

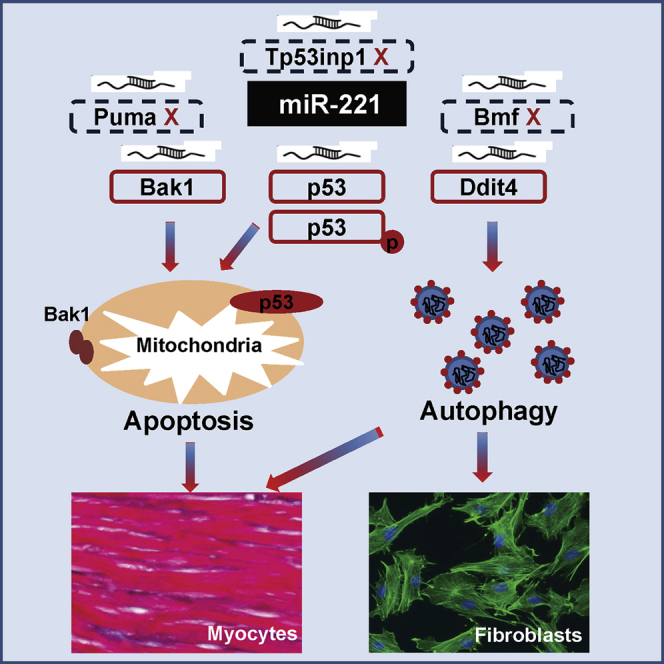
Abstract
Reduced myocardial miR-221 expression is associated with severe cardiac fibrosis in heart failure patients. We aimed to demonstrate its mechanisms in cardioprotection and remodeling following myocardial infarction (MI). Using in vitro hypoxia and reoxygenation (H/R) of H9c2 and rat cardiac fibroblast (cFB) models, we found that miR-221 protects H9c2 through combined anti-apoptotic and anti-autophagic effects and cFB via anti-autophagic effects alone in H/R. It inhibits myofibroblast (myoFB) activation as indicated by lowering α-smooth muscle actin (α-SMA) expression, gel contraction, and collagen synthesis (Sircol assay). In vivo, following left coronary artery ligation (MI), rats were treated with miR-221 mimics (intravenous [i.v.], 1 mg/kg). With treatment, miR-221 increased by ∼15-fold in infarct and peri-infarct zones at day 2 post-MI. At days 7 and 30 post-MI, miR-221 reduced infarct size, fibrosis, and α-SMA+ cells in both infarct and remote myocardium. Left ventricle (LV) function was preserved as indicated by ejection fraction, infarct thickness, LV developed pressure, ±dP/dt, and end diastolic pressure. We demonstrated the anti-apoptotic and anti-autophagic effects were due to combined mechanisms of direct targeting on Bak1 and P53 and inhibition of phosphorylation at Ser46 and direct targeting on Ddit4, respectively. miR-221 enhances cardiomyocyte survival and protects cardiac function post-MI. It enhances cFB survival yet inhibits their activation, thus reducing adverse cardiac fibrosis.
Keywords: myocardial infarction, microRNA, apoptosis, autophagy, cardioprotection, cardiac remodeling, p-53, Ddit4, microRNA-221
Graphical Abstract
Introduction
Coronary artery disease, mainly via acute myocardial infarction (MI) and consequent heart failure, is a leading cause of morbidity and mortality worldwide. Due to the extremely limited regenerative capacity of cardiomyocytes, cardioprotection against ischemia or ischemia and reperfusion (I/R) injury is critical to salvaging ischemic myocardium. MicroRNA (miRNA) regulation is emerging as a promising therapeutic target. miR-221 is a well-known onco-miRNA promoting cell survival in tumorigenesis.1 The dysregulation of miR-221 has been reported in numerous clinical studies. Circulating miR-221 was found to be elevated in patients with acute MI.2 In myocardial biopsies from patients of dilated cardiomyopathy, significant loss of miR-221 was associated with cardiac fibrosis.3 Loss of miR-221 in atheroma might cause plaque rupture in the atherosclerotic patients.4 Serum miR-221 is lower in patients with spinal cord I/R, and miR-221 mimics protect neuronal cells against I/R-induced inflammation and apoptosis.5 These data suggest that miR-221 might be an important regulator of cell survival in cardiovascular diseases.
An early study of cardiac progenitor cell transplantation included miR-221 in a cocktail of three miRNA mimics, and it demonstrated pro-survival effects on the engrafted stem cells.6 However, the underlying mechanisms remain unexplored. Our previous work demonstrated miR-221 is upregulated by two different cardioprotective drugs, suggesting that upregulation of miR-221 might mediate aspects of cardioprotection. In vitro studies using miR-221 mimic transfection demonstrated improved cardiomyocyte survival after hypoxia and reoxygenation (H/R). Cardiomyocyte apoptosis was reduced through miR-221 targeting of pro-apoptotic genes of the bcl-2 family.7 miR-221 also directly targets Ddit4 and Tp53inp1, offering cardioprotection through an anti-autophagic mechanism.8 Consistent with our findings, other investigators have demonstrated that miR-221 protects cardiac myocytes by targeting PUMA.9 Constitutive cardiac-specific overexpression of miR-221 inhibits autophagy through targeting the p27/mammalian target of rapamycin (mTOR) pathway. Notwithstanding these findings, they believe that this regulation is associated with cardiac hypertrophy.10 In contrast to the abundant in vitro data, the cardiac therapeutic potential of miR-221 has not been investigated in models of MI in vivo.
Compared to cardiomyocytes, the role of cardiac fibroblasts (cFBs) in the response to cardiac ischemic injury is controversial. It is generally believed that cFBs produce extracellular matrix (ECM) and play a critical role in wound healing after myocardium infarction. Blockade of an appropriate fibrotic response may cause cardiac rupture after acute MI.11 Preservation of fibroblasts through the inhibition of apoptosis in the infarct area ameliorates post-MI remodeling and promotes functional recovery.12 Phagocytosis of dead myocytes by cFBs is beneficial in post-MI repair.13 However, many studies show that anti-fibrotic therapy may offer benefits by opposing excessive fibrosis and the associated increased wall stiffness, loss of normal systolic and diastolic functions, and consequent progression to heart failure.14, 15, 16, 17 Although there is a knowledge gap in the post-MI cFB function, it is believed that post-MI cFBs proliferate and transdifferentiate into myofibroblasts (myoFBs), which exhibit dynamic phenotypical alterations.18 The sensitivity of cFBs in responding to environmental stresses creates certain difficulties for the study of cFBs ex vivo.18, 19 We have characterized cFB responses to different stimuli ex vivo, and we developed comprehensive standardized protocols to quantify cFB phenotypical changes.20 This previous work led us to investigate the roles of cFBs in miR-221-mediated effects both in vitro and in myocardial repair following infarction.
