

Summary
DNA-damaging compounds, commonly used as chemotherapeutic drugs, are known to trigger cells to undergo programmed cell death such as apoptosis and necroptosis. However, the molecular mechanism of DNA damage-induced cell death is not fully understood. Here, we report that RARγ has a critical role in DNA damage-induced programmed cell death, specifically in necroptosis. The loss of RARγ abolishes the necroptosis induced by DNA damage. In addition, cells that lack RARγ are less susceptible to extrinsic apoptotic pathway activated by DNA-damaging agents whereas the intrinsic apoptotic pathway is not affected. We demonstrate that RARγ is essential for the formation of RIPK1/RIPK3 death complex, known as Ripoptosome, in response to DNA damage. Furthermore, we show that RARγ plays a role in skin cancer development by using RARγ1 knockout mice and human squamous cell carcinoma biopsies. Hence, our study reveals that RARγ is a critical component of DNA damage-induced cell death.
Subject Areas: Biological Sciences, Biochemistry, Molecular Biology, Cell Biology
Graphical Abstract
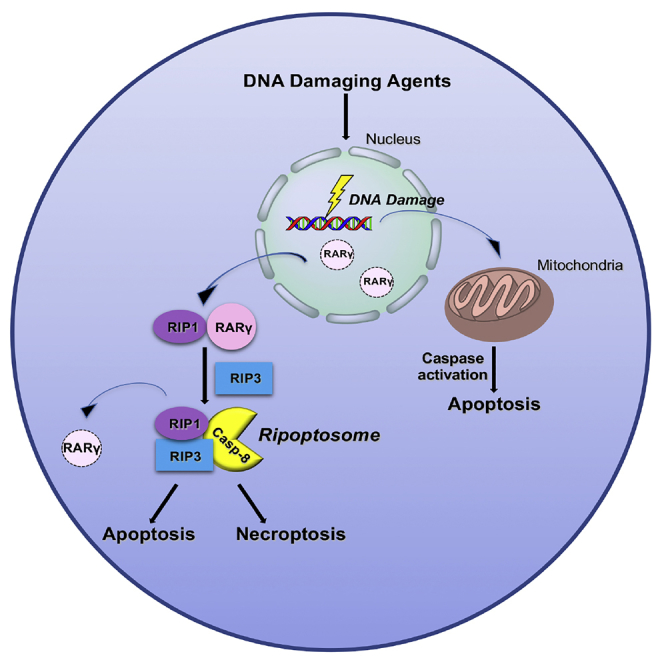
Highlights
-
•
RARγ plays a key role in DNA damage-induced cell death
-
•
RARγ is essential for RIPK1-mediated necroptosis and apoptosis following DNA damage
-
•
RARγ is required for the formation of Ripoptosome in response to DNA damage
-
•
Loss of RARγ correlates with skin cancer development
Biological Sciences; Biochemistry; Molecular Biology; Cell Biology
Introduction
DNA-damaging agents are a widely used group of prominent drugs of cancer treatment (Bozko et al., 2017). Among the commonly used DNA-damaging agents, which covalently modify DNA, for instance, cisplatin and oxaliplatin cross-link DNA bases (Kelland, 2007), whereas other agents, such as inhibitors of topoisomerases (camptothecin, etoposide, and doxorubicin) or antimetabolites (pyrimidine analog 5-fluorouracil), cause DNA damage by changing chromatin structure and topology or by interfering with DNA replication (Longley et al., 2003, Nitiss, 2009, Pommier, 2006). It has been well documented that DNA damage elicits diverse cellular responses such as DNA repair, cell-cycle arrest, and cell death (Mills et al., 2018, O'Connor, 2015). Although the underlying molecular mechanisms of these cellular responses have been extensively studied, many aspects of DNA damage responses are not fully understood (Cheung-Ong et al., 2013).
Following DNA damage, a predominant outcome of affected cells is apoptosis (Roos and Kaina, 2006, Roos et al., 2016). DNA-damaging agents induce apoptosis by blocking DNA replication, which leads to the collapse of replication forks and double-strand breaks (DSBs). DSBs result in p53-dependent caspase activation and subsequent apoptosis. However, it is known that DNA damage could also trigger p53-independent, death domain kinase, RIPK1 (receptor-interacting protein kinase 1)-mediated cell death (Biton and Ashkenazi, 2011, Hur et al., 2003, Hur et al., 2006). Recent evidence suggests that DNA-damaging agents induce not only apoptosis but also non-apoptotic death such as necroptosis and pyroptosis (Wang et al., 2017). Particularly, although the mechanism of DNA damage-induced apoptosis has been well studied, the molecular regulation of other forms of DNA damage-induced cell death are still elusive (Norbury and Zhivotovsky, 2004, Roos et al., 2016).
Necroptosis is a programmed, caspase-independent cell death that is morphologically similar to necrosis. For tumor necrosis factor (TNF)-induced necroptosis, the receptor interacting protein kinases, RIPK1 and RIPK3, and the mixed lineage kinase domain-like (MLKL) are the key mediators (Cho et al., 2009, He et al., 2009, Sun et al., 2012, Zhang et al., 2009). It is known that, after being phosphorylated by RIPK3, MLKL translocates to the plasma membrane and mediates the execution of necroptosis (Cai et al., 2014, Cai and Liu, 2014, Wang et al., 2014, Weinlich et al., 2017). It has been shown that DNA damage could induce necroptosis and that RIPK1 and RIPK3 are essential for the process (Moriwaki et al., 2015, Pasparakis and Vandenabeele, 2015, Vandenabeele et al., 2010, Weinlich et al., 2017). However, although it has been shown that RIPK1 mediates the formation of the cytosolic cell death complex, known as Ripoptosome (Tenev et al., 2011), it is not clear how RIPK1 is engaged in response to DNA damage.
