determined for 8 clades by FISH) either in the euphotic zone or below the euphotic zone (Fig. 3, Table 5, and Fig. S3). In the euphotic zone (0 to 150 m), members of SAR86, SAR11 surface groups 4 and 1, SAR116, Rickettsiales S25.593, Ascidiaceihabitans, Prochlorococcus, Rhodobacteraceae, and the AEGEAN-169 marine group had high relative read abundances (Fig. 3b). The AEGEAN-169 marine group had an abundance of 3 to 6% (1.6 × 104 cells ml−1, determined by FISH) throughout the surface water (top 100 m), with a particularly high relative abundance in the top 20 m of the center of the gyre (Fig. 3a). In contrast, the SAR86 group was more abundant (3 to 5%; 1.7 × 104 cells ml−1, determined by FISH) in the surface waters (top 100 m) outside the central gyre (Fig. 3a). SAR11 was enumerated using a clade-specific FISH probe and therefore exhibited a relative abundance of 10 to 50% (average of 2 × 105 cells ml−1) throughout the upper water column, which decreased slightly with depth and toward the eastern end of the transect (Fig. 3a and Fig. S3).
FIG 3.

Niche partitioning of the bacterial community of the SPG. (a) Contour plots of absolute cellular abundances of SAR86, SAR11, AEGEAN-169, and Prochlorococcus (cells per milliliter) enumerated by FISH with depths from 0 to 500 m. (b) Bubble plot showing the relative read abundance (determined by tag sequencing) and depth distribution of the 20 dominant bacterial clades (relative read abundance of >5%) in the SPG. All samples were sorted first by depth and then by irradiance zone before plotting. The samples of the 1% irradiance zone are plotted by station. The euphotic, 1% irradiance, and aphotic zones are represented by blue shading. (c) Contour plots of relative cellular abundances of SAR324, SAR406, and SAR202 (percentage of total cell counts determined by FISH) with depths from 0 to 5,000 m.
TABLE 5.
Specific oligonucleotide probes for fluorescence in situ hybridization applied in this studya
| Probe | Target organism or specificity | Sequence (5′→3′) | FA concn (%) | Reference |
|---|---|---|---|---|
| SAR11-152R | SAR11 clade | ATTAGCACAAGTTTCCYCGTGT | 25 | 52 |
| SAR11-441R(ori) | SAR11 clade | TACAGTCATTTTCTTCCCCGAC | 25 | 52 |
| SAR11-441Rmod | SAR11 clade | TACCGTCATTTTCTTCCCCGAC | 25 | 52 (modified) |
| SAR11-487mod | SAR11 clade | CGGACCTTCTTATTCGGG | 25 | 53 (modified) |
| SAR11-487-h3 | Helper to SAR11-487mod | CGGCTGCTGGCACGAAGTTAGC | 25 | 72 |
| SAR11-542R | SAR11 clade | TCCGAACTACGCTAGGTC | 25 | 52 |
| SAR11-732R | SAR11 clade | GTCAGTAATGATCCAGAAAGYTG | 25 | 52 |
| PRO405 | Prochlorococcus | AGAGGCCTTCGTCCCTCA | 15 | 73 |
| AEGEAN169-395 | AEGEAN-169 clade | GTCACTCACGCTGCATTG | 20 | This study |
| AEGEAN169-395-comp | Competitor to AEGEAN169-395 | GTCACTCACGCGGCATTG | 20 | This study |
| AEGEAN169-395-h1 | Helper to AEGEAN169-395 | CTGGATCAGGGTTTCCCC | 20 | This study |
| AEGEAN169-395-h2 | Helper to AEGEAN169-395 | TACTTCCCTAAGGCCTTC | 20 | This study |
| AEGEAN169-744 | AEGEAN-169 clade | ATCTCAGCGTCAAAAATGG | 20 | This study |
| AEGEAN169-744-h1 | Helper to AEGEAN169-744 | CCTAGTTAGTCGCCTTCG | 20 | This study |
| AEGEAN169-744-h2 | Helper to AEGEAN169-744 | TGCTACCCACGCTTTCGT | 20 | This study |
| SAR86-1245 | SAR86 clade | TTAGCGTCCGTCTGTAT | 35 | 74 |
| SAR86-1245-h3 | Helper to SAR86 | GGATTRGCACCACCTCGCGGC | 35 | 74 |
| SAR86-1245-h5 | Helper to SAR86 | CCATTGTAGCACGTGTGTAGC | 35 | 74 |
| SAR202-312R | SAR202 clade | TGTCTCAGTCCCCCTCTG | 40 | 20 |
| SAR324-1412 | SAR324 clade | GCCCCTGTCAACTCCCAT | 35 | 23 |
| SAR406-97 | SAR406 clade | CACCCGTTCGCCAGTTTA | 40 | 75 |
For the detection of members of the SAR11 clade, the following probes were mixed according to methods described previously (72): SAR11-152R, SAR11-441R(ori), SAR11-441Rmod, SAR11-487mod, SAR11-542R, SAR11-732R, and helper SAR11-487-h3. For the detection of members of the AEGEAN-169 clade, the following probes were mixed: AEGEAN169-395 and AEGEAN169-744; their helpers AEGEAN169-395-h1, AEGEAN169-395-h2, AEGEAN169-744-h1, and AEGEAN169-744-h2; and the competitor AEGEAN169-395-comp. For the SAR86 clade, the probe SAR86-1245 was mixed with the helpers SAR86-1245-h3 and SAR86-1245-h5. All probes were mixed in equimolar concentrations. FA, formamide.
