

FAST in association with the fluorogenic ligand HMBR is characterized as a successful, highly fluorescent reporter system in C. acetobutylicum. FAST can be used to distinguish between promoters in live cells using flow cytometry or a fluorescence microplate reader and can be used to tag and examine protein localization in live, anaerobically grown cells. Given that FAST is highly fluorescent under anaerobic conditions, it can be used in several applications of this and likely many Clostridium organisms and other strict anaerobes, including studies involving cell sorting, sporulation dynamics, and population characterization in pure as well as mixed cultures, such as those in various native or synthetic microbiomes and syntrophic cultures.
KEYWORDS: anaerobic reporter, Clostridium acetobutylicum, cell biology, cell division, divisome, flow cytometry, fusion protein, population dynamics, protein localization, sporulation
ABSTRACT
Visualizing protein localization and characterizing gene expression activity in live Clostridium cells is limited for lack of a real-time, highly fluorescent, oxygen-independent reporter system. Enzymatic reporter systems have been used successfully for many years with Clostridium spp.; however, these assays do not allow for real-time analysis of gene expression activity with flow cytometry or for visualizing protein localization through fusion proteins. Commonly used fluorescent reporter proteins require oxygen for chromophore maturation and cannot be used for most strictly anaerobic Clostridium organisms. Here we show that the fluorescence-activating and absorption-shifting tag protein (FAST), when associated with the fluorogenic ligand 4-hydroxy-3-methylbenzylidene-rhodanine (HMBR; now commercially available) and other commercially available ligands, is highly fluorescent in Clostridium acetobutylicum under anaerobic conditions. Using flow cytometry and a fluorescence microplate reader, we demonstrated FAST as a reporter system by employing the promoters of the C. acetobutylicum thiolase (thl), acetoacetate decarboxylase (adc), and phosphotransbutyrylase (ptb) metabolic genes, as well as a mutant Pthl and modified ribosome binding site (RBS) versions of Padc and Pptb. Flow cytometry-based sorting was efficient and fast in sorting FAST-expressing cells, and positively and negatively sorted cells could be effectively recultured. FAST was also used to tag and examine protein localization of the predicted cell division FtsZ partner protein, ZapA, to visualize the divisome localization in live C. acetobutylicum cells. Our findings suggest that FAST can be used to further investigate Clostridium divisomes and more broadly the localization and expression levels of other proteins in Clostridium organisms, thus enabling cell biology studies with these organisms.
IMPORTANCE FAST in association with the fluorogenic ligand HMBR is characterized as a successful, highly fluorescent reporter system in C. acetobutylicum. FAST can be used to distinguish between promoters in live cells using flow cytometry or a fluorescence microplate reader and can be used to tag and examine protein localization in live, anaerobically grown cells. Given that FAST is highly fluorescent under anaerobic conditions, it can be used in several applications of this and likely many Clostridium organisms and other strict anaerobes, including studies involving cell sorting, sporulation dynamics, and population characterization in pure as well as mixed cultures, such as those in various native or synthetic microbiomes and syntrophic cultures.
INTRODUCTION
Fluorescent proteins (FPs) are an essential tool in molecular biology but are challenging to use in anaerobic bacteria like Clostridium organisms. Traditional FPs like green fluorescent protein (GFP) and mCherry require oxygen for chromophore maturation (1), which limits FP use for live observation of gene activity in anaerobic bacteria. Traditional FPs can be used in some (e.g., Clostridium perfringens) (2), but not all, Clostridium cells with aerobic fluorescence recovery (AFR) to allow for chromophore maturation in traditional FPs by incubation with oxygen after anaerobic growth (3). These Clostridium organisms must be aerotolerant for AFR, or they must be fixed before incubation with oxygen to preserve protein localization (4). However, traditional FPs have not been successful in Clostridium acetobutylicum, and AFR can be limiting for high-throughput applications like library screening and when tracking cells or protein localization in live cells (5).
Enzymatic reporter assays are traditionally used for gene expression analysis in Clostridium spp. (6–9), but lysing cells to isolate the protein of interest (POI) and the low-throughput enzyme assay can be tedious and time-consuming and cannot be used for microscopy studies or high-throughput screening by flow cytometry.
Anaerobic fluorescent proteins, like flavin-binding fluorescent proteins (FbFPs), which do not require oxygen for chromophore maturation (10), have been examined as possible effective reporters. For example, the FbFP phiLOV2.1 has been used as a reporter and fluorescent tag in Clostridioides difficile (formerly Clostridium difficile) (11). However, while FbFPs show strong fluorescence in Escherichia coli, in C. acetobutylicum, FbFPs like phiLOV2.1 and CreiLOV did not show fluorescence above background levels of the control (12).
A promising alternative to traditional FPs is the fluorescence-activating and absorption-shifting tag protein (FAST) (13), a 14-kDa variant of the photoactive yellow protein (PYP) that does not require oxygen for fluorescence activation. FAST is a fluorescence-activating protein (FAP) that binds to a fluorogenic ligand (such as 4-hydroxy-3-methylbenzylidene-rhodanine [HMBR]) to activate fluorescence. FAST has been used successfully in HeLa cells, zebrafish embryos, Saccharomyces cerevisiae, and E. coli but has not previously been tested in a strict anaerobe. Recently, FAST was used to study the low-oxygen environment of E. coli bacterial biofilms, and it was found to be superior to GFP and mCherry in such environments (14).
Here we demonstrate that a codon-optimized FAST is a strong fluorescing protein suitable as a reporter and for protein tagging in strictly anaerobic clostridia such as C. acetobutylicum. Because of the lack of strongly fluorescing proteins in anaerobic bacteria, localization of proteins in prokaryotic anaerobes has not been as widely investigated as in aerobic bacteria. In this study, we have chosen the cell division protein ZapA as a proof of concept for utilizing FAST as a protein tag in live C. acetobutylicum cells. The “Z ring” scaffold is made up of many different proteins involved in the recruitment of components of the cell division machinery, the divisome (15). A central component of the Z ring is the tubulin homolog, FtsZ (16), a self-activating GTPase widely conserved among prokaryotes (17, 18). FtsZ was previously used to characterize the phiLOV fluorescent system in C. difficile (11). ZapA, one of several components of the Z ring assembly in model prokaryotes, interacts with FtsZ during Z ring assembly (19) to enhance the polymerization and stability of FtsZ (16, 19–21). ZapA has previously been fused with mCherryOpt to show ZapA localization in fixed C. difficile cells (22). The predicted C. acetobutylicum ZapA (encoded by CA_C2355) (23) and its function have not been previously investigated.
RESULTS
Expression of FAST off the native and modified C. acetobutylicum thiolase promoter in E. coli and C. acetobutylicum shows promising fluorescence intensity using flow cytometry, a microplate reader, and confocal microscopy.
FAST was successfully expressed in E. coli (13). Before examining FAST expression in C. acetobutylicum, we wanted to confirm and characterize FAST expression in E. coli NEB 5-alpha using the E. coli-C. acetobutylicum pSOS95 shuttle vector under the control of the strong C. acetobutylicum thiolase promoter (Pthl) (7). A codon optimized for the C. acetobutylicum FAST gene was used (see Table S1 in the supplemental material). Flow cytometry analysis showed that expression of FAST in E. coli (strain termed E. coli FAST) when 20 μM HMBR was added showed high fluorescence during exponential growth and higher fluorescence in the stationary phase of culture, thus suggesting that the FAST protein is stable in E. coli. E. coli FAST without HMBR showed no fluorescence (Fig. S1A). E. coli FAST with 20 μM HMBR showed a large shift in fluorescein isothiocyanate (FITC) fluorescence compared to that of E. coli FAST in filtered phosphate-buffered saline (PBS) (no-HMBR control) after 22 h (Fig. S1B). Similarly, using a fluorescence microplate reader, E. coli FAST with 20 μM HMBR showed a higher fluorescence intensity in stationary phase, consistent with the flow cytometry data (Fig. S1C and S2A). E. coli FAST showed cells in exponential growth at around 4 h and cells in stationary phase at 22 h of growth (Fig. S1D). E. coli FAST with 20 μM HMBR showed a linear fluorescence intensity correlation with cell mass concentration (optical density at 600 nm [OD600]) (Fig. S2A) and a bright fluorescent signal using confocal microscopy compared to E. coli FAST in filtered PBS (Fig. S2B). There is no fluorescent signal in the absence of HMBR.