In this study, we found that miR-221 enhances cardiomyocyte survival through the inhibition of apoptosis and autophagy via actions upon P53 (Tp53 gene), Bak1, and Ddit4, respectively. It protects cFBs through the inhibition of autophagy by targeting Ddit4 only. Strikingly, it preserves cFBs through the inhibition of autophagy, without exaggerating injury-stimulated cardiac fibrosis in the infarct zone or adverse fibrosis in the peri-infarct zone. The net effect of miR-221 is to ameliorate adverse post-ischemic left ventricle (LV) remodeling and preserve cardiac function. Our results also support the important concept that miRNA effects are cell type and condition dependent. Better understanding of these phenomena and their underlying mechanisms will help to optimize the therapeutic potential and minimize the off-target effects of manipulating miR-221.
Results
miR-221 Protects H9c2 against H/R through the Inhibition of Apoptosis
miR-221 protected H9c2 against H/R injury, as indicated by increased cell number, reduced lactate dehydrogenase (LDH) release, and decreased apoptosis, and it prevented the loss of mitochondrial membrane potential (MMP) (Figures 1A–1C). miR-221 significantly downregulated total P53 protein expression and phosphorylation at serine46 (P-P53 Ser46) in both normoxia and H/R (Figure 1D). However, Tp53 mRNA expression was unchanged by miR-221 (Figure S1A). Although not predicted as a target for miR-221 (http://mirdb.org), p53 has two potential unconventional binding sites (BS1 and BS2) for this miRNA. Psicheck2 luciferase reporter activity of p53 3′ UTR was suppressed by miR-221 mimics, supporting a direct targeting of miR-221 to the p53 3′ UTR (Figures 1E and 1F). Mutagenesis of either or both of the binding sites confirmed a specific role of BS1, but not BS2, as critically responsible for this interaction (Figures 1E and 1F).
Figure 1.

miR-221 Protects H9c2 against H/R through the Inhibition of Apoptosis
H9c2 cells were transfected with miR-221 mimic or control (miR-221 and MC) and subjected to 16-h hypoxia followed by 2-h reoxygenation (H/R). (A) Phase-contrast images of H9c2 cells at the end of H/R. Scale bar, 100 μm. Cell injury was measured by cell number count and lactate dehydrogenase (LDH) release. (B) Cell apoptosis was assessed by Annexin V and 7-AAD double staining followed by flow cytometry measurements. Representative apoptosis profiles are shown for each group. Results are expressed as the percentage of cells in each quadrant. (C) Mitochondrial membrane potential (MMP) was measured by MMP-activated fluorescence dye staining and flow cytometry analysis. Results are expressed as the percentage of depolarized cells. (D) Western blot of P53 phosphorylated at Serine 46 (P-P53 Ser46), total P53 (T-P53), and the loading control β-actin. (E) Putative miR-221-binding sites in the p53 3′ UTR and their mutations. (F) Luciferase reporter assay using co-transfection of the psicheck2 dual luciferase reporter plasmid carrying the specified 3′ UTR inserts and miR-221 mimics or mimic control. Sensor, a miR-221 complementary sequence, was used as a positive control. Results were expressed as Renilla:Firefly luminescence ratios normalized to empty vector control. *p < 0.05, **p < 0.01, ***p < 0.001 versus MC. n = 3 in triplicates.
miR-221 Protects cFBs against H/R Injury through the Inhibition of Autophagy
Similar experiments with miR-221 were performed in cFBs. miR-221 increased cell number and reduced LDH release (Figure 2A), but it did not reduce apoptosis (Figure 2B). miR-221 did not downregulate the expressions of Tp53 mRNA (Figure S1B), protein, or P-P53 Ser46 in cFBs (Figure 2C). Previously reported pro-apoptotic genes of Bak1, Puma, and Bmf7, 21, 22 were examined in cFBs; miR-221 did not downregulate them at the protein level under normoxia and H/R conditions (Figures S1C and S1D).
Figure 2.

miR-221 Protects cFBs against H/R Injury through the Inhibition of Autophagy
Adult rat cardiac fibroblasts (cFBs) were transfected with miR-221 mimic or control (miR-221 and MC) and subject to 24-h hypoxia followed by 2-h re-oxygenation (H/R). (A) Phase-contrast images of cFBs at the end of H/R. Scale bar, 100 μm. Cell injury was measured by cell number count and lactate dehydrogenase (LDH) release. (B) Cell apoptosis was assessed by Annexin V and 7-AAD double staining followed by flow cytometry measurements. Representative apoptosis profiles are shown for each group. Results are expressed as the percentage of cells in each quadrant. Western blot to detect (C) P53 phosphorylated at Serine 46 (P-P53 Ser46) and total P53 (T-P53); (D) P62, LC3-I, and LC3-II; and (E) TP53INP1 and DDIT4. The expressions were normalized to their loading control of β-actin.*p < 0.05, **p < 0.01, ***p < 0.001 versus MC. n = 3 in triplicates.
As the anti-autophagic role of miR-221 in the protection of cardiomyocytes has been reported previously,8 we investigated autophagy in cFBs. The activation of autophagy, as signaled by an increased LC3-II:I ratio and decreased P62, was induced by H/R. miR-221 suppressed the increase in LC3-II:I ratio, an indication of reduced autophagosome formation (Figure 2D). The protein expressions of TP53INP1 and DDIT4, direct targets of miR-221, were upregulated under H/R and significantly suppressed by miR-221 (Figure 2E). The mRNA expressions of Tp53inp1 and Ddit4 were downregulated by miR-221 as well (Figure S2A). An autophagy inhibitor, 3-methyladenine (3-MA), was similarly protective as miR-221 against H/R injury (Figure S2B). Overexpression of DDIT4 in cFBs exacerbated H/R injury, as indicated by a further reduction in cell number. Overexpression of TP53INP1 had no obvious impact (Figures S2C and S2D).
miR-221 Inhibits myoFB Activation and Collagen Synthesis
While miR-221 increased cFB number, with respect to fibrotic effects, mRNA expression and protein levels of α-smooth muscle actin (α-SMA), a myoFB activation marker, were significantly downregulated by miR-221 (Figures 3A and 3B). This is consistent with published results.3 Accordingly, miR-221 reduced gel contraction, an indication of cFB-mediated mechanical contraction (Figure 3C). miR-221 downregulated gene expressions of collagen type I (Col1a1 and Col1a2) and type III (Col3a) and fibronectin (Fn1) and its fibrotic isoform with extra domain A (Fn-EDA), and it inhibited collagen synthesis (Figures 3D and 3E). This effect was supported by reduced P-SMAD2/3 (Ser 423/425). miR-221 did not affect the expressions of total Smad2/3 at the mRNA and protein levels. (Figures 3F and 3G).
Figure 3.