The most significant changes in bacterial composition occurred in the 1% irradiance zone (Fig. 3b), where there was a decrease in the abundance in the euphotic clades and an increase in the abundance of the mesopelagic clades. Additionally, the phototrophic bacteria Prochlorococcus and Synechococcus exhibited a distinct distribution profile around the 1% irradiance zone. Prochlorococcus was present at a high abundance (5 to 30%; 7.9 × 104 cells ml−1, determined by FISH) throughout the top 250 m (Fig. 3a) and remained at a high abundance within the 1% irradiance zone but showed a decreased abundance just below it (150 to 200 m) (Fig. 3a). Comparatively, the read abundance of Synechococcus was low (not counted by FISH) in the surface waters and the 1% irradiance zone and increased only below the peak of Prochlorococcus at a 150- to 250-m depth (Fig. 3b).
Below the euphotic and 1% irradiance layers (aphotic, 150 to 5,125 m), the relative and absolute abundances of well-known mesopelagic bacterial clades, SAR324, SAR406, SAR202, Sulfitobacter, and the SVA0996 marine group, increased (Fig. 3b and c) (20–22). In addition to the bacterial clades with high read abundances, there was also a large rare bacterial community throughout the SPG (relative read abundance of <0.1%). About 550 clades had a low relative abundance (<0.5%) and were detectable at only a few sites (3 or fewer), whereas 120 clades had a low abundance (<0.5%) but were ubiquitously present. These ubiquitous but rare clades were predominantly from the Verrucomicrobia (Puniceicoccaceae), Planctomycetes, Deltaproteobacteria, and Bacteroidetes (Flavobacteriaceae) (Fig. S5).
DISCUSSION
Oligotrophic gyres cover vast areas of the Earth’s surface and contribute, due to microbial carbon and nitrogen cycling, significantly to global biogeochemical cycles (4, 5, 7). However, our current understanding of the abundance and distribution patterns of the microbial community of the largest of these gyres, the SPG, is still limited due to both infrequent sampling and a lack of on-site community analysis. Therefore, during the SO-245 cruise, we developed an onboard microbial community analysis pipeline which enabled the on-site sequencing of 147 samples and the enumeration of 275 samples by FISH. The outcome of our method developments is a readily applicable system for efficient, cost-effective, field-based, comprehensive microbial community analysis.
Picoplankton community of the SPG.
Using our newly established pipeline, we discovered that the microbial community of the SPG showed a pronounced vertical distribution pattern. The composition of the community changed significantly with depth, which was directly correlated with the availability of light (Fig. 2 and Table 4). Such a noticeable vertical distribution has also been observed in other oceanic gyres (North Pacific, South Atlantic, and Northern Atlantic Gyres) (23–27) and was linked to significant changes in the physicochemical conditions related to depth: changes in temperature, nutrient concentrations, availability of light, and availability of labile organic matter (28–30).
The euphotic surface waters of the central gyre were extremely limited in inorganic macronutrients and especially in nitrogen salts (3, 7). The low nutrient availability restricts growth to specialist oligotrophic organisms, which was reflected by the low cellular abundance in the surface mixed layer (4 × 105 cells ml−1) (Fig. 1c). Dominant clades were Prochlorococcus, SAR11, SAR116, SAR86, and the AEGEAN-169 marine group (Fig. 3b), all of which, except for the AEGEAN-169 marine group, are well documented to be optimized for an oligotrophic lifestyle (31–34). Cultured and genome-sequenced representatives of these clades are also reported to have streamlined genomes and specialized resource acquisition abilities (35–37).
Additionally, Prochlorococcus, SAR11, SAR86, and SAR116 are equipped with the genetic potential for photosynthesis or phototrophy via proteorhodopsins (31, 38, 39). Although our current knowledge of the genetic potential of members of the AEGEAN-169 marine group is limited (27, 40–42), their high cellular abundance, of up to 3 × 104 cells ml−1 in the surface waters of the central gyre, indicates a specialized oligotrophic lifestyle. Previous studies have highlighted some of the potential factors affecting the AEGEAN-169 marine group distribution patterns (41) but contrastingly found them to have a high relative abundance in deeper waters (500 m) (27). A possible explanation for these dissimilarities is the presence of multiple ecological species of the AEGEAN-169 marine group (41). Future metagenomic studies of these organisms are required to examine the importance of this abundant clade in the most oligotrophic surface waters in the SPG.
Prochlorococcus, the dominant primary producer in oligotrophic ocean regions (43, 44), was also the most abundant autotrophic organism in the surface waters of the SPG. However, in comparison to studies in the Atlantic gyres, its absolute abundance in surface waters was low (Fig. 3a) (23, 45, 46). Interestingly, the abundance of Prochlorococcus increased with depth and peaked at between 100 and 150 m in and around the 1% irradiance zone. The low abundance of Prochlorococcus in the surface waters of the SPG could be an indication that low levels of nutrients, high solar irradiance, or a combination of both inhibits its growth (9, 47, 48).
The measured chlorophyll fluorescence in the surface waters of the SPG was below the detection limit in our study (Fig. 1c), although previous studies measured up to 0.017 μg liter−1 (49). Chlorophyll fluorescence peaked deep in the water column around 200 m and could be measured down to nearly a 300-m depth (see Fig. S2f in the supplemental material). Light availability, as indicated by the 1% irradiance layer, was maximal in the central waters of the SPG, reaching down to 162 m. The deep penetration of blue light in the water column of the SPG, down to 210 m, indicated that the light conditions were suitable for photosynthetic activity even at these depths. Similar chlorophyll measurements taken in the North and South Atlantic Gyres show comparable fluorescence profiles, although the depth of the DCM in the Atlantic is considerably higher in the water column (120 to 165 m) than in the SPG, and surface waters are not entirely depleted of chlorophyll (23, 50).
In the mesopelagic zone of the SPG, where light became limiting (51), there was a distinct change in the microbial community dominated by SAR11 surface clade 1, SAR86, and Prochlorococcus to one dominated by SAR324, SAR406, and SAR202. Although there are currently no cultured representatives of these three bacterial groups, metagenomic analyses have revealed some insight into their possible metabolic capabilities. SAR202 and SAR324 have been associated with carbon and with sulfur oxidation (52–55) and are likely chemolithoautotrophs ubiquitous in the dark oceans. In particular, SAR324 was also hypothesized to degrade the lipid chains of chlorophyll a, which may explain its increased abundance below the DCM (56). Interestingly, in the mesopelagic zone, a novel and so-far-undescribed group called SVA0996 of the Actinobacteria was found to be highly represented in the 16S rRNA tag reads. Because of its abundance, this group could be of interest in future studies.
