

Abstract
Progressive remodeling of the bone marrow microenvironment is recognized as an integral aspect of leukemogenesis. Expanding acute myeloid leukemia (AML) clones not only alter stroma composition, but also actively constrain hematopoiesis, representing a significant source of patient morbidity and mortality. Recent studies revealed the surprising resistance of long‐term hematopoietic stem cells (LT‐HSC) to elimination from the leukemic niche. Here, we examine the fate and function of residual LT‐HSC in the BM of murine xenografts with emphasis on the role of AML‐derived extracellular vesicles (EV). AML‐EV rapidly enter HSC, and their trafficking elicits protein synthesis suppression and LT‐HSC quiescence. Mechanistically, AML‐EV transfer a panel of miRNA, including miR‐1246, that target the mTOR subunit Raptor, causing ribosomal protein S6 hypo‐phosphorylation, which in turn impairs protein synthesis in LT‐HSC. While HSC functionally recover from quiescence upon transplantation to an AML‐naive environment, they maintain relative gains in repopulation capacity. These phenotypic changes are accompanied by DNA double‐strand breaks and evidence of a sustained DNA‐damage response. In sum, AML‐EV contribute to niche‐dependent, reversible quiescence and elicit persisting DNA damage in LT‐HSC.
Keywords: AML, DNA damage, extracellular vesicles, hematopoiesis
Subject Categories: Cancer, Membrane & Intracellular Transport, Stem Cells
Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous disease that arises from mutations in hematopoietic stem and progenitor cells (HSPC) 1. The characteristic, and often disproportionate, suppression of native hematopoiesis that develops in the bone marrow (BM) during disease progression and post‐treatment relapse accounts for significant morbidity and mortality 2, 3, 4, 5, 6. Substantial experimental evidence supports the malignant transition of BM function during AML invasion, and its role in disrupting hematopoiesis and sustaining AML 7, 8, 9, 10, 11, 12. Several prior studies of BM niche‐conversion emphasize leukemia‐induced alterations in stromal and vascular function 8, 13, 14, 15. Cell–cell interactions also target the hematopoietic components in the BM and contribute to the functional suppression and displacement of the hematopoietic progenitors predominantly responsible for steady‐state hematopoiesis 15, 16, 17, 18, 19, 20.
The fate of residual hematopoietic stem cells (HSC) under leukemic stress, however, has been more elusive. Unlike the depletion of highly susceptible HSPC, HSC have proved to be more resilient during leukemic invasion, and multiple groups reported the relative accumulation of primitive hematopoietic cells in both murine models and xenograft studies 8, 15, 16, 21, 22, 23. Intriguingly, HSC in the leukemic niche enter quiescence through an unidentified process, yet appear to retain their repopulation capacity upon subsequent re‐transplantation 22.
Extracellular vesicles (EV) comprise multiple populations of nano‐sized vesicles, which carry protein and nucleic acids, participate in the regulation of BM function 24, 25, 26. We recently showed that AML‐EV, including exosomes, are highly abundant in microRNA (miR)‐150 and miR‐155, which both target the transcription factor c‐Myb to suppress HSPC clonogenicity 17, 27, 28, 29. Here, we test the hypothesis that EV impact the fate of residual HSC in the AML niche via a distinct mechanism, since HSC function does not rely on c‐Myb expression at high levels 30.
Our studies in immunodeficient mice confirm the relative accumulation and quiescence of residual HSC previously observed 16, 18, 22, 23, and reveal that AML‐EV suppress protein synthesis in LT‐HSC. Mechanistically, AML‐EV transfer miR‐1246 to LT‐HSC to cause the translational suppression of the mTOR subunit Raptor, which in turn facilitates the hypo‐phosphorylation of S6RP with ensuing deficits in protein synthesis. Intriguingly, while these changes are resolved upon transfer to a naïve BM niche, we show that AML‐EV elicit DNA damage that persists in vitro and in vivo through serial progenitor replating and transplantation, respectively.
Results
AML‐EV are taken up by hematopoietic cells, including LT‐HSC
We previously showed 17, 28, 29, 31 and herein confirmed that AML cells (Molm‐14 and U‐937) predominantly release nano‐sized, lipid bilayer vesicles with a diameter of 50–130 nm, as demonstrated by Cryo‐TEM imaging (Fig 1A). To investigate the quantitative uptake of AML‐EV in HSC, we relied on a set of AML cell lines (Molm‐14, U‐937, and HL‐60) that were transduced with a lentiviral vector to constitutively express green fluorescence protein with a myristoyl group (mGFP) (Fig 1B). The resulting GFP‐tag was incorporated into the lipid bilayer of both the cell and the released EV, allowing measurement of uptake in vivo and in vitro, as previously reported 17. As modeled in Fig 1C, we then injected these engineered AML cells into NSG mice for 3–6 weeks to allow the AML cells to reach to 20–40% of the BM. We targeted low levels of chimerism to minimize cell–cell contact driving the AML‐HSC crosstalk. GFP+ EV purified from the peripheral blood plasma of Molm‐14 and the U‐937 xenografts were visualized by fluorescence microscopy (Fig 1D). Live‐cell imaging of xenograft‐derived KSL and LT‐HSC demonstrated the uptake of mGFP+ EV into the intracellular space (Fig 1E). Next, we measured the kinetics of EV uptake by exposing KSL and LT‐HSC to EV harvested from Molm‐14‐mGFP or U‐937‐mGFP cells in vitro. By capturing live‐cell 3D z‐stacks at 0, 30, and 150 min, we found that both KSL and LT‐HSC bind and internalize numerous mGFP+ EV within 30 min of exposure with continued accumulation at 150 min (Fig EV1A). We also analyzed KSL from wild‐type Molm‐14 xenografts to rule out confounding autofluorescence. HSPC harvested from non‐transduced Molm‐14 xenografts contained no mGFP+ foci and exhibited relative background fluorescence similar to non‐xenografted controls (Fig EV1B), confirming that mGFP+ foci were membrane‐derived vesicles originating from Molm‐14‐mGFP and U‐937‐mGFP cells.
Figure 1. In vivo uptake of AML‐EV in hematopoietic stem cells.

- Cryo‐TEM images demonstrate the lipid bilayer EV purified from Molm‐14 and U‐937 cells. Scale bars are 100 nm.
- A schematic diagram of the myristoylated GFP (mGFP)‐expressing lentiviral construct and its incorporation into the cell membrane and EV. Long terminal repeat (LTR), poly‐adenylate (pA), cytomegalovirus (CMV).
- Schematic diagram of the workflow. Cells were injected via tail‐vein injection into NSG mice. After 21 days, bone marrow (BM) cells were flushed to sort GFP+ cells by flow cytometry and perform imaging of sorted HSC.
- Peripheral blood (PB) plasma of control animals contains no mGFP+ foci (top); however, Molm‐14‐mGFP (middle) and U‐937‐mGFP (bottom) xenografts contain numerous mGFP‐labeled EV (green) detectible without vesicle concentration. Scale bars are 5 μm.
- Live‐cell microscopy of KSL cells (left panel) LT‐HSC cells (right panel). Cells were sorted from control mice (top), Molm‐14‐mGFP (middle), and U‐937‐mGFP (bottom), stained with Cell Mask (red) and nuclear stain Hoechst (blue), imaged using the GE/API Deltavision (DV) widefield microscope (60× objective) to show the uptake of the GFP+ EV (green). Scale bars are 5 μm.
Figure EV1. The kinetics of AML‐EV interaction with BM hematopoietic cells.
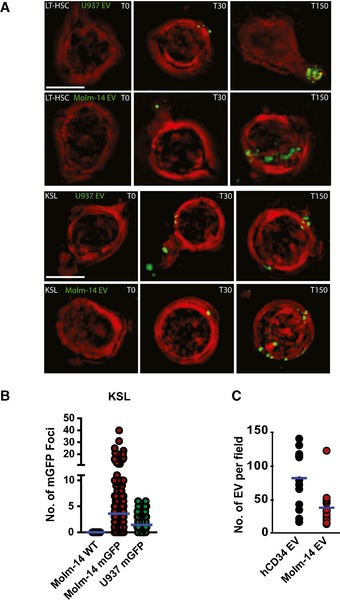
- Microscopic images using the GE/API Deltavision widefield microscope (60× objective) of live LT‐HSC (top rows), or KSL (bottom rows), in vitro exposed to EV from Molm‐14‐mGFP and U‐937‐mGFP cells for 0, 30, and 150 min. Green: mGFP+ EV, red: plasma membrane surface. Scale bars are 5 μm.
- Quantification of mGFP+ EV foci in KSL FACS purified from AML xenografts: wild‐type Molm‐14 (n = 40), Molm‐14‐mGFP (n = 247), and U‐937‐mGFP (n = 107). The background autofluorescence was measured in non‐engrafted controls, and this threshold value was then subtracted from xenografted mice. Individual mGFP+ foci were counted in individual KSL from xenografted animals using Imaris software.
- Microscopic quantification of EV from hCD34 (3 × 107 cells) or Molm‐14 (3 × 107 cells). The purified EV were stained with the lipid dye, FM1‐43, imaged using the Yokogawa CSU‐W1 spinning disk microscope (100× objective) and quantified using Imaris software. Separate measurements for hCD34, 11; Molm14 EV, 8.
AML‐EV increase the relative frequency of LT‐HSC, upregulate P53, and confer quiescence
To study the effect of AML‐EV on residual HSC fate, we relied on the Molm‐14‐xenograft model complemented with intrafemoral (IF) injection of EV from Molm‐14, HL‐60, and U‐937 cell lines versus EV from expanded healthy donor BM CD34+ cell (hCD34) followed by flow cytometric analysis (Fig 2A). To ensure a valid comparison of the injected EV, we normalized the numbers of the EV‐producing cells (EV from 3 × 107 cells/femur) and confirmed their EV concentration by microscopic quantification using a lipid dye, FM1‐43, finding no significant differences (Fig EV1C).