miR-221 Inhibits myoFB Activation and Collagen Synthesis
Adult rat cFB was serum starved for 24 h, then transfected with miR-221 mimic or control (miR-221 and MC) and incubated for a specified period of time. (A) qRT-PCR and (B) western blot measurements of α-SMA mRNA and protein expressions. (C) Collagen gel contraction was measured as the percentage reduction of gel area over the culture surface after 48-h pre-stress and 48-h free contraction. (D) qRT-PCR measurement of mRNA expression of collagen I (Col1a1 and Col1a2), collagen III (Col3a), fibronectin (Fn1), and its extra domain A isoform (Fn-EDA). (E) Sircol collagen assay of cFB supernatants 7 days after mimic transfection. (F) qRT-PCR and (G) western blot measurement of Smad2 and Smad3 mRNA and protein expressions. Antibodies recognized phosphorylated (P-SMAD2/3, Serine 423/425) and total (T-SMAD2/3), respectively. *p < 0.05, **p < 0.01, ***p < 0.001 versus MC. n = 3 in triplicates.
miR-221 Reduces Apoptosis and Attenuates Autophagy at Day 2 Post-MI
At 2 days post-MI and miR-221 mimic injection, miR-221 levels were upregulated by ∼15-, 14-, and 8-fold in infarct, border, and remote areas, respectively, compared to MI control (Figure 4A). Notably, compared to sham, the endogenous expression of miR-221 was elevated in the infarct and border areas but unchanged in the remote myocardium. Our in vitro data indicated that miR-221 was enriched in cFBs compared to cardiomyocytes (data not shown), and whether its observed upregulation post-MI was induced by ischemia or reflected change in cell type composition was uncertain.
Figure 4.

miR-221 Reduces Apoptosis and Attenuates Autophagy at Day 2 Post-MI
Rats with myocardial infarction (MI) received 1 mg/kg miR-221 mimic or PBS injection (miR-221 or MI +miR-221 and MI-Ctrl) right after MI. Hearts were harvested 48 h later, and they were dissected according to infarct, border, and remote areas. RNA and protein were extracted for analysis. (A) miRNA Taqman qPCR measurement of miR-221 levels in different regions. Western blot and densitometry for (B) blots, (ii) cleaved caspase-3, (iii) P53 phosphorylated at Serine 46 (P-P53 Ser46) and total P53 (T-P53); (C) P62 and LC3-I and LC3-II; and (D) TP53INP1 and DDIT4. β-actin was used as a loading control. *p < 0.05, **p < 0.01, ***p < 0.001 versus sham; #p < 0.05, ##p < 0.01 versus MI-Ctrl. n = 5 each group.
Cleaved Caspase-3 was readily detected in MI hearts, indicating activated apoptosis. With miR-221 treatment, cleaved Caspase-3 was significantly reduced (Figure 4B, i and ii). Tp53 mRNA (Figure S3A) and P-P53 were significantly upregulated in infarct tissue. miR-221 treatment did not alter mRNA levels, but it significantly reduced the P-P53 and T-P53 protein levels (Figure 4B, i and iii). miR-221 dramatically reduced LC3-II in the infarct area, indicating the inhibition of autophagy. No parallel accumulation of P62 was observed; instead it was decreased (Figure 4C). In the peri-infarct border zone, LC3-I and P62 were significantly increased by miR-221, a sign of inhibited autophagy. No significant changes were detected in the remote area (Figure 4C). In the infarct area, DDIT4 was significantly reduced by miR-221, but TP53INP1 was not changed (Figure 4D). Gene expression of Ddit4 showed a trend toward downregulation (Figure S3B).
Previously reported miR-221 targets were tested in the in vivo heart. miR-221 only downregulated the mRNA and protein expressions of BAK1 in the infarct area. Protein levels of BMF and PUMA were unchanged (Figures S4A and S4B).
miR-221 Reduces Post-infarct Fibrosis and Inhibits myoFB Activation Post-MI
As early as 2 days after MI, Col1a1 mRNA expression was significantly increased by ∼14- and ∼8-fold in infarct and border zones versus sham (Figure 5A). This change was significantly suppressed by miR-221. Concurrently, transforming growth factor β (TGF-β)-signaling P-SMAD2/3 was upregulated in the infarct zone, which was also inhibited by miR-221 (Figure 5B, ii and iii). Changes of Smad2/3 mRNA and total protein in the heart were not significant (Figure 5B, i and ii).
Figure 5.

miR-221 Reduces Post-infarct Fibrosis and Inhibits myoFB Activation at Days 2 and 7 Post-MI
Rats with MI received 1 mg/kg miR-221 mimic or PBS injection right after coronary artery ligation and were harvested at day 2 post-MI, or they received another injection after 72 h and were harvested at day 7 post-MI (miR-221 or MI +miR-221 and MI-Ctrl). qRT-PCR measurements of (A) Col1a1 mRNA and (B, i) Smad3 mRNA; (B, ii and iii) western blot measurement of SMAD2/3 at day 2 post-MI. (C) miRNA Taqman qPCR measurement of miR-221 levels at day 7 post-MI. (D) Picro-sirius red staining to quantify tissue collagen content at day 7 post-MI. Representative images of heart cross-sections are shown for each group. Results are expressed as the percentage of positive staining over the area of LV. (E) Immunohistochemistry with anti-α-SMA (green) and DAPI (blue) nuclei staining at day 7 post-MI. Scale bar, 150 μm. Results are expressed as the percentage of α-SMA+ cells over the total number of nuclei. *p < 0.05, **p < 0.01, ***p < 0.001 versus sham; #p < 0.05 versus MI-Ctrl. n = 4, 6, and 4 for sham, MI-Ctrl, and MI +miR-221, respectively.
At day 7 post-MI, levels of miR-221 remained high with miR-221 treatment, ∼2-fold and ∼1.5-fold in the infarct and border zones versus MI control (Ctrl), respectively (Figure 5C). Collagen deposition was significantly decreased by miR-221 (11.2% ± 4.3% versus 18.5% ± 3.0% MI-Ctrl of LV area, p < 0.05; Figure 5D). α-SMA+ myoFBs were abundant in the infarct and border zones and somewhat increased in the remote zone as well. α-SMA+ myoFBs were clearly reduced by miR-221 in all areas (Figure 5E). Areas were demarcated by intact cardiomyocyte morphology from a bright-field image (Figure S5A). Accumulation of fibrosis was not apparent in remote myocardium at this time frame (Figure S5B).
miR-221 Reduces MI, Attenuates LV Remodeling, and Improves LV Function at Days 7 and 30 Post-MI
At day 7 post-MI, infarcted hearts showed a striking loss of myocardium reflected in reduced wall thickness. Tissue loss was significantly ameliorated by miR-221 (Figure 6A). Echocardiography corroborated significantly greater wall thickness (systolic and diastolic) in the infarct zone with miR-221 treatment. Non-infarcted wall thickness (septum) was unchanged. The post-MI enlargement of the LV chamber size (LV internal dimensions [LVIDs]) was partially prevented by miR-221. miR-221 treatment significantly improved ejection fraction (EF) compared to MI-Ctrl (Figure 6B).
