

Abstract
Background
Traumatic brain injury (TBI) is recognized as a metabolic disease, characterized by acute cerebral glucose hypo-metabolism. Adaptive metabolic responses to TBI involve the utilization of alternative energy substrates, such as ketone bodies. Cerebral microdialysis (CMD) has evolved as an accurate technique allowing continuous sampling of brain extracellular fluid and assessment of regional cerebral metabolism. We present the successful application of a combined hypothesis- and data-driven metabolomics approach using repeated CMD sampling obtained routinely at patient bedside. Investigating two patient cohorts (n = 26 and n = 12), we identified clinically relevant metabolic patterns at the acute post-TBI critical care phase.
Methods
Clinical and CMD metabolomics data were integrated and analysed using in silico and data modelling approaches. We used both unsupervised and supervised multivariate analysis techniques to investigate structures within the time series and associations with patient outcome.
Findings
The multivariate metabolite time series exhibited two characteristic brain metabolic states that were attributed to changes in key metabolites: valine, 4-methyl-2-oxovaleric acid (4-MOV), isobeta-hydroxybutyrate (iso-bHB), tyrosyine, and 2-ketoisovaleric acid (2-KIV). These identified cerebral metabolic states differed significantly with respect to standard clinical values. We validated our findings in a second cohort using a classification model trained on the cerebral metabolic states. We demonstrated that short-term (therapeutic intensity level (TIL)) and mid-term patient outcome (6-month Glasgow Outcome Score (GOS)) can be predicted from the time series characteristics.
Interpretation
We identified two specific cerebral metabolic patterns that are closely linked to ketometabolism and were associated with both TIL and GOS. Our findings support the view that advanced metabolomics approaches combined with CMD may be applied in real-time to predict short-term treatment intensity and long-term patient outcome.
Keywords: Traumatic brain injury, Cerebral microdialysis, Ketometabolism, Metabolic state
Abbreviation: 2-KIV, 2-ketoisovaleric acid; 4-MOV, 4-methyl-2-oxovaleric acid; 8-OH-C8, 8-hydroxyoctanoic acid; 10-OH-C10, 10-hydroxydecanoic acid; AcAc, acetoacetate; AU, arbitrary units; BCAA, branched-chain amino acids; bHB, beta-hydroxybutyrate; CMD, cerebral microdialysis; GABA, γ-aminobutyric acid; GCS, Glasgow coma scale; GOS, Glasgow outcome score; ICP, intracranial pressure; ICU, intensive care unit; iso-bHB, isobeta-hydroxybutyrate; KAA, ketogenic amino acids; LC-MS, liquid chromatography-mass spectrometry; LSTM, long-short-term memory network; MCFA, medium chain fatty acids; MCT, monocarboxylate transporter; PLS, partial least squares; PbtO2, brain tissue oxygen tension; RMSE, root mean square error; TBI, traumatic brain injury; TIL, therapeutic intensity level; UTC, coordinated universal time
Research in context.
Evidence before this study
Traumatic brain injury (TBI) is recognized as a metabolic disease with acute impaired glucose metabolism. The utilization of alternative energy substrates, such as ketone bodies could be beneficial for the patients. Cerebral microdialysis (CMD) is an accurate technique for continuous sampling of brain extracellular fluid and assessment of cerebral metabolism.
Added value of this study
Metabolomics (metabolic profiling) of brain extracellular fluid can identity different states of brain metabolism during the acute post-TBI critical care phase. The identified metabolic states are associated with routinely measured parameters used in clinical settings to monitor the patients' outcome. Ketometabolism was identified as one of the key components associated to the patients' outcome, suggesting an implication of brain metabolism in TBI pathophysiology and the potential for interventions targeting ketometabolism for TBI neuro-repair. The identified metabolic signature could predict short- and long-term patient outcome.
Implication of all the available evidence
The prediction of short- and long-term patient outcome could potentially be executed in near real-time using a streamlined workflow encompassing sample collection followed by metabolomics analysis. This approach could deliver insightful data and potentially have a big impact on patient well-being.
We also believe that these metabolic states can be modified during their time at the ICU using appropriate nutritional intervention to alter ketometabolism. Further clinical studies are required to investigate the flexibility and adaptation of these metabolic states to enable optimal clinical intervention.
Alt-text: Unlabelled Box
1. Introduction
Traumatic Brain Injury (TBI) is a significant cause of death, as each year, >50 million TBIs occur worldwide. TBI is associated with large economic costs to the healthcare systems, as patients often require intensive care for prolonged periods followed by even longer rehabilitation periods [[1], [2], [3]].
From a pathophysiological standpoint, TBI can be subdivided into primary and secondary injuries. Primary injuries result as an immediate effect of the impacts, whereas secondary injuries often develop within hours or days after the primary event and are characterized by a complex cascade of events, including oedema [4], hypoxia/ischemia, epileptic seizures. Another important pathological determinant of TBI is metabolic dysfunction, and indeed TBI may be considered a metabolic disorder, characterized by a state of cerebral glucose hypo-metabolism [2,3,[5], [6], [7], [8]].
The cerebral microdialysis (CMD) technique has recently been introduced to capture the subtle alterations of regional cerebral energy metabolism following TBI, allowing sampling of patient brain extracellular fluid and the monitoring of various energy metabolites involved in cerebral glucose and oxidative metabolism (lactate, glucose, pyruvate) [9] and, more recently, in cerebral ketone metabolism (beta-hydroxybutyrate (bHB), acetoacetate (AcAc)) [7,10]. Cerebral metabolic sampling also allows the calculation of the lactate/pyruvate ratio, to assess brain cell redox state, and the sampling of other molecules, such as glutamate, as a marker of brain excitotoxicity [11]. CMD is therefore well suited to monitor the cerebral metabolic state at the acute phase of TBI in humans [12].
Keto-metabolism may play a key role in TBI neuro-repair. In that respect, ketogenic diets [10] and branched-chain amino acids (BCAA) [13] supplementation are used as nutritional supports and have been shown to be more efficient than glucose in terms of ATP yields [14], improvement of cognitive performance [15,16], reduction in seizure activity [15], enhancement of mitochondrial biogenesis and reduction in reactive oxygen species production [14,17]. The beneficial aspects of these alternative energy substrates might thus not only be restricted to ketone bodies production, but rather to overall ketometabolism. This includes several pathways involved in the metabolism of medium-chain fatty acids (MCFA), BCAA, ketogenic amino acids (KAA) that contribute directly or indirectly to the production of ketones or acetyl-CoA and succinyl-CoA. Therefore, we hypothesized that ketometabolism is one of the main components affecting the brain metabolic state during the course of TBI as well as one potential therapeutic path for intervention during and post-trauma.
We report for the first time the successful application of a combined hypothesis-driven and data-driven metabolomics approach conducted on CMD samples obtained routinely during patient monitoring. We combined metabolomics with data modelling and in silico pathway analysis to define the metabolic state of the patients during the acute post-TBI critical care phase (Fig. 1).
Fig. 1.

Schematic diagram outlining the workflow of the study. (Panel a was reproduced with friendly permission of https://doi.org/10.1007/s00134-017-5031-6).
2. Materials and methods
2.1. Patient cohorts
We initially retrospectively assessed brain ketometabolism in a cohort of 26 TBI patients (Study 1) admitted to the Department of Intensive Care Medicine (ICU), at the Centre Hospitalier Universitaire Vaudois (CHUV)-Lausanne University Hospital, in Lausanne/Switzerland between 2013 and 2017. We then used a cohort of 12 patients (validation cohort) to validate our findings (Study 2). A total of 38 TBI subjects from 2 clinical studies were therefore included in the present analysis. All subjects used for this study were severe TBI patients with a post-resuscitation Glasgow Coma Scale (GCS) <9 and an abnormal CT scan (defined by the presence of intracranial lesions [contusions, hematoma]), who underwent brain monitoring with CMD, intracranial pressure (ICP; Codman®, Raynham, MA, USA) and brain tissue oxygen tension (PbtO2; Licox®, Integra Neurosciences, Plainsboro, NJ, USA) probes, as part of standard patient care. Approval for the study was obtained by the Ethical Committee of the University of Lausanne, informed consent was obtained from each patient's next-of-kin.
In Study 1, we captured the brain metabolims of 26 patients in an average sample rate of approximately 4 h and across the time of their ICU stay (116 ± 71 h). The average Glasgow Outcome Score (GOS) and therapeutic intensity scores (TIL) across Study 1 are 3.1 (±1.4) and 2.5 (±1.1) respectively. In Study 2, a smaller set of patients were monitored for shorter periods of time (maximum 7 days) in a predefined sampling frequency of 12 h. A summary of the patient characteristics and underlying demographics is given in Table 1.
Table 1.