We designed and optimized an onboard sequencing and data analysis pipeline that enabled us to obtain on-site microbial community diversity results for the SPG within 34 h of sampling. In surface waters, the community was dominated by a few key oligotrophic organisms, which are adapted to extreme physicochemical conditions. The ability to obtain “direct” insights into the microbial diversity, even at extremely remote oligotrophic sampling sites, enables the close examination of novel discovered microbial clades, such as the AEGEAN-169 marine group in the surface waters or the SVA0996 group (this study) in the deeper water layers. Additionally, and most importantly, it allows microbial ecologists to perform more-targeted sampling, thereby furthering our understanding of the diversity and metabolic capabilities of key microorganisms.
MATERIALS AND METHODS
Sampling.
Seawater samples were collected aboard the R/V Sonne during the UltraPac cruise (SO-245) from Antofagasta, Chile (17 December 2015), to Wellington, New Zealand (28 January 2016). Water samples were taken from a total of 15 stations (see Table S1 in the supplemental material) using a Seabird sbe911+ CTD (Seabird Scientific, WA, USA) attached to an SBE32 carousel water sampler containing 24 12-liter bottles. Two types of stations were sampled: main stations and intermediate stations. On main stations, the CTD was cast through the entire water column to 50 to 100 m above the seafloor, and samples were taken at various depths throughout the water column (57). Generally, 4 to 5 CTDs were cast to reduce the time between sampling at depth and processing of the samples. Intermediate stations consisted of a single CTD cast down to 500 m, and samples were taken from various depths (57). For diversity analysis, a total of 1 liter of seawater was sampled. The water was directly filtered onto a 47-mm polycarbonate filter (0.2-μm pore size) using a bottle-top Nalgene filter holder (Thermo Fisher, MA, USA) and a vacuum pump. After filtration, samples were immediately used for DNA extraction.
Physicochemical data.
Physicochemical characteristics were examined using a CTD (Sbe911+ probe; Seabird Electronics Inc.). The system was equipped with double temperature (SBE 3) and conductivity (SBE 4) probes, a pressure sensor (Digiquartz), an oxygen sensor (SBE 43), an altimeter (Bentos), and a chlorophyll fluorometer combined with a turbidity sensor (ECO-AFL/FL; Wet Labs). The sensors were precalibrated by the manufacturers. The data were recorded with Seasave V7.23.1 software and processed using Seabird SBE data processing software. Data were despiked and also visually checked. The ship position was derived from the shipboard GPS system linked to the CTD data. The time zone is given in coordinated universal time (UTC). Salinity was quality checked by reference samples (n = 30), measured with an Optimare precision salinometer (OPS S/N 004) 5 months after the cruise. All CTD data were obtained from and are available from Pangaea (www.pangaea.de) (51). The physicochemical data were visualized using ODV4 software (https://odv.awi.de/).
The underwater light field was measured utilizing a HyperPro II profiler (Satlantic Inc., Canada) according to procedures described previously (58). For these measurements, the profiler was lowered into the water at least 30 m behind the vessel to avoid ship shadowing when free-falling. A downward irradiance reference sensor was mounted at an elevated, nonshaded location. Profiler data processing and calculation of photosynthetic available radiation were performed with ProSoft 7.7.16 (Satlantic Inc., Canada).
DNA extraction, PCR, size selection, and quantification.
Each step from DNA extraction to data processing was selected and optimized to achieve a high level of high-quality reads in the shortest possible processing time. The advantages and disadvantages of each processing step are highlighted in Table S2. The final optimal protocol for the SPG study is described below.
DNA extractions were done using the MoBio Power Water DNA extraction kit (MoBio Laboratories Inc., CA, USA), as recommended by the manufacturer. PCR was carried out using the Platinum PCR SuperMix high-fidelity polymerase kit (Thermo Fisher), using the primers S-D-Bact-0341-b-S-17 and S-D-Bact-0785-a-A-21 targeting the V3-V4 variable region of the 16S rRNA, evaluated previously (59). Both primers were fusion primers with additional adaptor and barcode sequences at the 5′ end to allow sequencing and separation of samples in downstream analyses. The reverse primers contained the Ion tr-P1 adaptor at the 5′ end of the primer, and the forward primers contained both the Ion A adaptor and one of 40 IonXpress barcodes (Ion Xpress 1 to 40) as well as the key sequence (GAT) before the primer. The reverse fusion primer sequence was 5′-CCTCTCTATGGGCAGTCGGTGAT-GACTACHVGGGTA-TCTAATCC-3′. The forward fusion primer sequence was 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-XXXXXXXXXX-GAT-CCTACGGGNGGCWGCAG-3′ (where XXXXXXXXXX indicates barcode sequences 1 to 40). After amplification, the PCR amplicons were size selected using Agencourt AMPure XP (Beckman Coulter, Krefeld, Germany).
A successful sequencing reaction requires precise quantities of the template library to ensure clonal amplification on individual ion sphere particles (ISPs) (see below). A fragment analyzer (AATI) was used to determine the quality and quantity of the extracted DNA, the size-selected PCR products, and the final sequencing pools. Genomic DNA was analyzed using the DNF-488 high-sensitivity genomic DNA analysis kit (size range from 50 bp to 40,000 bp; AATI). All template libraries and final sequencing pools were analyzed using DNF-472 standard-sensitivity NGS kit sizing DNA (size range from 25 bp to 5,000 bp and up to a minimum of 0.1 ng μl−1; AATI), as recommended by the manufacturer. The fragment was adapted to ship movements by adding magnets to the individual sample trays, thereby preventing the accidental dropping of a sampling tray caused by ship pitches, during plate movement, or at the “on-hold” position inside the tray drawers. The internal plate lift was mechanically stabilized for ship movement and vibration by the installation of an additional guide rail on the upper side connected via rubber mounts. Additionally, a specialized stand with transport handles and attachments was applied for easy manual transport and to allow for secure attachment to a surface (Fig. S6a to e).