Retinoic acid receptors (RARs), RARα, RARβ, and RARγ belong to the superfamily of nuclear hormone receptors and act as transcription factors after activation by retinoic acid (De Luca, 1991, Kastner et al., 1995). RARs regulate the expression of a large number of genes that are critical for cell growth, differentiation, and cell death (Lohnes et al., 1993, Marill et al., 2003). Although the localization of these RARs is predominantly nuclear, cytoplasmic localizations of RARs have been reported in some types of cells, but the function of the cytosolic RARs is unknown (Han et al., 2009). Our previous work demonstrated that cytoplasmic RARγ was essential for TNF-induced RIPK1-initiated apoptosis and necroptosis. RARγ is released from the nucleus to orchestrate the formation of the cytosolic death complexes and functions as a critical checkpoint of RIPK1-initiated cell death.
RARγ is the major RAR expressed in human and mouse epidermis, whereas very little expression of RARα and no expression of RARβ is seen in skin (Duong and Rochette-Egly, 2011). RARγ has been shown to be a tumor suppressor, as loss of RARγ predisposed skin to tumors (Chen et al., 2004, Darwiche et al., 1995, Darwiche et al., 1996). In addition, there is evidence that UV radiation from the sun induces a dramatic decrease in RARγ by proteasomal degradation resulting in premature skin aging and skin cancer (Chen et al., 2004, Wang et al., 1999). However, the role of RARγ in skin cancer development is not well studied.
Here, we report that RARγ has a critical role in DNA damage-induced necroptotic cell death. We show that loss of RARγ abolishes the necroptosis induced by DNA-damaging agents. Furthermore, RARγ-null MEFs are less susceptible to apoptosis induced by DNA-damaging agents. We demonstrate that the involvement of RARγ in DNA damage-induced necroptosis and apoptosis is mediated through RIPK1 and that RARγ does not play a role in the intrinsic apoptosis triggered by DNA damage. This study reveals that RARγ is a critical component of DNA damage-induced cell death. Furthermore, we demonstrate that significant decrease in RARγ expression in the human squamous cell carcinoma (SCC) compared with normal skin and the loss of RARγ in keratinocytes leads to resistance to DNA-damaging drugs. Therefore, RARγ could be used as a chemotherapeutic marker for cancers.
Results
DNA-Damaging Compounds Induce RIPK1-Dependent Necroptosis in MEFs
It has been reported that DNA-damaging agents could induce necroptosis and trigger both extrinsic and intrinsic apoptotic pathways in certain types of cells (Fulda and Debatin, 2006, Jing et al., 2018, Koo et al., 2015, Matt and Hofmann, 2016, Nowsheen and Yang, 2012). To study the mechanisms of DNA damage-induced cell death, we first examined if these cell death pathways are activated in mouse embryonic fibroblasts (MEFs) in response to DNA damage by checking the commonly used markers for these pathways, MLKL phosphorylation for necroptosis, Caspase-9 (Casp-9) for intrinsic apoptosis, and Caspase-8 (Casp-8) for extrinsic apoptosis (Hengartner, 2000, Li and Yuan, 2008, Pasparakis and Vandenabeele, 2015, Wang et al., 2014). MEF cells were treated with DNA-damaging agents, cisplatin or etoposide and were collected to examine these proteins by immunoblotting at the indicated times (Figure 1A). As shown in Figure 1A, all three cell death pathways are activated by cisplatin or etoposide. Interestingly, among these three cell death pathways, necroptosis appears to be engaged first, followed by the activation of extrinsic apoptosis. The engagement of intrinsic apoptosis is significantly slower compared with the other two. These results suggest that DNA damage induced both necroptosis and apoptosis, with necroptosis and extrinsic apoptosis preceding intrinsic apoptosis, in MEF cells.
Figure 1.

DNA-Damaging Compounds Induce Both RIPK1-Dependent Apoptotic and Necroptotic Cell Death
(A) MEF WT cells were treated with cisplatin (left panel) or etoposide (right panel) for the indicated time. Cell lysates were analyzed by immunoblotting as indicated.
(B) Ripk1+/+ and Ripk1−/− cells were treated with cisplatin (left panel) or etoposide (right panel) for the indicated time. Cell lysates were analyzed by immunoblotting as indicated.
(C) Ripk1+/+ and Ripk1−/− cells were treated with cisplatin 50 μM (left panel) or etoposide 50 μM (right panel) for the indicated time period, and cell death analysis was determined by propidium iodide staining and analyzed by flow cytometry. See also Figure S1.
All blots above are representative of one of three experiments. Results shown are averages ± SEM from three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001.