Patient demographics in the development and validation cohort.
| Study 1 | Study 2 | |
|---|---|---|
| Subjects (M/F) | 26 (6/20) | 12 (9/3) |
| Average Age [y] (SD) | 44.1 (17.0) | 56.7 (18.1) |
| Average Weight [kg] (SD) | 80.7 (17.1) | 84.6 (12.8) |
| Average Height [cm] (SD) | 175.2 (7.5) | 172.7 (6.5) |
| Average Glasgow Outcome Score [1](SD) | 3.14 (1.39) | N/A |
| Average Therapeutic Intensity Score [1](SD) | 2.54 (1.07) | 2.93 (0.75) |
| Number of deceased patients | 5 | N/A |
2.2. Sample collection and liquid-chromatography mass spectrometry (LC-MS) analysis
CMD collection consisted of an intra-parenchymal (sub-cortical white matter, visually normal brain) catheter (20 kDa cut-off CMA 70®, CMA Microdialysis AB, Solna, Sweden) that was inserted in the operating room following standard of care procedures. The catheter was constantly perfused (rate: 0.3 μL/min) with a sterile solution mimicking cerebrospinal fluid content through a pump (CMA 106®, CMA Microdialysis AB), as described previously [18,19] and in line with recent consensus guidelines [20].
For Study 1, samples were collected every 60 min, and immediately analysed at the bedside using a colorimetric enzymatic assay system (ISCUS FLEX) for glucose, lactate pyruvate, glutamate, and glycerol. The remaining sample was frozen in liquid nitrogen and kept at −80 °C until the retrospective analysis, representing a total of 1111 samples. For Study 2, two time points per day over a maximum of 7 days were prospectively collected representing 128 samples.
2.3. Chemicals and reagents
Liquid-chromatography mass spectrometry (LC-MS) grade acetonitrile and isopropanol were purchased from VWR Internationals (Leuven, Belgium) and Merck (Darmstadt, Germany), respectively. Water was purified in-house using a Milli-Q Advantage A10 system from Merck Millipore (Billerica, MA, USA). Acetic acid was supplied by Sigma-Aldrich (St-Louis, MO, USA). Pure chemicals used as external standards were purchased from Sigma-Aldrich (St-Louis, MO, USA), Larodan Fine Chemicals AB (Malmoe, Sweden), Toronto Research Chemicals (Toronto, ON, Canada), and Cambridge Isotope Laboratories Inc. (Tewksbury, MA, USA). 2H and 13C labelled compound were obtained from Sigma-Aldrich (St-Louis, MO, USA), Larodan Fine Chemicals AB (Malmoe, Sweden), Toronto Research Chemicals (Toronto, ON, Canada), Cambridge Isotope Laboratories Inc. (Tewksbury, MA, USA), and CDN Isotopes (Pointe Claire, QC, Canada) and used as internal standards. Perfusion Fluid CNS was bought from Harvard Apparatus (Holliston, MA, USA) and used as a blank matrix.
2.4. Preparation of internal and external standards
Individual 100 μM stock solutions of each internal standard were prepared from the corresponding commercially available powder and stored at −20 °C. On the day of analysis, a 0.1–0.5 μM internal standards mixture in mobile phase A was prepared and used for calibration standards and samples preparation. External standards were prepared similarly: individual 25–100 mM stock solutions of each analyte were prepared from the corresponding commercially available powder and stored at −20 °C. Then, a mixture of all analytes (concentration 0.06–1 mM) in methanol/water (1:1) was prepared. Different volumes of the analytes and internal standards solutions were then mixed, and the volume of each standard was manually adjusted to 0.5 mL with mobile phase A. A series of 8 calibration standards was obtained and further diluted by mixing 15 μL of each solution with 15 μL of perfusion fluid CNS in a PCR plate. The plate was sealed, placed in a Thermomixer Comfort C maintained at 4 °C and shaken for 5 min at 1000 rpm. After this mixing step, the calibration standards (approximate concentrations of 0.005–100 μM) were placed in the autosampler and analysed.
2.5. Sample preparation
CMD samples from consecutive time points were pooled until a volume of 15 μL was obtained. 15 μL of sample were pipetted and mixed with 15 μL of dilution solution (0.1–0.5 μM internal standards solution in mobile phase A) in a PCR plate. The plate was sealed, placed in a Thermomixer Comfort C (Eppendorf AG, Hamburg, Germany) maintained at 4 °C and shaken for 5 min at 1000 rpm. The plate was placed in the autosampler and samples immediately analysed.
2.6. Liquid chromatographic separation and mass spectroscopic detection
LC-MS was performed on a I-Class UPLC system (Waters Corporation, Milford, MA, USA) combining a binary pump, a FTN autosampler and a column oven. Chromatographic separation was achieved on a Waters ACQUITY UPLC BEH C8 Column, (100 × 2.1 mm, 1.7 μm) with binary solvent system at a flow rate of 450 μL/min. Mobile phase A was 0.1% acetic acid in water and B was 0.1% acetic acid in acetonitrile/isopropanol (1:1). The binary solvent gradient was as follow: 0.0–1.0 min at 0% B, 1.0–6.5 min from 0% to 100% B, 6.5–8.5 min 100% B, followed by 2 min of equilibration at initial conditions. Column oven temperature was set to 55 °C and the autosampler injection volume to 1 μL.
High-resolution mass spectrometric analysis was performed on a Thermo Scientific Q Exactive Plus instrument (ThermoFisher Scientific, Bremen, Germany). Detection was performed in data dependent mode (top 3) in negative and positive ionization modes in two separated injections. Instrument parameters were identical for both ionization modes and were as follow: for MS1, mass range m/z 65–600, resolving power of 35′000 (at m/z = 200), automatic gain control (AGC) target 5e6, maximum injection time 120 ms. For MS2, resolving power of 17′500 (at m/z = 200), AGC target 1e5, maximum injection time 50 ms, isolation window 2 Da, normalized collision energy (NCE) 40, intensity threshold 3e5, dynamic exclusion 5 s.
The mass spectrometer was interfaced to the UPLC system using a HESI probe. The spray voltage was set to −4.3 kV or + 4 kV depending on the ionization mode. For both positive and negative ionization modes, heater and capillary temperatures were set to 350 °C, sheath gas flow rate to 45 arbitrary units (AU), auxiliary gas to 15 AU and sweep gas to 1 AU. The instrument was calibrated every four days according to manufacturer specifications.
2.7. Mass spectroscopic data analysis
MS data analyses were performed with Xcalibur software 4.0 (ThermoFisher Scientific, Bremen, Germany). MS1chromatograms were extracted using a mass tolerance of 5 ppm and signals were integrated with the ISIS algorithm.
2.8. Hypothesis-driven in silico modelling approach to define the ketometabolic impact space
We first defined a search space of potential key metabolic components impacting TBI in a hypothesis-driven manner. Building on the clinically known relevance of AcAc, bHB, lactate and pyruvate, we used the RECON 2.2 (human genome scale metabolic model) to search the neighbourhood metabolic space [21,22]. This was defined by identifying metabolites connected to those mentioned above by less than four metabolic reactions, using the information in the stoichiometric matrix. The entire space obtained (illustrated in S1 Figure), consisted of 555 metabolites, spread across different compartments. Amongst these, we selected 57 unique metabolites (highlighted in yellow in S1 Figure) based on four criterions; a) less than equal to three steps upstream from the key metabolites of interest (bHB and AcAc), b) interconnected to greater than five other metabolites identified in the search globally, c) previous literature evidence of implication in ketometabolism, and d) feasibility to measure using MS-based metabolomics. In total 57 metabolites relevant in the context of brain metabolism were detectable. Following quality control procedures 40 out of the 57 metabolites were used for subsequent statistical data analysis. The metabolites are mostly related to metabolism of KAA, BCAA and MCFA, and represent notably a source of energy by either providing acetyl-CoA, succinyl-CoA or ketones to the TCA cycle. The final list of species selected for further measurement and analysis is tabulated in S1 Table Supplementary_Table_Hypothesis_Driven_List_Metabolites_1.xlxs.
2.9. Data aggregation across multiple sources
Alongside the metabolomics data a wide array of additional time-resolved metadata variables was available. These notably included systemic physiologic parameters (e.g. temperature, and oxygen saturation), cerebral physiologic and metabolic variables (e.g. ICP, CMD-derived metabolites). On a per-subject level, clinically relevant demographic data such as age, gender, weight, height, type of brain injury, days of hospital stay and the ultimate patient outcome, were collected. A summary of all available metadata variables is given in S2 table. We developed a unified data warehousing system that aligned the heterogeneous datasets via stable identifiers and unique timestamps allowing subsequent data pre-processing and analysis, which could be executed in a straightforward way.
2.10. Data pre-processing
The acquired metabolomics data resulted in a feature vector representing 40 molecules. For every patient, each feature vector was annotated with the exact timestamp of sample acquisition in coordinated universal time (UTC) and were concatenated into multivariate time series. Due to the variable nature of patient treatment and sample acquisition, the obtained time series were of different length and temporal resolution for each subject.
2.11. Statistical data analysis
In a first step, we investigated the entirety of the metabolic time series using an unsupervised approach with the goal of identifying time-segments of similar metabolic behaviour across patients. As we were primarily interested in the dynamic aspects of brain metabolism, we scaled the individual time series to unit variance before combining all data to a master matrix. We then performed Ward's algorithm on the master matrix to calculate similarities between the metabolic snapshots and to construct a dendrogram. We then secondly investigated the significant metabolic differences between the temporal segments (called “brain metabolic states”) as well as the associations with available clinical metadata using univariate statistical approach.
Thirdly, to test the feasibility of building predictive model capable of classifying metabolic states in new patient data, we mathematically described the metabolic states with the help of a classification model, which we constructed using an extreme gradient boosting approach [23] trained using a 10 × 10 cross-validation procedure.