Figure 2. AML‐EV increase the relative frequency of LT‐HSC, upregulate P53 expression, and confer quiescence.

-
ASchematic diagram of methods: the in vivo AML xenografts (tail‐vein injection of 105 Molm‐14 cells or vehicle per mouse) and the intrafemoral (IF) injection of AML‐EV into one femur with a contralateral control vehicle‐injected femur of the same mouse. EV were isolated by serial high‐speed centrifugation (at 2×, 10×, 100 × 103 g) from AML cell‐line culture media or AML patient plasma. Bone marrow was flushed from long bones at the indicated time points, and immunophenotypic analysis was performed by flow cytometry.
-
B, CFlow cytometric analysis showing the frequency of LK and KSL cells in lineage‐negative cells (left panel) and LT‐HSC in KSL (right panel) in: (B) Molm‐14 xenograft (red, n = 10) versus control (black, n = 10). Data were obtained from at least two independent experiments. (C) IF injection of EV from Molm‐14 cells (red, n = 8), AML plasma EV (orange, n = 6), and human CD34 EV (blue, n = 4) relative to the vehicle‐injected contralateral femurs. Data were obtained from at least two independent experiments. Statistics: Student's t‐test (*P < 0.05, ***P < 0.001).
-
DCell‐cycle histograms of KSL (upper panel) and LT‐HSC (lower panel) from Molm‐14 xenografts or control using Hoechst‐33342 staining.
-
EFlow cytometric analysis of the Ki67‐ve percentage of LT‐HSC representing the G0 phase of cell cycle in: (left panel) Molm‐14 xenografts (red, n = 7) versus control (black, n = 9); (right panel) IF injection of Molm‐14‐EV, U‐937 EV, HL‐60 EV (red, n = 5,4,4) versus human CD34 EV (blue, n = 3) versus controls (black). Data were obtained from at least two independent experiments Statistics: Student's t‐test (*P < 0.05, **P < 0.01).
-
F, GFlow cytometric analysis of intracellular P53 levels shown in histograms (F) and MFI (G) of LT‐HSC in: (left panel) Molm‐14 xenograft, n = 6 red versus non‐engrafted control, n = 4 black, or (right panel) IF injection of EV from Molm‐14, U‐937, HL‐60 (red, n = 5,5,3) or human CD34 cells (blue, n = 6) normalized to vehicle‐injected contralateral femurs. Data were obtained from at least two independent experiments. Statistics: Student's t‐test (*P < 0.05, **P < 0.01).
We first assessed the frequencies of myeloid progenitor LK (lineage−/cKit+); the early progenitor/stem pool KSL (Lin−/cKit+/Sca‐1+); and LT‐HSC (KSL/CD48−/CD150+) (Fig 2A). While there was no change in KSL frequency or absolute cell number, AML‐EV caused a relative reduction in myeloid progenitor LK cells (Figs 2B and C, and EV2A). More importantly, we observed a significant increase in LT‐HSC frequency and absolute cell number in Molm‐14 xenografts and after IF injection of Molm‐14‐EV, but not after control CD34+ EV (Figs 2B and C, and EV2A). In addition to cell‐line‐derived EV, we also tested EV from the plasma of six AML patients (Appendix Table S2). IF injection of patient plasma EV confirmed the observed reduction in LK and a concomitant increase in LT‐HSC (Fig 2C). Together, the data suggest suppressed progenitor differentiation with proportional accumulation in LT‐HSC after exposure to AML‐EV.
Figure EV2. AML‐EV induce a p53‐dependent quiescence in hematopoietic cells with no evidence of apoptosis.
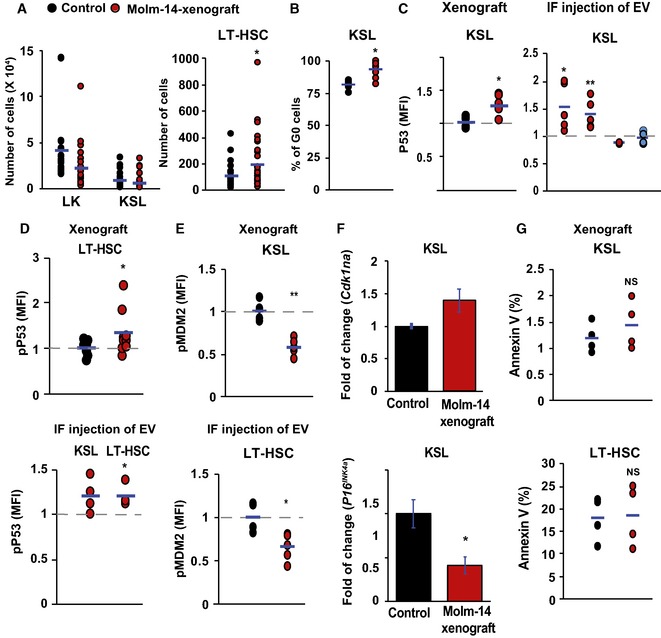
- Flow cytometric analysis showing the absolute cell number of LK and KSL cells in lineage‐negative cells (left panel) and LT‐HSC (right panel) in Molm‐14 xenograft (red, n = 10) versus control (black, n = 10). Statistics: Student's t‐test (*P < 0.05).
- Flow cytometric analysis of KSL percentage in G0 phase from Molm‐14 xenografts (red, n = 7) versus control (black, n = 9). Statistics: Student's t‐test (*P < 0.05).
- Flow cytometric analysis of P53 MFI in KSL in: (left panel) Molm‐14 xenograft (red, n = 6) versus control (black, n = 4), or (right panel) IF injection of EV from Molm‐14, U‐937, HL‐60 (red, n = 5,5,3), or CD34 cells (blue, n = 6) normalized to vehicle‐injected femurs. Statistics: Student's t‐test (*P < 0.05, **P < 0.01).
- Flow cytometric analysis of pP53ser15 MFI of LT‐HSC in: (upper panel) Molm‐14 xenografts, n = 9 red versus control, n = 7 black, or (lower panel) LT‐HSC and KSL after IF injection of Molm‐14 EV (red, n = 4) normalized to vehicle‐injected contralateral femurs. Data are presented after subtracting the background fluorescence. Statistics: Student's t‐test (*P < 0.05).
- Flow cytometric analysis of the pMDM2 MFI in KSL (upper panel) and LT‐HSC (lower panel) in Molm‐14 xenografts (red, n = 5) versus control (black, n = 4). Statistics: Student's t‐test (*P < 0.05, **P < 0.01).
- qRT–PCR showing the fold change of Cdkn1a (upper panel) and P16 INK4a (lower panel) in KSL from Molm‐14 xenografts relative to control mice and normalized to Gapdh endogenous control. Data are mean ± SEM from at least three independent experiments with technical replicates. Statistics: one‐way ANOVA with Bonferroni post hoc correction (*P < 0.05).
- Annexin V+ analysis of KSL (upper panel) and LT‐HSC (lower panel), in Molm‐14 xenograft (red, n = 4) versus control (black, n = 4). Statistics: Student's t‐test (NS = not significant).&!#6;
To further test this hypothesis, we investigated the impact of AML‐EV on cell‐cycle status in the xenograft model and after direct injection of AML‐EV. We found that AML‐EV consistently induced quiescence in LT‐HSC as shown in the histograms after Hoechst staining (Fig 2D), as well as the fraction of cells in G0 cell‐cycle phase determined by Ki67 staining (Fig 2E). AML‐EV‐mediated quiescence was also observed in the bulk KSL population (Fig EV2B) and occurred after IF injection of AML‐EV, but not after injection of EV from healthy hCD34 control cultures. Owing to its crucial role in regulating HSC quiescence 32, we evaluated the P53 activation in residual HSC. We found a significant upregulation of P53 in LT‐HSC and KSL from the xenografted mice as well as the IF‐injected mice with EV from AML cells or patient plasma (Figs 2F–G and EV2C). We also observed hyper‐phosphorylation of P53Ser15 in LT‐HSC influenced by AML‐EV (Fig EV2D). We found a significant hypo‐phosphorylation of MDM2ser166, a negative regulator of P53, in both KSL and LT‐HSC from xenografted mice and after IF injection of Molm‐14‐EV (Fig EV2E). In further agreement, we observed transcriptional upregulation of the Cdkn1a (p21), a P53‐effector, in KSL from Molm‐14 xenografts relative to control (Fig EV2F). We found no evidence of senescence induction via P16INK4a expression in KSL from Molm‐14 xenografts (Fig EV2F). Likewise, no evidence of apoptosis was observed in KSL or in LT‐HSC from Molm‐14 xenografts (Fig EV2G). Together, our data thus far demonstrated a proliferative defect in LT‐HSC by AML‐EV.
AML‐EV induces ribosome biogenesis suppression in hematopoietic cells
To understand the mechanism by which AML‐EV may enforce quiescence, we performed tandem mass tag proteomic profiling of in vitro‐cultured cKit+ HSPC (to obtain the minimum required amount of protein lysates) treated with EV from HL‐60 or Molm‐14 for 48 h versus vehicle‐treated controls. We used the differential expression statistical package EdgeR 33 with multiple testing corrections to calculate the false discovery rate (FDR) of differentially expressed proteins between EV‐ and vehicle‐treated controls. Numbers of quantifiable proteins and biological replicates differed between the Molm‐14 experiments (4,407 proteins, 2 vehicle replicates, and 4 EV replicates) and the HL‐60 experiments (4883 proteins, 4 vehicle replicates, and 4 EV replicates). We chose FDR cutoffs for each experiment (FDR < 10−2 for the Molm‐14 experiment and FDR < 10−9 for the HL‐60 experiment) to produce lists of differentially abundant candidates that contained ~10% of the quantifiable proteins (Datasets EV1 and EV2). There were 394 differentially regulated proteins in response to the Molm‐14‐EV (221 upregulated and 173 downregulated), with 491 differentially regulated in response to the HL‐60‐EV (325 upregulated and 166 downregulated). While there were 111 commonly upregulated proteins, 54 proteins were consistently downregulated in HSPC after treatment with both HL‐60‐EV and Molm‐14‐EV relative to vehicle‐treated controls (Fig 3A). Functional analysis of these proteins using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) platform identified ribosomal biogenesis as the most highly enriched functionally related gene group among downregulated targets in EV‐exposed cells (Fig 3B). This category comprised ribosomal proteins involved in RNA‐binding, RNA‐splicing, and translation initiation (Fig 3C).