Figure 6.

miR-221 Attenuates LV Remodeling and Improves LV Function at Day 7 Post-MI
Rats with MI received 1 mg/kg miR-221 mimic or PBS injections right after and 72 h post-MI. Hearts were harvested at day 7 post-MI for histological analysis. LV function was measured by echocardiography. (A) Masson trichrome staining of heart cross-sections. Representative images are shown for each group. Infarct area was measured as the percentage of blue + purple staining over the area of LV. (B) Ejection fraction (EF), LV internal dimension (LVID) at systole (s), and infarct and septum thickness at diastole (d) and systole (s), as measured by echocardiography. *p < 0.05, **p < 0.01, ***p < 0.001 versus sham; #p < 0.05, ##p < 0.01 versus MI-Ctrl. n = 4, 6, and 4 for histology; n = 10, 15, and 11 for echocardiography, for sham, MI-Ctrl, and MI +miR-221, respectively.
At day 30 post-MI, the accumulation of LV fibrosis was pronounced in the control MI hearts, and it was significantly suppressed by miR-221 in both infarct and border zones (Figure 7A, i–iii). Importantly, fibrosis also accumulated in remote myocardium, and this increase was also abrogated by miR-221 (Figure 7A iii and iv). LV catheterization demonstrated that miR-221 significantly improved contractility (LV developing pressure [LVDP]) and relaxation (±dP/dt and Tau) and reduced end diastolic pressure (EDP) (Figure 7B). Two-way ANOVA repeated measurement with Bonferroni adjustment indicated a significant improvement of cardiac performance over the 30 days post-MI by echocardiography assessment (Figure S6). Capillary density at day 30 post-MI was assessed by isolectin B4 staining (Figure S7).
Figure 7.

miR-221 Attenuates LV Remodeling and Improves LV Function at Day 30 Post-MI
Rats with MI received 1 mg/kg miR-221 mimic or PBS injections right after and 72 h post-MI. At day 30 post-MI, LV function was measured terminally by LV catheterization and heart tissues were harvested for histology. (A) Picro-sirius red staining of collagen. (i) Complete cross-sections of representative hearts. (ii) Quantification of collagen content over the area of LV. (iii) Representative images of infarct, border, and remote areas. Scale bar, 200 μm. (iv) Quantification of collagen content in the remote areas. (B) (i) Representative Labchart data acquisition: upper panel, right carotid artery blood pressure and LV developing pressure; lower panel, ±dP/dt. (ii) LVDP, left ventricle developing pressure; EDP, end diastolic pressure (EDP); ±dP/dt, maximum and minimum pressure changes. *p < 0.05, **p < 0.01, ***p < 0.001 versus sham; #p < 0.05 versus MI-Ctrl. n = 6, 7, and 7 for sham, MI-Ctrl, and MI +miR-221, respectively.
Discussion
We have undertaken a multi-faceted investigation of the mechanisms underlying cardioprotection from miR-221 by comparing myocyte versus cFB and normoxia versus H/R in both in vitro and in vivo settings. Our results clearly demonstrate that gene regulation by miR-221 is cell type and condition dependent, with results observed in vitro not necessarily translating to in vivo effects. miR-221 enhances cardiomyocyte survival through both reduced apoptosis and autophagy and cFB survival through reduced autophagy alone. miR-221 increases cFB survival while inhibiting the expression of α-SMA and gel contraction, expression of collagen genes, and collagen secretion. In a rat MI model, miR-221 mimics reduce infarct size and cardiac fibrosis in the infarct zone and adverse fibrosis in the peri-infarct zone, with amelioration of adverse LV remodeling and preservation of cardiac function. The novel findings are as follows: (1) miR-221 significantly inhibits P53 phosphorylation at Ser46 and downregulates P53 expression through direct binding to the 3′ UTR of p53; (2) regulation of P53 and Bak1 along with anti-apoptotic effects of miR-221 occur in myocytes, but not in cFBs; and (3) aspects of the cFB phenotype are differentially regulated by miR-221, with increased cFB survival but less myoFB activation and reduced collagen secretion producing a net anti-fibrotic effect.
miR-221 significantly inhibited ischemia-induced apoptosis both in vitro and in vivo. Puma, Bmf, and Bak1 are known as pro-apoptotic factors,21, 22 and our earlier work indicated that the anti-apoptotic effect of miR-221 reflected the downregulation of these genes in cardiac myocytes.8 Here we could only confirm the downregulation of Bak1, but not Puma and Bmf, by miR-221 in the post-MI heart. Therefore, other anti-apoptotic mechanisms of miR-221 might be involved in cardioprotection.