Ion Torrent sequencing and raw sequence processing.
The Ion Torrent PGM platform was adapted for onboard use by securing it to a 2-cm-thick polyethylene baseplate, and the internal hard drives were replaced by solid-state drives (SSDs). The base was equipped with handles that could be used for manual transportation of the sequencer and to fix it to the surface (Fig. S6f to h). A similar base was fastened to the Ion OneTouch2 instrument (Thermo Fisher) and the Ion OneTouch ES instrument (Thermo Fisher). The Torrent server (Thermo Fisher) was also adapted to withstand shipboard vibration and transport by placing it in a custom-made metal frame using rubber mounts (Fig. S6f to h).
Sequencing was carried out as recommended by the manufacturer, using an Ion Torrent PGM sequencer (Thermo Fisher). Emulsion PCR and enrichment of template-positive ISPs were done using the Ion PGM Hi-Q OT2 kit (Thermo Fisher) on the Ion OneTouch 2 instrument (Thermo Fisher) and the Ion OneTouch ES instrument (Thermo Fisher) according to the Ion Torrent user manual. Subsequently, the library fragments (attached to the ISP) were sequenced using the Ion PGM Hi-Q sequencing kit (Thermo Fisher) according to the user manual on an Ion PGM system (Thermo Fisher). Sequencing was done with Ion 314, 316, and 318 chip kit v2 (Thermo Fisher), with a total of 1,200 flows per sequencing run. The chips vary in their capacity (number of sensors) and, therefore, total output, run time, and processing time. Specifically, the Ion 314 chip has 1.2 million sensors, a total output of up to 100 Mb, and a run time of 2 to 4 h. The Ion 316 chip has 6.1 million sensors, an output of up to 1 Gb, and a run time of 3 to 5 h. The Ion 318 chip has 11 million sensors, a total output of up to 2 Gb, and a run time of 4 to 7 h.
Torrent Suite software, which converts the raw signals (raw pH values) into incorporation measurements and ultimately into base calls for each read, was used for initial quality trimming. The standard Torrent Suite settings and more stringent settings were applied. The standard settings and stringent settings were defined in the BaseCaller arguments of the Torrent Suite software (standard, BaseCaller –barcode-filter 0.01 –barcode-filter-minreads 20 –barcode-mode 1 –barcode-cutoff 3 –trim-qual-cutoff 10 –trim-qual-window-size 20 –trim-min-read-len 100; stringent, BaseCaller –barcode-mode 1 –barcode-cutoff 0 –trim-qual-cutoff 15 –trim-qual-window-size 10 –trim-min-read-len 250). Finally, the reads were exported as .sff files using the file exporter plug-in in the Torrent Suite software. The .sff files were split into individual sample FASTA files using mothur version 1.35.1 (60) [sffinfo()] and analyzed using the offline SILVAngs pipeline called lab on a ship (see below).
Offline SILVAngs pipeline “lab on a ship.”
The computer cluster “lab on a ship” was developed to facilitate offline 16S rRNA sequence classification using the SILVAngs pipeline. Previously, this was available only using the online platform (14). The benefit of having an offline version is the potential to use it on board a research vessel. To ensure a quick classification system, an efficient computing cluster was obtained for the offline analysis and consisted of 4× Intel Xeon E5-4607 6-core, 2.6-GHz, 256-GB RAM mounted on a Supermicro X9QR7-TF+ main board (Supermicro, CA, USA), 3 480-GB SATA/600 hard disks (Samsung) for fast data read/write processes, and 5 2-TB SATA3 server RAID hard disks (Ultrastar; HGST, USA) for data storage. The server was installed in a portable 19-in standard rack and placed in the ship’s server room.
The server was preinstalled with an offline copy of the SILVAngs pipeline, including BLAST (61), the ARB software package (62), as well as the SINA aligner (v1.2.11) (63). The complete SILVAngs pipeline can be run using a single command line argument. Additionally, mothur software (version 1.35.1) (60) and R-Studio with all required packages (64) were installed to offer users further analysis and graphing options. Version 123 of the SILVA (small-subunit [SSU]) data set was used as the classification reference by both the offline and online pipelines. The standard SILVAngs settings for alignment (minimum alignment identity of 50%, minimum alignment score of 40, and minimum base pair score of 30%), quality trimming (minimum sequence quality of 30%, minimum length of 250, maximum ambiguities of 2%, and maximum homopolymers of 2%), clustering (CD-Hit version 4.6; minimum operational taxonomic unit [OTU] identity of 98%), and classification (BLAST version SINA v1.2.10-pre [revision 24275 M]; similarity of 86%) were applied. To increase the speed of the alignment stage, a custom-alignment SEED was used by the offline version of the pipeline, which uses an alignment trimmed to match the sequence region of the SSU gene. The offline server cluster enables the classification of 40 samples with an average of 18,000 reads per sample within 3 h. To test the lab-on-a-ship server and ensure that similar community composition results are obtained using different quality trimming methods, a mock community analysis was done. The mock community samples consisted of a defined number of quality-trimmed reads (10,000) obtained from 10 Ion Torrent-sequenced marine samples from the Atlantic Ocean. The community classification outputs from the two servers were then compared using cluster analysis.
Statistical analysis.