As RIPK1 is known to be a key player in DNA damage-induced necroptosis and apoptosis (Biton and Ashkenazi, 2011, Hur et al., 2003, Hur et al., 2006), we then examined the involvement of RIPK1 in the phosphorylation of MLKL and the activation of the extrinsic and intrinsic apoptotic pathways using wild-type (WT) (Ripk1+/+) and RIPK1-null (Ripk1−/−) MEFs. Cells were treated with cisplatin or etoposide and were then collected for western blot. As shown in Figure 1B, consistent with previous findings, RIPK1 is essential for cisplatin or etoposide-induced MLKL phosphorylation. Interestingly, we found that whereas Casp-8 activation was significantly impaired and delayed, activation of Casp-9 by these two drugs was not affected by the deletion of RIPK1 (Figure 1B). These results suggest that the engagement of necroptosis and extrinsic apoptosis were both mediated by RIPK1, which does not play any role in the intrinsic apoptotic pathway. These conclusions were further supported by measuring DNA damage-induced cell death in these cells. As shown in Figure 1C, loss of RIPK1 only partially blocked DNA damage-induced cell death because deletion of RIPK1 has no effect on Casp-9 activation. Consistently, RIPK3 and MLKL deletion also partially blocked DNA damage-induced cell death and the phosphorylation of MLKL (Figure S1).
RARγ Plays a Role in DNA Damage-Induced Necroptosis and Extrinsic Apoptosis
Recently, we reported that the nuclear receptor, RARγ, plays a key role in RIPK1-mediated tumor necrosis factor (TNF)-induced cell death (Xu et al., 2017a). As RIPK1 mediates DNA damage-induced necroptosis and extrinsic apoptosis, we then tested the role of RARγ in DNA damage-induced cell death. To do so, the WT (Rarγ+/+) and RARγ-null (Rarγ−/−) MEFs (Figure S2A) were treated with cisplatin or etoposide and collected for examining the phosphorylation of MLKL and the activation of Casp-8 and Casp-9. As shown in Figures 2A and 2B, similar to that observed with RIPK1 deficiency, deletion of RARγ led to the abolishment of MLKL phosphorylation and the impaired activation of Casp-8, but had no effect on Casp-9 activation in response to DNA damage. Consistent with the results of MLKL phosphorylation and the activation of Casp-8 and Casp-9, loss of RARγ partially protected cells against DNA damage-induced cell death when compared with that observed in WT MEFs by cisplatin or etoposide (Figure 2C) or other DNA-damaging agents (Figure S2B). To confirm that the resistance of Rarγ−/− MEFs to DNA-damaging agents is due to the loss of RARγ, RARγ was reintroduced back to Rarγ−/− MEFs. The ectopic expression of RARγ restored the sensitivity of the cells to cisplatin or etoposide (Figure 2D). We also examined the role of RARγ in DNA damage-induced cell death in human HT29 cancer cells. As shown in Figure S3, we found that DNA damage-induced cell death, the phosphorylation of MLKL, and the activation of Casp-8 are significantly decreased in RARγ knockdown HT29 (RARγ-short haprpin RNA [shRNA]) cells compared with WT control-shRNA HT29 cells following cisplatin treatment, whereas Casp-9 activation is not affected. These results are similar to what we observed in Rarγ+/+ and Rarγ−/− MEFs (Figures 2A–2C). We next tested if the partial protection against DNA damage-induced cell death in RARγ-null cells is due to the activation of caspases. To do so, we pre-treated Rarγ+/+ and Rarγ−/− cells with the caspase inhibitor z-VAD-fmk (zVAD) and a Casp-9-specific inhibitor, zLEHD, because zVAD does not block Casp-9 activation efficiently (Rodriguez-Enfedaque et al., 2012), followed by cisplatin or etoposide treatment. As shown in Figure 3A, although zVAD treatment did increase the survival of RARγ-null cells, the combined treatment of zVAD and zLEHD almost completely blocked cell death triggered by cisplatin or etoposide in RARγ−/− cells. However, these caspase inhibitors only partially protected WT cells against cell death induced by cisplatin and etoposide (Figure 3A). The remaining DNA damage-induced cell death of the WT MEF cells with pre-treatment of both zVAD and zLEHD is due to the activation of the necroptotic pathway because treating these WT cells with the specific RIPK1 inhibitor, necrostatin-1, completely blocked cell death induced by cisplatin or etoposide (Figures 3B and S4). In addition, as shown in Figures 3C and S4C, treatment with caspase inhibitors blocked caspase activation, but had no effect on MLKL phosphorylation. Taken together, these results suggest that RARγ is essential for DNA damage-induced necroptosis and is involved in extrinsic, but not intrinsic, apoptosis induced by DNA-damaging compounds.
Figure 2.

RARγ Is Required for DNA Damage-Induced Necroptosis and Extrinsic Apoptosis
(A and B) Rarγ+/+ and Rarγ−/− cells were treated with cisplatin (left panels) or etoposide (right panels) for the indicated time. Cell lysates were analyzed by immunoblotting as indicated. See also Figure S2.
(C) Rarγ+/+ and Rarγ−/− cells were treated with cisplatin 50 μM (left panel) or etoposide 50 μM (right panel) for the indicated time period, and cell death analysis was determined by propidium iodide staining and analyzed by flow cytometry. See also Figure S3.
(D) Rarγ+/+, Rarγ−/−, and Rarγ−/− + WT-RARγ cell lysates were analyzed by immunoblotting as indicated (upper panel). Rarγ+/+, Rarγ−/−, and Rarγ−/− + WT-RARγ cells were treated with cisplatin 50 μM (lower left panel) or etoposide 50 μM (lower right panel) for the indicated time period, and cell death analysis was determined by popidium iodide staining and analyzed by flow cytometry.