When FAST was expressed in C. acetobutylicum under the control of the native Pthl using p95thlFAST, only a low-fluorescence signal was detected (see, e.g., Fig. 1B). We hypothesized that this was due to low FAST expression. Thus, a mutant Pthl promoter, 1200-9-9 (24), which we here refer to as super Pthl (Pthlsup), was used to replace the native Pthl. This strain was termed C. acetobutylicum thlsupFAST. Using 20 μM HMBR in filtered PBS, FAST expression in this strain resulted in increased fluorescence compared to that of C. acetobutylicum thlFAST or the wild-type (WT) C. acetobutylicum control (Fig. 1A). Using confocal microscopy, C. acetobutylicum thlsupFAST with 20 μM HMBR had more frequent and brightly fluorescent cells than C. acetobutylicum thlFAST, which had fewer brightly fluorescent cells and an overall dimmer fluorescent signal (Fig. 1B). Control WT C. acetobutylicum with 20 μM HMBR showed slight autofluorescence (Fig. 1B). In the absence of HMBR and in filtered PBS, no fluorescent signal was detected in any of the three strains examined, WT C. acetobutylicum, C. acetobutylicum thlFAST, or C. acetobutylicum thlsupFAST, using confocal microscopy (Fig. S3).
FIG 1.

C. acetobutylicum thlsupFAST shows improved fluorescence compared to C. acetobutylicum thlFAST and C. acetobutylicum ATCC 824 using flow cytometry and confocal microscopy. (A) Geometric mean fluorescence of C. acetobutylicum ATCC 824, C. acetobutylicum thlFAST, and C. acetobutylicum thlsupFAST after ∼10 h at an OD600 of ∼1.0 (n = 2). Error bars show SD. *, P < 0.05 (Student t test). (B) Confocal microscopy of C. acetobutylicum ATCC 824 (top), C. acetobutylicum thlFAST (middle), and C. acetobutylicum thlsupFAST (bottom) with 20 μM HMBR (left), brightfield (middle), and merge (right) after ∼30 h. (C) Confocal microscopy of C. acetobutylicum thlsupFAST vegetative cells (left) and clostridial-form cells (top and bottom right) with 20 μM HMBR after ∼20 h.
Plamont and colleagues have demonstrated the reversibility of HMBR binding to FAST (13). We wanted to test if this is also the case for the Gram-positive C. acetobutylicum. C. acetobutylicum thlsupFAST cells were washed twice with filtered PBS after being exposed to 20 μM HMBR and showed a low mean fluorescence intensity (MFI) after each wash. The low fluorescence is apparently due to residual ligand retained in the cells. The same C. acetobutylicum thlsupFAST cells were exposed again to HMBR, and this restored fluorescence to the original high levels. When cells were washed twice again, fluorescence was reduced to low levels after each wash. These data demonstrate the reversibility of HMBR binding to FAST in Gram-positive cells (Fig. S4A) and also that HMBR appears to freely permeate the cells in both directions. We also wanted to test the linear fluorescence stability of C. acetobutylicum thlsupFAST over time. To do this, we created dilutions of C. acetobutylicum thlsupFAST cultures in filtered PBS with 20 μM HMBR from an OD600 of 0.078 to an OD600 of 5. Fluorescence intensity was linear with cell density (OD600). It increased from 7 h to 11 h of growth and from 11 h to 24 h of growth but stayed consistently fluorescent after 24 h of growth (Fig. S4B). C. acetobutylicum thlsupFAST showed fluorescence (FAST expression) in both vegetative and clostridial-form cells (Fig. 1C).
We also wanted to compare our synthesized HMBR to the newly commercially available version of HMBR (TFLime) and another FAST ligand, TFCoral, among five now commercially available (Twinkle Bioscience). We also compared the fluorescence levels of the cells with and without washing with PBS before exposure to the ligand. There was no statistically significant difference between our synthesized HMBR versus its commercial version, TFLime (Fig. S5A). Fluorescence from using the TFCoral ligand was strong as well (Fig. S5). TFCoral showed low overlapping signal using the FITC filter (Fig. S5A). The phycoerythrin-Texas Red (PE-Tx Red) filter, HMBR, and TFLime showed no overlapping signal with TFCoral (Fig. S5B). Washing the cells with PBS prior to ligand addition decreased somewhat the fluorescence (Fig. S5A). The higher fluorescence from the unwashed cells is likely due to the remaining growth medium on the cells. Importantly, though, these data show that the ligands can be used on unwashed cells directly for effortless fluorescence detection.
Development of a FAST-based fluorescent reporter system for C. acetobutylicum.
Tummala et al. have previously established a beta-galactosidase activity-based reporter assay for C. acetobutylicum using the early-growth-stage promoter Pptb, the more broadly expressed Pthl, and the late-growth-stage promoter Padc (7). The ptb promoter controls the gene encoding the phosphotransbutyrylase, which is involved in the production of butyrate in C. acetobutylicum during exponential-phase-based acidogenesis (25, 26). The thl promoter controls the gene encoding the thiolase which carries out a condensation reaction between 2 molecules of acetyl coenzyme A (acetyl-CoA), a reaction utilized throughout active culture (including the stationary-phase-associated solvent production) as long as there is active sugar metabolism (26, 27). The adc promoter controls the gene encoding the acetoacetate decarboxylase, which catalyzes the decarboxylation of acetoacetate to form acetone during solvent production, and is thus expressed late in culture (26, 28). In addition to the C. acetobutylicum thlFAST and thlsupFAST strains discussed above, we constructed strains C. acetobutylicum adcFAST and ptbFAST using the native Padc and native Pptb promoters to express FAST. Inspired by the optimized spacer between the RBS and start codon developed by Yang and colleagues for the Pthlsup promoter (24), we used the same spacer to modify the native Padc and Pptb promoters, thus generating plasmids p95adcmodFAST and p95ptbmodFAST for expressing FAST in C. acetobutylicum.
Using flow cytometry and a fluorescence microplate reader, WT control C. acetobutylicum with 20 μM HMBR background fluorescence was measured over 37 h and was found to be minimal (Fig. S6A and B). We then examined FAST fluorescence by flow cytometry and the plate reader for the various promoters. In flow cytometry histograms of C. acetobutylicum thlFAST, only a small portion of cells are fluorescent between 7 and 15.5 h, with a small shift in fluorescent populations at 31 h (Fig. 2A). C. acetobutylicum thlsupFAST showed a larger shift in green fluorescence than did C. acetobutylicum thlFAST, with a bimodal cell population for most of the culture (Fig. 2B). When the whole population was used to calculate the MFI by flow cytometry, C. acetobutylicum thlFAST showed consistently low MFI over the 36 h of culture (Fig. 2C). When MFI was measured based only on the fluorescent population, C. acetobutylicum thlFAST showed a higher but still rather low MFI (Fig. 2D). When MFI was measured based on the whole cell population, C. acetobutylicum thlsupFAST showed the highest MFI early, with MFI decreasing thereafter (Fig. 2E). When only the fluorescent population was used to calculate MFI, C. acetobutylicum thlsupFAST showed an overall high MFI increasing with culture progression (Fig. 2F).
FIG 2.