In a fourth and last step, we investigated possibility of building supervised multivariate models to associate the brain-specific TIL composite scale, a reliable measurement instrument with a high degree of validity for assessing therapeutic intensity and the aggressiveness of care necessary to control elevated ICP in severe TBI patients [24] as well as the 6-month patient outcome using the GOS – using the time series data. As the time series were of unequal length across the patients, we calculated secondary variables (so called time series descriptors or time series proxies) to extract relevant time series characteristics. This approach allowed us to obtain equal number of variables per metabolite and patient whilst retaining most of the relevant time series characteristics. In total 10 descriptors were calculated for each time series which are summarised in S1 table. For the modelling we predominantly used a partial least squares (PLS) regression approach [25]. In addition, we also tested the feasibility of using a more advanced Long-Short-Term Memory Network (LSTM) approach [ 26] for use on multivariate metabolic time series data.
3. Results
3.1. The stratification of brain metabolic states using unsupervised multivariate analysis
We used an unbiased data-driven approach to investigate the patient ketometabolic states during their ICU stay. As stated, around 43 time points were available for each patient describing 116 ± 71 h of monitoring on average. Hierarchical clustering revealed the presence of two distinct metabolic states (Fig. 2) which were projected back on the original patient time lines resulting in characteristic barcode patterns depicted in Fig. 4. Statistical analysis revealed that valine, 4-methyl-2-oxovaleric acid (4-MOV), isobeta-hydroxybutyrate (iso-bHB), tyrosyine, 2-KIV, leucine, threonine, phenylalanine, methionine, and butyrylcarnitine are significantly higher in state “A” compared to “B”. The complete list of metabolites and their corresponding p-values are shown in Table 2. We also trained a metabolic state classification model on the data. Using 10 × 10 cross-validation we were able to achieve AUC value of 0.9943 for the hold-out data (Fig. 3a). We obtained the top 10 variables with the highest gain contribution from the model as visualized in Fig. 3b.
Fig. 2.
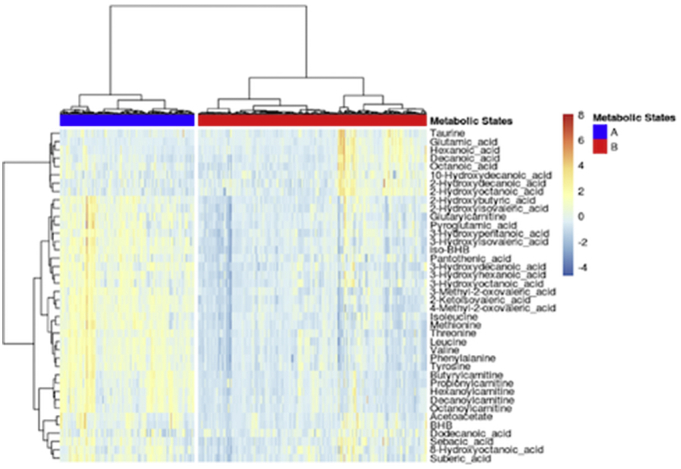
Metabolic states derived using hierarchical cluster analysis.
Fig. 4.

Temporal distribution of identified metabolic states (blue = “A”, red = ”B”) across patient trajectories. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
Significantly different metabolites between metabolic states.
| Metabolite | P-value after FDR correction | Significant |
|---|---|---|
| Valine | 1.896E-142 | Yes |
| 4-Methyl-2-oxovaleric acid | 9.782E-138 | Yes |
| iso-BHB | 1.629E-131 | Yes |
| Tyrosine | 4.966E-130 | Yes |
| 2-Ketoisovaleric acid | 2.701E-124 | Yes |
| Leucine | 9.875E-118 | Yes |
| Threonine | 1.058E-116 | Yes |
| Phenylalanine | 5.296E-116 | Yes |
| Methionine | 1.883E-110 | Yes |
| Butyrylcarnitine | 9.731E-101 | Yes |
| 3-Hydroxyisovaleric acid | 6.713E-94 | Yes |
| Isoleucine | 1.699E-90 | Yes |
| 3-Methyl-2-oxovaleric acid | 7.877E-89 | Yes |
| Propionylcarnitine | 1.803E-86 | Yes |
| 3-Hydroxydecanoic acid | 3.331E-84 | Yes |
| 3-Hydroxyhexanoic acid | 9.483E-84 | Yes |
| Glutarylcarnitine | 9.236E-71 | Yes |
| Hexanoylcarnitine | 9.288E-68 | Yes |
| Octanoylcarnitine | 1.600E-63 | Yes |
| Decanoylcarnitine | 3.754E-63 | Yes |
| 2-Hydroxybutyric acid | 5.106E-60 | Yes |
| 3-Hydroxypentanoic acid | 8.771E-48 | Yes |
| Pyroglutamic acid | 9.028E-47 | Yes |
| 2-Hydroxyisovaleric acid | 1.229E-46 | Yes |
| Pantothenic acid | 6.759E-44 | Yes |
| 3-Hydroxyoctanoic acid | 4.141E-41 | Yes |
| Suberic acid | 3.607E-32 | Yes |
| Glutamic acid | 5.747E-27 | Yes |
| Acetoacetate | 2.480E-24 | Yes |
| 2-Hydroxydecanoic acid | 7.342E-20 | Yes |
| Sebacic acid | 1.185E-16 | Yes |
| 8-Hydroxyoctanoic acid | 9.649E-16 | Yes |
| Hexanoic acid | 7.387E-14 | Yes |
| 2-Hydroxyoctanoic acid | 2.015E-12 | Yes |
| BHB | 1.257E-09 | Yes |
| Decanoic acid | 6.882E-09 | Yes |
| 10-Hydroxydecanoic acid | 6.510E-04 | Yes |
| Taurine | 1.418E-03 | Yes |
| Octanoic acid | 6.721E-02 | No |
| Dodecanoic acid | 5.791E-01 | No |
Fig. 3.
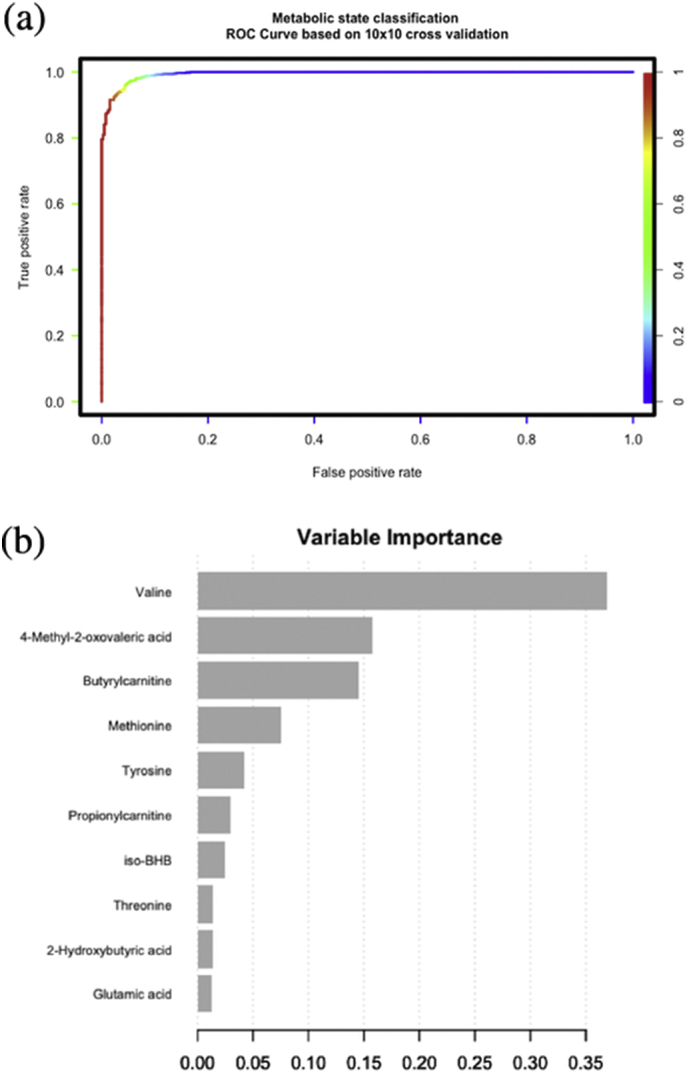
a. Receiver Operating Characteristic (ROC) curve for the metabolic state gradient boost classification model (top 10 variables). b. Variable importance (based on gain contribution) for the metabolic state gradient boost classification model (top 10 variables).
3.2. Association between brain metabolic states and clinical outcomes
Here, we further investigated if the found metabolic states were associated with brain metabolic and physiological variables. We observed that the clusters were significantly associated with the clinical standard values of CMD glucose, lactate, pyruvate and glutamate (p < .0001) (Fig. 6). This means that metabolic state “A” has higher levels of CMD lactate, pyruvate and glucose compared to metabolic state “B” while CMD glutamate and ICP is lower in state “A”. Differences in ICP were significant (p < .0001) between metabolic states; on the contrary however, the differences in brain tissue oxygen tension (PbtO2) as well as the lactate/pyruvate ratio were not significant between the metabolic states. Based on the currently available evidence for clinical practice, we conclude that the metabolic state “A” overall is a healthier state compared to the metabolic state “B”. We also investigated whether the average duration of a patient in a given state can be associated to patient outcome. On average patients spent 46.8 h (±29.7 h) in state “A” and 70.2 h (±49.8 h) in state “B”; individuals transitioned between states at different frequency and with different duration. The patterns of transitions between metabolic states vary considerably across patients as shown in Fig. 4. For some patients the duration in a given metabolic state only lasts a few hours whereas others can stretch up to numerous days. We argue that additional investigation is necessary to better understand the underlying manifestation of the brain metabolic states as they allow a real-time window into the brains metabolic status.