All blots above are representative of one of three experiments. Results shown are averages ± SEM from three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001.
Figure 3.

Caspase Inhibitors Block DNA Damage-Induced Cell Death in RARγ-KO Cells
(A) Rarγ+/+ and Rarγ−/− cells were pre-treated for 30 min with or without z-VAD-fmk (zVAD), zLEHD, or combined treatments as indicated followed by treatment with cisplatin 50 μM (left panel) or etoposide 50 μM (right panel) for 36 h. Cell survival was determined by propidium iodide (PI) staining and analyzed by flow cytometry.
(B) Rarγ+/+ and Rarγ−/− cells were pre-treated for 30 min with or without zVAD, zLEHD, NEC1, or combined treatments as indicated followed by treatment with cisplatin 50 μM (left panel) or etoposide 50 μM (right panel) for 36 h. Cell survival was determined by PI staining and analyzed by flow cytometry.
(C) Rarγ+/+ and Rarγ−/− cells were pre-treated for 30 min with or without combined z-VAD-fmk (zVAD) and zLEHD followed by treatment with cisplatin (left panel) or etoposide (right panel) for 20 h. Cell lysates were analyzed by immunoblotting as indicated. All blots above are representative of one of three experiments. See also Figure S4. Results shown are averages ± SEM from three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
It is known that DNA-damaging compounds could trigger the autocrine production of TNFα in cells (Biton and Ashkenazi, 2011, Xu et al., 2015, Xu et al., 2017b). It has been suggested that the secreted TNFα contributes to DNA damage-induced necroptosis in mouse L929 cells (Xu et al., 2017b). Therefore, to examine the effect of the secreted TNFα in DNA damage-induced necroptosis in MEFs, we examined if TNFα is produced in response to DNA damage in Rarγ+/+ and Rarγ−/− MEFs. We found that cisplatin or etoposide treatment leads to the production of TNFα at similar levels in Rarγ+/+ and Rarγ−/− MEFs (Figure S5A). These results suggest that DNA damage induces the production of TNFα in MEFs and RARγ is not involved in the production of TNFα. To investigate the involvement of autocrine TNFα in DNA damage-induced cell death, we then examined the cisplatin- or etoposide-induced cell death, MLKL phosphorylation and the activation of Casp-8 and Casp-9 in Tnfr1+/+ and Tnfr1−/− MEFs. As shown in Figures S5B and S5C, there is no difference in cell death, MLKL phosphorylation and the activation of Casp-8 and Casp-9 in Tnfr1+/+ and Tnfr1−/− MEFs. Therefore, these results suggest that although DNA damage induces TNFα production, TNF signaling does not contribute to DNA damage-induced cell death in MEFs.
RARγ Is Required for RIPK1 to Initiate Necroptosis in Response to DNA Damage
To further investigate the involvement of RARγ in DNA damage-induced necroptosis, we next examined whether RARγ is required for RIPK1 to initiate the formation of the necrosome complex (Ripoptosome) in response to DNA damage. We previously reported that RARγ is critical for RIPK1 to initiate the formation of Ripoptosome triggered by TNF (Xu et al., 2017a). Specifically, we found that RARγ and RIPK1 formed an intermediate complex initially in response to induction of necroptosis by treatment with TSZ (TNF, Smac mimetic, z-VAD-fmk) and that RARγ was not present in the final necrosome complex when RIPK3 was recruited (Xu et al., 2017a). Therefore, we first tested if RARγ and RIPK1 form a complex in response to DNA damage by immunoprecipitating RARγ. As shown in Figure 4A, immunoprecipitating RARγ efficiently pulled RIPK1, but not RIPK3, following cisplatin treatment. To examine the recruitment of RIPK3 to the necrosome by RIPK1, we performed immunoprecipitation experiments with an anti-Casp-8 antibody as we and others have reported previously (Cho et al., 2009, Wang et al., 2008, Xu et al., 2017a). As shown in Figure 4B, immunoprecipitating caspase 8 from lysates of cisplatin-treated WT MEFs efficiently pulled down both RIPK1 and RIPK3. However, Casp-8 immunoprecipitation failed to pull down either of RIPK1 and RIPK3 from lysates of cisplatin-treated Rarγ−/− cells. However, as we found previously, RARγ was not present in the necrosome complex pulled down by immunoprecipitating Casp-8. Therefore, these results suggest that RARγ is essential for RIPK1 to initiate the formation of the necrosome induced by DNA-damaging agents.
Figure 4.

Cytosolic RARγ Is Required for RIPK1 to Initiate Necroptosis in Response to DNA Damage
(A) Rarγ+/+ cells were treated with cisplatin for the indicated time period. Cell lysates were collected and immunoprecipitated with anti-RARγ antibody. The immunoprecipitated complexes were immunoblotted with the indicated antibodies.
(B) Rarγ+/+ and Rarγ−/− cells were treated with cisplatin for the indicated time period. Cell lysates were collected and immunoprecipitated with anti-caspase 8 antibody. The immunoprecipitated complexes were immunoblotted with the indicated antibodies. See also Figure S5.
(C) Confocal microscopy of WT MEFs transfected with GFP- RARγ plasmid and treated with cisplatin, and images were captured in the indicated time period (blue, DAPI; green, RARγ). Images are representative of one of three experiments. Scale bar, 10 μm.