The interpretation and visualization of the microbial diversity data were done using normalized genus abundance-to-site matrices in R software with the packages Vegan (community ecology package [64]) and Rioja (Analysis of Quaternary Science Data [65]). Normalization was done using the decostand (method = “total”) function of the Vegan software package. For beta diversity analysis and related hypothesis testing, Bray-Curtis dissimilarity matrices of the normalized read abundances of all samples were constructed. Differences in the community structures between sampling sites were analyzed by comparing all samples by analysis of similarity (ANOSIM) and visualized in nonmetric multidimensional scaling (NMDS) plots. To test for significant changes in the community composition by longitude, depth, and irradiance, ANOSIMs were performed and visualized using NMDS plots. Subsequently, permutation multivariate analysis of variance (PERMANOVA) with pairwise analysis was performed to identify the amount of variance associated with individual factors.
Total cell counts and FISH.
DAPI (4′,6-diamidino-2-phenylindole) staining and catalyzed reporter deposition (CARD)-FISH were carried out as described previously (12, 66). DAPI- and FISH-stained cells were visualized and counted automatically using a fully automated image acquisition and cell enumeration system (12). FISH probe sequences are listed in Table 5, along with their corresponding competitors and helper oligonucleotides, their specificity, and formamide concentrations in the hybridization buffer. For this study, a new probe specific for the AEGEAN-169 clade was designed and tested, based on the latest SILVA 16S rRNA database (refnr 128). Total cellular abundances were also determined by flow cytometry (FACSort; Becton, Dickinson) as described previously by Zubkov and Tarran (67).
Data availability.
All sequence data were deposited in the European Nucleotide Archive (ENA) (68) using the data brokerage service of the German Federation for Biological Data (GFBio) (69), in compliance with the MIxS standard (70). The INSDC accession number for the data is PRJEB39460.
Supplementary Material
ACKNOWLEDGMENTS
We thank Captain Mallon and the crew of the R/V Sonne for assistance at sea. We also thank Andreas Ellrott, Jörg Wulf, Rohan Henkel, Gabriele Klockgether, and Gaute Lavik for technical assistance in the laboratory and at sea. We thank Anke Meyerdierks for helpful discussions.
This project was funded by the Max Planck Society, and the UltraPac Expedition (SO-245) was funded by the Federal Ministry of Education and Research of Germany (grant 03G0245A).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00184-19.
REFERENCES
- 1.Longhurst A, Sathyendranath S, Platt T, Caverhill C. 1995. An estimate of global primary production in the ocean from satellite radiometer data. J Plankton Res 17:1245–1271. doi: 10.1093/plankt/17.6.1245. [DOI] [Google Scholar]
- 2.Morel A, Gentili B, Claustre H, Babin M, Bricaud A, Ras J, Tièche F. 2007. Optical properties of the “clearest” natural waters. Limnol Oceanogr 52:217–229. doi: 10.4319/lo.2007.52.1.0217. [DOI] [Google Scholar]
- 3.Raimbault P, Garcia N, Cerutti F. 2008. Distribution of inorganic and organic nutrients in the South Pacific Ocean—evidence for long-term accumulation of organic matter in nitrogen-depleted waters. Biogeosciences 5:281–298. doi: 10.5194/bg-5-281-2008. [DOI] [Google Scholar]
- 4.Van Wambeke F, Obernosterer I, Moutin T, Duhamel S, Ulloa O, Claustre H. 2008. Heterotrophic bacterial production in the eastern South Pacific: longitudinal trends and coupling with primary production. Biogeosciences 5:157–169. doi: 10.5194/bg-5-157-2008. [DOI] [Google Scholar]
- 5.Halm H, Lam P, Ferdelman TG, Lavik G, Dittmar T, LaRoche J, D’Hondt S, Kuypers MM. 2012. Heterotrophic organisms dominate nitrogen fixation in the South Pacific Gyre. ISME J 6:1238–1249. doi: 10.1038/ismej.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh EA, Smith DC, Sogin ML, D’Hondt S. 2015. Bacterial and archaeal biogeography of the deep chlorophyll maximum in the South Pacific Gyre. Aquat Microb Ecol 75:1–13. doi: 10.3354/ame01746. [DOI] [Google Scholar]
- 7.Letscher RT, Knapp AN, James AK, Carlson CA, Santoro AE, Hansell DA. 2015. Microbial community composition and nitrogen availability influence DOC remineralization in the South Pacific Gyre. Mar Chem 177:325–334. doi: 10.1016/j.marchem.2015.06.024. [DOI] [Google Scholar]
- 8.Claustre H, Huot Y, Obernosterer I, Gentili B, Tailliez D, Lewis M. 2008. Gross community production and metabolic balance in the South Pacific Gyre, using a non intrusive bio-optical method. Biogeosciences 5:463–474. doi: 10.5194/bg-5-463-2008. [DOI] [Google Scholar]
- 9.Lami R, Cottrell MT, Ras J, Ulloa O, Obernosterer I, Claustre H, Kirchman DL, Lebaron P. 2007. High abundances of aerobic anoxygenic photosynthetic bacteria in the South Pacific Ocean. Appl Environ Microbiol 73:4198–4205. doi: 10.1128/AEM.02652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West NJ, Lepere C, Manes CL, Catala P, Scanlan DJ, Lebaron P. 2016. Distinct spatial patterns of SAR11, SAR86, and Actinobacteria diversity along a transect in the ultra-oligotrophic South Pacific Ocean. Front Microbiol 7:234. doi: 10.3389/fmicb.2016.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin Q, Fu B, Li B, Shi X, Inagaki F, Zhang XH. 2013. Spatial variations in microbial community composition in surface seawater from the ultra-oligotrophic center to rim of the South Pacific Gyre. PLoS One 8:e55148. doi: 10.1371/journal.pone.0055148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennke CM, Reintjes G, Schattenhofer M, Ellrott A, Wulf J, Zeder M, Fuchs BM. 2016. Modification of a high-throughput automatic microbial cell enumeration system for shipboard analyses. Appl Environ Microbiol 82:3289–3296. doi: 10.1128/AEM.03931-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim YW, Cuevas DA, Silva GG, Aguinaldo K, Dinsdale EA, Haas AF, Hatay M, Sanchez SE, Wegley-Kelly L, Dutilh BE, Harkins TT, Lee CC, Tom W, Sandin SA, Smith JE, Zgliczynski B, Vermeij MJ, Rohwer F, Edwards RA. 2014. Sequencing at sea: challenges and experiences in Ion Torrent PGM sequencing during the 2013 Southern Line Islands Research Expedition. PeerJ 2:e520. doi: 10.7717/peerj.