Fig. 6.

Associations of external time-resolved variables with brain metabolic states. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Prediction of patient outcome based on the longitudinal brain metabolic signature
In order to investigate whether the information contained in the brain metabolic time series can be utilised to predict near-term and mid-term patient outcome, we constructed regression models for the TIL and GOS. For the TIL model, the root mean square error (RMSE) of the PLS prediction model was 0.1436, which corresponded to a r2 value of 0.9851 between the actual and predicted TIL values (see Fig. 7a). Inspecting the variable contribution for the PLS revealed that 10-hydroxydecanoic acid (10-OH-C10), suberic acid, bHB, 8-hydroxyoctanoic acid (8-OHC8), 2-hydroxyoctanoic acid, dodecanoic acid, glutamic acid, acetoacetate, butyrylcarnitine and isoleucine were the 10 most relevant metabolites for the model. The fact that a model with a high r2 value could be built indicates that metabolic time properties (described via time series descriptors) can serve as useful input variables for TIL score prediction since they ultimately capture altered metabolic pathway characteristics.
Fig. 7.
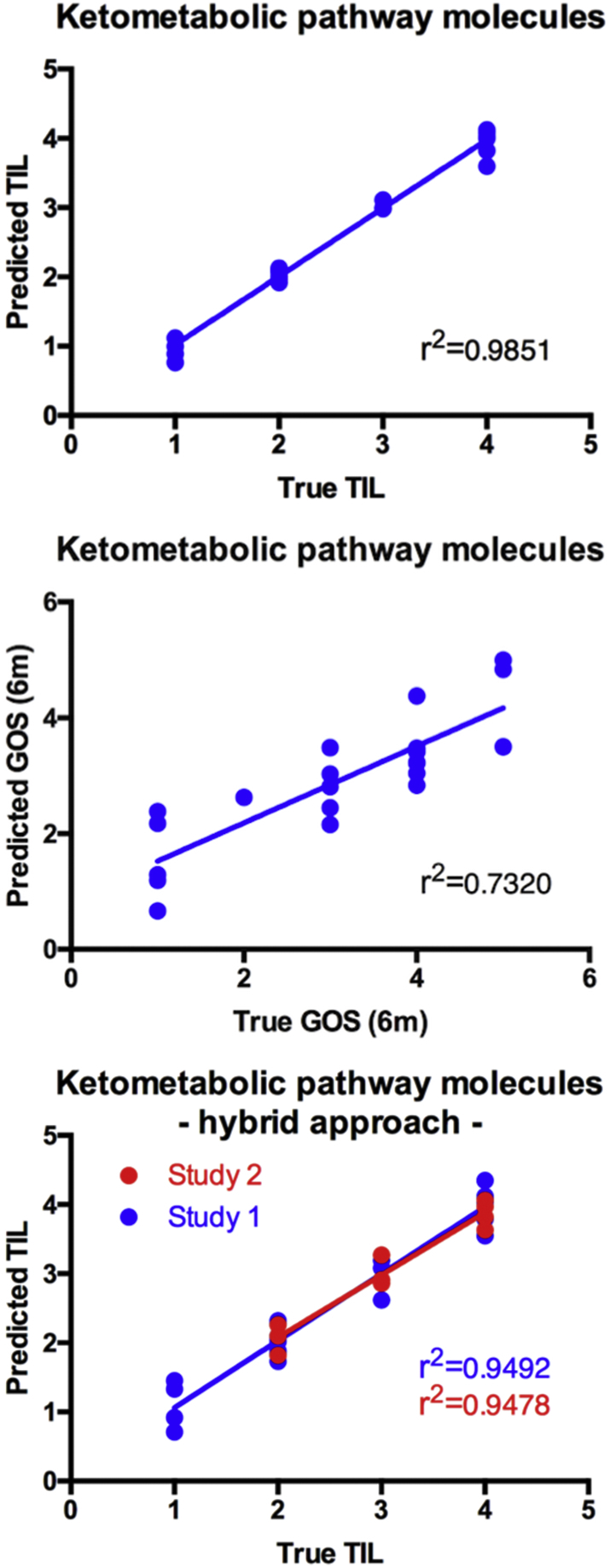
a. Predicted vs. actual TIL using ketometabolic pathway molecules as input data for PLS regression with leave-one-out validation (Study 1). b. Predicted vs. actual GOS (6 m) using ketometabolic pathway molecules as input data for neural net with leave-one-out validation. c. Predicted vs. actual TIL using ketometabolic pathway molecules as input data for hybrid PLS regression with leave-one-out validation for Study 1 (blue) and Study 2 (red).
We also tried to use the PLS approach to model the GOS, but it proved to be inefficient. We therefore tested the potential of a LSTM to model the GOS. Since the dataset is comparably small for training a neural network architecture, we multiplexed our training data set by adding random noise to the raw temporal trajectories. Again, cross-validation was carried out in leave-one-patient validation method. The r2 value of the best neural prediction model was 0.7320 (see Fig. 7b).
3.4. Further validation of findings with a second clinical cohort
To evaluate the hypothesis derived from Study 1, we used the data from Study 2 comprised of 12 patients. First, we performed Principal Component Analysis as well as Hierarchical Cluster Analysis. This primary investigation revealed that no systematic difference exists between both data sets, which allowed the additional samples to be utilised for the validation of our models (data not shown). We then applied the “metabolic state classification model” that we developed on Study 1 data to the new data from Study 2. Results showed that the model was capable to recognise the metabolic states in unseen patients with high level of confidence (89.8% of the samples were assigned class membership probabilities of 90% and higher). Following the prediction, we then mapped the metabolic states onto the patient time series (Fig. 5). Visual comparison with the sequence patterns of two different states was obtained from the Study 1 patient set (Fig. 4).
Fig. 5.

Temporal distribution of identified metabolic states (blue = “A”, red = ”B”) across external patient trajectories.
In a second phase we also investigated the possibility of testing the TBI prediction model on the validation cohort. As the validation cohort was acquired at a much lower sampling frequency (every 12 h) compared to the development cohort (~4 h on average) we could not directly apply the existing model as such. To overcome this limitation, we resampled the time series from Study 1 with the help of an interpolation procedure to match the characteristics of the development cohort. This approach made the time series and as such the descriptor calculation comparable but still could not compensate for the lack of more detailed temporal features in the validation cohort. To address whether a TIL prediction is nevertheless possible despite the bias in sample acquisition we build a new (hybrid) PLS model combining both data sets and with a leave-one-out validation approach. The r2 value for the development cohort samples was 0.9492 and 0.9478 for the validation cohort respectively (Fig. 7c).
4. Discussion
Here we report for the first time the successful application of a metabolomics approach conducted on CMD samples obtained routinely during patient monitoring. Given the limitation on the amount of CMD samples and in order to optimize the outcome, we used the combination of hypothesis-driven and data-driven metabolomics to capture the most relevant metabolites that are important for defining the metabolic state of the TBI patients upon their admission and during their stay in critical care unit. We were able to quantify the temporal trajectories of 40 brain related metabolites throughout the course of the monitoring. The cerebral metabolic patterns predominantly identified in our study include several metabolites that are crucially involved in ketone metabolism, thereby suggesting an implication of human brain ketometabolism in TBI pathophysiology and the potential for interventions targeting ketometabolism (e.g. supplemental bHB, MCFA, BCAA) for TBI neuro-repair.
When analysing the entirety of the available time series data in unsupervised way, we showed that patients undergo different brain metabolic states, which (in some cases) can last up to several days (Fig. 4). The groupings – obtained through hierarchical cluster analysis – are statistically significant with respect to many metabolites with valine, 4-MOV, iso-bHB, tyrosine, 2-KIV, and leucine amongst the most significant variables (Fig. 2; Table 2). Investigating the external time-resolved data revealed that lactate, pyruvate, glucose and glutamate measured in the CMD samples are significantly different between the brain metabolic states (Fig. 6) whereas blood glucose is not significantly different.
We further investigated the association between the identified metabolic and parameters that are routinely measured in clinical settings to monitor the patients' outcome, namely lactate, pyruvate, glutamate, and ICP. In particular, results revealed increased brain lactate, glucose and pyruvate, and decreased brain glutamate, arterial haemoglobin and ICP with state “A” as compared to state “B”. There are other markers such as UCH-L1 and GFAP which have been associated with TBI [27,28]; however we have not looked into association with these markers as they are not currently used in our center to monitor TBI patients.
The neuroprotective role of lactate has been extensively studied during the past few years. In particular, intracerebroventricular injection of lactate was shown to reduce lesion size and improve neurologic deficits after middle cerebral artery occlusion in mice [29]. Interestingly, lesion size was positively correlated with ICP in TBI patients [30], and reduced ICP was associated with reduced brain swelling and water content in a rat TBI model [31]. Moreover, administration of hypertonic sodium lactate increased cerebral blood flow by causing vasodilation of cerebral resistance vessels [32,33]. In an in vitro ischemia model, lactate was shown to increase TREK1 expression, and therefore to protect against excitotoxicity by promoting efficient potassium buffering and glutamate clearance [34].