(D) Cellular fractionation of Rarγ+/+ cells treated with DMSO or cisplatin 50 μM for 6 h was performed. Total (T), cytosolic (C), and nuclear (N) fractions were analyzed by immunoblotting with the indicated antibodies. All blots and images above are representative of one of three experiments.
In our earlier study, we demonstrated that RARγ was released to the cytoplasm from the nucleus during TSZ-induced necroptosis (Xu et al., 2017a). We then examined the cellular localization of RARγ during the process of DNA damage-induced necroptosis. First, we transfected WT MEFs cells with a green fluorescent protein (GFP)-tagged RARγ plasmid and found that the GFP-RARγ protein almost exclusively localized in the nucleus in non-treated cells (Figure 4C). Interestingly, cisplatin treatment caused a dramatic increase of GFP-RARγ presence in the cytoplasm after 6-h treatment and this release of RARγ to the cytosol occurs parallel with the formation RARγ and RIPK1 complex (Figure 4C). Furthermore, we confirmed the release of the endogenous RARγ from the nucleus by cell fractionation and western blot (Figure 4D). These results indicated that RARγ was released to the cytoplasm from the nucleus and facilitates RIPK1 to initiate necroptosis in response to DNA damage.
Loss of RARγ Blocks DNA Damage-Induced Necroptosis in Keratinocytes and Promotes Carcinogen-Induced Papilloma
RARγ was found to be highly expressed in keratinocytes and its expression is decreased in skin cancers (Darwiche et al., 1995, Finzi et al., 1992, Xu et al., 2001). Early studies also suggested that RARs may play a role in skin tumor development and that RARγ may function as a tumor suppressor (Chen et al., 2004, Duong and Rochette-Egly, 2011). To study the role of RARγ-mediated cell death in skin cancer development, we first checked RARγ and RARα expression in human SCC samples and found that the expression of RARγ, but not RARα, is dramatically decreased in all cancer samples compared with normal adjacent tissues (Figure 5A). Our findings are consistent with previous reports and support that RARγ may function as a tumor suppressor in skin cancer development. However, the underlying mechanism of RARγ′s role in skin tumorigenesis is largely unknown.
Figure 5.

RARγ Required for DNA Damage-Induced Necroptosis in Keratinocytes and Promotes Carcinogen-Induced Papilloma
(A) Immunostaining of paraffin-embedded human SCC tumors and adjacent normal skin sections from human biopsy samples. Sections were stained with H&E or immunohistochemically stained with anti-RARγ or anti-RARα antibodies.
(B) Primary keratinocytes from Rarγ 1+/+ and Rarγ 1−/− mice were treated with DMBA or vehicle (acetone) for 5 days. Popidium iodide-positive population of cells mentioned above was determined by flow cytometry.
(C) Primary keratinocytes from RARγ1+/+ and RARγ1−/− mice were treated with DMBA or acetone for the indicated time. Cell lysates were analyzed by immunoblotting as indicated.
(D) Rarγ 1+/+ and Rarγ1−/− mice were treated with a single topical application of DMBA for 24 h, and the skin was collected. Skin sections were stained with H&E or immunohistochemically stained with anti-p-MLKL antibody. Right panel, the p-MLKL-positive cells were counted for 1 mm linear length of the epidermis.
(E) Rarγ1+/+ and Rarγ 1−/− mice were treated with a single topical application of DMBA followed 2 weeks later by twice weekly topical applications of TPA for 33 weeks. The number and size of papillomas on each mouse were recorded every 1 week. The average number of papillomas (more than 2 mm in diameter) per mouse is plotted versus the number of weeks post-initiation (left panel). Average papilloma size (in mm) was recorded for Rarγ1+/+ and Rarγ1−/− mice (right panel). See also Figure S6.
All blots and images above are representative of one of three experiments. Scale bar, 50 μm. Results shown in graphs are averages ± SEM from three independent experiments. ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
To explore the possible involvement of RARγ-mediated necroptosis in tumorigenesis, we employed the well-established initiation and promotion two-stage skin carcinogenesis mouse model. In this model, mice are first subjected to a single topical application of the carcinogen 7, 12-dimethylbenz[a]-anthracene (DMBA), which initiates the formation of tumors. Initiation stage is followed by the promotion stage during which animals are treated twice a week with a pro-inflammatory agent 12-0-tetradecanoylphorbol 13-acetate (TPA). As DMBA is a known DNA adduct agent, we first examined if DMBA induces necroptosis in primary keratinocytes. To do so, we tested the effect of DMBA treatment on WT and RARγ1-knockout (KO) cells in vitro. As shown in Figure 5B, loss of RARγ resulted in significant protection of the cells against DMBA-induced cell death. Importantly, DMBA triggers necroptosis in WT keratinocytes, but not the RARγ-KO cells (Figure 5C). Then, we checked if DMBA induced necroptosis in keratinocytes in vivo by examining the effect of the topical treatment of DMBA in the epidermal layer in WT and RARγ1-KO mice. Both WT and RARγ1-KO littermates were treated with a single topical dose of DMBA for 1 day, and skin samples were collected for MLKL phosphorylation with an anti-phosphoryl-MLKL antibody (Jiao et al., 2018). As shown in Figure 5D, the epidermis of RARγ1-KO mice had no phosphoryl-MLKL-positive cells, whereas abundant positive cells of MLKL phosphorylation in the epidermis of WT mice were observed, suggesting that loss of RARγ protected skin epidermal cells from DMBA-induced necroptosis. To be sure that RARγ deletion does not affect the promotion of epidermal hyperplasia by TPA, we treated WT and RARγ1-KO littermates with three topical applications of TPA over 1 week, and skin was collected 48 h after the last treatment. The epidermal thickness, measured as the distance from the basal to the upper granular layer, was similar between WT and RARγ1-KO, confirming that loss of RARγ does not affect TPA-promoted epidermal growth (Figure S3). Finally, we examined the effect of RARγ deletion on papilloma formation by treating both WT and RARγ1-KO littermates, generated in our previous study (Xu et al., 2017a), with a single topical dose of DMBA followed by twice weekly application of TPA for 32 weeks. As shown in Figures 5E and S6B, papillomas appeared after 10 weeks of TPA promotion. However, by 17 weeks, RARγ1-KO mice had a significant increase in papilloma size when compared with WT mice and by 22 weeks, in papilloma number as well. By 32 weeks, the size and number of papillomas on RARγ1-KO mice are twice as large and more numerous compared with WT mice. Taken together, the above results imply that RARγ-mediated necroptosis may play a key role in limiting DMBA/TPA-induced papilloma formation and growth.