Surprisingly, miR-221 significantly suppressed total P53 in H9c2 and in cardiac infarcts at day 2 post-MI. As P53 is not an in silico-predicted target of miR-221 (http://www.targetscan.org/vert_72/; http://www.mirdb.org/), we undertook manual alignment of the miR-221 seed sequence with P53 3′ UTR, which clearly indicated two possible binding sites with low energy scores of −10.63 and −9.14 kcal/mol. This method has been used previously to discover potential RNA-RNA interactions based on binding energy.23 Subsequently, our luciferase reporter assay demonstrated a direct interaction between miR-221 and the P53 3′ UTR. A similar situation has been reported with miR-150 targeting CXCR4.24 Mutagenesis of the two binding sites of the P53 3′ UTR reinforced that binding site 1 is crucial for miR-221 and P53 3′ UTR interaction. For the first time we have demonstrated P53 to be a direct target of miR-221. Downregulation by miR-221 was evident at the protein rather than the mRNA level. Discrepancy in mRNA and protein abundance has been frequently observed, with mechanisms usually remaining elusive.25 It was demonstrated that a perfect complementary sequence binding between the miRNA seed region and the 3′ UTR leads to mRNA degradation, while the non-perfect binding usually leads to transcriptional suppression.26
P53 is a well-known tumor suppressor through the regulation of cell-cycle arrest and apoptosis.27 Total P53 expression and activation of P53 phosphorylation at Ser46 increases in cardiac ischemia28 and in human failing myocardium.29 Genetic knockdown of P53 expression in the heart increases survival after MI due to the inhibition of apoptosis.30 Consistent with this background information, we found that P-P53 Ser46 was significantly increased in H9c2 following H/R and in infarcted myocardium at day 2 post-MI. miR-221 reduced both total P53 and P-P53 Ser46. It is reported that the translocation of P-P53 Ser46 to the mitochondrial membrane causes cytochrome c leakage and caspase activation.31 P53 promotes MPP by forming complexes with anti-apoptotic Bcl-XL and Bcl-2 proteins.32 Bak1, a known pro-apoptotic regulator, undergoes conformation changes and oligomerization on the mitochondrial outer member upon ischemic stress. This change leads to apoptosis.33 This working hypothesis is strongly supported by the results that miR-221 significantly reduces the loss of MMP under H/R. Taken together, these data suggest that miR-221 reduces apoptosis through the inhibition of P53 and Bak1. Another group has reported that miR-221 has pro-proliferative, pro-migration, and anti-apoptotic effects on vascular smooth muscle cells, with the opposite effects on endothelial cells.34
cFBs actively proliferate upon stimulation. It was believed cFBs accumulating in infarcts were transdifferentiated and migrated from multiple sources, e.g., bone marrow-derived progenitors, epicardial cells, endothelial cells, pericytes, and resident cFBs.35, 36 With the advance of in vivo lineage-tracing techniques, now it is clear that myoFBs in the infarct zone are mainly derived from resident cFBs.19, 37, 38 Therefore, the enhanced survival of residential cFBs in infarcted hearts is important. As reported previously, miR-221 inhibits autophagy in cardiomyocytes through directly targeting Ddit4 and Tp53inp1.8 In an in vitro cFB H/R model, we observed that miR-221 significantly downregulated the expressions of Ddit4 and Tp53inp1 at both the mRNA and protein levels. Accordingly, the overexpression of Ddit4 was detrimental to cFBs under I/R, while Tp53inp1 had no effect. Importantly, Ddit4 expression in the infarct area at day 2 post-MI was greatly inhibited by miR-221, while Tp53inp1 was unaffected. These data strongly support the proposition that miR-221 targeting of Ddit4 plays a major role in the inhibition of autophagy. This finding is consistent with another group’s work in which the overexpression of miR-221 reduced autophagy flux in the heart.39 Although we did not observe both reduced ratio of LC3-II:I along with increased P62 in cultured cFBs and MI heart, we repeatedly observed the inhibition of LC3-II by miR-221. Taken together, we concluded that the anti-autophagic effect of miR-221 is due to direct targeting of Ddit4. We therefore suggest that miR-221 protects cFBs from ischemic death due to the inhibition of autophagy.
An important question to be addressed is whether an increased cFB number could at some point promote adverse cardiac fibrosis and, therefore, impair cardiac functional recovery. Many publications report concurrent changes in cFB proliferation, myoFB activation, and collagen synthesis. For example, miR-125a positively regulates while miR-101a negatively regulates cFB numbers, myoFB activation, and collagen synthesis.40, 41, 42 Whether miR-221 could preserve cFBs via increased cell survival as indicated in in vitro study, it is clear that miR-221 is anti-fibrotic via the suppression of myoFB activation and reduced collagen secretion. MI-induced adverse cardiac fibrosis is associated with progression to heart failure and an increased incidence of arrhythmias.43 miR-221 reduced myoFB activation in the infarct, peri-infarct, and remote myocardium as early as day 7 post-MI. It inhibited fibrosis in the infarct area and interstitial fibrosis in the remote zone at day 30 post-MI. With this improved cardiac remodeling, miR-221 improved cardiac contractility and relaxation. The differential regulation of cFB phenotype is a novel and under-appreciated phenomenon. On careful review of the literature, we found one study showing that cFB-specific p38α deletion could dramatically inhibit myoFB activation and reduce cardiac fibrosis while increasing cFB proliferation.38
Interesting observations in this study warrant further studies. With miR-221 mimic treatment, miR-221 increased in all areas in the heart but accumulated preferentially in the infarct area. The underlying mechanisms are unknown. A recent study demonstrated that anti-miR-92a delivered in vivo is largely taken up by macrophages.44 We postulate that the increase of miR-221 in the infarct and border might be caused by an accumulation of macrophages and a change in cellular composition. The regulation of T-P53 expression and P-P53 at Ser46 by miR-221 was only observed in myocytes and not in cFBs. Context- and cell type-dependent miRNA function has been observed by other groups.34, 45 The mechanisms are unknown. Cell type- and condition-dependent miRNA effects and differentially regulated cFB phenotype warrant further in-depth investigation. A full understanding of the mechanisms will help to optimize the therapeutic potential of miRNA treatments and minimize the off-target effects. Smad-3 phosphorylation is the key step in TGF-β-signaling activation leading to fibrosis.46, 47 Our results showed that miR-221 significantly inhibited the phosphorylation of Smad-3 in cFBs and MI heart. Again, the underlying mechanisms mandate further study.
There are some limitations to this study. (1) Based on van Rooij’s48 study on miR-29b mimics’ pharmacokinetics, the miRNA mimics remained elevated only for 2–3 days after delivery. In our own prior experience with miR-101a mimics, we confirmed that the miRNA level had returned to baseline 1 week after the last injection.42 In this study, the treatment strategy was carefully designed as 2 injections at day 0 and day 3 post-MI. The rationale was to keep miR-221 levels elevated throughout the first week post-MI, which could induce beneficial changes in the acute phase of MI. The long-term benefits would be secondary effects of improved cardiomyocyte survival and favorable fibrotic remodelling. However, miR-122 antagomiRNA, the first miRNA clinical trials of the treatment of hepatitis C, was given at a weekly interval.49 To optimize the miRNA therapeutic effects, the miRNA level in the heart should be monitored. (2) Cell number of cFBs could be a mixed effect of cell proliferation and survival. In this study we demonstrated that miR-221 protected cFBs against H/R injury. Whether miR-221 regulates cFB proliferation and what are the anti-fibrogenic mechanisms are unknown. (3) miR-221 is known as an oncomiRNA. High levels of miR-221 are associated with cancer cell proliferation and metastasis, due to the direct targeting of p27, a well-known cell cycle inhibitor.50 However, data also show that the expression of p27 in healthy tissue does not correlate with the expression of miR-221.51 Therefore, a clear understanding of the miR-221 regulation on cell proliferation and possibly oncogenesis is clinical relevant. (4) To fully understand the effects of miR-221 in the regulation of autophagy, autophagy flux should be studied. All of these topics warrant further investigation.
In conclusion, we have demonstrated that miR-221 enhances cardiomyocyte survival, in the setting of H/R, through reduced apoptosis and autophagy, while reducing cFB death through the inhibition of autophagy alone. miR-221 promoted increased numbers of cFBs showing a unique anti-fibrotic profile, that is, reduced α-SMA expression and collagen secretion. miR-221 therapy reduces infarct size and cardiac fibrosis, with amelioration of adverse LV remodeling and preservation of cardiac function in a rat MI model.