Histograms of thlFAST and thlsupFAST show improvement in fluorescence shift over time, and thlsupFAST shows a pattern consistent with promoter activity when fluorescence of the whole population is measured. (A) Histograms showing C. acetobutylicum thlFAST (left column) after 7 h, 11 h, 15.5 h, 31 h, and 35 h. (B) Histograms showing C. acetobutylicum thlsupFAST (right column) after 6 h, 10 h, 14.5 h, 30 h, and 34 h, compared with WT C. acetobutylicum (gray) after 9 h of growth. (C) C. acetobutylicum thlFAST geometric means of entire population and growth curve over ∼35 h (n = 4). (D) C. acetobutylicum thlFAST geometric mean fluorescence of the fluorescent population and growth curve over ∼35 h in Turbo CGM plus erythromycin using flow cytometry (n = 4). (E) C. acetobutylicum thlsupFAST geometric means of entire population and growth curve over ∼36 h (n = 3). (F) C. acetobutylicum thlsupFAST geometric mean fluorescence of the fluorescent population and growth curve in Turbo CGM plus erythromycin or Turbo CGM over ∼36 h using flow cytometry (n = 3). Error bars indicate SD. Lag times were standardized between fermentations by normalizing an OD600 of 1.0 at hour 10 of growth as previously described (49).
Both C. acetobutylicum adcFAST and adcmodFAST showed a low green fluorescence shift early in culture, with a dramatic shift after 30 h of culture consistent with the expectation that this is a transition and stationary-phase promoter (Fig. 3A and B). C. acetobutylicum adcmodFAST had a slightly larger shift in the fluorescent population after 30 h, a more pronounced bimodal population after 34 h (Fig. 3B), and an overall higher MFI. Whether MFI is based on the whole cell population or the fluorescent population (Fig. 3C to F), the overall trend is the same, showing increasing MFI with time during a standard batch culture, which is consistent with the fact that the adc gene is expressed late in culture.
FIG 3.

Histograms of adcFAST and adcmodFAST over time show slight improvement in fluorescence shift over time, and both adcFAST and adcmodFAST show patterns consistent with promoter activity when fluorescence of the whole population is measured and when only the fluorescent population is measured. (A) Histograms showing C. acetobutylicum adcFAST (left column) after 6 h, 10 h, 14.5 h, 30 h, and 34 h. (B) Histograms showing C. acetobutylicum adcmodFAST (right column) after 6 h, 10 h, 14.5 h, 30 h, and 34 h compared with WT C. acetobutylicum (gray) after 9 h of growth. (C) C. acetobutylicum adcFAST geometric means of entire population and growth curve over ∼34 h (n = 2). (D) C. acetobutylicum adcFAST geometric mean fluorescence of the fluorescent population and growth curve over ∼34 h in Turbo CGM plus erythromycin using flow cytometry (n = 2). (E) C. acetobutylicum adcmodFAST geometric means of entire population and growth curve over ∼34 h (n = 3). (F) C. acetobutylicum adcmodFAST geometric mean fluorescence of the fluorescent population and growth curve over ∼34 h in Turbo CGM plus erythromycin using flow cytometry (n = 2). Error bars indicate SD. Lag times were standardized between fermentations by normalizing an OD600 of 1.0 at hour 10 of growth as previously described (49).
C. acetobutylicum ptbFAST (Fig. S7A) and C. acetobutylicum ptbmodFAST (Fig. S7B) showed comparable green fluorescence shift patterns in early culture using flow cytometry and differed only slightly after 30 h, with C. acetobutylicum ptbmodFAST showing a more pronounced shift in the fluorescent population at 30 h than the nonfluorescent control. Overall, the MFI values were lower than those of the Pthl and Padc promoters (native and modified RBS), with MFI based on the fluorescent population being, as expected, higher and with somewhat different patterns, reflecting errors from the population gating (and thus the MFI values) when the fluorescence intensity is relatively low.
Fluorescence microplate readers are some of the most widely available instruments for fluorescence detection, so we sought to further validate the FAST using such a microplate reader. The data for the six promoters are shown in Fig. 4. These data are largely consistent with the data from flow cytometry. Specifically, they confirm that the super thiolase promoter results in fluorescences ca. 2.5 times higher than that with the native thiolase promoter, that Padc shows the highest fluorescence intensity among all promoters and is expressed at higher levels late in culture, and that in contrast, the thl- and ptb-based promoters show higher expression during exponential growth, with the exception of the phosphotransbutyrylase promoter with the modified RBS.
FIG 4.

C. acetobutylicum thlsupFAST shows improved fluorescence over time compared to C. acetobutylicum thlFAST, but C. acetobutylicum adcFAST, and C. acetobutylicum ptbFAST show higher fluorescence intensity than C. acetobutylicum adcmodFAST and C. acetobutylicum ptbmodFAST over time as determined with a fluorescence microplate reader. (A) C. acetobutylicum thlFAST fluorescence intensity and growth curve over ∼35 h in Turbo CGM plus erythromycin using a fluorescence microplate reader (n = 2). (B) C. acetobutylicum thlsupFAST fluorescence intensity and growth curve in Turbo CGM plus erythromycin or Turbo CGM over ∼36 h using a fluorescence microplate reader (n = 3). (C) C. acetobutylicum adcFAST fluorescence intensity and growth curve over ∼36 h in Turbo CGM plus erythromycin using a fluorescence microplate reader (n = 2). (D) C. acetobutylicum adcmodFAST fluorescence intensity and growth curve over ∼36 h in Turbo CGM plus erythromycin using a fluorescence microplate reader (n = 2). (E) C. acetobutylicum ptbFAST fluorescence intensity and growth curve over ∼37 h in Turbo CGM plus erythromycin using a fluorescence microplate reader (n = 2). (F) C. acetobutylicum ptbmodFAST fluorescence intensity and growth curve over ∼36 h in Turbo CGM plus erythromycin using a fluorescence microplate reader (n = 2). Error bars indicate SD. Lag times were standardized between fermentations by normalizing an OD600 of 1.0 at hour 10 of growth as previously described (49).
Fluorescence-assisted cell sorting of C. acetobutylicum using FAST.
Flow cytometry and fluorescence-assisted cell sorting (FACS) of C. acetobutylicum have been used previously to study microbial heterogeneity of endospore formation (29). Because FAST is a promising fluorescent reporter, we investigated FAST for use in FACS using C. acetobutylicum thlsupFAST and adcFAST. For cell sorting using FAST, we tested cells in exponential growth, cells in early stationary phase, and cells in late stationary phase to examine the morphology of cells expressing FAST and test if positively or negatively sorted cells (based on FAST fluorescence) could be recultured. Each population was gated for the lowest 25% fluorescence and the highest 25% fluorescence and sorted based on these gates. As expected, exponentially growing cultures of both C. acetobutylicum thlsupFAST and adcFAST showed only vegetative cells in both high-fluorescence and low-fluorescence gates (data not shown). C. acetobutylicum thlsupFAST in early stationary phase showed vegetative-type cells in the low-fluorescence population and both vegetative and clostridial-form cells in the high-fluorescence population (Fig. 5A). In later stationary phase, the C. acetobutylicum thlsupFAST low-fluorescence population showed vegetative morphology as well as cell debris and dying cells, while the high-fluorescence population showed cells in clostridial morphology and clumping vegetative cells (Fig. 5B). Sorting cells quickly (about 5 to 10 min) under aerobic conditions (see Materials and Methods for details), both low-fluorescence and high-fluorescence sorted C. acetobutylicum thlsupFAST cell populations (about 300,000 cells in each population) could be recultured in standard medium when returned to the anaerobic chamber, with growth being observed after about 2 days, which is similar to the time it would take to observe growth from a small number of cells not exposed to oxygen. The recultured cells from both the low-fluorescence and high-fluorescence populations demonstrated typical C. acetobutylicum thlsupFAST fluorescence activity and could not be distinguished from each other after reculturing. The C. acetobutylicum adcFAST low-fluorescence population in early stationary phase showed only vegetative cells, while the high-fluorescence population showed both vegetative and clostridial morphologies (Fig. S8A). In late stationary phase, the low-fluorescence C. acetobutylicum adcFAST population contained vegetative cells and cell debris, while the high-fluorescence population showed both clumping vegetative and clostridial morphologies (Fig. S8B). The entire C. acetobutylicum adcFAST population displayed a decrease in fluorescence in late stationary phase. Overall, highly fluorescent cells for both strains had higher forward scatter (FSC) and side scatter (SSC), which means larger (higher FSC) and more granular (higher SSC) cells. Assuming that the amount of FAST per cell is, on average, the same for all cells in a given physiological state, then the larger cells have more total FAST protein, and this may be why they may have stronger fluorescence. This would be corroborated by the fact that following cell sorting, upon subculturing, low- and high-fluorescence sorted cells give rise to similar cell populations in terms of FAST fluorescence intensity.