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and Web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Props R, Kerckhof FM, Rubbens P, De Vrieze J, Hernandez Sanabria E, Waegeman W, Monsieurs P, Hammes F, Boon N. 2017. Absolute quantification of microbial taxon abundances. ISME J 11:584–587. doi: 10.1038/ismej.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeLong EF. 2009. The microbial ocean from genomes to biomes. Nature 459:200–206. doi: 10.1038/nature08059. [DOI] [PubMed] [Google Scholar]
- 17.Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. 2012. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335:587–590. doi: 10.1126/science.1212665. [DOI] [PubMed] [Google Scholar]
- 18.Lam P, Lavik G, Jensen MM, van de Vossenberg J, Schmid M, Woebken D, Gutierrez D, Amann R, Jetten MS, Kuypers MM. 2009. Revising the nitrogen cycle in the Peruvian oxygen minimum zone. Proc Natl Acad Sci U S A 106:4752–4757. doi: 10.1073/pnas.0812444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinti DL. 2014. Oxygen-minimum zone In Amils R, Gargaud M, Cernicharo Quintanilla J, Cleaves HJ, Irvine WM, Pinti D, Viso M (ed), Encyclopedia of astrobiology. Springer, Berlin, Germany. [Google Scholar]
- 20.Morris RM, Rappe MS, Urbach E, Connon SA, Giovannoni SJ. 2004. Prevalence of the Chloroflexi-related SAR202 bacterioplankton cluster throughout the mesopelagic zone and deep ocean. Appl Environ Microbiol 70:2836–2842. doi: 10.1128/AEM.70.5.2836-2842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheik CS, Jain S, Dick GJ. 2014. Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics. Environ Microbiol 16:304–317. doi: 10.1111/1462-2920.12165. [DOI] [PubMed] [Google Scholar]
- 22.Milici M, Vital M, Tomasch J, Badewien TH, Giebel H-A, Plumeier I, Wang H, Pieper DH, Wagner-Döbler I, Simon M. 2017. Diversity and community composition of particle-associated and free-living bacteria in mesopelagic and bathypelagic Southern Ocean water masses: evidence of dispersal limitation in the Bransfield Strait. Limnol Oceanogr 62:1080–1095. doi: 10.1002/lno.10487. [DOI] [Google Scholar]
- 23.Schattenhofer M, Fuchs BM, Amann R, Zubkov MV, Tarran GA, Pernthaler J. 2009. Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ Microbiol 11:2078–2093. doi: 10.1111/j.1462-2920.2009.01929.x. [DOI] [PubMed] [Google Scholar]
- 24.Friedline CJ, Franklin RB, McCallister SL, Rivera MC. 2012. Bacterial assemblages of the eastern Atlantic Ocean reveal both vertical and latitudinal biogeographic signatures. Biogeosciences 9:2177–2193. doi: 10.5194/bg-9-2177-2012. [DOI] [Google Scholar]
- 25.DeLong EF, Preston CM, Mincer T, Rich V, Hallam SJ, Frigaard NU, Martinez A, Sullivan MB, Edwards R, Brito BR, Chisholm SW, Karl DM. 2006. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 311:496–503. doi: 10.1126/science.1120250. [DOI] [PubMed] [Google Scholar]
- 26.Agogue H, Lamy D, Neal PR, Sogin ML, Herndl GJ. 2011. Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol Ecol 20:258–274. doi: 10.1111/j.1365-294X.2010.04932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cram JA, Chow CE, Sachdeva R, Needham DM, Parada AE, Steele JA, Fuhrman JA. 2015. Seasonal and interannual variability of the marine bacterioplankton community throughout the water column over ten years. ISME J 9:563–580. doi: 10.1038/ismej.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pakulski JD, Benner R. 1994. Abundance and distribution of carbohydrates in the ocean. Limnol Oceanogr 39:930–940. doi: 10.4319/lo.1994.39.4.0930. [DOI] [Google Scholar]
- 29.Osterholz H, Niggemann J, Giebel HA, Simon M, Dittmar T. 2015. Inefficient microbial production of refractory dissolved organic matter in the ocean. Nat Commun 6:7422. doi: 10.1038/ncomms8422. [DOI] [PubMed] [Google Scholar]
- 30.Moran MA, Kujawinski EB, Stubbins A, Fatland R, Aluwihare LI, Buchan A, Crump BC, Dorrestein PC, Dyhrman ST, Hess NJ, Howe B, Longnecker K, Medeiros PM, Niggemann J, Obernosterer I, Repeta DJ, Waldbauer JR. 2016. Deciphering ocean carbon in a changing world. Proc Natl Acad Sci U S A 113:3143–3151. doi: 10.1073/pnas.1514645113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giovannoni SJ, Bibbs L, Cho JC, Stapels MD, Desiderio R, Vergin KL, Rappe MS, Laney S, Wilhelm LJ, Tripp HJ, Mathur EJ, Barofsky DF. 2005. Proteorhodopsin in the ubiquitous marine bacterium SAR11. Nature 438:82–85. doi: 10.1038/nature04032. [DOI] [PubMed] [Google Scholar]
- 32.Molloy S. 2012. Marine microbiology. SAR86: streamlined for success. Nat Rev Microbiol 10:82. doi: 10.1038/nrmicro2736. [DOI] [PubMed] [Google Scholar]
- 33.Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, Gonzalez JM, Luo H, Wright JJ, Landry ZC, Hanson NW, Thompson BP, Poulton NJ, Schwientek P, Acinas SG, Giovannoni SJ, Moran MA, Hallam SJ, Cavicchioli R, Woyke T, Stepanauskas R. 2013. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci U S A 110:11463–11468. doi: 10.1073/pnas.1304246110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown MV, Ostrowski M, Grzymski JJ, Lauro FM. 2014. A trait based perspective on the biogeography of common and abundant marine bacterioplankton clades. Mar Genomics 15:17–28. doi: 10.1016/j.margen.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, Halpern AL, Lasken RS, Nealson K, Friedman R, Venter JC. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6:1186–1199. doi: 10.1038/ismej.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tripp HJ. 2013. The unique metabolism of SAR11 aquatic bacteria. J Microbiol 51:147–153. doi: 10.1007/s12275-013-2671-2. [DOI] [PubMed] [Google Scholar]