Discussion
DNA-damaging compounds induce programmed cell death, such as apoptosis and necroptosis in diverse types of mammalian cells (Fulda and Debatin, 2006, Koo et al., 2015, Matt and Hofmann, 2016, Roos and Kaina, 2006, Roos et al., 2016). Although the molecular mechanism of DNA damage-induced apoptosis is well studied, the regulation of DNA damage-induced necroptosis is not fully understood. Our previous study defined a role for RARγ in RIPK1-initiated apoptosis and necroptosis (Xu et al., 2017a). Here, we found that the loss of RARγ rendered cells resistant to both RIPK1-initiated apoptosis and necroptosis induced by DNA-damaging compounds. Furthermore, we showed that treatment with DNA-damaging compounds leads to RARγ being released from the nucleus to the cytosol, which mediates the formation of the death complexes. We showed that RARγ is essential for RIPK1 and RIPK3 to form complex and the recruitment of pMLKL, which is activated by RIPK3 during necroptotic cell death in response to DNA damage.
Ours and others' early studies demonstrated that RIPK1 is crucial for DNA damage-induced cell death (Biton and Ashkenazi, 2011, Hur et al., 2003, Hur et al., 2006). In our current study, our results indicate that RARγ is critical for DNA damage-induced necroptosis and partial apoptotic pathway. We found that RIPK1 is essential for DNA damage-induced necroptosis and partially affects extrinsic apoptosis but not intrinsic apoptosis. Therefore, our work extended our understanding about the role of RIPK1 in DNA damage-induced cell death. Our current study suggests that whereas DNA damage induced both apoptosis and necroptosis, necroptosis and extrinsic apoptosis precede the intrinsic apoptotic pathway as the loss of RARγ leads to the delayed activation of caspase 8, the extrinsic apoptosis marker (Fulda and Debatin, 2006, Hengartner, 2000, Li and Yuan, 2008), but has no effect on the activation of the intrinsic apoptosis marker caspase 9. These conclusions are supported by the result that the combined treatment of zVAD and zLEHD completely prevent the DNA damage-induced caspase activation.
An earlier study reported that TNFα was produced in response to DNA damage and contributed to DNA damage-induced necroptosis (Xu et al., 2017b). In this study, L929 cells were used that the secreted TNFα contributes to DNA damage-induced necroptosis. L929 cells are known to undergo necroptosis following TNFα treatment alone. However, TNFα alone does not trigger any cell death in MEF cells, which undergo TNF-induced necroptosis in the presence of Smac mimetic and z-VAD (Vercammen et al., 1998, Zhang et al., 2011, Zhu et al., 2018). Although we also observed the production of TNFα following DNA damage, we did not observe any difference of cell death between WT and TNFR1 KO cells in response to DNA damage (Figure S5). Our finding in MEFs is similar to that reported by Xu et al., in which they found that cisplatin-induced production of autocrine TNFα has no effect in cell death in the mouse proximal tubule cells (Xu et al., 2015). In addition, we found that RARγ deletion does not affect the production of TNFα following DNA damage (Figure S5A). Previous studies found that the cytosolic death complex mediated by RIPK1, known as RIPoptosome, is responsible for DNA damage-induced necroptosis independently of all death factor and receptors (Biton and Ashkenazi, 2011, Tenev et al., 2011). Our current study provides insight about the formation of the RIPoptosome and demonstrated the critical role of RARγ in the formation of this cytosolic death complex in response to DNA damage. Therefore, DNA damage-induced necroptosis is mainly mediated by RARγ-orchestrated, cytosolic RIPK1/RIPK3 complex.
Previous studies suggest that RARγ may have a tumor suppressor role in skin tumor development (Chen et al., 2004, Wang et al., 1999). Our study with human SCC biopsy samples supported this possibility as a significant decrease in RARγ expression is observed in cancer samples compared with normal skin tissues that express high levels of RARγ. Furthermore, our use of RARγ1-KO mice in a model of skin cancer showed that the lack of RARγ leads to a significant increase in the number and size of papillomas. Importantly, from the examination of the epidermal layer after treatment with of DMBA, we found that loss of RARγ protected keratinocytes and skin epidermal cells from DMBA-induced necroptosis in vitro and in vivo, respectively. These results imply that RARγ may function as a tumor suppressor by mediating necroptosis to limit tumor growth during skin cancer development. However, when solid tumors grow to certain sizes, the cells in the core of solid tumors will undergo necroptosis because of hypoxia and nutrient depletion. Our recent work found that necroptosis at this stage of solid tumors following the reprogramming of RIPK3 expression has a promoting effect on tumor metastasis (Jiao et al., 2018).The underlying mechanism of this metastasis-promoting effect of tumor necroptosis is under investigation.