Figure 3. AML‐EV impair ribosome biogenesis in hematopoietic cells and suppress protein synthesis in LT‐HSC.
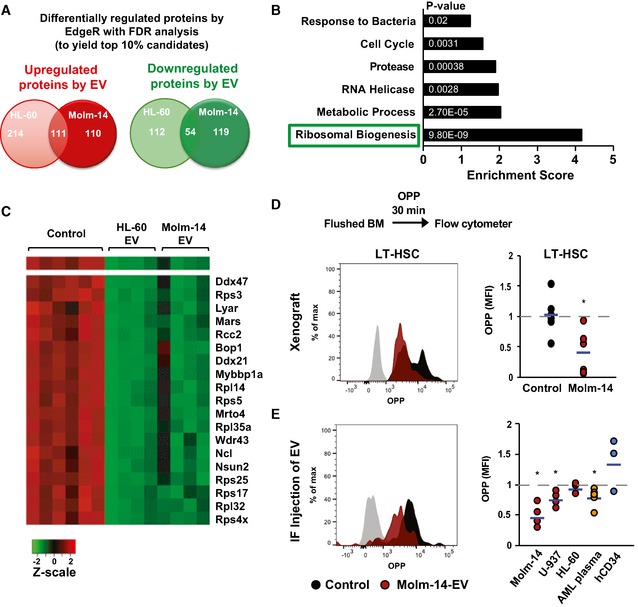
-
AA Venn diagram showing the most highly up‐ and downregulated proteins between Molm‐14‐EV and HL‐60 EV‐treated cKit+ HSPC determined by isobaric TMT labeling and edgeR analyses to calculate the false discovery rate (FDR) analysis to obtain ˜10% of the differentially regulated proteins. We obtained 325 and 221 upregulated proteins in the HL‐60 dataset and the Molm‐14 dataset, respectively. We also obtained 166 and 173 downregulated protein in the HL‐60 dataset and the Molm‐14 dataset, respectively. Among them, 54 proteins were commonly downregulated.
-
BA bar graph showing the functional annotation enrichment analysis by DAVID biostatistical plate form of the 54 commonly downregulated protein identified the indicated pathways; among them, the ribosomal biogenesis pathway showed the highest enrichment. P‐values are indicated in white. Modified Fisher's exact test.
-
CA heatmap showing the highly deregulated ribosomal proteins in AML‐EV‐exposed cells.
-
D, EFlow cytometric analysis showing the histograms and MFI of O‐propargyl‐puromycin (OPP) incorporation in LT‐HSC in: (D) Molm‐14 xenografts, red n = 6 versus non‐engrafted controls, black n = 6, or (E) IF injection of EV from Molm‐14, U‐937, HL‐60 (red, n = 5,4,3), AML patient plasma (orange, n = 6) or human CD34+ cells (blue, n = 3) normalized to vehicle‐injected contralateral femurs after subtracting the background fluorescence. Data were obtained from at least two independent experiments. Statistics: Student's t‐test (*P < 0.05).
AML‐EV suppress protein synthesis only in LT‐HSC
Ribosomal biogenesis is a principal regulatory step for protein homeostasis 34. This prompted us to test the effect of AML‐EV on protein synthesis rates among HSPC populations. We used the recently validated OPP Click‐iT assay 35 that relies on the incorporation and labeling of a modified puromycin analogue in newly generated proteins, and thus positively correlates gains in fluorescence intensity with protein synthesis. After animal sacrifice, cells were cultured in RPMI with 10% FBS and treated with OPP for 30 min to measure the fluorescently labeled OPP by flow cytometry. Among the different HSPC populations, only the LT‐HSC from Molm‐14 xenografts showed significant suppression in their protein synthesis, as represented in the histogram and MFI quantification (Figs 3D and EV3A and B). Protein synthesis was similarly suppressed in LT‐HSC and KSL cells from IF‐injected mice with EV from Molm‐14, U‐937, and AML patient plasma, but not the hCD34‐EV, and more modestly by HL‐60‐EV (Figs 3E and EV3C). These data suggest that the global EV‐mediated ribosome biogenesis impairment in all HSPC most profoundly suppresses protein synthesis in LT‐HSC, but not KSL or other progenitors. Together, the data demonstrate protein synthesis suppression and quiescence induction in LT‐HSC by AML‐EV.
Figure EV3. AML do not suppress protein synthesis in other hematopoietic populations.
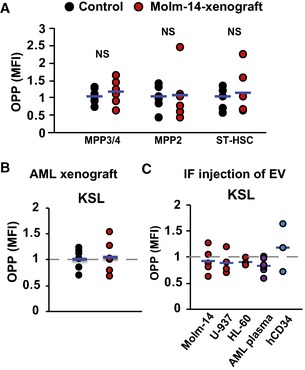
-
AFlow cytometric analysis showing MFI of OPP incorporation in MPP3/4, MPP2, and ST‐HSC in the Molm‐14 xenografts (red, n = 6) versus non‐engrafted mice (black, n = 6). Statistics: Student's t‐test (not significant “NS”).
-
B, COPP flow cytometric analysis of KSL in: (B) Molm‐14 xenografts (red, n = 6) versus non‐engrafted mice (black, n = 6), or (C) IF injection of EV from Molm‐14, U‐937, HL‐60 (red, n = 5,4,3), AML patient plasma (orange, n = 6), or CD34+ cells (blue, n = 3) normalized to contralateral femurs after subtracting background fluorescence.
AML‐EV impair protein synthesis in LT‐HSC via the mTOR pathway
The mTOR pathway is critical for translating extrinsic signals into cell‐intrinsic events and, among other functions, governs ribosome biogenesis and protein synthesis 36, 37, 38. Because we found a global reduction of ribosome biogenesis in HSPC (Fig 3), we evaluated the EV‐mediated regulation of the mTOR pathway using the bulk HSPC, KSL. Our qPCR transcriptional analysis indicated the downregulation of several mTOR‐associated targets, including the mTORC1 subunit Raptor, in KSL from Molm‐14 xenografts relative to controls (Fig 4A). KSL cells from femurs injected with Molm‐14‐EV confirmed the suppression of Raptor, and other mTOR‐associated genes, relative to cells from vehicle‐injected contralateral femurs (Fig 4B). To assess mTOR activity, we tested the effect of AML‐EV on S6RP phosphorylation, a downstream mTOR effector 38, and found a consistent reduction of pS6RP in LT‐HSC in the xenografts relative to control mice (Fig 4C and D) and after injection of AML‐EV but not hCD34‐EV (Fig 4C and E). Similarly, AML‐EV decreased pS6RP in KSL, suggesting that the AML‐EV‐mediated mTOR suppression affects the HSPC pool more broadly (Fig EV4A). Consistent with the reported role of Raptor in regulating HSC quiescence 36, we found that siRNA‐targeted silencing of both Raptor and rapamycin (a pharmacological Raptor inhibitor) suppressed LT‐HSC activation and increased the percentage of cells at G0 phase (Fig EV4B and C). Indeed, KSL and LT‐HSC from Molm‐14 xenografts showed significant suppression of the Raptor protein assessed by flow cytometry (Fig EV4D). To test whether mTOR suppression requires the continued uptake of AML‐EV, we examined mTOR expression after 7 days of ex vivo culture of HSPC derived from Molm‐14 xenografts or control mice. Interestingly, we found a complete reversion of the pS6RP suppression in the xenograft‐derived KSL and LT‐HSC relative to Day 0 (Fig EV4E). We specifically ruled out significant changes in cMyc levels, another critical protein synthesis regulator, in KSL as well as LT‐HSC of AML xenograft and following IF injection of AML‐EV (Fig EV4F and G). The data thus far indicate that AML‐EV impair mTOR‐dependent ribosome biogenesis in the pooled HSPC populations. Important from a functional point of view, LT‐HSC reveal an exaggerated sensitivity to protein synthesis suppression consistent with previous reports 35, 39, 40.
Figure 4. AML‐EV‐contained miR‐1246 suppresses protein synthesis in LT‐HSC via the mTOR pathway.

-
A, BqRT–PCR gene expression analysis showing the fold change of PI3K/mTOR‐associated gene panel in KSL cells sorted from: (A) Molm‐14‐xenografted mice or (B) IF‐injected mice with Molm‐14‐EV relative to their controls and normalized to Gapdh endogenous control. Data are expressed as mean ± SEM from at least three independent experiments, performed in technical replicates. Statistics: One‐way ANOVA with Bonferroni post hoc correction (*P < 0.05, **P < 0.01).
-
C–EFlow cytometric analysis showing the histograms (C) and MFI of intracellular pS6KRP in LT‐HSC in: (D) Molm‐14 xenograft (red, n = 9) versus non‐engrafted control (black, n = 7), or (E) IF injection of EV from Molm‐14, U‐937, HL‐60 (red, n = 7,3,3), or human CD34 cells (blue, n = 4) normalized to the control contralateral femur with subtraction of background fluorescence. Data were obtained from at least two independent experiments. Statistics: Student's t‐test (*P < 0.05, ***P < 0.001).