The exact mechanisms causative of the observed temporal patterns are unclear at the moment and warrant further investigations on larger clinical cohorts. However; we suggest that it could potentially serve as a tool to assess the real-time (i.e. during monitoring) brain metabolic status in TBI patients. We proved the feasibility of recognising the described metabolic states in unseen patient data with the help of a mathematical model which in theory could facilitate a real-time patient assessment. Also, overlaying the differentiating metabolites for the two states (“A”/“B”) with the human genome scale metabolic model, we can hypothesize that the most impacted/impacting pathways could include fatty acid oxidation, valine, leucine and isoleucine metabolism, folate metabolism, methionine and cysteine metabolism, urea cycle, and glutamate metabolism (S2 Figure).
In an early clinical study, it was shown that TBI causes a nitrogen imbalance in which nitrogen excretion exceeds nitrogen uptake [35], suggesting increased protein breakdown. Indeed, the principal source of excreted nitrogen is urea, which is generated from deamination of amino acids, such as valine, tyrosine, leucine, threonine, phenylalanine, and methionine.
Valine, leucine and isoleucine (which precursor is threonine [KEGG database]) [[36], [37], [38]] are part of the BCAA. After crossing the blood-brain barrier, BCAA are notably involved in the synthesis of the excitatory neurotransmitter glutamate, which is in turn a precursor of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (through glutamate decarboxylase). This suggests that, besides being involved in the nitrogen balance, BCAA might play a key role in excitatory-inhibitory circuitry in the brain of TBI patients [39]. In rodent models of TBI, opposite shifts in circuit excitability and disruption in inhibitory synaptic function [40,41], which were associated with a decrease in brain BCAA levels, have been observed [41]. Upon dietary supplementation of BCAA, hippocampal synaptic efficacy was restored leading to the improvement of cognitive performance [41,42]. Noteworthy, such a decrease in BCAA (as well as their derivative metabolites, such as 4-MOV) was observed in plasma of TBI patients [16,43], and not only was found to correlate with elevated ICP [43], but supplemental BCAA also enhanced cognitive recovery [16] and increased plasma tyrosine levels [16,44]. In this respect, phenylalanine, which only differ by one hydroxy group from tyrosine, was found elevated in plasma of TBI patients and was associated with beneficial decrease in ICP [45]. In addition, both tyrosine and phenylalanine are precursors of catecholamine [46] and involved in folate metabolism, which have been linked to TBI [47,48].
Glutamate molecules in the synaptic cleft are prone to induce excitotoxicity if not adequately cleared [49]. In line with this, acute posttraumatic glutamate release has been implicated in being responsible for excitotoxicity following TBI leading to neuronal injury, cell death and dysfunction of surviving neurons [50].
Alternatively, valine can also be converted into 2-KIV, which can be released from glia through monocarboxylate transporter 1 (MCT1) [51] and transported into neurons, where it could either modulate the metabolism of glutamate [52] or be used as an alternative energy substrate via formation of succinyl-CoA [52,53]. Interestingly, succinate was recently proposed to support brain energy metabolism in TBI patients, as its focal administration potentiated brain metabolism [54,55]. It was also suggested that, as a ketoacid, 2-KIV can protect against oxidative stress in reacting with hydrogen peroxide [52,56]. Iso-bHB, similarly as its precursor 2-KIV, can be released in the extracellular space through MCT1, re-enter valine catabolic pathway via 3-hydroxyisobutyrate dehydrogenase and further be used as an alternative energy substrate or serve as an anaplerotic function for the TCA cycle (i.e. new synthesis of glutamate/GABA molecules) [52,57]. Finally, methionine was identified as a key differentiator between the metabolic states “A” and “B”. Reduced availability of methionine in the plasma of severe TBI patients [58] and in the brain of TBI rats [59] were reported and suggested to alter multiple cellular processes, including protein synthesis, epigenetic regulation of gene expression, cytoprotection against oxidative stress and cellular transport of amino acids in multiple organs (including the injured brain).
When we analysed the available time series data with supervised modelling techniques, we showed that a PLS approach can predict TIL with high accuracy (r2 = 0.9851 on the development cohort and r2 = 0.9478 on the validation cohort). The TIL score is an aggregate measurement taking several patient parameters such as ICP, hypocapnia levels, levels of CSF drainage as well as body temperature [24] into consideration and is widely used in clinical practice. Predicting the TIL score from metabolomics data in clinical practice would facilitate an alternative way to assess patients within a very short time frame. In particular, in this analysis we found important contribution from ketone bodies, as well as MCFA and BCAA.
In their rat model, Prins et al., demonstrated an increased in MCT2 expression after TBI, suggesting enhanced ketones uptake and utilization under such conditions [60]. The correlation between brain and plasma bHB (with larger plasma bHB than brain bHB) observed in the present study is thus in line with potential utilization of ketones from the circulation. Yet, bHB oxidation result in higher ATP production, as compared to glucose [61] and to an increase in NAD+/NADH ratio, which notably protects against oxidative stress [14]. Other beneficial effects of bHB administration were notably associated to a decrease in cerebral edema formation through the improvement of energy metabolism following ischemia in rats [62] and an increase in cerebral blood flow [63]. AcAc also resulted in a reduction of glutamate-induced toxicity both in vivo and in vitro [64]. Interestingly, it was shown that plasma bHB and AcAc, which correlated with CSF levels although not significantly, were low in acute brain injury patients, and that ketone supplementation was required to increase their concentrations to clinically relevant levels [65].
Regarding fatty acids, 10-OH-C10, suberic acid and 8-OH-C8 are metabolites of ω-oxidation. W-oxidation is prominent in the brain, and, unlike β-oxidation, occurs in both neurons and glia [66]. Products of ω-oxidation are oxidized in the cytosol to their dicarboxylic analogs, and then β-oxidized in peroxisomes, stimulating therefore peroxisome proliferation and modulating fatty acid binding proteins [66,67].
Regarding GOS prediction, we were able only to detect a moderate (r2 = 0.7320) association with the metabolomics data. Predicting the GOS from the time series was more difficult and did not yield the same degree of predictive performance compared to the TIL model. LSTM are currently one of the most powerful approaches in artificial intelligence and machine learning but really excel when trained on larger training data sets. We therefore suggest to further investigate the robustness of this approach using data obtained from a larger patient cohort.
We would like to emphasize that the available study data was affected by a variety of limitations that can be summarised as follows: firstly, the overall cohort sizes are comparably small with only 26 subjects in the first and only 12 subjects in the second study. We retrospectively calculated a power analysis for both cohorts in context of the investigation with the TIL. Assuming an effect size f2 of 0.15 and an error probability of 0.05 and further setting the number of predictors to 4 (representing the number of PLS components used in the model) and using a one-tailed linear multiple regression model, a power of 0.60 was determined for the first and 0.33 for the second cohort. The combined power of 0.75 was determined for the combination of both cohorts. Furthermore the type, location, and severity of the brain injuries as well as the underlying demographics vary amongst the individuals. In addition, the number of available time points as well as their corresponding temporal resolution varied considerably amongst patients. Thirdly, the characteristics of this study reflect the reality in an ICU environment with sample collection merely conducted as an add-on to a very demanding patient care environment.
Despite the limitations, we strongly believe that investigating brain metabolism using comprehensive metabolomics approach coupled to the microdialysis sampling described in this study can provide valuable and novel insights into the brain metabolic states, which is currently not possible with other technologies. A streamlined sample collection and subsequent metabolomics workflow could deliver data within a short timeframe and potentially having a big impact on patient well-being. To the best of our knowledge, this is the first report of the discovery and characterisation of metabolic states in the TBI patient. The prediction of short-term and long-term patient outcome using advanced metabolomics approach can be potentially executed in real-time as new samples become available and be used to monitor the brain metabolic states.
In addition to monitoring the patients, our working hypothesis is that these metabolic states can be modified during their time at the ICU using appropriate nutritional intervention to alter ketometabolism. Further clinical studies are required to investigate the flexibility and adaptation of these metabolic states to enable optimal clinical intervention (Fig. 8).
Fig. 8.

Schematic working hypothesis to alter brain metabolic states in TBI patients using nutritional intervention.
Declaration of interest
There is no competing interest
Author contribution
MO was responsible clinician for designing both clinical studies and provided clinical data. JPM collected the samples. MM designed the metabolomics study, NC developed and performed the LC-MS analysis. ME developed algorithm for data treatment and perform statistical analysis. AC performed in silico modelling. BC contributed to the design of the validation cohort. MM and ME performed data analysis and interpretation. MM and ME wrote the manuscript. SS performed literature search, contributed to writing and formatting the manuscript. All authors revised the manuscript, contributed and approved the final version.
Acknowledgement
This study was sponsored by Nestle Health Science.
Mojgan Masoodi had full access to all the data in the study and had full responsibility for the quality of the data generated, interpretation and submission for publication.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2019.05.054.