FIG 5.

Sorting low (25%)- and high (25%)-fluorescence populations of C. acetobutylicum thlsupFAST shows that FAST is expressed in both vegetative and clostridial morphologies and low-fluorescence cells remain in a vegetative state. (A) C. acetobutylicum thlsupFAST histogram (top) of low fluorescence (P1) and high fluorescence (P2) and dot plots (bottom) with accompanying phase-contrast images of sorted cell populations after 32 h of growth. (B) C. acetobutylicum thlsupFAST histogram (top) of low fluorescence (P1) and high fluorescence (P2) and dot plots (bottom) with accompanying phase-contrast images of sorted cell populations after 56 h of growth. Scale bars in insets represent 5 μm.
Development of a successful bacterial cell division (ZapA) fusion protein using FAST to examine the Z-ring formation in the C. acetobutylicum divisome.
There are currently not many options for creating fluorescent fusion proteins in Clostridium organisms. Cell fixation and aerobic fluorescence recovery can be time-consuming (22) and do not seem to work for all Clostridium organisms. Previously established anaerobic fluorescent proteins have been shown to have low fluorescence expression in clostridia (12). FAST has previously been used to create fusion proteins and can be used to successfully observe protein localization in eukaryotic cells (13, 30), but there are no reports of FAST-based fusion proteins in bacteria. To test whether FAST can be used to tag cell proteins in C. acetobutylicum, we designed and expressed a FAST-based fusion protein of the hypothetical FtsZ interacting cell division protein ZapA (CAC2355). The C-terminal end of C. acetobutylicum ZapA was fused to the N-terminal end of FAST using a small protein linker (GGGS) aiming to avoid any steric hindrances of the N-terminal, bundling-associated head of ZapA (16, 31). Expression was driven by the native Pthl. ZapA-FAST was first expressed in E. coli, where fluorescently labeled ZapA protein appears to be localized at the center of dividing native components of the E. coli divisome. Most cells were highly elongated, with some of such elongated cells showing three fluorescent ZapA loci apparently from cells that aborted cell division, thus leading to cell elongation. Cell elongation is observed when the native ZapA is overexpressed in E. coli (32), and this is consistent with the cell elongation phenotype observed in this study (Fig. 6). Expression of the ZapA FAST fusion in C. acetobutylicum proved equally successful, with fluorescent ZapA loci observed in the center of cell but also at the opposite poles of dividing cells (Fig. 7). This localization (center and poles) is consistent with Z-ring formation in the model Gram-positive endospore-forming Bacillus subtilis (33): the polar Z ring is necessary for establishing cellular asymmetry during commitment to sporulation. Cells committing to sporulation form polar Z rings, with some cells showing both polar and middle-cell Z rings at a transient cellular state. Although the sporulation programs of B. subtilis and Clostridium organisms are not identical (34), this model appears to apply to C. acetobutylicum at least, based on the images in Fig. 7.
FIG 6.
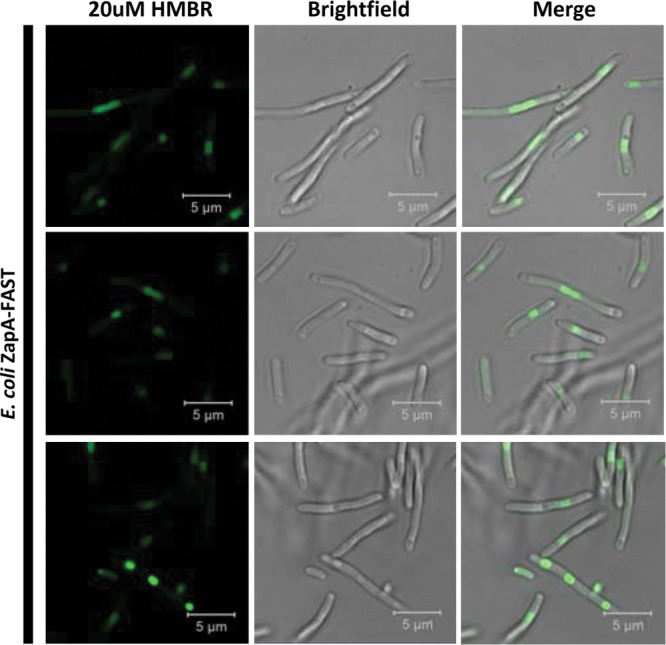
FAST and HMBR are an efficient system to view protein localization within E. coli cells. Shown is E. coli ZapA-FAST with 20 μM HMBR (left), brightfield (middle), and merge (right) using confocal microscopy. Each row represents a different field of view.
FIG 7.
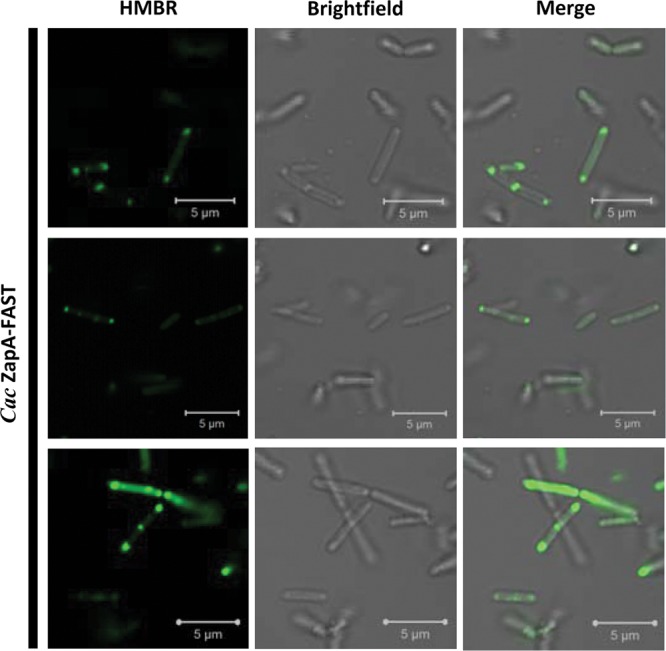
FAST and HMBR are an efficient system to view protein localization within C. acetobutylicum cells. Shown is C. acetobutylicum ZapA-FAST with 20 μM HMBR (left), brightfield (middle), and merge (right) using confocal microscopy. Each row represents a different field of view.
DISCUSSION
The goals of this study were to establish a highly fluorescent oxygen-independent reporter system using FAST and to show that FAST fusion proteins can be functional in live C. acetobutylicum cells. Tummala et al. (7) have previously established a beta-galactosidase reporter system using the promoters Pthl, Padc, and Pptb, with Pptb and Pthl showing higher activity in exponential growth and Padc showing higher activity in the stationary phase of batch cultures without pH control. The FAST-based promoter activity data presented here are consistent with the data of Tummala et al. (7), although not identical, apparently due to likely different translational efficiencies and stability of the two reporter proteins, as well as the fact that one is based on cell lysates and the other on whole-cell fluorescence.