-
F, GFlow cytometric assessment of protein synthesis showing the MFI of: (F) OPP or (G) pS6RP in NIH‐3T3 cells 72 h after transfection with the indicated miRNA mimics. The results were calculated relative to control (miR‐scramble) with the background fluorescence subtracted and performed with at least three independent experiments, in technical replicates. Statistics: one‐way ANOVA with Bonferroni post hoc correction (*P < 0.05, **P < 0.01).
-
HFlow cytometric analysis showing the MFI of Raptor in NIH‐3T3 cells 72 h after transfection with the indicated miRNA mimics. The data were calculated relative to miR‐scramble and are presented as mean ± SEM, and the background fluorescence was subtracted. Performed with at least three independent experiments, in technical replicates. Statistics: one‐way ANOVA with Bonferroni post hoc correction (*P < 0.05, **P < 0.01).
-
IDual‐luciferase reporter assay. NIH‐3T3 was transfected with the miRNA mimics. Three hours later, the cells were transfected with the Raptor 3′UTR cloned into the psiCheck‐2 vector for a total of 48 h. Data are presented as %RLU (relative luciferase units) of the miR‐scramble control as mean ± SEM from at least three independent experiments, performed in technical replicates. Statistics: one‐way ANOVA with Bonferroni post hoc correction (*P < 0.05).
-
J, KCell‐cycle flow cytometric analysis using Ki67/Hoechst‐33342 staining of the percentage of LT‐HSC in the G0 phase after nucleofection of cKit+ cells using the Amaxa™ P3 Primary Cell 4D‐Nucleofector Kit, (J) Cells were nucleofected with miR‐scramble (CTRL) or miR‐1246 mimic (n = 3) for 72 h, (K) cells were nucleofected with anti‐miR‐scramble (CTRL) or anti‐miR‐1246 (n = 5) and 1 h later co‐treatment with Molm‐14 EV for 72 h. Statistics: Student's t‐test (*P < 0.05, **P < 0.01).
Figure EV4. AML‐EV suppress mTOR pathway, but not cMyc, in hematopoietic cells.

-
AFlow cytometric analysis of pS6KRP MFI in KSL in: (left panel) Molm‐14 xenografts, red, n = 4 versus control mice, black, n = 3; or (right panel) IF injection of EV from Molm‐14, U‐937, HL‐60 (red, n = 7,3,3), or CD34 cells (blue, n = 4) normalized to vehicle‐injected contralateral femurs. The background fluorescence was subtracted. Statistics: Student's t‐test (*P < 0.05).
-
B, CCell‐cycle flow cytometric analysis using Ki67/Hoechst‐33342 staining of the percentage of LT‐HSC in the G0 phase after (B) nucleofection of cKit+ cells with siControl or siRaptor for 72 h using the Amaxa™ P3 Primary Cell 4D‐Nucleofector Kit. (C) Treatment of ex vivo‐cultured cKit+ cells with vehicle or rapamycin (100 nM) for 72 h. Statistics: Student's t‐test (**P < 0.01).
-
DFlow cytometric analysis of intracellular Raptor MFI levels in KSL (upper panel) and LT‐HSC (lower panel) derived from Molm‐14 xenografts (red, n = 5) versus control (black, n = 3). Statistics: Student's t‐test (**P < 0.01, ***P < 0.001).
-
EFlow cytometric analysis of pS6RP MFI in KSL (upper panel) and LT‐HSC (lower panel) obtained from Molm‐14 xenografts (red, n = 5) versus non‐engrafted mice (black, n = 3) at Day 0 (freshly isolated HSPC cells) or Day 7 (after 7 days of ex vivo culture in RPMI with 50 ng/ml IL‐3/SCF and 10% FBS at 37°C, 5% CO2). Statistics: Student's t‐test (not significant “NS”, **P < 0.01 and ***P < 0.001).
-
F, GFlow cytometric analysis of cMyc MFI in KSL (left panel) and LT‐HSC (right panel) in: (F) Molm‐14 xenografts (red, n = 3 and 5) versus control (black, n = 3 and 3), or (G) IF injection of EV from Molm‐14, U‐937, and HL‐60 (n = 4,3, and 3). Statistics: Student's t‐test (not significant “NS”).
-
HqRT–PCR gene expression showing the fold change of Raptor, Rictor, or Pten genes in NIH‐3T3 transfected with the indicated miRNA mimics for 48 h. The fold change was calculated relative to miR‐scramble after normalizing to Gapdh endogenous control. Data are mean ± SEM from at least three independent experiments, performed in technical replicates. Statistics: one‐way ANOVA with Bonferroni post hoc correction (*P < 0.05 and **P < 0.01).
-
IFlow cytometric analysis showing the histograms of Raptor in NIH‐3T3 cells 72 h after transfection with miR‐scramble or miR‐1246 mimics.
EV miRNAs target Raptor and suppress protein synthesis
We recently demonstrated that AML‐EV highly incorporate specific cellular miRNAs capable of altering bystander cell function 17, 26, 31. Those studies identified a panel of miRNAs that were highly abundant in AML cell‐line EV and plasma EV from AML patients, but not those from healthy donors. The panel includes miR‐27a‐5p, miR‐155‐5p, miR‐181b‐5p, miR‐1246, and miR‐1290, some previously reported as potential AML biomarkers 31, 41, 42, 43. We hypothesized that one or more of these candidate AML‐EV miRNAs might cooperate in the mTOR‐dependent suppression of LT‐HSC protein synthesis. To test this, we used synthetic miRNA mimics and functionally screened their ability to suppress protein synthesis in murine NIH‐3T3 cells relative to a scramble control miRNA. Several mimics, including miR‐1246, showed a significant reduction in OPP fluorescence and pS6RP (Fig 4F and G). Transcriptional analysis of the mTOR‐associated genes showed that among the miRNAs, miR‐1246 mimic significantly downregulates Raptor (Fig EV4H). Flow cytometric analysis confirmed that miR‐1246 mimic suppressed Raptor protein levels (Figs 4H and EV4I). Notably, experiments with the luciferase reporter system containing the Raptor 3`UTR indicated that the miR‐1246 mimic most profoundly suppresses the Raptor gene (Fig 4I). We further confirmed that miR‐1246 levels were in fact higher in EV from AML patient versus healthy donor plasma, and increased alongside AML disease burden in Molm‐14 xenografts (Appendix Fig S1). As expected, nucleofection of miR‐1246 mimics into LT‐HSC ex vivo increased their quiescence (Fig 4J), whereas nucleofection of LT‐HSC with anti‐miR‐1246 rescued the Molm‐14 EV‐mediated quiescence induction (Fig 4K). Altogether, AML‐EV deliver miR‐1246 and other miRNAs that downregulate the Raptor/mTOR pathway to suppress protein synthesis and induce quiescence in HSC.
LT‐HSC restore serial repopulation capacity and protein synthesis rates
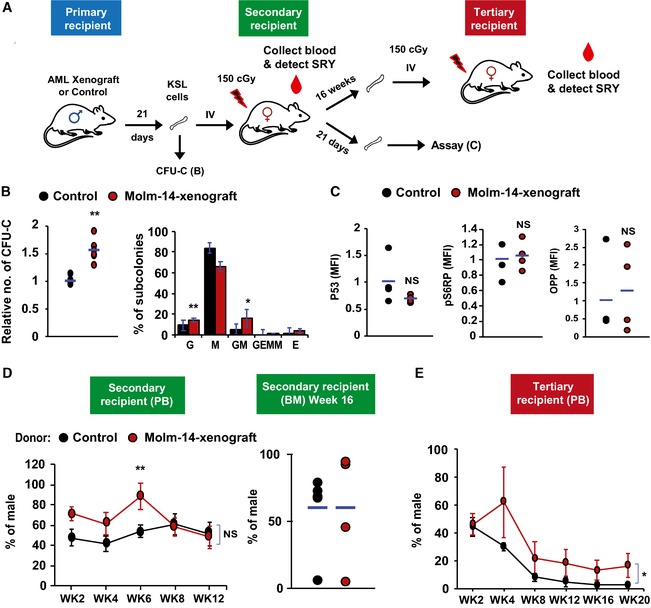
Others previously noted the reversibility of quiescence once hematopoietic cells are removed from the leukemic niche 16, 23. To test this possibility in our model, we first examined the serial colony‐forming unit assay (CFU‐C) as a correlate of potential functional impairment of residual HSPC pool by AML‐EV (Fig 5A). Surprisingly, we observed a consistent increase in colony growth compared with controls (Fig 5B). Further subtyping of the colonies revealed that the observed increase in overall CFU‐C was due to gains in the less‐committed bi‐potential granulocyte/monocyte progenitors (Fig 5B). We then tested the reversibility of protein synthesis suppression and quiescence in vivo. We transplanted male donor KSL from AML xenografts or controls into sublethally irradiated (150 cGy) female recipients (Fig 5A). Following validation of the sensitivity in defined mixtures of cells from both sexes at different ratios (Fig EV5A and B), we evaluated the donor chimerism by qPCR quantification of the sex‐determining region Y (SRY) in the recipients’ peripheral blood (PB) leukocytes 44. Twenty‐one days after transplantation, the animals were sacrificed and LT‐HSC revealed normalized OPP incorporation, pS6RP, and P53 levels (Fig 5C). These data echoed the pS6RP activity recovery that we observed in xenograft‐derived KSL and LT‐HSC after ex vivo culturing in AML‐free media (Fig EV4E). Consistent with the CFU‐C results, xenograft‐derived donor cells showed initial levels of PB chimerism (SRY) that exceeded those from control donors (Fig EV5C).
Figure 5. LT‐HSC restore protein synthesis and proliferative function upon transplantation to a non‐leukemic host.
-
AA diagram showing the scheme of repopulation experiments on non‐leukemic host. KSL cells were purified from Molm‐14‐xenografted male mice (red) or control males (black) and transplanted into 150 cGy sublethally irradiated female recipients (104 cells per mouse) and used for subsequent repopulation studies described in (D, E).