Materials and Methods
Animals
Female Sprague-Dawley rats (240–330 g) were used for primary cFB isolation. Male Sprague-Dawley rats (170–220 g) were used for MI in vivo studies. All rats were kept in a temperature-controlled room (21°C ± 2°C) with 12-h light and dark cycle. Water and diet were available ad libitum. Protocols were approved by the Institutional Animal Care and Use Committee of the National University of Singapore, and they complied with the Guide for the Care and Use of Laboratory Animals published by the NIH (Publication 85-23, Revised 1996). Rat cardiac myoblast H9c2 cells were purchased from ATCC (Bio-REV, Singapore). The 25th–30th passage cells were used in this study. Chemicals and reagents were purchased from Sigma-Aldrich (Singapore) if not otherwise indicated.
H/R and Cell Injury Assessment
The method of isolating cFBs from adult rat hearts has been reported previously.20 2.5 mg/mL collagenase II and 80 U/mL DNase I (Worthington Biochemical, i-DNA Biotechnology, Singapore) were used to digest the tissue. Only passage 1 (p1) cells were used in this study. H/R injury was modeled by incubation of cells in serum-free DMEM with low glucose (1 g/L) and 0.2% oxygen (BioSpherix) for 24 h, followed by 2-h re-oxygenation in growth medium. 25 nM miR-221 mimic (miR-221) or mimic control (MC) was transfected using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Singapore) 24 h before the start of H/R.7
LDH activity was measured in hypoxia culture supernatants as a marker of cell injury (TOX-7 kit, Sigma-Aldrich, Singapore). Cell number, apoptosis, and MMP were assessed using MUSE cell analyzer with Annexin V and 7-aminoactinomycin D (7-AAD) double staining and Mitopotential assay kit (Merck Millipore, Singapore).7, 52
The Assessment of Collagen Secretion and Collagen Gel Contraction
In brief, following serum starvation and miR-221 or MC transfection, cFBs were further cultured in serum-free medium with 1% insulin-transferrin-selenium and 0.22 mM ascorbic acid supplements for 7 days. Collagen secretion into the culture medium was measured by the Sircol collagen assay (Biocolor, Axil Scientific, Singapore).20
cFB suspension and transfection complexes were mixed with collagen gel solution (ibidi, Sciencewerke, Singapore). After solidification, the gel was incubated in 2% fetal bovine serum (FBS) DMEM for 48 h. The gels were released from the culture well for photography. Contraction was presented as the percentage reduction of gel area over the entire culture surface.
The 3′ UTR Luciferase Reporter Assay
cDNA of the 3′ UTR of Tp53 was prepared by qPCR and cloned into psiCHECK-2 dual luciferase reporter plasmid (Promega, Singapore). The primer sequences are as follows: forward, 5′-CCGCTCGAG CAGCCTCTGCATCCTGTC-3′; and reverse, 5′-ATAAGAATGCGGCCGC ACAAGTGGTAACAAAAGTTTATTGT-3′. This 3′ UTR sequence has two non-conventional binding sites of miR-221. miR-221 sensor, the complementary binding sequence to miR-221, served as a positive control. Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Singapore) was used to mutate the binding sites and confirm the specificity. The luciferase reporter plasmid was co-transfected with the miR-221 or MC into HeLa cells. Results were expressed as the ratio of Renilla:Firefly luciferase activities using Dual-Glo luciferase assay system (Promega, Singapore).8
In Vivo MI Model and miR-221 Mimic Delivery
Rats were anesthetized with ketamine and xylazine (75 and 10 mg/kg, respectively, intraperitoneally [i.p.]), intubated, and ventilated. The chest was opened by an incision between the left third and fourth ribs. The left anterior descending coronary artery (LAD) was ligated using a 7/0 polypropylene suture. The chest was closed. An identical surgical procedure without LAD ligation was carried out to generate a sham control group (sham).
The miR-221 and Invivofectamine 3.0 (Thermo Fisher Scientific, Singapore) complex of 1 mg/kg miR-221 in 800 μL volume or equal volume of PBS was given via a tail vein injection. Each rat received 2 injections immediately after LAD ligation and 3 days post-surgery (MI +miR-221). PBS was injected as a control (MI-Ctrl). Hearts were collected at days 2, 7, and 30 post-MI.
Cardiac function was monitored by echocardiography (MS250 transducer, Vevo 2100, FUJIFILM VisualSonics, ITS Science and Medical, Singapore) before and 2, 7, 14, and 30 days post-MI under isoflurane (1.5%–2%) anesthesia. At day 30, rats were anesthetized with pentobarbital (60 mg/kg, i.p.) for LV function assessment. A Millar Mikro-Tip pressure catheter (Millar, Houston, TX, USA) was inserted through the right carotid artery into the LV to assess cardiac function, e.g., LV developing pressure, end diastolic pressure, ±dP/dt, and heart rate (Powerlab data acquisition system, ADInstruments, Advanced Tech, Singapore).
Histology
Hearts of day 2 and day 7 post-MI were dissected to collect infarct, border-zone, and remote myocardium, which was snap frozen in liquid nitrogen for protein and RNA analysis. Hearts at day 7 and day 30 post-MI were fixed in 10% neutral buffered formalin for histology. The mid-myocardium was cut into 5-μm sections and stained with Masson trichrome or picro-sirius red (Sigma-Aldrich, Singapore) plus immunofluorescence staining for α-SMA (clone 1A4; Sigma-Aldrich, Singapore) and DAPI (Thermo Fisher Scientific, Singapore). Bright-field and fluorescent images were captured (Nikon Eclipse Ti inverted microscope with C-HGFIE pre-centered fiber illuminator, Nikon, Singapore) and analyzed using ImageJ 1.45 s.37
miRNA Stem-Loop qPCR and mRNA qRT-PCR
RNA was extracted with TRIzol Reagent followed by DNase I treatment (Invitrogen, Life Technologies, Thermo Fisher Scientific, Singapore). miRNA expression was determined by stem-loop qPCR (TaqMan MicroRNA Assay, QuantStudio 7 Flex Real-Time PCR System, Thermo Fisher Scientific, Singapore) and normalized to U6B. Gene expressions were measured by universal reverse transcription and real-time PCR (iScript RT Supermix, iTaq Universal SYBR Green Supermix, Bio-Rad, Singapore), and they were normalized by β-actin. Primers are listed in Table S1.
Western Blots
Protein expression in cell or tissue lysates was analyzed using SDS-PAGE and immunoblotting with the following different antibodies: phospho-p53 (Ser46), Bmf (GeneTex, Axil Scientific, Singapore), β-actin, t-p53, p- and t-Smad 2/3, Bak1, Puma (Santa Cruz Biotechnology, Axil Scientific, Singapore), Tp53inp1, Ddit4 (Sigma-Aldrich, Singapore), P62, LC3, and cleaved caspase-3 (Casp-3) (Cell Signaling Technology, Research Biolabs, Singapore).