Flow cytometry is a high-throughput method for detecting and measuring several physical, chemical, and biological characteristics of single cells (3). We showed that FAST and the fluorogen HMBR can be used to examine real-time gene activity using flow cytometry, demonstrating promoter activity patterns consistent with what was expected from three well-known promoters, but also to identify population heterogeneity in terms of gene expression. The microplate reader measures an average MFI based on the whole population, while flow cytometry allows one to examine different subpopulations in terms of expression activity. It has been shown that due to differences in cell differentiation and morphogenesis, sporulating bacterial populations, including Clostridium organisms, have heterogeneous populations when examined by flow cytometry (3, 29) and consequently, one can couple fluorescence from FAST alone or FAST-based fusion proteins to examine expression in different stages of sporulation of C. acetobutylicum and other Clostridium organisms. The origin of replication in the vectors used in this study, pIM13, has been shown to be very stable even without antibiotic use (35, 36). Even with the additional erythromycin added at 24 h of growth, we still saw a nonfluorescent population. This heterogeneity in the fluorescent population is what is expected from the sporulating population of C. acetobutylicum as previously captured by flow cytometric analysis of forward (FSC) versus side (SSC) scatter (29) over the culture duration. In essence, this means that all cell dimensions change over the culture period (see, e.g., the different cell sizes in the microscopy image insets in Fig. 5), and this will affect the fluorescence intensity of individual cells even if the average concentration of the fluorescent protein is the same, which is generally not the case. FAST is not evenly distributed in the sporulating cells, as shown in the images in Fig. 1C: there is indeed an asymmetric distribution of FAST in the differentiating cells.
Here we demonstrated that FAST can also be used for FACS-based cell sorting and that FAST is expressed in both vegetative and clostridial morphologies (Fig. 5). As we have shown that cells sorted quickly aerobically can be effectively recultured when returned to an anaerobic environment, flow cytometry can also be used to quickly and efficiently screen promoter libraries or any type of DNA sequence that imparts differential expression. Rohlhill et al. have used a sorting-sequencing (sort-seq) method for sorting variants of a formaldehyde-inducible E. coli promoter (Pfrm) (37). Using FAST, this method could also be adapted to quickly sort promoter libraries in Clostridium organisms and other anaerobes but also to develop more advanced genomic and metagenomics library screening technologies, such as those based on the fluorescent-protein trap concept (38).
We chose a predicted cell division protein to view protein localization in C. acetobutylicum because the localization pattern has already been established in other systems (16, 19, 32, 39). Overexpression of ZapA-FAST in E. coli appears to localize to the Z ring and may potentially replace the native ZapA. This could be what caused the elongated cell morphology in E. coli (Fig. 6), which is consistent with overexpression of the native E. coli ZapA (32). C. acetobutylicum ZapA-FAST appeared to localize in the center of dividing cells, as well as to opposite poles of dividing cells (Fig. 7), which is consistent with the B. subtilis model of cells committing to sporulation (33). The Clostridium divisome has not been well characterized due to the limitation of traditional FPs for viewing anaerobic cell division (40). It has been shown that the cell division-associated Min homolog in anaerobic Clostridioides difficile shows an oscillating pattern when tagged with yellow fluorescent protein (YFP) and expressed in B. subtilis (41). It was also shown that the DivIVA and MinD proteins show direct interaction in both C. difficile and Clostridium beijerinckii (40). The FAST-ZapA data presented here suggest the exciting potential of using FAST to tag other cell division proteins for live cell division imaging in C. acetobutylicum, other Clostridium spp., and, more broadly, other anaerobic prokaryotes.
The development of a fluorescent reporter system in C. acetobutylicum using FAST has allowed us to distinguish between autofluorescence and fluorescent signal, which previously was shown to not be possible using other oxygen-independent fluorescent proteins (12). Using FAST provides a solution to previous challenges faced by other aerobic and anaerobic fluorescent proteins. FAST could also be utilized in developing fluorescent reporter systems in other Clostridia spp. as well as other anaerobic bacteria. FAST fluorogenic ligands are now commercially available through Twinkle Bioscience in four TFFluorogen colors. Improved FAST variants have been recently reported, namely, the brighter FAST2, obtained by further optimization of FAST, as well as tandem versions of both FAST (tdFAST1) and FAST2 (tdFAST2) (42). The strong fluorescence of these new FAST variants could potentially help characterize expression of weak promoters and the function of low- expression uncharacterized proteins in Clostridium spp. and other anaerobes.
MATERIALS AND METHODS
Chemicals.
All chemicals were purchased from Sigma-Aldrich unless noted otherwise. E. coli NEB 5-alpha, Q5 DNA polymerase, and NEBuilder HiFi DNA assembly master mix were purchased from New England BioLabs (NEB). Restriction endonucleases were purchased from Thermo Fisher Scientific. TFLime and TFCoral were purchased from Twinkle Biosciences.
HMBR synthesis.
Rhodanine (200 mg; Acros), 4-hydroxy-3-methylbenzaldehyde (213 mg; Aldrich), and distilled water (130 ml) were combined in a screw-cap vial and stirred at 125 rpm in a water bath at 65°C for 8 days. The mixture was cooled to room temperature overnight, then filtered over a 47-mm fiberglass filter, and dried overnight in a desiccator with phosphorous pentoxide (13). The synthesis resulted in a yellow powder (191 mg; 51%). 1H nuclear magnetic resonance (NMR) (600 MHz, dimethyl sulfoxide [DMSO]-d6) specifications were as follows: δ 13.69 (s, 1H) 10.38 (s, 1H), 7.53 (s, 1H), 7.37 to 7.29 (m, 2H), 6.95 (d, J = 8.4 Hz, 1H), 2.18 (s, 3H).
Bacterial strains, media, and growth conditions.
All strains and plasmids used in this study are listed in Table 1. E. coli NEB 5-alpha was used to propagate all plasmids. E. coli strains were grown aerobically at 37°C at 250 rpm in liquid LB medium or solid LB agar medium. Appropriate antibiotics were added at the following concentrations: ampicillin, 100 μg/ml, and kanamycin, 25 μg/ml. E. coli and C. acetobutylicum strains were stored at −85°C in 20% glycerol. C. acetobutylicum ATCC 824 was grown anaerobically in either liquid or solid 2xYTG medium (43) or Turbo CGM medium (44). Erythromycin was added at the following concentrations where appropriate: 40 μg/ml for plates and 100 μg/ml for liquid cultures. Spore-forming strains were grown on 2xYTG for at least 5 days, and colonies were inoculated in 10 ml of Turbo CGM and heat shocked for 10 min at 70 to 80°C to kill vegetative cells that may have lost the megaplasmid pSOL1 (45). Optical density at 600 nm (OD600) was measured using a Beckman-Coulter DU370 spectrophotometer.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli | ||
| NEB 5-alpha | fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | NEB |
| ER2275 | hsdR mcrA recA1 endA1 | NEB |
| C. acetobutylicum | ||
| ATCC 824 | Wild-type strain | ATCC |
| Plasmids | ||
| pAN3 | Kmr; Φ3T I gene | 46 |
| pSOS94_MCS | ptb promoter; Ampr MLSr ColE1 Ori repL MCS | 46 |
| pSOS95_MCS | thl promoter; Ampr MLSr ColE1 Ori repL MCS | 43 |
| p95thlFAST | thl promoter; Ampr MLSr ColE1 Ori repL FAST | This study |
| p95thlsupFAST | thlsup promoter; Ampr MLSr ColE1 Ori repL FAST | This study |
| p95adcFAST | adc promoter; Ampr MLSr ColE1 Ori repL FAST | This study |
| p95adcmodFAST | adcmod promoter; Ampr MLSr ColE1 Ori repL FAST | This study |
| p95ptbFAST | ptb promoter; Ampr MLSr ColE1 Ori repL FAST | This study |
| p95ptbmodFAST | ptbmod promoter; Ampr MLSr ColE1 Ori repL FAST | This study |
| p95ZapA-FAST | thl promoter; Ampr MLSr ColE1 Ori repL ZapA FAST | This study |
Genetic manipulations.