-
BCFU‐C assay showing the relative number of colonies (left panel) and the colony subtypes (right panel) from KSL cells derived from Molm‐14‐xenografted mice (red, n = 6) versus non‐engrafted control mice (black, n = 5) and in vitro‐cultured for 7 days in methylcellulose media. Experiments were performed in technical triplicates. “G”—granulocyte, “M”—monocyte, and “E”—erythrocytes. Statistics: Student's t‐test (*P < 0.05, **P < 0.01).
-
CLT‐HSC assessment after repopulation, recipient female mice (n = 4 per group) were sacrificed after 21 days and their LT‐HSC were compared using P53, pS6RP, and OPP flow cytometric assays. Statistics: Student's t‐test.
-
D, EEvaluation of the in vivo long‐term repopulation capacity. (D) For secondary transplantation (n = 6 per group), PB samples were collected twice weekly for 12 weeks (WK), and the donor chimerism was measured by qPCR of sex‐determining region Y (SRY) normalized to Gapdh gatekeeper as shown in the left panel. Sixteen weeks later, mice were sacrificed and BM male chimerism percentage was assessed as shown in the right panel. (E) For tertiary transplantation, unfractionated BM cells from the secondary recipients (n = 6 per group, 106 cells per mouse) were injected into tertiary 150 cGy sublethally irradiated female recipients and blood SRY levels were assessed for 20 weeks. Statistics: Student's t‐test for two‐sample comparison and regression analysis for longitudinal comparison (NS = not significant, *P < 0.05 and **P < 0.01).
Figure EV5. AML and short‐term HSC repopulation.
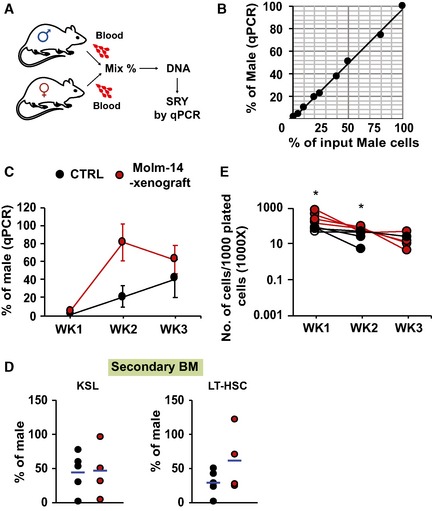
- Schematic diagram of the workflow. PB samples were collected from male and female mice. Blood leukocytes were counted and mixed at different proportions. DNA from the mixtures was extracted, and SRY was quantified by qRT–PCR and normalized to Gapdh.
- Correlation between measured male DNA determined by qPCR on the Y‐axis and the corresponding input male cells on the X‐axis (n = 1, R 2 = 0.9965).
- qRT–PCR of the PB donor male DNA from Molm‐14 xenografts (red, n = 4), or controls (black, n = 4) transplanted into secondary female recipients for 3 weeks. Data were normalized to Gapdh and are presented as mean ± SEM
- qRT–PCR of male chimerism in: (left panel) KSL or (right panel) LT‐HSC, from mice transplanted by either Molm‐14 xenografts (red, n = 4) or controls (black, n = 4) for 16 weeks.
- Total number of cells per 1,000 input cells, serially replated into methylcellulose media for three successive weeks. Cells derived from Molm‐14 xenografts and control mice were represented in red and black, respectively. Data were performed in technical triplicates. Statistics: Student's t‐test (*P < 0.05).
To evaluate the long‐term repopulation performance, we transplanted KSL from male Molm‐14 xenografts or controls into secondary sublethally irradiated female recipients and serially tracked the PB donor chimerism for 12 weeks. This was followed by a second round of transplantation using unfractionated BM from the secondary recipients into tertiary sublethally irradiated females, while tracking the original (male) donor cell chimerism for another 20 weeks. The results showed increased short‐term (until Week 6 in the secondary recipients, Fig 5D) and long‐term (tertiary recipients, Fig 5E) repopulation potential of the AML‐xenograft‐derived groups by t‐test and regression analysis, respectively. We also observed an increase in the BM LT‐HSC levels after secondary transplantation (Fig EV5D).
Enhanced replating potential and DNA‐damage accrual in AML‐EV‐exposed cells
Our sex‐mismatched xenograft approach did not permit selective recovery of purified HSC after transplantation. To understand the in vivo resistance to LT‐HSC depletion and persistent, if modest, repopulation advantage, we therefore performed serial ex vivo CFU‐C replating and evaluated the clonogenicity of stem and early progenitor subsets from Molm‐14 xenografts or control mice (Fig 6A). To insure a valid comparison, the serially plated cells were normalized prior to each replating. Our results showed persistently higher CFU‐C counts and average number of cells per colony (Fig 6B and C) as well as relative number of cells (Fig EV5E) for all xenograft‐derived progenitors relative to the controls. The data also showed that the xenograft‐derived cells were more resistant than control‐derived cells to exhaustion.
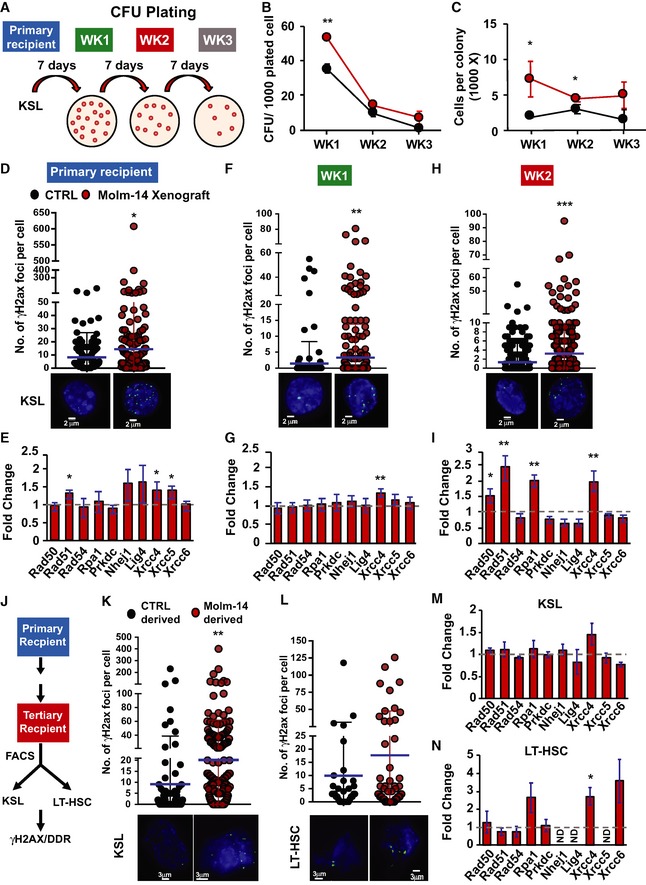
Figure 6. AML microenvironment conditioned HSC accrue persistent DDR and gain a proliferative advantage.
-
AA schematic diagram demonstrating the serial replating experiment. KSL cells were sorted from primary recipient control mice (black, n = 4) or Molm‐14 xenografts (red, n = 4) and plated in triplicate (500 cells/plate/mouse) in methylcellulose media. Seven days later, colonies were counted and serially replated in triplicate (5,000 cells/plate/mouse). The replating process was repeated twice for a total of three rounds (labeled week “WK” 1, 2, and 3).
-
B, CSerial replating CFU‐C analysis presented as mean ± SEM showing (B) the number of colonies per 1,000 plated cells and (C) the number of cells per colony, for each week time point. Statistics: Student's t‐test (*P < 0.05, **P < 0.01).
-
D–IDNA‐damage assessment in KSL from primary recipients and after two turns of replating. (D, F, H) Number of γH2AX foci in control‐derived and Molm‐14‐xenograft‐derived mean ± SD (D) primary recipient KSL, n = 211 and 321, (F) cells from W1 plating, n = 218 and 369, and (H) cells from W2 plating, n = 770 and 853. All cells were stained with phosphoH2axser139 and the nuclear stain Hoechst (blue), imaged using the GE/API DV widefield microscope (60× objective) to show the γH2AX foci (green) and counted using Imaris software. Scale bars are 2 μm. Statistics: Student's t‐test (*P < 0.05, **P < 0.01, and ***P < 0.001). (E, G, I) qRT–PCR gene expression analysis showing the fold change of select DNA‐damage response‐gene panel in Molm‐14‐xenograft‐derived (E) primary recipient KSL, n = 6, (G) cells from W1 plating, n = 4, and (I) cells from W2 plating, n = 4. Data indicated by bars of fold change calculated relative to matching controls were normalized to the endogenous control Gapdh and are presented as mean ± SEM. Statistics: one‐way ANOVA with Bonferroni post hoc correction (*P < 0.05, **P < 0.01).
-
JScheme of DDR assessment in KSL and LT‐HSC derived from serial in vivo repopulation.
-
K, LNumber of γH2AX foci, respectively, in control‐ and Molm‐14‐xenograft‐derived (K) KSL, n = 101/204, and (L) LT‐HSC, n = 33/53. Images were processed similar to (D–F). Scale bars are 3 μm. Statistics: Student's t‐test (**P < 0.01). Mean ± SD.
-
M, NqRT–PCR analysis of the DNA‐damage gene panel in Molm‐14‐xenograft‐derived (M) KSL and (N) LT‐HSC, relative to matched controls. Data depicted as fold change relative to matching controls were normalized to the endogenous control Gapdh and are presented as mean ± SEM. Statistics: one‐way ANOVA with Bonferroni post hoc correction (*P < 0.05).
Source data are available online for this figure.