- 37.Luo H, Moran MA. 2013. Assembly-free metagenomic analysis reveals new metabolic capabilities in surface ocean bacterioplankton. Environ Microbiol Rep 5:686–696. doi: 10.1111/1758-2229.12068. [DOI] [PubMed] [Google Scholar]
- 38.Beja O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich SB, Gates CM, Feldman RA, Spudich JL, Spudich EN, DeLong EF. 2000. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- 39.Oh HM, Kwon KK, Kang I, Kang SG, Lee JH, Kim SJ, Cho JC. 2010. Complete genome sequence of “Candidatus Puniceispirillum marinum” IMCC1322, a representative of the SAR116 clade in the Alphaproteobacteria. J Bacteriol 192:3240–3241. doi: 10.1128/JB.00347-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang C, Li Y, Zhou B, Zhou Y, Zheng W, Tian Y, Van Nostrand JD, Wu L, He Z, Zhou J, Zheng T. 2015. Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguine bloom in Xiamen Sea, China. Sci Rep 5:8476. doi: 10.1038/srep08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cram JA, Xia LC, Needham DM, Sachdeva R, Sun F, Fuhrman JA. 2015. Cross-depth analysis of marine bacterial networks suggests downward propagation of temporal changes. ISME J 9:2573–2586. doi: 10.1038/ismej.2015.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bougouffa S, Yang JK, Lee OO, Wang Y, Batang Z, Al-Suwailem A, Qian PY. 2013. Distinctive microbial community structure in highly stratified deep-sea brine water columns. Appl Environ Microbiol 79:3425–3437. doi: 10.1128/AEM.00254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flombaum P, Gallegos JL, Gordillo RA, Rincon J, Zabala LL, Jiao N, Karl DM, Li WK, Lomas MW, Veneziano D, Vera CS, Vrugt JA, Martiny AC. 2013. Present and future global distributions of the marine cyanobacteria Prochlorococcus and Synechococcus. Proc Natl Acad Sci U S A 110:9824–9829. doi: 10.1073/pnas.1307701110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Partensky F, Hess WR, Vaulot D. 1999. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol Mol Biol Rev 63:106–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zwirglmaier K, Jardillier L, Ostrowski M, Mazard S, Garczarek L, Vaulot D, Not F, Massana R, Ulloa O, Scanlan DJ. 2008. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol 10:147–161. doi: 10.1111/j.1462-2920.2007.01440.x. [DOI] [PubMed] [Google Scholar]
- 46.Gomez-Pereira PR, Hartmann M, Grob C, Tarran GA, Martin AP, Fuchs BM, Scanlan DJ, Zubkov MV. 2013. Comparable light stimulation of organic nutrient uptake by SAR11 and Prochlorococcus in the North Atlantic subtropical gyre. ISME J 7:603–614. doi: 10.1038/ismej.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Partensky F, Hoepffner N, Li W, Ulloa O, Vaulot D. 1993. Photoacclimation of Prochlorococcus sp. (Prochlorophyta) strains isolated from the North Atlantic and the Mediterranean Sea. Plant Physiol 101:285–296. doi: 10.1104/pp.101.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore LR, Rocap G, Chisholm SW. 1998. Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature 393:464–467. doi: 10.1038/30965. [DOI] [PubMed] [Google Scholar]
- 49.Ras J, Claustre H, Uitz J. 2008. Spatial variability of phytoplankton pigment distributions in the subtropical South Pacific Ocean: comparison between in situ and predicted data. Biogeosciences 5:353–369. doi: 10.5194/bg-5-353-2008. [DOI] [Google Scholar]
- 50.Robinson C, Poulton AJ, Holligan PM, Baker AR, Forster G, Gist N, Jickells TD, Malin G, Upstill-Goddard R, Williams RG, Woodward EMS, Zubkov MV. 2006. The Atlantic Meridional Transect (AMT) programme: a contextual view 1995–2005. Deep Sea Res Part 2 Top Stud Oceanogr 53:1485–1515. doi: 10.1016/j.dsr2.2006.05.015. [DOI] [Google Scholar]
- 51.Zielinski O, Henkel R, Voß D, Ferdelman TG. 2017. Physical oceanography during SONNE cruise SO245 (UltraPac). Institute for Chemistry and Biology of the Marine Environment, Carl von Ossietzky University of Oldenburg, Pangaea, Oldenburg, Germany. [Google Scholar]
- 52.Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. 2002. Cultivation of the ubiquitous SAR 11 marine bacterioplankton clade. Nature 418:630–633. doi: 10.1038/nature00917. [DOI] [PubMed] [Google Scholar]
- 53.Morris RM, Rappé MS, Connon SA, Vergin KL, Siebold WA, Carlson CA, Giovannoni SJ. 2002. SAR 11 clade dominates ocean surface bacterioplankton communities. Nature 420:806–810. doi: 10.1038/nature01240. [DOI] [PubMed] [Google Scholar]
- 54.Biers EJ, Sun S, Howard EC. 2009. Prokaryotic genomes and diversity in surface ocean waters: interrogating the global ocean sampling metagenome. Appl Environ Microbiol 75:2221–2229. doi: 10.1128/AEM.02118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Swan BK, Martinez-Garcia M, Preston CM, Sczyrba A, Woyke T, Lamy D, Reinthaler T, Poulton NJ, Masland ED, Gomez ML, Sieracki ME, DeLong EF, Herndl GJ, Stepanauskas R. 2011. Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 333:1296–1300. doi: 10.1126/science.1203690. [DOI] [PubMed] [Google Scholar]
- 56.Chitsaz H, Yee-Greenbaum JL, Tesler G, Lombardo MJ, Dupont CL, Badger JH, Novotny M, Rusch DB, Fraser LJ, Gormley NA, Schulz-Trieglaff O, Smith GP, Evers DJ, Pevzner PA, Lasken RS. 2011. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotechnol 29:915–921. doi: 10.1038/nbt.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reintjes G, Tegetmeyer H, Wulf J, Bürgisser M, Fuchs BM. 2017. On-board sequencing of the microbial community of the South Pacific Gyre. Pangaea https://doi.pangaea.de/10.1594/PANGAEA.882015.