All primers used in this study are listed in Table 2. A schematic of plasmid constructs for this study is shown in Fig. S8. The vector p95thlFAST was constructed to express FAST in C. acetobutylicum using the strong Pthl promoter native to C. acetobutylicum. An Integrated DNA Technologies Gblock was created for FAST (13) and was codon optimized for C. acetobutylicum (Table S1) using the Integrated DNA Technologies Codon Optimization Tool. The pSOS95_MCS vector (43) was digested using BamHI and the codon-optimized FAST gene was cloned into the vector using Gibson Assembly (NEB). Vector p95thlsupFAST was constructed to express FAST in C. acetobutylicum using the optimized Pthl promoter 1200-9-9 (24). An Integrated DNA Technologies Gblock was created for the Pthl promoter 1200-9-9 (Pthlsup). The p95thlFAST backbone was amplified using primers p95_thlFAST_F and p95_thlFAST_R, digested with DpnI in order to reduce the methylated plasmid background during transformation, and PCR purified using the QIAquick PCR purification kit (Qiagen). Pthlsup was cloned into p95thlFAST using Gibson Assembly. The vector p95ptbFAST was constructed to express FAST in C. acetobutylicum using the Pptb promoter native to C. acetobutylicum. The p95thlFAST backbone was amplified using primers p95_thlFAST_F and p95_thlFAST_R, and Pptb was amplified from the pSOS94_MCS vector (43, 46) using primers ptb_F and ptb_R. Pptb was cloned into p95thlFAST using Gibson Assembly. p95ptbmodFAST was constructed to express FAST in C. acetobutylicum using the Pptb promoter and the p95thlsupFAST backbone. The p95thlsupFAST backbone was amplified using primers p95_thlsupFAST_F and p95_thlsupFAST_R, and Pptb was amplified from the pSOS94_MCS vector using primers mod_ptb_F and mod_ptb_R. Pptb was cloned into p95thlsupFAST using Gibson Assembly. The vector p95adcFAST was constructed to express FAST in C. acetobutylicum using the Padc promoter native to C. acetobutylicum. The p95thlFAST backbone was amplified using primers p95_thlFAST_F and p95_thlFAST_R, and Padc was amplified from the pSOS95_MCS vector using primers adc_F and adc_R. The vector p95adcmodFAST was constructed to express FAST in C. acetobutylicum using the Padc promoter native to C. acetobutylicum. The p95thlsupFAST backbone was amplified using primers p95_thlsupFAST_F and p95thlsupFAST_R, and Padc was amplified from the pSOS95_MCS vector using primers mod_adc_F and mod_adc_R. The plasmid p95ZapA-FAST was created using the E. coli-Cac shuttle vector pSOS95_MCS. An Integrated DNA Technologies Gblock was created using the native thl promoter and fusing the C-terminal end of ZapA with the N-terminal end of FAST with a short fusion protein linker (GGGS) using the sequence GGAGGTGGAAGC. The p95thlsupFAST vector backbone was digested using SacI and EcoRI enzymes, and the thlZapA-FAST Gblock fragment was cloned into the backbone using Gibson Assembly.
TABLE 2.
Primers used in this study
| No. | Primer identifier | Sequence (5′–3′)a |
|---|---|---|
| 1 | p95_thlFAST_F | CAGGAGGTAGTCTATATGGAACAC |
| 2 | p95_thlFAST_R | AAAAAATAAAGAGGGTTATAATGAAC |
| 3 | p95_thlsupFAST_F | AGGAGGTTAGGATCCATGG |
| 4 | p95_thlsupFAST_R | AAAAAATAAAGAGGGTTATAATGAAC |
| 5 | ptb_F | ttcattataaccctctttattttttCCTCCTTATAAAATTAGTATAATTATAGC |
| 6 | ptb_R | cgtgttccatatagactacctcctgCATTTATATTTTAACAAACTTTTCACATG |
| 7 | mod_ptb_F | ttcattataaccctctttattttttCCTCCTTATAAAATTAGTATAATTATAGC |
| 8 | mod_ptb_R | cgtgttccatggatcctaacctcctCATTTATATTTTAACAAACTTTTCACATG |
| 9 | adc_F | ttcattataaccctctttattttttCCTCCTTATAAAATTAGTATAATTATAGC |
| 10 | adc_R | cgtgttccatatagactacctcctgAAAAGTCACCTTCCTAAATTTAATAATG |
| 11 | mod_adc_F | ttcattataaccctctttattttttCCTCCTTATAAAATTAGTATAATTATAGC |
| 12 | mod_adc_R | cgtgttccatggatcctaacctcctAAAAGTCACCTTCCTAAATTTAATAATG |
Binding regions are indicated by uppercase letters. Homology regions are indicated by lowercase letters.
Cell transformation.
Vectors were transformed into E. coli (NEB 5-alpha) and isolated using the QIAprep spin miniprep kit (Qiagen). Before transformation into C. acetobutylicum, vectors were transformed into electrocompetent E. coli ER2275(pAN3) (46) for in vivo methylation by the Bacillus subtilis phage Φ3T I methyltransferase (47) encoded on the pAN3 vector. Vectors were isolated using the QIAprep spin miniprep kit (Qiagen) and transformed into C. acetobutylicum as described previously (48).
Flow cytometry and sorting.
Cell fluorescence was analyzed and C. acetobutylicum cells were sorted with a BD FACSAria IIu flow cytometer (Becton, Dickinson). A blue solid-state laser (488-nm excitation) and a 530/30-nm filter were used to measure FAST. Flow Cytometry Standard (FCS) files were analyzed and histograms were created using Flowing Software v2.5.1 (Cell Imaging Core, Turku Centre for Biotechnology, Turku, Finland). For E. coli flow cytometry sampling, the geometric mean of the FITC-A fluorescence for 10,000 events was taken as the MFI (in arbitrary units [AU]). For C. acetobutylicum flow cytometry sampling, fluorescent populations of 10,000 events were normalized to C. acetobutylicum autofluorescence by creating a gate where C. acetobutylicum ATCC 824 control without HMBR had fluorescence of <1% unless otherwise specified and the geometric mean of the FITC-A fluorescence was measured for each fluorescent population as the MFI (in AU). E. coli and C. acetobutylicum cells were grown until early to mid-exponential phase of growth (OD600 of 0.1 to 1). Samples were pelleted aerobically at 2,400 × g for 15 to 30 min and washed with filtered PBS. Samples were resuspended to an OD600 of 1 in either filtered PBS or filtered 20 μM HMBR in PBS. A total of 5 μl of sample was then added to 500 μl of filtered PBS or filtered 20 μM HMBR in PBS for immediate flow cytometry analysis. Time courses were standardized by normalizing an OD600 at hour 10 of growth as previously reported (49).
For sorting, C. acetobutylicum thlsupFAST and C. acetobutylicum adcFAST were sorted into two gates of the highest 25% of fluorescent cells and the lowest 25% of fluorescent cells in the population, and 200,000 to 300,000 events (cells or small cell aggregates) were collected in each gate and sorted directly into anaerobic PBS. These cells were pelleted at 5,000 × g, the supernatant was carefully removed, and the pellet was resuspended in the remaining ∼10 μl of anaerobic PBS and prepared for imaging.
Fluorescence measurements in SpectraMax i3x microplate reader.