Statistics
Data were compared for differences by one-way ANOVA followed by Bonferroni post hoc analysis or unpaired two-tailed t test as appropriate (Prism; Graph Pad, La Jolla, CA, USA). Two-way ANOVA repeated measurement with Bonferroni adjustment was used for analysis of serial measurement of echocardiography. Values were expressed as mean ± SD. Differences between groups were regarded as significant at the p < 0.05 probability level.
Author Contributions
Conceptualization, P.W.; Methodology, Formal Analysis, Investigation, and Writing – Original Draft Review, Y.Z. and P.W.; Editing and Supervision, P.W. and A.M.R.; Funding Acquisition, A.M.R.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Ms. Ailing Yang, an exchange student from Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China, and Thiam Chien Shiok for their contributions in performing some of the experiments. This work was supported by grants from National Medical Research Council Center Grant (Ministry of Health, Singapore; PI: A.M.R.) and the CVRI Operating Fund (National University of Singapore, PI A.M.R.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.05.018.
Supplemental Information
References
- 1.Quintavalle C., Garofalo M., Zanca C., Romano G., Iaboni M., del Basso De Caro M., Martinez-Montero J.C., Incoronato M., Nuovo G., Croce C.M., Condorelli G. miR-221/222 overexpession in human glioblastoma increases invasiveness by targeting the protein phosphate PTPμ. Oncogene. 2012;31:858–868. doi: 10.1038/onc.2011.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coskunpinar E., Cakmak H.A., Kalkan A.K., Tiryakioglu N.O., Erturk M., Ongen Z. Circulating miR-221-3p as a novel marker for early prediction of acute myocardial infarction. Gene. 2016;591:90–96. doi: 10.1016/j.gene.2016.06.059. [DOI] [PubMed] [Google Scholar]
- 3.Verjans R., Peters T., Beaumont F.J., van Leeuwen R., van Herwaarden T., Verhesen W., Munts C., Bijnen M., Henkens M., Diez J. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload-Induced Heart Failure. Hypertension. 2018;71:280–288. doi: 10.1161/HYPERTENSIONAHA.117.10094. [DOI] [PubMed] [Google Scholar]
- 4.Bazan H.A., Hatfield S.A., O’Malley C.B., Brooks A.J., Lightell D., Jr., Woods T.C. Acute Loss of miR-221 and miR-222 in the Atherosclerotic Plaque Shoulder Accompanies Plaque Rupture. Stroke. 2015;46:3285–3287. doi: 10.1161/STROKEAHA.115.010567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao D., Deng S.C., Ma Y., Hao Y.H., Jia Z.H. miR-221 alleviates the inflammatory response and cell apoptosis of neuronal cell through targeting TNFAIP2 in spinal cord ischemia-reperfusion. Neuroreport. 2018;29:655–660. doi: 10.1097/WNR.0000000000001013. [DOI] [PubMed] [Google Scholar]
- 6.Hu S., Huang M., Nguyen P.K., Gong Y., Li Z., Jia F., Lan F., Liu J., Nag D., Robbins R.C., Wu J.C. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation. 2011;124(11 Suppl):S27–S34. doi: 10.1161/CIRCULATIONAHA.111.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y., Chen Q., Lew K.S., Richards A.M., Wang P. Discovery of Potential Therapeutic miRNA Targets in Cardiac Ischemia-Reperfusion Injury. J. Cardiovasc. Pharmacol. Ther. 2016;21:296–309. doi: 10.1177/1074248415604463. [DOI] [PubMed] [Google Scholar]
- 8.Chen Q., Zhou Y., Richards A.M., Wang P. Up-regulation of miRNA-221 inhibits hypoxia/reoxygenation-induced autophagy through the DDIT4/mTORC1 and Tp53inp1/p62 pathways. Biochem. Biophys. Res. Commun. 2016;474:168–174. doi: 10.1016/j.bbrc.2016.04.090. [DOI] [PubMed] [Google Scholar]
- 9.Yu B., Gong M., Wang Y., Millard R.W., Pasha Z., Yang Y., Ashraf M., Xu M. Cardiomyocyte protection by GATA-4 gene engineered mesenchymal stem cells is partially mediated by translocation of miR-221 in microvesicles. PLoS ONE. 2013;8:e73304. doi: 10.1371/journal.pone.0073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su M., Wang J., Wang C., Wang X., Dong W., Qiu W., Wang Y., Zhao X., Zou Y., Song L. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015;22:986–999. doi: 10.1038/cdd.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ichihara S., Senbonmatsu T., Price E., Jr., Ichiki T., Gaffney F.A., Inagami T. Targeted deletion of angiotensin II type 2 receptor caused cardiac rupture after acute myocardial infarction. Circulation. 2002;106:2244–2249. doi: 10.1161/01.cir.0000033826.52681.37. [DOI] [PubMed] [Google Scholar]
- 12.Hayakawa K., Takemura G., Kanoh M., Li Y., Koda M., Kawase Y., Maruyama R., Okada H., Minatoguchi S., Fujiwara T., Fujiwara H. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation. 2003;108:104–109. doi: 10.1161/01.CIR.0000074225.62168.68. [DOI] [PubMed] [Google Scholar]
- 13.Nakaya M., Watari K., Tajima M., Nakaya T., Matsuda S., Ohara H., Nishihara H., Yamaguchi H., Hashimoto A., Nishida M. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Invest. 2017;127:383–401. doi: 10.1172/JCI83822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thai H.M., Van H.T., Gaballa M.A., Goldman S., Raya T.E. Effects of AT1 receptor blockade after myocardial infarct on myocardial fibrosis, stiffness, and contractility. Am. J. Physiol. 1999;276:H873–H880. doi: 10.1152/ajpheart.1999.276.3.H873. [DOI] [PubMed] [Google Scholar]
- 15.Silvestre J.S., Heymes C., Oubénaïssa A., Robert V., Aupetit-Faisant B., Carayon A., Swynghedauw B., Delcayre C. Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation. 1999;99:2694–2701. doi: 10.1161/01.cir.99.20.2694. [DOI] [PubMed] [Google Scholar]
- 16.Okada H., Takemura G., Kosai K., Li Y., Takahashi T., Esaki M., Yuge K., Miyata S., Maruyama R., Mikami A. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–2437. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- 17.Pan Z., Sun X., Shan H., Wang N., Wang J., Ren J., Feng S., Xie L., Lu C., Yuan Y. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway. Circulation. 2012;126:840–850. doi: 10.1161/CIRCULATIONAHA.112.094524. [DOI] [PubMed] [Google Scholar]
- 18.Weber K.T., Sun Y., Gerling I.C., Guntaka R.V. Regression of Established Cardiac Fibrosis in Hypertensive Heart Disease. Am. J. Hypertens. 2017;30:1049–1052. doi: 10.1093/ajh/hpx054. [DOI] [PubMed] [Google Scholar]
- 19.Kanisicak O., Khalil H., Ivey M.J., Karch J., Maliken B.D., Correll R.N., Brody M.J., J Lin S.C., Aronow B.J., Tallquist M.D., Molkentin J.D. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y., Richards A.M., Wang P. Characterization and Standardization of Cultured Cardiac Fibroblasts for Ex Vivo Models of Heart Fibrosis and Heart Ischemia. Tissue Eng. Part C Methods. 2017;23:422–433. doi: 10.1089/ten.TEC.2017.0169. [DOI] [PubMed] [Google Scholar]