Taken together, our present results provide evidence that RARγ plays a critical role in DNA damage-induced necroptosis and extrinsic apoptosis by modulating RIPK1-initiated formation of the death complexes. This study reveals that RARγ is a critical regulatory component of the DNA damage-induced apoptotic and necroptotic cell death. Finally, our work further supported the possible role of RARγ as a tumor suppressor and demonstrated that RARγ could potentially serve as a clinical marker for chemotherapy.
Limitations of the Study
In this study, we examined the role of RARγ as a critical regulatory component of the DNA damage-induced apoptotic and necroptotic cell death. Our work examined the effect of RARγ in human and mouse skin cancer. However, a limitation of this study is that we did not examine the tumor suppressor function of RARγ in other types of cancer.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank R. Lake and CCR, LGCB microscopy core facility. This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Author Contributions
C.K. and Z.-G.L. conceived the study and designed the experiments. C.K. and S.C. did most of the experimental work. S.C., Q.X., and C. K. designed and performed animal experiments. C.K., S.C., and Z.-G.L. wrote the paper. Z.-G.L. supervised the study. All authors discussed the results and implications and commented on the manuscript at all stages.
Declaration of Interests
The authors declare no competing interests.
Published: July 26, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.06.019.
Supplemental Information
References
- Biton S., Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- Bozko M., Bozko A., Scholta T., Malek N.P., Bozko P. Editorial: DNA damage as a strategy for anticancer chemotherapy. Curr. Med. Chem. 2017;24:1487. doi: 10.2174/092986732415170630115722. [DOI] [PubMed] [Google Scholar]
- Cai Z., Liu Z.G. Execution of RIPK3-regulated necrosis. Mol. Cell. Oncol. 2014;1:e960759. doi: 10.4161/23723548.2014.960759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Jitkaew S., Zhao J., Chiang H.C., Choksi S., Liu J., Ward Y., Wu L.G., Liu Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.F., Goyette P., Lohnes D. RARgamma acts as a tumor suppressor in mouse keratinocytes. Oncogene. 2004;23:5350–5359. doi: 10.1038/sj.onc.1207682. [DOI] [PubMed] [Google Scholar]
- Cheung-Ong K., Giaever G., Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem. Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., Chan F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwiche N., Celli G., Tennenbaum T., Glick A.B., Yuspa S.H., De Luca L.M. Mouse skin tumor progression results in differential expression of retinoic acid and retinoid X receptors. Cancer Res. 1995;55:2774–2782. [PubMed] [Google Scholar]
- Darwiche N., Scita G., Jones C., Rutberg S., Greenwald E., Tennenbaum T., Collins S.J., De Luca L.M., Yuspa S.H. Loss of retinoic acid receptors in mouse skin and skin tumors is associated with activation of the ras(Ha) oncogene and high risk for premalignant progression. Cancer Res. 1996;56:4942–4949. [PubMed] [Google Scholar]
- De Luca L.M. Retinoids and their receptors in differentiation, embryogenesis, and neoplasia. FASEB J. 1991;5:2924–2933. [PubMed] [Google Scholar]
- Duong V., Rochette-Egly C. The molecular physiology of nuclear retinoic acid receptors. From health to disease. Biochim. Biophys. Acta. 2011;1812:1023–1031. doi: 10.1016/j.bbadis.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Finzi E., Blake M.J., Celano P., Skouge J., Diwan R. Cellular localization of retinoic acid receptor-gamma expression in normal and neoplastic skin. Am. J. Pathol. 1992;140:1463–1471. [PMC free article] [PubMed] [Google Scholar]
- Fulda S., Debatin K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- Han Y.H., Zhou H., Kim J.H., Yan T.D., Lee K.H., Wu H., Lin F., Lu N., Liu J., Zeng J.Z. A unique cytoplasmic localization of retinoic acid receptor-gamma and its regulations. J. Biol. Chem. 2009;284:18503–18514. doi: 10.1074/jbc.M109.007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Hengartner M.O. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hur G.M., Lewis J., Yang Q., Lin Y., Nakano H., Nedospasov S., Liu Z.G. The death domain kinase RIP has an essential role in DNA damage-induced NF-kappa B activation. Genes Dev. 2003;17:873–882. doi: 10.1101/gad.1062403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur G.M., Kim Y.S., Won M., Choksi S., Liu Z.G. The death domain kinase RIP has an important role in DNA damage-induced, p53-independent cell death. J. Biol. Chem. 2006;281:25011–25017. doi: 10.1074/jbc.M605577200. [DOI] [PubMed] [Google Scholar]
- Jiao D., Cai Z., Choksi S., Ma D., Choe M., Kwon H.J., Baik J.Y., Rowan B.G., Liu C., Liu Z.G. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018;28:868–870. doi: 10.1038/s41422-018-0058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L., Song F., Liu Z., Li J., Wu B., Fu Z., Jiang J., Chen Z. MLKL-PITPalpha signaling-mediated necroptosis contributes to cisplatin-triggered cell death in lung cancer A549 cells. Cancer Lett. 2018;414:136–146. doi: 10.1016/j.canlet.2017.10.047. [DOI] [PubMed] [Google Scholar]