E. coli and C. acetobutylicum cells were grown until the early to mid-exponential phase of growth (OD600 of 0.1 to 1). Samples were pelleted at 2,400 × g for 15 to 30 min and washed with filtered PBS. Samples were resuspended to an OD600 of 1 in either filtered PBS or filtered 20 μM HMBR in PBS and 100 μl was transferred to black conical-bottom 96-well plates (BRANDplates) for immediate fluorescence measurement. Fluorescence was measured at an excitation wavelength of 485 nm and emission wavelength of 535 nm using a SpectraMax i3x microplate reader (Molecular Devices, San Jose, CA). C. acetobutylicum samples were normalized to C. acetobutylicum ATCC 824 resuspended to an OD600 of 1 in filtered PBS. E. coli samples were normalized to E. coli FAST resuspended in filtered PBS. Time courses were standardized by normalizing the OD600 at hour 10 of growth as previously reported (49).
Confocal microscopy.
Eight-well μ-Slides (ibidi, Martinsried, Germany) were incubated with 0.1% (wt/vol) poly-l-lysine in H2O overnight, then washed with double-distilled water (ddH2O), and dried. E. coli and C. acetobutylicum cells were grown in an overnight culture, and samples were pelleted at 5,000 rpm for 15 to 30 min and washed with filtered PBS. Samples were resuspended to an OD600 of 1 to 2 in filtered PBS, and 200 to 300 μl was transferred to poly-l-lysine-coated 8-well μ-Slides and incubated for 1 h at room temperature. Cells were washed aerobically with sterile PBS, and 300 μl of either filtered PBS or filtered 20 μM HMBR in PBS was added to each well. Cells were imaged using confocal microscopy (Zeiss 880 Multiphoton confocal microscope with Airyscan) at the Delaware Biotechnology Institute Bioimaging Center. Images were acquired and processed using Zeiss ZEN digital imaging for light microscopy.
Phase-contrast microscopy.
Phase-contrast microscopy slides were prepared by pipetting 5 to 10 μl of cell suspension from concentrated sorted cells onto glass slides and covering with a glass coverslip. Coverslips were then sealed with nail polish and imaged using a Zeiss Axioplan 2 upright light microscope with a 100× objective.
Statistical analysis.
Statistical analysis was preformed using a two-tailed Student t test or means ± standard deviations (SD) using Microsoft Excel software. Significant differences were considered when P values were <0.05.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Army Research Office (ARO; award no. W911NF-17-1-0343) and the U.S. Department of Energy (DOE; award no. DE-SC0019155). Microscopy equipment used in this project was acquired with an NIH shared-instrumentation grant (S10 OD016361), and access was supported by the NIH-NIGMS (P20 GM103446), the NSF (IIA-1301765) and the state of Delaware. The NMR facilities used for HMBR characterization are supported by the Delaware COBRE program, supported by a grant from NIH-NIGMS (5 P30 GM110758-02).
We thank Kamil Charubin for providing the high-resolution confocal images used in Fig. 1C. We also thank Thomas Hanson (University of Delaware) for bringing the FAST system to our attention and Rujin Cheng from the Rozovsky Lab for facilitating the use of the University of Delaware NMR Core Facility.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00622-19.
REFERENCES
- 1.Remington SJ. 2006. Fluorescent proteins: maturation, photochemistry and photophysics. Curr Opin Struct Biol 16:714–721. doi: 10.1016/j.sbi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Hartman AH, Liu HL, Melville SB. 2011. Construction and characterization of a lactose-inducible promoter system for controlled gene expression in Clostridium perfringens. Appl Environ Microbiol 77:471–478. doi: 10.1128/AEM.01536-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tracy BP, Gaida SM, Papoutsakis ET. 2010. Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Curr Opin Biotechnol 21:85–99. doi: 10.1016/j.copbio.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Ransom EM, Williams KB, Weiss DS, Ellermeier CD. 2014. Identification and characterization of a gene cluster required for proper rod shape, cell division, and pathogenesis in Clostridium difficile. J Bacteriol 196:2290–2300. doi: 10.1128/JB.00038-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charubin K, Bennett RK, Fast AG, Papoutsakis ET. 2018. Engineering Clostridium organisms as microbial cell-factories: challenges & opportunities. Metab Eng 50:173–191. doi: 10.1016/j.ymben.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Bullifent HL, Moir A, Titball RW. 1995. The construction of a reporter system and use for the investigation of Clostridium perfringens gene expression. FEMS Microbiol Lett 131:99–105. doi: 10.1111/j.1574-6968.1995.tb07761.x. [DOI] [PubMed] [Google Scholar]
- 7.Tummala SB, Welker NE, Papoutsakis ET. 1999. Development and characterization of a gene expression reporter system for Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 65:3793–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ravagnani A, Jennert KCB, Steiner E, Grunberg R, Jefferies JR, Wilkinson SR, Young DI, Tidswell EC, Brown DP, Youngman P, Morris JG, Young M. 2000. Spo0A directly controls the switch from acid to solvent production in solvent-forming clostridia. Mol Microbiol 37:1172–1185. doi: 10.1046/j.1365-2958.2000.02071.x. [DOI] [PubMed] [Google Scholar]
- 9.Girbal L, Mortier-Barriere I, Raynaud F, Rouanet C, Croux C, Soucaille P. 2003. Development of a sensitive gene expression reporter system and an inducible promoter-repressor system for Clostridium acetobutylicum. Appl Environ Microbiol 69:4985–4988. doi: 10.1128/AEM.69.8.4985-4988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drepper T, Eggert T, Circolone F, Heck A, Krauss U, Guterl JK, Wendorff M, Losi A, Gartner W, Jaeger KE. 2007. Reporter proteins for in vivo fluorescence without oxygen. Nat Biotechnol 25:443–445. doi: 10.1038/nbt1293. [DOI] [PubMed] [Google Scholar]
- 11.Buckley AM, Jukes C, Candlish D, Irvine JJ, Spencer J, Fagan RP, Roe AJ, Christie JM, Fairweather NF, Douce GR. 2016. Lighting up Clostridium difficile: reporting gene expression using fluorescent Lov domains. Sci Rep 6:23463. doi: 10.1038/srep23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mordaka P, Heap JT. 2018. Stringency of synthetic promoter sequences in Clostridium revealed and circumvented by tuning promoter library mutation rates. ACS Synth Biol 7:672–681. doi: 10.1021/acssynbio.7b00398. [DOI] [PubMed] [Google Scholar]
- 13.Plamont M-A, Billon-Denis E, Maurin S, Gauron C, Pimenta FM, Specht CG, Shi J, Quérard J, Pan B, Rossignol J, Moncoq K, Morellet N, Volovitch M, Lescop E, Chen Y, Triller A, Vriz S, Le Saux T, Jullien L, Gautier A. 2016. Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc Natl Acad Sci U S A 113:497–502. doi: 10.1073/pnas.1513094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monmeyran A, Thomen P, Jonquiere H, Sureau F, Li CE, Plamont MA, Douarche C, Casella JF, Gautier A, Henry N. 2018. The inducible chemical-genetic fluorescent marker FAST outperforms classical fluorescent proteins in the quantitative reporting of bacterial biofilm dynamics. Sci Rep 8:10336. doi: 10.1038/s41598-018-28643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goehring NW, Beckwith J. 2005. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr Biol 15:R514–R526. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 16.Adams DW, Errington J. 2009. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol 7:642–653. doi: 10.1038/nrmicro2198. [DOI] [PubMed] [Google Scholar]
- 17.Margolin W. 2000. Themes and variations in prokaryotic cell division. FEMS Microbiol Rev 24:531–548. doi: 10.1111/j.1574-6976.2000.tb00554.x. [DOI] [PubMed] [Google Scholar]
- 18.de Boer P, Crossley R, Rothfield L. 1992. The essential bacterial cell-division protein FtsZ is a GTPase. Nature 359:254–256. doi: 10.1038/359254a0. [DOI] [PubMed] [Google Scholar]