- 21.Nakano K., Vousden K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 22.Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 23.Mann M., Wright P.R., Backofen R. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017;45(W1):W435–W439. doi: 10.1093/nar/gkx279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rolland-Turner M., Goretti E., Bousquenaud M., Léonard F., Nicolas C., Zhang L., Maskali F., Marie P.Y., Devaux Y., Wagner D. Adenosine stimulates the migration of human endothelial progenitor cells. Role of CXCR4 and microRNA-150. PLoS ONE. 2013;8:e54135. doi: 10.1371/journal.pone.0054135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maier T., Güell M., Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 26.Brodersen P., Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat. Rev. Mol. Cell Biol. 2009;10:141–148. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- 27.Poyurovsky M.V., Prives C. Unleashing the power of p53: lessons from mice and men. Genes Dev. 2006;20:125–131. doi: 10.1101/gad.1397506. [DOI] [PubMed] [Google Scholar]
- 28.Crow M.T. Revisiting p53 and its effectors in ischemic heart injury. Cardiovasc. Res. 2006;70:401–403. doi: 10.1016/j.cardiores.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Song H., Conte J.V., Jr., Foster A.H., McLaughlin J.S., Wei C. Increased p53 protein expression in human failing myocardium. J. Heart Lung Transplant. 1999;18:744–749. doi: 10.1016/s1053-2498(98)00039-4. [DOI] [PubMed] [Google Scholar]
- 30.Matsusaka H., Ide T., Matsushima S., Ikeuchi M., Kubota T., Sunagawa K., Kinugawa S., Tsutsui H. Targeted deletion of p53 prevents cardiac rupture after myocardial infarction in mice. Cardiovasc. Res. 2006;70:457–465. doi: 10.1016/j.cardiores.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 31.D’Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002;4:11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 32.Mihara M., Erster S., Zaika A., Petrenko O., Chittenden T., Pancoska P., Moll U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 33.Westphal D., Dewson G., Menard M., Frederick P., Iyer S., Bartolo R., Gibson L., Czabotar P.E., Smith B.J., Adams J.M., Kluck R.M. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc. Natl. Acad. Sci. USA. 2014;111:E4076–E4085. doi: 10.1073/pnas.1415142111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X., Cheng Y., Yang J., Xu L., Zhang C. Cell-specific effects of miR-221/222 in vessels: molecular mechanism and therapeutic application. J. Mol. Cell. Cardiol. 2012;52:245–255. doi: 10.1016/j.yjmcc.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lajiness J.D., Conway S.J. Origin, development, and differentiation of cardiac fibroblasts. J. Mol. Cell. Cardiol. 2014;70:2–8. doi: 10.1016/j.yjmcc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Amerongen M.J., Bou-Gharios G., Popa E., van Ark J., Petersen A.H., van Dam G.M., van Luyn M.J., Harmsen M.C. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J. Pathol. 2008;214:377–386. doi: 10.1002/path.2281. [DOI] [PubMed] [Google Scholar]
- 37.Ivey M.J., Kuwabara J.T., Pai J.T., Moore R.E., Sun Z., Tallquist M.D. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell. Cardiol. 2018;114:161–174. doi: 10.1016/j.yjmcc.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molkentin J.D., Bugg D., Ghearing N., Dorn L.E., Kim P., Sargent M.A., Gunaje J., Otsu K., Davis J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation. 2017;136:549–561. doi: 10.1161/CIRCULATIONAHA.116.026238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su M., Wang J., Wang C., Wang X., Dong W., Qiu W., Wang Y., Zhao X., Zou Y., Song L. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015;22:986–999. doi: 10.1038/cdd.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Souders C.A., Bowers S.L., Baudino T.A. Cardiac fibroblast: the renaissance cell. Circ. Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagpal V., Rai R., Place A.T., Murphy S.B., Verma S.K., Ghosh A.K., Vaughan D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation. 2016;133:291–301. doi: 10.1161/CIRCULATIONAHA.115.018174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Y., Shiok T.C., Richards A.M., Wang P. MicroRNA-101a suppresses fibrotic programming in isolated cardiac fibroblasts and in vivo fibrosis following trans-aortic constriction. J. Mol. Cell. Cardiol. 2018;121:266–276. doi: 10.1016/j.yjmcc.2018.07.251. [DOI] [PubMed] [Google Scholar]
- 43.Assomull R.G., Prasad S.K., Lyne J., Smith G., Burman E.D., Khan M., Sheppard M.N., Poole-Wilson P.A., Pennell D.J. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J. Am. Coll. Cardiol. 2006;48:1977–1985. doi: 10.1016/j.jacc.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 44.Lucas T., Schäfer F., Müller P., Eming S.A., Heckel A., Dimmeler S. Light-inducible antimiR-92a as a therapeutic strategy to promote skin repair in healing-impaired diabetic mice. Nat. Commun. 2017;8:15162. doi: 10.1038/ncomms15162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eding J.E., Demkes C.J., Lynch J.M., Seto A.G., Montgomery R.L., Semus H.M., Jackson A.L., Isabelle M., Chimenti S., van Rooij E. The Efficacy of Cardiac Anti-miR-208a Therapy Is Stress Dependent. Mol. Ther. 2017;25:694–704. doi: 10.1016/j.ymthe.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegel P.M., Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 47.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 48.Montgomery R.L., Yu G., Latimer P.A., Stack C., Robinson K., Dalby C.M., Kaminski N., van Rooij E. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 2014;6:1347–1356. doi: 10.15252/emmm.201303604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janssen H.L., Reesink H.W., Lawitz E.J., Zeuzem S., Rodriguez-Torres M., Patel K., van der Meer A.J., Patick A.K., Chen A., Zhou Y. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 50.Chu I.M., Hengst L., Slingerland J.M. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 51.le Sage C., Nagel R., Egan D.A., Schrier M., Mesman E., Mangiola A., Anile C., Maira G., Mercatelli N., Ciafrè S.A. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao X.F., Zhou Y., Wang D.Y., Lew K.S., Richards A.M., Wang P. Urocortin-2 suppression of p38-MAPK signaling as an additional mechanism for ischemic cardioprotection. Mol. Cell. Biochem. 2015;398:135–146. doi: 10.1007/s11010-014-2213-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.