Given the gains in AML‐EV‐mediated quiescence in HSC and gains in replating efficiency, we decided to evaluate their DNA‐damage response (DDR) 45, 46, 47. We observed a consistent increase in γH2AX foci (Fig 6D), indicating the presence of DNA double‐strand breaks, along with the induction of canonical DDR transcripts (including the non‐homologous end‐joining pathway transcripts) in KSL selected HSPC from AML xenografts relative to controls (Fig 6E). Interestingly, the observed DDR (γH2AX foci and DDR transcripts) in xenograft‐derived cells persisted through subsequent replating versus controls (Week 1 in Fig 6F and G and Week 2 in Fig 6H and I). Next, we attempted to corroborate this observation in vivo and assessed the DDR in HSPC after sex‐mismatched serial transplantations (Fig 6J). Again, we found a consistent increase in γH2AX foci and DDR transcripts, in both KSL and LT‐HSC from xenograft‐derived serially transplanted mice relative to the control ones (Fig 6K–N). Together, the data indicate the accrual of DNA damage and a sustained DNA‐damage response in residual progenitor and stem cells in the AML niche that, unlike the protein synthesis suppression, is not reversible. Based on the aggregate data, we propose a model whereby AML‐EV transfer of miRNA leads to suppression of the mTOR subunit Raptor and promotes quiescence in LT‐HSC. However, while protein synthesis suppression is functionally relieved upon transfer to naïve microenvironment, LT‐HSC in the AML niche accrue and retain DNA damage.
Discussion
A series of studies over the past decade has shown that the presence of AML blasts in bone marrow leads to functional alterations in stromal components with emergence of a leukemia‐permissive niche 15, 27, 48, 49, 50. Recently, investigators have also begun to provide detailed analyses of residual hematopoietic cells in the AML‐BM, revealing the initially surprising, and seemingly selective, quiescence and preservation of LT‐HSC 16, 18, 22, 23, 51. At the same time, the mechanisms by which residual HSC function in the AML niche is altered 16 and reports of mutation accrual in phenotypically normal cells of AML patients in remission 52, 53 remain unexplained. Our studies herein suggest a model of cell–cell crosstalk involving AML‐derived EV that shape residual HSC function, eliciting reversible quiescence in the short term, but persistent DNA damage.
EV trafficking serves a broad range of constitutive cellular functions, whereby several subclasses of nano‐sized vesicles transfer protein and nucleic acids between cells, including the BM niche 18, 26, 28, 29, 48, 54. We previously reported the loss of BM progenitors as a result of AML‐EV trafficking 17. Here, we hypothesized that EV also regulate residual HSC and contribute to their relative resistance to elimination, observed by others 16, 22, 23, 51. To test this hypothesis, we combined in vivo modeling in a non‐irradiated niche, high‐resolution immunophenotyping of the hematopoietic compartment, and an EV‐labeling system using palmytoylated/myristoylated fluorescence tagging 17, 55. Differential uptake of EV into hematopoietic cells has not been previously explored, and we began by showing that cells at different levels of hematopoietic differentiation, including immunophenotypically defined stem cells, were susceptible to AML‐EV entry, without significant differences in entry kinetics into HSC compared with progenitors.
In a series of xenografts, we confirmed cell‐cycle quiescence and relative enrichment of LT‐HSC 15, 18, but also replicated those outcomes using IF injection of AML‐EV. EV signaling relies on transfer of protein or nucleic acids, but rather than pursuing an EV content survey we took an outcome‐driven approach to resolve the underlying mechanism. Proteomic screening pointed to the possibility that AML‐EV exposure conferred quiescence through deregulation of ribosome biogenesis, consistent with the observed P53 upregulation and cell‐cycle arrest 34, 56. The idea that compromised ribosomal protein synthesis can erode HSC homeostasis is well established in models of myelodysplasia 57 and inherited BM disorders, such as Diamond‐Blackfan anemia 58, 59. Experimentally, we combined flow cytometric analysis for cell identity (HSPC versus LT‐HSC) and OPP incorporation. Although we observed a consistent ribosomal biogenesis impairment in all the HSPC cells, we found the most profound suppression of protein synthesis in LT‐HSC using AML xenografts. These results were independently confirmed after injection of EV isolated from AML cell‐line culture media as well as AML patient plasma.
To understand how EV target HSC protein synthesis, we explored dysregulation of key components of the mTOR pathway, a critical gatekeeper of cellular proliferation and transmission of extracellular signals. Given the role of miRNA in regulating hematopoiesis on the one hand, and miRNA cell–cell transfer via AML‐EV on the other 54, 60, we next focused on a specific panel of highly incorporated miRNA as potential regulators of mTOR. Based on the candidate panel we previously identified, we included miR‐155‐5p, miR‐1246, and miR‐1290 31, as well as miR‐27a‐5p and miR‐181b‐5p as putative AML biomarkers 41, 42, 43. In a series of experiments, we observed that among these candidates, miR‐1246 specifically targeted the mTORC1 subunit Raptor, causing a downstream loss in protein synthesis that coincided with cell quiescence. The data align well with the loss of known Raptor functions in the mTOR pathway, namely HSC activation, proliferation, and ribosome biogenesis 36, 39, 61, 62. Moreover, Raptor was previously shown to be required for HSC regeneration and its loss led to accumulation of monocytoid cells, pancytopenia, and splenomegaly in mice 63. Our data were further consistent with observations of reversible HSC quiescence by others 16, 23 and support the direct action of AML‐EV miRNA on mTOR and protein synthesis suppression that is readily reversed in a naïve niche, i.e., following transplantation to a healthy host. Thus, our results fit a model of translational suppression by AML‐EV miRNA 64 and find support in existing reports of miRNA suppression of protein synthesis 65, 66 and Raptor deregulation in hematopoietic cells 67, 68, 69. Although miR‐1246 was previously identified as a regulator of cancer progression, drug resistance 70, 71, and a putative biomarker 31, 72, our data suggest a role in regulating other tissue compartments, including BM HSC.
While enforced quiescence is a plausible protective response to leukemic stress in the BM, it also renders HSC susceptible to DNA damage through reliance on non‐homologous end joining for double‐strand lesion repair 45, 73. Unrepaired DNA breaks in HSC in turn pose a significant risk to long‐term functional integrity 74 or accrual of additional mutations. In AML patients, the acquisition of mutations in residual HSC can promote clonal expansion, malignant transformation, and overt relapse 52, 53, 75. These studies provide an intriguing context for the double‐strand breaks we observed here, and which persisted through serial in vitro replating and in vivo transplantation, respectively. While the identified gains in proliferative fitness are consistent across all experiments, and echo observations by others whereby AML‐exposed HSC outcompete controls 16, future studies will have to address the question of specific genomic lesions.
In summary, we propose a model whereby AML use EV miRNAs to suppress protein synthesis via targeting of the mTOR pathway in LT‐HSC to elicit their quiescence. Despite the functional LT‐HSC recovery, these cells acquire long‐lasting DNA damage. Whereas HSPC are generally susceptible to EV entry and utilize the mTOR pathway, HSC selectively enter quiescence in this model owing to their unique sensitivity to protein synthesis disruption 35, 39, 40. This model readily accommodates previously unexplained observations by other groups that indicate the selective preservation of residual HSC in the AML niche. It is tempting to speculate that the complex interplay of cells in the AML niche that leads to a selective advantage and subsequent transformation of residual HSC may involve EV. Finally, given the reversibility of hematopoiesis suppression in the context of AML, already noted by others 15, 16, 22, 23, our data reveal potential therapeutic targets to accelerate hematopoietic recovery. We believe that this study deepens our understanding of cell–cell communication and reveals unexpected collateral damage among hematopoietic stem cells in the AML niche.
Materials and Methods
Animals
All animal studies were approved by the OHSU Institutional Animal Care and Use Committee (IACUC). Mice were purchased from the Jackson Laboratory. For Molm‐14 xenografts, cells (0.1 × 106/animal) were engrafted into 6‐ to 8‐week‐old NOD Cg‐Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice by tail‐vein injection. For intrafemoral (IF) injections, NSG and C57BL/6J mice of 6–10 weeks old were used and IF injection was performed as previous 17, 29. Animals were anesthetized, the injection site disinfected, and the femur was positioned at 90° with the tibia. The femur cavity was cored, and EV in 50 μl PBS (HyClone) were slowly injected.
Cell culture
Molm‐14 and HL‐60 cells were provided by Dr. Jeffery Tyner, OHSU. U‐937 cells were purchased from ATCC. Cells were cultured in RPMI (Gibco) with 10% fetal bovine serum (FBS, GemCell) and 1× penicillin/streptomycin (Gibco) at 37°C, 5% CO2, and > 95% humidity. Mycoplasma co‐precipitate with EV, and contamination was excluded using the MycoAlert Plus Kit (Lonza). Healthy donor CD34+ were purchased from ATCC. Cells were cultured at 37°C, 5% CO2, and > 95% humidity in the X‐vivo media (Lonza) with BIT 9500 Serum Substitute (StemCell Technologies), FMS‐like tyrosine kinase 3 ligand (50 ng/ml), G‐CSF (10 ng/ml), IL‐3 (10 ng/ml), IL‐6 (10 ng/ml), stem cell factor (SCF) (50 ng/ml), and thrombopoietin (25 ng/ml) (Peprotech).
EV preparation
EV were purified by serial high‐speed centrifugation as previously described 17. For culture media EV, cells were cultured for 48 h in RPMI media with vesicle‐free FBS. For plasma EV, peripheral blood was obtained under IRB‐approved protocols at Texas Children's Cancer and Hematology Center, Baylor College of Medicine, Houston TX. Culture media or plasma was serially spun at 300 g, 2,000 g, and 10,000 g. The supernatants were then centrifuged at 100,000 g using the SW32Ti rotor (Beckman Coulter) to pellet down the EV.