- 19.Gueiros FJ, Losick R. 2002. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev 16:2544–2556. doi: 10.1101/gad.1014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Low HH, Moncrieffe MC, Lowe J. 2004. The crystal structure of ZapA and its modulation of FtsZ polymerisation. J Mol Biol 341:839–852. doi: 10.1016/j.jmb.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 21.Small E, Marrington R, Rodger A, Scott DJ, Sloan K, Roper D, Dafforn TR, Addinall SG. 2007. FtsZ polymer-bundling by the Escherichia coli ZapA orthologue, YgfE, involves a conformational change in bound GTP. J Mol Biol 369:210–221. doi: 10.1016/j.jmb.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 22.Ransom EM, Ellermeier CD, Weiss DS. 2015. Use of mCherry red fluorescent protein for studies of protein localization and gene expression in Clostridium difficile. Appl Environ Microbiol 81:1652–1660. doi: 10.1128/AEM.03446-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nolling J, Breton G, Omelchenko MV, Makarova KS, Zeng QD, Gibson R, Lee HM, Dubois J, Qiu DY, Hitti J, Wolf YI, Tatusov RL, Sabathe F, Doucette-Stamm L, Soucaille P, Daly MJ, Bennett GN, Koonin EV, Smith DR. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol 183:4823–4838. doi: 10.1128/JB.183.16.4823-4838.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang G, Jia D, Jin L, Jiang Y, Wang Y, Jiang W, Gu Y. 2017. Rapid generation of universal synthetic promoters for controlled gene expression in both gas-fermenting and saccharolytic Clostridium species. ACS Synth Biol 6:1672–1678. doi: 10.1021/acssynbio.7b00155. [DOI] [PubMed] [Google Scholar]
- 25.Wiesenborn DP, Rudolph FB, Papoutsakis ET. 1989. Phosphotransbutyrylase from Clostridium acetobutylicum ATCC 824 and its role in acidogenesis. Appl Environ Microbiol 55:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papoutsakis ET. 2008. Engineering solventogenic clostridia. Curr Opin Biotechnol 19:420–429. doi: 10.1016/j.copbio.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Wiesenborn DP, Rudolph FB, Papoutsakis ET. 1988. Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl Environ Microbiol 54:2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomas CA, Alsaker KV, Bonarius HPJ, Hendriksen WT, Yang H, Beamish JA, Paredes CJ, Papoutsakis ET. 2003. DNA array-based transcriptional analysis of asporogenous, nonsolventogenic Clostridium acetobutylicum strains SKO1 and M5. J Bacteriol 185:4539–4547. doi: 10.1128/JB.185.15.4539-4547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tracy BP, Gaida SM, Papoutsakis ET. 2008. Development and application of flow-cytometric techniques for analyzing and sorting endospore-forming clostridia. Appl Environ Microbiol 74:7497–7506. doi: 10.1128/AEM.01626-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li CG, Plamont MA, Sladitschek HL, Rodrigues V, Aujard I, Neveu P, Le Saux T, Jullien L, Gautier A. 2017. Dynamic multicolor protein labeling in living cells. Chem Sci 8:5598–5605. doi: 10.1039/c7sc01364g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roach EJ, Kimber MS, Khursigara CM. 2014. Crystal structure and site-directed mutational analysis reveals key residues involved in Escherichia coli ZapA function. J Biol Chem 289:23276–23286. doi: 10.1074/jbc.M114.561928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galli E, Gerdes K. 2012. FtsZ-ZapA-ZapB interactome of Escherichia coli. J Bacteriol 194:292–302. doi: 10.1128/JB.05821-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margolin W. 2002. Bacterial sporulation: FtsZ rings do the twist. Curr Biol 12:R391–R392. doi: 10.1016/S0960-9822(02)00882-5. [DOI] [PubMed] [Google Scholar]
- 34.Al-Hinai MA, Jones SW, Papoutsakis ET. 2015. The Clostridium sporulation programs: diversity and preservation of endospore differentiation. Microbiol Mol Biol Rev 79:19–37. doi: 10.1128/MMBR.00025-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SY, Mermelstein LD, Bennett GN, Papoutsakis ET. 1992. Vector construction, transformation, and gene amplification in Clostridium acetobutylicum ATCC 824. Ann N Y Acad Sci 665:39–51. doi: 10.1111/j.1749-6632.1992.tb42572.x. [DOI] [PubMed] [Google Scholar]
- 36.Lee SY, Mermelstein LD, Papoutsakis ET. 1993. Determination of plasmid copy number and stability in Clostridium acetobutylicum ATCC 824. FEMS Microbiol Lett 108:319–323. doi: 10.1111/j.1574-6968.1993.tb06122.x. [DOI] [PubMed] [Google Scholar]
- 37.Rohlhill J, Sandoval NR, Papoutsakis ET. 2017. Sort-Seq approach to engineering a formaldehyde-inducible promoter for dynamically regulated Escherichia coli growth on methanol. ACS Synth Biol 6:1584–1595. doi: 10.1021/acssynbio.7b00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaida SM, Sandoval NR, Nicolaou SA, Chen YL, Venkataramanan KP, Papoutsakis ET. 2015. Expression of heterologous sigma factors enables functional screening of metagenomic and heterologous genomic libraries. Nat Commun 6:7045. doi: 10.1038/ncomms8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galli E, Gerdes K. 2010. Spatial resolution of two bacterial cell division proteins: ZapA recruits ZapB to the inner face of the Z-ring. Mol Microbiol 76:1514–1526. doi: 10.1111/j.1365-2958.2010.07183.x. [DOI] [PubMed] [Google Scholar]
- 40.Valencikova R, Krascsenitsova E, Labajova N, Makroczyova J, Barak I. 2018. Clostridial DivIVA and MinD interact in the absence of MinJ. Anaerobe 50:22–31. doi: 10.1016/j.anaerobe.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 41.Makroczyova J, Jamroskovic J, Krascsenitsova E, Labajova N, Barak I. 2016. Oscillating behavior of Clostridium difficile Min proteins in Bacillus subtilis. Microbiologyopen 5:387–401. doi: 10.1002/mbo3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tebo AG, Pimenta FM, Zhang Y, Gautier A. 2018. Improved chemical-genetic fluorescent markers for live cell microscopy. Biochemistry 57:5648–5653. doi: 10.1021/acs.biochem.8b00649. [DOI] [PubMed] [Google Scholar]
- 43.Fast AG, Papoutsakis ET. 2018. Functional expression of the Clostridium ljungdahlii acetyl-coenzyme A synthase in Clostridium acetobutylicum as demonstrated by a novel in vivo CO exchange activity en route to heterologous installation of a functional Wood-Ljungdahl pathway. Appl Environ Microbiol 84:e02307-17. doi: 10.1128/AEM.02307-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charubin K, Papoutsakis ET. 2019. Direct cell-to-cell exchange of matter in a synthetic Clostridium syntrophy enables CO2 fixation, superior metabolite yields, and an expanded metabolic space. Metab Eng 52:9–19. doi: 10.1016/j.ymben.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Cornillot E, Nair RV, Papoutsakis ET, Soucaille P. 1997. The genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 reside on a large plasmid whose loss leads to degeneration of the strain. J Bacteriol 179:5442–5447. doi: 10.1128/jb.179.17.5442-5447.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Hinai MA, Fast AG, Papoutsakis ET. 2012. Novel system for efficient isolation of clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl Environ Microbiol 78:8112–8121. doi: 10.1128/AEM.02214-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mermelstein LD, Papoutsakis ET. 1993. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 59:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mermelstein LD, Welker NE, Bennett GN, Papoutsakis ET. 1992. Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Nat Biotechnol 10:190–195. doi: 10.1038/nbt0292-190. [DOI] [PubMed] [Google Scholar]
- 49.Sillers R, Al-Hinai MA, Papoutsakis ET. 2009. Aldehyde-alcohol dehydrogenase and/or thiolase overexpression coupled with CoA transferase downregulation lead to higher alcohol titers and selectivity in Clostridium acetobutylicum fermentations. Biotechnol Bioeng 102:38–49. doi: 10.1002/bit.22058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.