- Kastner P., Mark M., Chambon P. Nonsteroid nuclear receptors: what are genetic studies telling us about their role in real life? Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- Koo G.B., Morgan M.J., Lee D.G., Kim W.J., Yoon J.H., Koo J.S., Kim S.I., Kim S.J., Son M.K., Hong S.S. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–725. doi: 10.1038/cr.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- Lohnes D., Kastner P., Dierich A., Mark M., LeMeur M., Chambon P. Function of retinoic acid receptor gamma in the mouse. Cell. 1993;73:643–658. doi: 10.1016/0092-8674(93)90246-m. [DOI] [PubMed] [Google Scholar]
- Longley D.B., Harkin D.P., Johnston P.G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Marill J., Idres N., Capron C.C., Nguyen E., Chabot G.G. Retinoic acid metabolism and mechanism of action: a review. Curr. Drug Metab. 2003;4:1–10. doi: 10.2174/1389200033336900. [DOI] [PubMed] [Google Scholar]
- Matt S., Hofmann T.G. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016;73:2829–2850. doi: 10.1007/s00018-016-2130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills C.C., Kolb E.A., Sampson V.B. Development of chemotherapy with cell-cycle inhibitors for adult and pediatric cancer therapy. Cancer Res. 2018;78:320–325. doi: 10.1158/0008-5472.CAN-17-2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K., Bertin J., Gough P.J., Orlowski G.M., Chan F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015;6:e1636. doi: 10.1038/cddis.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury C.J., Zhivotovsky B. DNA damage-induced apoptosis. Oncogene. 2004;23:2797–2808. doi: 10.1038/sj.onc.1207532. [DOI] [PubMed] [Google Scholar]
- Nowsheen S., Yang E.S. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012;34:243–254. [PMC free article] [PubMed] [Google Scholar]
- O'Connor M.J. Targeting the DNA damage response in cancer. Mol. Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enfedaque A., Delmas E., Guillaume A., Gaumer S., Mignotte B., Vayssiere J.L., Renaud F. zVAD-fmk upregulates caspase-9 cleavage and activity in etoposide-induced cell death of mouse embryonic fibroblasts. Biochim. Biophys. Acta. 2012;1823:1343–1352. doi: 10.1016/j.bbamcr.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Roos W.P., Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Roos W.P., Thomas A.D., Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer. 2016;16:20–33. doi: 10.1038/nrc.2015.2. [DOI] [PubMed] [Google Scholar]
- Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Tenev T., Bianchi K., Darding M., Broemer M., Langlais C., Wallberg F., Zachariou A., Lopez J., MacFarlane M., Cain K. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Vercammen D., Beyaert R., Denecker G., Goossens V., Van Loo G., Declercq W., Grooten J., Fiers W., Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Boudjelal M., Kang S., Voorhees J.J., Fisher G.J. Ultraviolet irradiation of human skin causes functional vitamin A deficiency, preventable by all-trans retinoic acid pre-treatment. Nat. Med. 1999;5:418–422. doi: 10.1038/7417. [DOI] [PubMed] [Google Scholar]
- Wang L., Du F., Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- Wang H., Sun L., Su L., Rizo J., Liu L., Wang L.F., Wang F.S., Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Wang Y., Gao W., Shi X., Ding J., Liu W., He H., Wang K., Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- Weinlich R., Oberst A., Beere H.M., Green D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- Xu X.C., Wong W.Y., Goldberg L., Baer S.C., Wolf J.E., Ramsdell W.M., Alberts D.S., Lippman S.M., Lotan R. Progressive decreases in nuclear retinoid receptors during skin squamous carcinogenesis. Cancer Res. 2001;61:4306–4310. [PubMed] [Google Scholar]
- Xu Y., Ma H., Shao J., Wu J., Zhou L., Zhang Z., Wang Y., Huang Z., Ren J., Liu S. A role for tubular necroptosis in cisplatin-induced AKI. J. Am. Soc. Nephrol. 2015;26:2647–2658. doi: 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Jitkaew S., Choksi S., Kadigamuwa C., Qu J., Choe M., Jang J., Liu C., Liu Z.G. The cytoplasmic nuclear receptor RARgamma controls RIP1 initiated cell death when cIAP activity is inhibited. Nat. Commun. 2017;8:425. doi: 10.1038/s41467-017-00496-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Ma H.B., Fang Y.L., Zhang Z.R., Shao J., Hong M., Huang C.J., Liu J., Chen R.Q. Cisplatin-induced necroptosis in TNFalpha dependent and independent pathways. Cell Signal. 2017;31:112–123. doi: 10.1016/j.cellsig.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C., Dong M.Q., Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhang D.W., Zheng M., Zhao J., Li Y.Y., Huang Z., Li Z., Han J. Multiple death pathways in TNF-treated fibroblasts: RIP3- and RIP1-dependent and independent routes. Cell Res. 2011;21:368–371. doi: 10.1038/cr.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K., Liang W., Ma Z., Xu D., Cao S., Lu X., Liu N., Shan B., Qian L., Yuan J. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018;9:500. doi: 10.1038/s41419-018-0524-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.