Appendix A. Supplementary data
Supplementary material
References
- 1.Dhandapani S., Manju D., Sharma B., Mahapatra A. Prognostic significance of age in traumatic brain injury. J Neurosci Rural Pract. 2012;3:131–135. doi: 10.4103/0976-3147.98208. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dhandapani S, Manju D, Sharma B, Mahapatra A. Prognostic significance of age in traumatic brain injury. J Neurosci Rural Pract. 2012; 3:131–5. [DOI] [PMC free article] [PubMed]
- 2.Stocchetti N., Carbonara M., Citerio G. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16:452–464. doi: 10.1016/S1474-4422(17)30118-7. [DOI] [PubMed] [Google Scholar]; Stocchetti N, Carbonara M, Citerio G, et al. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017; 16:452–64. [DOI] [PubMed]
- 3.Maas A.I.R., Menon D.K., Adelson P.D. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16:987–1048. doi: 10.1016/S1474-4422(17)30371-X. [DOI] [PubMed] [Google Scholar]; Maas AIR, Menon DK, Adelson PD, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017; 16:987–1048. [DOI] [PubMed]
- 4.Sharma D., Vavilala M.S. Perioperative management of adult traumatic brain injury. Anesthesiol Clin. 2012;30:333–346. doi: 10.1016/j.anclin.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sharma D and Vavilala MS. Perioperative management of adult traumatic brain injury. Anesthesiol Clin. 2012; 30:333–46 [DOI] [PMC free article] [PubMed]
- 5.Glenn T.C., Martin N.A., Horning M.A. Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J Neurotrauma. 2015;32:820–832. doi: 10.1089/neu.2014.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]; Glenn TC, Martin NA, Horning MA, et al. Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J Neurotrauma. 2015; 32:820–32 [DOI] [PMC free article] [PubMed]
- 6.Jalloh I., Carpenter K.L., Helmy A., Carpenter T.A., Menon D.K., Hutchinson P.J. Glucose metabolism following human traumatic brain injury: methods of assessment and pathophysiological findings. Metab Brain Dis. 2015;30:615–632. doi: 10.1007/s11011-014-9628-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jalloh I, Carpenter KL, Helmy A, Carpenter TA, Menon DK, Hutchinson PJ. Glucose metabolism following human traumatic brain injury: methods of assessment and pathophysiological findings. Metab Brain Dis. 2015; 30:615–32 [DOI] [PMC free article] [PubMed]
- 7.White H., Venkatesh B. Clinical review: ketones and brain injury. Crit Care. 2011;15:219. doi: 10.1186/cc10020. [DOI] [PMC free article] [PubMed] [Google Scholar]; White H and Venkatesh B. Clinical review: ketones and brain injury. Crit Care. 2011; 15:219 [DOI] [PMC free article] [PubMed]
- 8.Selwyn R., Hockenbury N., Jaiswal S., Mathur S., Armstrong R.C., Byrnes K.R. Mild traumatic brain injury results in depressed cerebral glucose uptake: an (18)FDG PET study. J Neurotrauma. 2013;30:1943–1953. doi: 10.1089/neu.2013.2928. [DOI] [PubMed] [Google Scholar]; Selwyn R, Hockenbury N, Jaiswal S, Mathur S, Armstrong RC, Byrnes KR. Mild traumatic brain injury results in depressed cerebral glucose uptake: An (18)FDG PET study. J Neurotrauma. 2013; 30:1943–53 [DOI] [PubMed]
- 9.Carpenter K.L., Jalloh I., Hutchinson P.J. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci. 2015;9:112. doi: 10.3389/fnins.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]; Carpenter KL, Jalloh I, Hutchinson PJ. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci. 2015; 9:112 [DOI] [PMC free article] [PubMed]
- 10.Bernini A., Masoodi M., Solari D. Modulation of cerebral ketone metabolism following traumatic brain injury in humans. J Cereb Blood Flow Metab. 2018 doi: 10.1177/0271678X18808947. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]; Bernini A, Masoodi M, Solari D, et al. Modulation of cerebral ketone metabolism following traumatic brain injury in humans. J Cereb Blood Flow Metab. 2018; doi: 10.1177/0271678X18808947 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 11.De Lima Oliveira M., Kairalla A.C., Fonoff E.T., Martinez R.C., Texeira M.J., Bor-Seng-Shu E. Cerebral microdialysis in traumatic brain injury and subarachnoid hemorrhage : state of the art. Neurocrit Care. 2014;21:152–162. doi: 10.1007/s12028-013-9884-4. [DOI] [PubMed] [Google Scholar]; De Lima Oliveira M, Kairalla AC, Fonoff ET, Martinez RC, Texeira MJ, Bor-Seng-Shu E. Cerebral microdialysis in traumatic brain injury and subarachnoid hemorrhage : state of the art. Neurocrit Care. 2014; 21:152–62. [DOI] [PubMed]
- 12.Oddo M., Hutchinson P.J. Understanding and monitoring brain injury: the role of cerebral microdialysis. Intensive Care Med. 2018;44:1945–1948. doi: 10.1007/s00134-017-5031-6. [DOI] [PubMed] [Google Scholar]; Oddo M and Hutchinson PJ. Understanding and monitoring brain injury: the role of cerebral microdialysis. Intensive Care Med. 2018; 44:1945–8 [DOI] [PubMed]
- 13.Sharma B., Lawrence D.W., Hutchison M.G. Branched chain amino acids (BCAA) and traumatic brain injury: y systematic review. J Head Trauma Rehabil. 2018;33:33–45. doi: 10.1097/HTR.0000000000000280. [DOI] [PubMed] [Google Scholar]; Sharma B, Lawrence DW, Hutchison MG. Branched chain amino acids (BCAA) and traumatic brain injury: y systematic review. J Head Trauma Rehabil. 2018; 33:33–45 [DOI] [PubMed]
- 14.Elamin M., Ruskin D.N., Masino S.A., Sacchetti P. Ketone-based metabolic therapy: is increased NAD+ a primary mechanism ? Front Mol Neurosci. 2017;10:377. doi: 10.3389/fnmol.2017.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]; Elamin M, Ruskin DN, Masino SA, Sacchetti P. Ketone-Based Metabolic Therapy: Is Increased NAD+ a primary mechanism ?. Front Mol Neurosci. 2017; 10:377. [DOI] [PMC free article] [PubMed]
- 15.Augustin K., Khabbush A., Williams S. Mechanisms of actions for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018;17:84–93. doi: 10.1016/S1474-4422(17)30408-8. [DOI] [PubMed] [Google Scholar]; Augustin K, Khabbush A, Williams S, et al. Mechanisms of actions for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018; 17:84–93 [DOI] [PubMed]
- 16.Aquilani R., Iadarola P., Contardi A. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86:1729–1735. doi: 10.1016/j.apmr.2005.03.022. [DOI] [PubMed] [Google Scholar]; Aquilani R, Iadarola P, Contardi A, et al. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005; 86:1729–35 [DOI] [PubMed]
- 17.Tajiri K., Shimizu Y. Branched-chain amino acids in liver diseases. World J Gastroenterol. 2013;19:7620–7629. doi: 10.3748/wjg.v19.i43.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tajiri K and Shimizu Y. Branched-chain amino acids in liver diseases. World J Gastroenterol. 2013; 19:7620–29 [DOI] [PMC free article] [PubMed]
- 18.Patet C., Quintard H., Suys T. Neuroenergetic response to prolonged cerebral glucose depletion after severe brain injury and the role of lactate. J Neurotrauma. 2015;32:1560–1566. doi: 10.1089/neu.2014.3781. [DOI] [PubMed] [Google Scholar]; Patet C, Quintard H, Suys T, et al. Neuroenergetic response to prolonged cerebral glucose depletion after severe brain injury and the role of lactate. J Neurotrauma. 2015; 32:1560–6 [DOI] [PubMed]
- 19.Sala N., Suys T., Zerlauth J.B. Cerebral extracellular lactate increase is predominantly nonischemic in patients with severe traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1815–1822. doi: 10.1038/jcbfm.2013.142. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sala N, Suys T, Zerlauth JB, et al. Cerebral extracellular lactate increase is predominantly nonischemic in patients with severe traumatic brain injury. J Cereb Blood Flow Metab. 2013; 33:1815–22 [DOI] [PMC free article] [PubMed]
- 20.Hutchinson P.J., Jalloh I., Helmy A. Consensus statement from the 2014 international microdialysis forum. Intensive Care Med. 2015;41:1517–1528. doi: 10.1007/s00134-015-3930-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hutchinson PJ, Jalloh I, Helmy A, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med. 2015; 41:1517–28 [DOI] [PMC free article] [PubMed]
- 21.Thiele I., Swainston N., Fleming R.M. A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013;31:419–425. doi: 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]; Thiele I, Swainston N, Fleming RM, et al., A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013; 31:419–25 [DOI] [PMC free article] [PubMed]
- 22.Swainston N., Smallbone K., Hefzi H. Recon 2.2: from reconstruction to model of human metabolism. Metabolomics. 2016;12:109. doi: 10.1007/s11306-016-1051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Swainston N, Smallbone K, Hefzi H, et al., Recon 2.2: from reconstruction to model of human metabolism. Metabolomics. 2016; 12:109 [DOI] [PMC free article] [PubMed]