- 58.Holinde L, Zielinski O. 2016. Bio-optical characterization and light availability parameterization in Uummannaq Fjord and Vaigat-Disko Bay (West Greenland). Ocean Sci 12:117–128. doi: 10.5194/os-12-117-2016. [DOI] [Google Scholar]
- 59.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 62.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, Forster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, Konig A, Liss T, Lussmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH. 2004. ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pruesse E, Peplies J, Glockner FO. 2012. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Wagner H. 2013. vegan: community ecology package. R package v2.0-10. http://CRAN.R-project.org/package=vegan.
- 65.Juggins S. 2016. Rioja: analysis of quaternary science data, v0.9-9. Cran. http://www.staff.ncl.ac.uk/stephen.juggins/.
- 66.Thiele S, Fuchs BM, Amann RI. 2011. Identification of microorganisms using the ribosomal RNA approach and fluorescence in situ hybridization. Treatise Water Sci 3:171–189. [Google Scholar]
- 67.Zubkov MV, Tarran GA. 2008. High bacterivory by the smallest phytoplankton in the North Atlantic Ocean. Nature 455:224–226. doi: 10.1038/nature07236. [DOI] [PubMed] [Google Scholar]
- 68.Toribio AL, Alako B, Amid C, Cerdeno-Tarraga A, Clarke L, Cleland I, Fairley S, Gibson R, Goodgame N, Ten Hoopen P, Jayathilaka S, Kay S, Leinonen R, Liu X, Martinez-Villacorta J, Pakseresht N, Rajan J, Reddy K, Rosello M, Silvester N, Smirnov D, Vaughan D, Zalunin V, Cochrane G. 2017. European Nucleotide Archive in 2016. Nucleic Acids Res 45:D32–D36. doi: 10.1093/nar/gkw1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Diepenbroek M, Glöckner F, Grobe P, Güntsch A, Huber R, König-Ries B, Kostadinov I, Nieschulze J, Seeger B, Tolksdorf R, Triebel D. 2014. Towards an integrated biodiversity and ecological research data management and archiving platform: the German Federation for the Curation of Biological Data (GFBio), p 1711–1721. In Plödereder E, Grunske L, Schneider E, Ull D (ed), Lecture notes in informatics (LNI)—proceedings, vol 232 Köllen Verlag, Bonn, Germany. [Google Scholar]
- 70.Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, Gilbert JA, Karsch-Mizrachi I, Johnston A, Cochrane G, Vaughan R, Hunter C, Park J, Morrison N, Rocca-Serra P, Sterk P, Arumugam M, Bailey M, Baumgartner L, Birren BW, Blaser MJ, Bonazzi V, Booth T, Bork P, Bushman FD, Buttigieg PL, Chain PSG, Charlson E, Costello EK, Huot-Creasy H, Dawyndt P, DeSantis T, Fierer N, Fuhrman JA, Gallery RE, Gevers D, Gibbs RA, San Gil I, Gonzalez A, Gordon JI, Guralnick R, Hankeln W, Highlander S, Hugenholtz P, Jansson J, Kau AL, Kelley ST, Kennedy J, Knights D, Koren O, et al. 2011. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol 29:415–420. doi: 10.1038/nbt.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlitzer R. 2017. Ocean Data View. http://odv.awi.de.
- 72.Gómez-Pereira PR, Schüler M, Fuchs BM, Bennke C, Teeling H, Waldmann J, Richter M, Barbe V, Bataille E, Glöckner FO, Amann R. 2012. Genomic content of uncultured Bacteroidetes from contrasting oceanic provinces in the North Atlantic Ocean. Environ Microbiol 14:52–66. doi: 10.1111/j.1462-2920.2011.02555.x. [DOI] [PubMed] [Google Scholar]
- 73.West NJ, Schönhuber WA, Fuller NJ, Amann RI, Rippka R, Post AF, Scanlan DJ. 2001. Closely related Prochlorococcus genotypes show remarkably different depth distributions in two oceanic regions as revealed by in situ hybridization using 16S rRNA-targeted oligonucleotides. Microbiology 147:1731–1744. doi: 10.1099/00221287-147-7-1731. [DOI] [PubMed] [Google Scholar]
- 74.Zubkov MV, Fuchs BM, Burkill PH, Amann R. 2001. Comparison of cellular and biomass specific activities of dominant bacterioplankton groups in stratified waters of the Celtic Sea. Appl Environ Microbiol 67:5210–5218. doi: 10.1128/AEM.67.11.5210-5218.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuchs BM, Woebken D, Zubkov MV, Burkill P, Amann R. 2005. Molecular identification of picoplankton populations in contrasting waters of the Arabian Sea. Aquat Microb Ecol 39:145–157. doi: 10.3354/ame039145. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data were deposited in the European Nucleotide Archive (ENA) (68) using the data brokerage service of the German Federation for Biological Data (GFBio) (69), in compliance with the MIxS standard (70). The INSDC accession number for the data is PRJEB39460.