Flow cytometry sorting and analysis of BM cells
For sorting, BM cells were flushed from long bones as previously reported 17, 29. Briefly, femur and tibia were isolated, and cells were flushed using RPMI with 2% FBS and spun at 300 g. Red blood cells (RBC) were hemolyzed, and cells were filtered through 35‐μm nylon mesh cell strainers followed by suspension in PBS with 2% FBS. BM cells were then stained with the appropriate antibodies (Appendix Table S1) at 4°C for 30 min before flow cytometric sorting using the Influx™ cell sorter (BD Biosciences). The immunophenotypic classification of HSPC populations was previously reported 76. For analytical flow cytometry, stained cells were analyzed using CANTO II flow cytometer (BD Biosciences). For intracellular staining, cells were fixed with 2% paraformaldehyde, permeabilized with 0.5% Triton X‐100, and intracellularly stained. Protein synthesis quantification was performed using the Click‐iT™ Plus O‐propargyl‐puromycin (OPP) Alexa Fluor™ 488 Kit according to the manufacturer's protocol (Thermo Fisher). Cells were treated with OPP, cultured at 37°C for 30 min, and labeled. All data were analyzed using FlowJo software (Tree Star) and are presented as mean fluorescence intensities (MFI).
RNA, DNA extraction, and RT–PCR
KSL cells were sorted into Buffer RLT (Qiagen), and total RNA was extracted using the RNeasy Kit (Qiagen). Total RNA was quantified using the NanoDrop 2000c spectrophotometer (Thermo Fisher). RNA (50 ng) samples were converted into complementary DNA using the SuperScript III First‐Strand Synthesis Kit (Invitrogen). For SRY male chimerism analysis, blood samples were collected, RBC were hemolyzed, and genomic DNA was extracted using the DNeasy Kit (Qiagen). All the RT–PCR analyses were performed using the FastStart PCR SYBR Mix and the LightCycler® 480 thermo cycler (Roche Diagnostics). Relative quantification was calculated using the ΔΔCT algorithm with GAPDH or ACTB as endogenous control when appropriate. For miRNA quantification, blood samples were collected from mice and spun at 100 g and plasma was extracted. EV were purified, and their total small RNA was extracted using the miRNeasy Kit (Qiagen). The TaqMan miR‐1246 Assay Kit (Applied Biosystems) was used for reverse transcription and qRT–PCR analyses, and the U6 snRNA was used as endogenous control.
Cells transduction, transfection, and nucleofection
AML cell lines were transduced with lentivirus harboring the GFP transgene fused in frame with a myristoylation peptide (Addgene) and then sorted to purity by flow cytometry (Molm‐14‐mGFP). For the miRNA mimic transfection studies, NIH‐3T3 cells were cultured in DMEM (Gibco) supplemented with 10% FBS and 1 × penicillin/streptomycin. Cells were transfected with the designated mirVana miRNA mimics (Thermo Fisher, Table S1) using the Lipofectamine RNAiMax (Thermo Fisher) and cultured for 48–72 h according to the downstream experiment. For siRNA nucleofection experiment, cKit+ cells selected from C57BL/6J mice were nucleofected with siRNA against control or Raptor (Thermo Fisher) using the Amaxa™ P3 Primary Cell 4D‐Nucleofector Kit (Lonza) according to the manufacturer's protocol for 72 h.
Dual‐Luciferase reporter system
The assay was performed as previously described 17. The 3′‐untranslated regions (3′UTR) of Raptor was PCR‐amplified from freshly isolated murine cKit+ genomic DNA and cloned into psiCHECK‐2 vector (Promega). NIH‐3T3 cells were first transfected with the miRNA mimics (50 nM). After 3 h, the cells were re‐transfected with 100 ng of the 3′Raptor_psiCHECK‐2 construct for additional 45 h. Cells were then washed in PBS, and the dual‐luciferase assay was measured according to the manufacturer's protocol (Promega). The Firefly luminescence and Renilla luminescence were quantified using the Synergy H1 Luminometer (BioTek), and the relative luminescence units (RLUs) were compared with the miR‐scramble.
CFU‐C assay
KSL (500 cells) were cultured in methylcellulose with cytokines (StemCell Technologies) into 35‐mm dishes (Thermo Fisher). Cells were incubated at 37°C with 5% CO2 for a week, and colonies were counted using a 4× magnification light microscope. For serial replating, methylcellulose was solubilized in PBS. Cells were then pelleted down at 300 g and plated (5,000 cells/plate) in methylcellulose for another week. The replating process was repeated for a total of three rounds.
Microscopic imaging
For γH2AX staining, cells were spun into microscope‐glass slides (Fisher Scientific) at 300 g, fixed, permeabilized, and stained with anti‐phosphoH2axSer139 (Biolegend) and Hoechst (Thermo Fisher). Images were taken by the GE/API Deltavision widefield microscope.
Live‐cell ex vivo imaging
LT‐HSCs and KSL cells were sorted into 2 ml Eppendorf tubes containing 1 ml of IMDM (as described above). Freshly sorted cells were centrifuged at 1,000 × g for 10 min at 4C and resuspended in fresh phenol‐free media, before being replated onto Matrigel (growth factor reduced/phenol‐free) in 35 mm live‐cell culture chambers with #1.5 polymer coverslip bottom (Ibidi). Cells were then incubated at 37°C 5% CO2 for 45 min to allow for attachment to Matrigel‐coated chamber. Cells were then stained with Hoechst (250 ng/ml; Thermo Fisher) and CellMask Deep Red (1 μl/ml; Thermo Fisher) for 15 min at 37C and washed twice before adding fresh media. Cells were then imaged using a Deltavision CoreDV/Olympus IX71 microscope, equipped with 60× Plan Apo N 1.49 objective, Nikon Coolpix HQ CCD camera, and live‐cell chamber supplying 37°C/5% CO2. 3D Z‐stacks were acquired in three channels using a 300 nm Z‐step through the entire cell volume to maximize capture speed while maintaining adequate axial resolution. Exposure times and laser intensity were held constant between conditions. To identify non‐specific background for the 488 channel (GFP), z‐stacks of SLAM and KSL from non‐xenografted mice were captured to determine baseline autofluorescence and thresholding value. Images were deconvolved using softWoRX and analyzed using Imaris software (Bitplane).
Live‐cell microscopy in vitro imaging
LT‐HSCs and KSLs were FACS purified from C57BL/6J mice and grown on Matrigel‐coated ibidi gridded culture slides with #1.5 coverslip for 45 min. Cells were imaged to assess background autofluorescence in the 488 channel (time 0) cells to determine thresholding value. Cells were then exposed to 1 × 108 Molm‐14‐mGFP EV and were imaged at 30 and 150 min following exposure. Images were captured using a Deltavision CoreDV/Olympus IX71 microscope, equipped with 60× Plan Apo N 1.49 objective, Nikon Coolpix HQ CCD camera, and live‐cell chamber supplying 37°C with 5% CO2. 3D Z‐stacks were acquired in 2 channels with a 300 nm Z‐step through the entire cell volume to maximize capture speed while maintaining adequate axial resolution. Exposure times and laser power were held constant across conditions. Images were analyzed using Imaris software (Bitplane).
EV imaging and quantification
EV from cell culture supernatant or peripheral blood were embedded into solid hydrogel matrices to minimize Brownian motion and allow for focused image acquisition. To embed vesicles, EV were resuspended in Matrigel, then added to a well of a 96‐well imaging plate with #1.5 bottom (ibidi). The mixture was incubated at 37°C for 4 h to allow for solidification. Following incubation, solid gels containing mGFP+ EV (or FM1‐43 stained EV) were imaged with a Nikon TiE microscope equipped with Yokogawa CSU‐W1 Spinning Disk confocal, 100× Plan Apo TIRF 1.49 objective, with a Nikon CCD camera. Concentrations are determined by acquiring 3D Z‐stacks with a fixed volume (100 × 100 × 10 μm = 1 × 10−7 ml) using 488 nm laser excitation. Individual mGFP foci are counted within 5 fields of fixed volume using Imaris Surfaces function and compared across conditions. To validate resolution of subdiffraction‐level structure, 34, 100 and 180 nm reference beads (Thermo Fisher) were utilized to determine detection limit and relative signal point spread. Using a 100× Plan Apo 1.49 TIRF objective and 50 nm pinhole, signals from all three beads were detectable with consistent size and intensity, with a point spread roughly fourfold larger than the actual size of the respective reference bead. This ensured that detection of particles in the exosomes size range is possible using this microscopy system.
Cryo‐TEM imaging
EV were purified from the culture media of Molm‐14 and U‐937 cells. From the EVs samples, 2.5–3 μl were pipetted onto glow‐discharged (120 s 15 mA, negative mode) copper Quantifoil holey carbon support grids (Ted Pella 658‐300‐CU) and then vitrified on liquid ethane using a Mark IV Vitrobot (Thermo Fisher). For cryopreservation, the vitrobot was set at 100% humidity, blot force 1, and 3‐s blotting time. We also used low‐dose conditions to acquire images on a FEI Talos Arctica at 200 kV equipped with a Ceta 16M camera. Cryo‐EM images were then collected with a defocus range of 2–4 μm.
Tandem mass tag (TMT) spectrometry analysis
C57BL/6J mouse selected cKit+ cells (1 × 106) were exposed to purified EV from the culture media (RPMI with 10% FBS and 50 ng/ml SCF and 50 ng/ml IL‐3) of Molm‐14 or HL‐60 cells versus controls for 48 h. Cell lysates from each of the two independent experiments were then prepared and labeled with TMT (Thermo Fisher Scientific) 77 as previously described 29. Total protein reporter ion intensities were normalized and tested for differential expression using the Bioconductor package edgeR 33. Each TMT experiment was analyzed independently, and edgeR default multiple testing corrections (Benjamini–Hochberg) were applied to generate false discovery rates (FDRs). Because of the different numbers of detected proteins and replicate numbers in each experiment, FDR cutoffs were selected to give similar proportions of differential expression candidates (about 10%) in each experiment. The full lists of proteins, reporter ion intensities, and edgeR results are supplied in the Datasets EV1 and EV2. The differential expression analyses are detailed in Jupyter notebook S1 and S2. Functional annotation enrichment analysis was done using the DAVID bioinformatics platform 78.