- 23.Chen T., Guestrin C. XGBoost: a scalable tree boosting system. KDD. 2016;16:785–794. [Google Scholar]; Chen T and Guestrin C. XGBoost: A scalable tree boosting system. KDD 16. 2016; 785–794
- 24.Zuercher P., Groen J.L., Aries M.J. Reliability and validity of the therapy intensity level scale : analysis of clinimetrics properties of a novel approach to assess management of intracranial pressure in traumatic brain injury. J Neurotrauma. 2016;33:1768–1774. doi: 10.1089/neu.2015.4266. [DOI] [PubMed] [Google Scholar]; Zuercher P, Groen JL, Aries MJ, et al. Reliability and validity of the therapy intensity level scale : analysis of clinimetrics properties of a novel approach to assess management of intracranial pressure in traumatic brain injury. J Neurotrauma. 2016; 33:1768–74 [DOI] [PubMed]
- 25.Martens H. Reliable and relevant modelling of real world data: a personal account of the development of PLS regression. Chem Int Lab Sys. 2001;58:85–95. [Google Scholar]; Martens H. Reliable and relevant modelling of real world data: a personal account of the development of PLS regression. Chem Int Lab Sys. 2001; 58:85–95
- 26.Hochreiter S., Schmidhuber J. Long short-term memory. Neural Comput. 1997;9:1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]; Hochreiter S and Schmidhuber J. Long short-term memory. Neural Comput. 1997; 9:1735–80 [DOI] [PubMed]
- 27.Takala R.S., Posti J.P., Runtti H. Glial fibrillary acidic protein and ubiquitin C-terminal hydrolase-L1 as outcome predictors in traumatic brain injury. World Neurosurg. 2016;87:8–20. doi: 10.1016/j.wneu.2015.10.066. [DOI] [PubMed] [Google Scholar]; Takala RS, Posti JP, Runtti H, et al., Glial fibrillary acidic protein and ubiquitin C-terminal hydrolase-L1 as outcome predictors in traumatic brain injury. World Neurosurg. 2016; 87:8-20 [DOI] [PubMed]
- 28.Lee J.Y., Lee C.Y., Kim H.R., Lee C.H., Kim H.W., Kim J.H. A role of serum-based neuronal and glial markers as potential predictors for distinguishing severity and related outcomes in traumatic brain injury. J Korean Neurosurg Soc. 2015;58:93–100. doi: 10.3340/jkns.2015.58.2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lee JY, Lee CY, Kim HR, Lee CH, Kim HW, Kim JH. A role of serum-based neuronal and glial markers as potential predictors for distinguishing severity and related outcomes in traumatic brain injury. J Korean Neurosurg Soc. 2015. 58:93-100 [DOI] [PMC free article] [PubMed]
- 29.Berthet C., Lei H., Thevenet J., Gruetter R., Magistretti P.J., Hirt L. Neuroprotective role of lactate after cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:1780–1789. doi: 10.1038/jcbfm.2009.97. [DOI] [PubMed] [Google Scholar]; Berthet C, Lei H, Thevenet J, Gruetter R, Magistretti PJ, Hirt L. Neuroprotective role of lactate after cerebral ischemia. J Cereb Blood Flow Metab. 2009; 29:1780–9. [DOI] [PubMed]
- 30.Tabaddor K., Danziger A., Wisoff H.S. Estimation of intracranial pressure by CT scan in closed head trauma. Surg Neurol. 1982;18:212–215. doi: 10.1016/0090-3019(82)90395-0. [DOI] [PubMed] [Google Scholar]; Tabaddor K, Danziger A, Wisoff HS. Estimation of intracranial pressure by CT scan in closed head trauma. Surg Neurol. 1982; 18:212–5 [DOI] [PubMed]
- 31.Soustiel J.F., Vlodavsky E., Milman F., Gavish M., Zaaroor M. Improvement of cerebral metabolism mediated by Ro5-4864 is associated with relief of intracranial pressure and mitochondrial protective effect in experimental brain injury. Pharm Res. 2011;28:2945–2953. doi: 10.1007/s11095-011-0463-0. [DOI] [PubMed] [Google Scholar]; Soustiel JF, Vlodavsky E, Milman F, Gavish M, Zaaroor M. Improvement of cerebral metabolism mediated by Ro5-4864 is associated with relief of intracranial pressure and mitochondrial protective effect in experimental brain injury. Pharm Res. 2011; 28:2945–53 [DOI] [PubMed]
- 32.Shackford S.R., Schmoker J.D., Zhuang J. The effect of hypertonic resuscitation on pial arteriolar tone after brain injury and shock. J Trauma. 1994;37:899–908. doi: 10.1097/00005373-199412000-00005. [DOI] [PubMed] [Google Scholar]; Shackford SR, Schmoker JD, Zhuang J. The effect of hypertonic resuscitation on pial arteriolar tone after brain injury and shock. J Trauma. 1994; 37:899–908. [DOI] [PubMed]
- 33.Gordon G., Choi H., Rungta R., Ellis-Davies G., MacVicar B. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gordon G, Choi H, Rungta R, Ellis-Davies G, MacVicar B. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008; 456:745–9. [DOI] [PMC free article] [PubMed]
- 34.Banerjee A., Ghatak S., Sikdar S.K. L-lactate mediates neuroprotection against ischemia by increasing TREK1 channels expressions in rat hippocampal astrocytes in vitro. J Neurochem. 2016;138:265–281. doi: 10.1111/jnc.13638. [DOI] [PubMed] [Google Scholar]; Banerjee A, Ghatak S, Sikdar SK. L-Lactate mediates neuroprotection against ischemia by increasing TREK1 channels expressions in rat hippocampal astrocytes in vitro. J Neurochem. 2016; 138:265–81. [DOI] [PubMed]
- 35.Clifton G.L., Robertson C.S., Contant C.F. Enteral Hyperalimentation in head injury. J Neurosurg. 1985;62:186–193. doi: 10.3171/jns.1985.62.2.0186. [DOI] [PubMed] [Google Scholar]; Clifton GL, Robertson CS, Contant CF. Enteral Hyperalimentation in head injury. J Neurosurg. 1985; 62:186–93. [DOI] [PubMed]
- 36.Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017; 45:D353–61. [DOI] [PMC free article] [PubMed]
- 37.Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457–62. [DOI] [PMC free article] [PubMed]
- 38.Kanehisa M., Goto S.K.E.G.G. Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kanehisa M and Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed]
- 39.Pinheiro J.L., Rocha A., Pinheiro J. Traumatic brain injury and branched-chain amino acids. J Clin Nutr Metabo. 2018;2:1. [Google Scholar]; Pinheiro JL, Rocha A, Pinheiro J. Traumatic Brain Injury and Branched-Chain Amino Acids. J Clin Nutr Metabo. 2018; 2:1
- 40.Witgen B.M., Lifshitz J., Smith M.L. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a system, network and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]; Witgen BM, Lifshitz J, Smith ML, et al. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a system, network and cellular evaluation. Neuroscience. 2005; 133:1–15 [DOI] [PubMed]
- 41.Cole J.T., Mitala C.M., Kundu S. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107:366–371. doi: 10.1073/pnas.0910280107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci USA. 2010; 107:366–71 [DOI] [PMC free article] [PubMed]
- 42.Elkind J.A., Lim M.M., Johson B.N. Efficacy, dosage and duration of action of branched chain amino acid therapy for traumatic brain injury. Front Neurol. 2015;6:73. doi: 10.3389/fneur.2015.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]; Elkind JA, Lim MM, Johson BN, et al. Efficacy, dosage and duration of action of branched chain amino acid therapy for traumatic brain injury. Front Neurol. 2015; 6:73. [DOI] [PMC free article] [PubMed]
- 43.Jeter C.B., Hergenroeder G.W., Ward H.N., Moore A.N., Dash P.K. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30:671–679. doi: 10.1089/neu.2012.2491. [DOI] [PubMed] [Google Scholar]; Jeter CB, Hergenroeder GW, Ward HN, Moore AN, Dash PK. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013; 30:671–9 [DOI] [PubMed]
- 44.Aquilani R., Iadarola P., Boschi F., Pistarini C., Arcidiaco P., Contardi A. Reduced plasma levels of tyrosine, precursor of brain catecholamines, and of essential amino acids in patients with severe traumatic brain injury after rehabilitation. Arch Phys Med Rehabil. 2003;84:1258–1265. doi: 10.1016/s0003-9993(03)00148-5. [DOI] [PubMed] [Google Scholar]; Aquilani R, Iadarola P, Boschi F, Pistarini C, Arcidiaco P, Contardi A. Reduced plasma levels of tyrosine, precursor of brain catecholamines, and of essential amino acids in patients with severe traumatic brain injury after rehabilitation. Arch Phys Med Rehabil. 2003; 84:1258–65. [DOI] [PubMed]
- 45.Vuille-Dit-Bille R.N., Ha-Huy R., Stover J.F. Changes in plasma phenylalanine, isoleucine, leucine and valine are associated with significant changes in intracranial pressure and jugular venous oxygen saturation in patients with severe traumatic brain injury. Amino Acids. 2012;43:1287–1296. doi: 10.1007/s00726-011-1202-x. [DOI] [PubMed] [Google Scholar]; Vuille-Dit-Bille RN, Ha-Huy R, Stover JF. Changes in plasma phenylalanine, isoleucine, leucine and valine are associated with significant changes in intracranial pressure and jugular venous oxygen saturation in patients with severe traumatic brain injury. Amino Acids. 2012. 43:1287–96 [DOI] [PubMed]
- 46.Fernstrom J.D. Branched-chain amino acids and brain function. J Nutr. 2005;135:46S–1539S. doi: 10.1093/jn/135.6.1539S. [DOI] [PubMed] [Google Scholar]; Fernstrom JD. Branched-chain amino acids and brain function. J Nutr. 2005; 135:1539S-–46S. [DOI] [PubMed]
- 47.Jenkins P.O., Mehta M.A., Sharp D.J. Catecholamines and cognition after traumatic brain injury. Brain. 2016;139:2345–2371. doi: 10.1093/brain/aww128. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jenkins PO, Mehta MA, Sharp DJ. Catecholamines and cognition after traumatic brain injury. Brain. 2016; 139:2345–71. [DOI] [PMC free article] [PubMed]
- 48.Endres M., Ahmadi M., Kruman I., Biniszkiewicz D., Meisel A., Gertz K. Folate deficiency increases postischemic brain injury. Stroke. 2005;36:321–325. doi: 10.1161/01.STR.0000153008.60517.ab. [DOI] [PubMed] [Google Scholar]; Endres M, Ahmadi M, Kruman I, Biniszkiewicz D, Meisel A, Gertz K. Folate deficiency increases postischemic brain injury. Stroke. 2005; 36:321–5. [DOI] [PubMed]
- 49.Lucas D., Newhouse J. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58:193–201. doi: 10.1001/archopht.1957.00940010205006. [DOI] [PubMed] [Google Scholar]; Lucas D, and Newhouse J. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957; 58:193–201. [DOI] [PubMed]
- 50.Guerriero R.M., Giza C.C., Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. 2015;15:27. doi: 10.1007/s11910-015-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. 2015; 15:27. [DOI] [PMC free article] [PubMed]
- 51.Bröer S., Schneider H.P., Bröer A., Rahman B., Hamprecht B., Deitmer J.W. Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem J. 1998;333:167–174. doi: 10.1042/bj3330167. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bröer S, Schneider HP, Bröer A, Rahman B, Hamprecht B, Deitmer JW. Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem J. 1998; 333:167–74. [DOI] [PMC free article] [PubMed]
- 52.Murìn R., Mohammadi G., Leibfritz D., Hamprecht B. Glial metabolism of valine. Neurochem Res. 2009;34:1195–1203. doi: 10.1007/s11064-008-9895-2. [DOI] [PubMed] [Google Scholar]; Murìn R, Mohammadi G, Leibfritz D, Hamprecht B. Glial metabolism of valine. Neurochem Res. 2009; 34:1195–203. [DOI] [PubMed]
- 53.Brand K. Metabolism of 2-oxoacid analogues of leucine, valine and phenylalanine by heart muscle, brain and kidney of the rat. Biochim Biophys Acta. 1981;677:126–132. doi: 10.1016/0304-4165(81)90153-7. [DOI] [PubMed] [Google Scholar]; Brand K. Metabolism of 2-oxoacid analogues of leucine, valine and phenylalanine by heart muscle, brain and kidney of the rat. Biochim Biophys Acta. 1981; 677:126–32 [DOI] [PubMed]
- 54.Jalloh I., Helmy A., Howe D.J. Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab. 2017;37:2626–2638. doi: 10.1177/0271678X16672665. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jalloh I, Helmy A, Howe DJ, et al., Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab. 2017; 37:2626–38. [DOI] [PMC free article] [PubMed]
- 55.Stovell M.G., Mada M.O., Helmy A. The effect of succinate on brain NADH/NAD+ redox state and high energy phosphate metabolism in acute traumatic brain injury. Sci Rep. 2018;8 doi: 10.1038/s41598-018-29255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; Stovell MG, Mada MO, Helmy A, et al., The effect of succinate on brain NADH/NAD+ redox state and high energy phosphate metabolism in acute traumatic brain injury. Sci Rep. 2018; 8:11140 [DOI] [PMC free article] [PubMed]
- 56.Desagher S., Glowinski J., Prémont J. Pyruvate protects neurons against hydrogen peroxide-induce toxicity. J Neurosci. 1997;17:9060–9067. doi: 10.1523/JNEUROSCI.17-23-09060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; Desagher S, Glowinski J Prémont J. Pyruvate protects neurons against hydrogen peroxide-induce toxicity. J Neurosci. 1997; 17:9060–7 [DOI] [PMC free article] [PubMed]
- 57.Murìn R., Schaer A., Kowtharapu B.S., Verleysdonk S., Hamprecht B. Expression of 3-hydroxyisobutyrate dehydrogenase in cultured neuronal cells. J Neurochem. 2008;105:1176–1186. doi: 10.1111/j.1471-4159.2008.05298.x. [DOI] [PubMed] [Google Scholar]; Murìn R, Schaer A, Kowtharapu BS, Verleysdonk S, Hamprecht B. Expression of 3-hydroxyisobutyrate dehydrogenase in cultured neuronal cells. J Neurochem. 2008; 105:1176–86. [DOI] [PubMed]
- 58.Dash P.K., Hergenroeder G.W., Jeter C.B., Choi H.A., Kobori N., Moore A.N. Traumatic brain injury alters methionine metabolism: implications for pathophysiology. Front Sys Neurosci. 2016;10:36. doi: 10.3389/fnsys.2016.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dash PK, Hergenroeder GW, Jeter CB, Choi HA, Kobori N, Moore AN. Traumatic brain injury alters methionine metabolism: implications for pathophysiology. Front Sys Neurosci. 2016; 10:36. [DOI] [PMC free article] [PubMed]
- 59.Arun P., Rittase W.B., Wilder D.M., Wang Y., Gist I.D., Long J.B. Defective methionine metabolism in the brain after repeated blast exposures might contribute to increased oxidative stress. Neurochem Int. 2018;112:234–238. doi: 10.1016/j.neuint.2017.07.014. [DOI] [PubMed] [Google Scholar]; Arun P, Rittase WB, Wilder DM, Wang Y, Gist ID, Long JB. Defective methionine metabolism in the brain after repeated blast exposures might contribute to increased oxidative stress. Neurochem Int. 2018; 112:234–8 [DOI] [PubMed]
- 60.Prins M.L., Giza C.C. Induction of monocarboxylate transporter 2 expression and ketone transport following traumatic brain injury in juvenile and adult rats. Dev Neurosci. 2006;28:447–456. doi: 10.1159/000094170. [DOI] [PubMed] [Google Scholar]; Prins ML and Giza CC. Induction of monocarboxylate transporter 2 expression and ketone transport following traumatic brain injury in juvenile and adult rats. Dev Neurosci. 2006; 28:447–56. [DOI] [PubMed]
- 61.Veech R.L. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70:309–319. doi: 10.1016/j.plefa.2003.09.007. [DOI] [PubMed] [Google Scholar]; Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004; 70:309–19 [DOI] [PubMed]
- 62.Suzuki M., Suzuki M., Kitamura Y. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn J Pharmacol. 2002;89:36–43. doi: 10.1254/jjp.89.36. [DOI] [PubMed] [Google Scholar]; Suzuki M, Suzuki M, Kitamura Y, et al. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn J Pharmacol. 2002; 89:36–43 [DOI] [PubMed]
- 63.Hasselbalch S.G., Madsen P.L., Hageman L.P. Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am J Physiol. 1996;270:E746–E751. doi: 10.1152/ajpendo.1996.270.5.E746. [DOI] [PubMed] [Google Scholar]; Hasselbalch SG, Madsen PL, Hageman LP, et al. Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am J Physiol. 1996; 270:E746–51. [DOI] [PubMed]
- 64.Massieu L., Haces M.L., Montiel T., Hernandez-Fonseca K. Acetoacetate protects hippocampal neurons against glutamate-mediated neuronal damage during glycolysis inhibition. Neuroscience. 2003;120:365–378. doi: 10.1016/s0306-4522(03)00266-5. [DOI] [PubMed] [Google Scholar]; Massieu L, Haces ML, Montiel T, Hernandez-Fonseca K. Acetoacetate protects hippocampal neurons against glutamate-mediated neuronal damage during glycolysis inhibition. Neuroscience. 2003; 120:365–78 [DOI] [PubMed]
- 65.White H., Venkatesh B., Jones M., Fuentes H. Serial changes in plasma ketone concentrations in patients with acute brain injury. Neurol Res. 2017;39:1–6. doi: 10.1080/01616412.2016.1251695. [DOI] [PubMed] [Google Scholar]; White H, Venkatesh B, Jones M, Fuentes H. Serial changes in plasma ketone concentrations in patients with acute brain injury. Neurol Res. 2017; 39:1–6 [DOI] [PubMed]
- 66.Alexander J.J., Snyder A., Tonsgard J.H. Omega-oxidation of monocarboxylic acids in rat brain. Neurochem Res. 1998;23:227–233. doi: 10.1023/a:1022441211177. [DOI] [PubMed] [Google Scholar]; Alexander JJ, Snyder A, Tonsgard JH. Omega-oxidation of monocarboxylic acids in rat brain. Neurochem Res. 1998; 23:227–33. [DOI] [PubMed]
- 67.Ockner R.K. Historic overview of studies on fatty acid-binding proteins. Mol Cell Biochem. 1990;98:3–9. doi: 10.1007/BF00231361. [DOI] [PubMed] [Google Scholar]; Ockner RK. Historic overview of studies on fatty acid-binding proteins. Mol Cell Biochem. 1990; 98:3–9 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material