Statistical analysis
Paired and unpaired Student's t‐tests were used to compare flow cytometric results among two groups for IF and xenograft experiments, respectively. One‐way ANOVA with Bonferroni correction was used to compare two or more groups for qRT–PCR. Statistical significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Author contributions
All the authors have approved their authorship in this work, gave input to the manuscript, and contributed as follows: Conceptualization: SA and PK; Methodology: SA, JTB, and BD; Investigation: SA, JTB, AH, and BD; Formal Analysis: SA, JTB, and PAW; Writing—Original Draft: SA and PK; Visualization: SA; Resources: PK, BHC, DLM, EN, and TH; Funding Acquisition: SA, BHC, and PK; Supervision: PK.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Figure 6
Acknowledgements
This study was supported by a Hyundai Hope on Wheels Scholar Grant, Max Blue Butterfly Campaign (P.K), and the OHSU Medical Research Foundation (S.A). The authors thank Dr. William H. Fleming, Dr. Devorah Goldman, and Dr. Mu‐shui Dai (OHSU) for their critical feedback; members of the OHSU flow cytometry core, Pamela Canaday and Dorian LaTocha, for cell sorting assistance; and Dr. Claudia Lopez for the electron microscopy analysis at the Multiscale Microscopy Core (MMC) at the OHSU Center for Spatial Systems Biomedicine (OCSSB). Mass spectrometric analysis was performed by the OHSU Proteomics Shared Resource with partial support from NIH grants P30EY010572, P30CA069533, and S10OD012246. We would also like to thank Dr. Noah Hornick, Dr. Santhosh C. Verghese, Dr. Makiko Mochizuki‐Kashio, and Lotte Tholen for helpful discussions.
EMBO Reports (2019) 20: e47546
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 79 partner repository with the dataset identifier PXD013688 (http://www.ebi.ac.uk/pride/archive/projects/PXD013688).
References
- 1. SEER (2008. –2014) Cases and deaths the surveillance, epidemiology and end results (SEER) database. Bethesda, MD: National Cancer Institute; [Google Scholar]
- 2. Pession A, Masetti R, Rizzari C, Putti MC, Casale F, Fagioli F, Luciani M, Lo Nigro L, Menna G, Micalizzi C et al (2013) Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 122: 170–178 [DOI] [PubMed] [Google Scholar]
- 3. Creutzig U, Zimmermann M, Dworzak MN, Gibson B, Tamminga R, Abrahamsson J, Ha SY, Hasle H, Maschan A, Bertrand Y et al (2014) The prognostic significance of early treatment response in pediatric relapsed acute myeloid leukemia: results of the international study Relapsed AML 2001/01. Haematologica 99: 1472–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sasine JP, Schiller GJ (2016) Acute myeloid leukemia: how do we measure success? Curr Hematol Malig Rep 11: 528–536 [DOI] [PubMed] [Google Scholar]
- 5. Kantarjian H, O'Brien S, Cortes J, Giles F, Faderl S, Jabbour E, Garcia‐Manero G, Wierda W, Pierce S, Shan J et al (2006) Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high‐risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer 106: 1090–1098 [DOI] [PubMed] [Google Scholar]
- 6. Estey E, Dohner H (2006) Acute myeloid leukaemia. Lancet 368: 1894–1907 [DOI] [PubMed] [Google Scholar]
- 7. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N et al (2007) Chemotherapy‐resistant human AML stem cells home to and engraft within the bone‐marrow endosteal region. Nat Biotechnol 25: 1315–1321 [DOI] [PubMed] [Google Scholar]
- 8. Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, Passegue E (2013) Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self‐reinforcing leukemic niche. Cell Stem Cell 13: 285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim JA, Shim JS, Lee GY, Yim HW, Kim TM, Kim M, Leem SH, Lee JW, Min CK, Oh IH (2015) Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Can Res 75: 2222–2231 [DOI] [PubMed] [Google Scholar]
- 10. Chandran P, Le Y, Li Y, Sabloff M, Mehic J, Rosu‐Myles M, Allan DS (2015) Mesenchymal stromal cells from patients with acute myeloid leukemia have altered capacity to expand differentiated hematopoietic progenitors. Leuk Res 39: 486–493 [DOI] [PubMed] [Google Scholar]
- 11. Huang JC, Basu SK, Zhao X, Chien S, Fang M, Oehler VG, Appelbaum FR, Becker PS (2015) Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J 5: e302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geyh S, Rodriguez‐Paredes M, Jager P, Khandanpour C, Cadeddu RP, Gutekunst J, Wilk CM, Fenk R, Zilkens C, Hermsen D et al (2016) Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 30: 683–691 [DOI] [PubMed] [Google Scholar]
- 13. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Duhrsen U, Frenette PS (2014) Acute myelogenous leukemia‐induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 15: 365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, Haltalli M, Ruivo N, Straszkowski L, Vervoort SJ et al (2018) Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell 22: 64–77.e66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boyd AL, Reid JC, Salci KR, Aslostovar L, Benoit YD, Shapovalova Z, Nakanishi M, Porras DP, Almakadi M, Campbell CJV et al (2017) Acute myeloid leukaemia disrupts endogenous myelo‐erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol 19: 1336–1347 [DOI] [PubMed] [Google Scholar]
- 16. Miraki‐Moud F, Anjos‐Afonso F, Hodby KA, Griessinger E, Rosignoli G, Lillington D, Jia L, Davies JK, Cavenagh J, Smith M et al (2013) Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc Natl Acad Sci USA 110: 13576–13581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hornick NI, Doron B, Abdelhamed S, Huan J, Harrington CA, Shen R, Cambronne XA, Chakkaramakkil Verghese S, Kurre P (2016) AML suppresses hematopoiesis by releasing exosomes that contain microRNAs targeting c‐MYB. Sci Signal 9: ra88 [DOI] [PubMed] [Google Scholar]
- 18. Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, McDonald T, Lin A, Kumar AR, DiGiusto DL et al (2018) Acute myeloid leukemia transforms the bone marrow niche into a leukemia‐permissive microenvironment through exosome secretion. Leukemia 32: 575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sawai CM, Babovic S, Upadhaya S, Knapp D, Lavin Y, Lau CM, Goloborodko A, Feng J, Fujisaki J, Ding L et al (2016) Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity 45: 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD (2014) Clonal dynamics of native haematopoiesis. Nature 514: 322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA (2008) Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 322: 1861–1865 [DOI] [PubMed] [Google Scholar]
- 22. Akinduro O, Weber TS, Ang H, Haltalli MLR, Ruivo N, Duarte D, Rashidi NM, Hawkins ED, Duffy KR, Lo Celso C (2018) Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat Commun 9: 519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheng H, Hao S, Liu Y, Pang Y, Ma S, Dong F, Xu J, Zheng G, Li S, Yuan W et al (2015) Leukemic marrow infiltration reveals a novel role for Egr3 as a potent inhibitor of normal hematopoietic stem cell proliferation. Blood 126: 1302–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kahlert C, Kalluri R (2013) Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med 91: 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tkach M, Thery C (2016) Communication by extracellular vesicles: where we are and where we need to go. Cell 164: 1226–1232 [DOI] [PubMed] [Google Scholar]
- 26. Butler JT, Abdelhamed S, Kurre P (2018) Extracellular vesicles in the hematopoietic microenvironment. Haematologica 103: 382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Viola S, Traer E, Huan J, Hornick NI, Tyner JW, Agarwal A, Loriaux M, Johnstone B, Kurre P (2015) Alterations in acute myeloid leukaemia bone marrow stromal cell exosome content coincide with gains in tyrosine kinase inhibitor resistance. Br J Haematol 172: 983–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huan J, Hornick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT Jr, Kurre P (2013) RNA trafficking by acute myelogenous leukemia exosomes. Can Res 73: 918–929 [DOI] [PubMed] [Google Scholar]
- 29. Huan J, Hornick NI, Goloviznina NA, Kamimae‐Lanning AN, David LL, Wilmarth PA, Mori T, Chevillet JR, Narla A, Roberts CT Jr et al (2015) Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia 29: 2285–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sakamoto H, Takeda N, Arai F, Hosokawa K, Garcia P, Suda T, Frampton J, Ogawa M (2015) Determining c‐Myb protein levels can isolate functional hematopoietic stem cell subtypes. Stem Cells 33: 479–490 [DOI] [PubMed] [Google Scholar]
- 31. Hornick NI, Huan J, Doron B, Goloviznina NA, Lapidus J, Chang BH, Kurre P (2015) Serum exosome MicroRNA as a minimally‐invasive early biomarker of AML. Sci Rep 5: 11295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S et al (2009) p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4: 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He X, Li Y, Dai MS, Sun XX (2016) Ribosomal protein L4 is a novel regulator of the MDM2‐p53 loop. Oncotarget 7: 16217–16226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Signer RA, Magee JA, Salic A, Morrison SJ (2014) Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509: 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gan B, DePinho RA (2009) mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle 8: 1003–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124: 471–484 [DOI] [PubMed] [Google Scholar]
- 38. Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB (2007) Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 25: 209–226 [DOI] [PubMed] [Google Scholar]
- 39. Signer RA, Qi L, Zhao Z, Thompson D, Sigova AA, Fan ZP, DeMartino GN, Young RA, Sonenberg N, Morrison SJ (2016) The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E‐BPs. Genes Dev 30: 1698–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jarzebowski L, Le Bouteiller M, Coqueran S, Raveux A, Vandormael‐Pournin S, David A, Cumano A, Cohen‐Tannoudji M (2018) Mouse adult hematopoietic stem cells actively synthetize ribosomal RNA. RNA 24: 1803–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhi F, Cao X, Xie X, Wang B, Dong W, Gu W, Ling Y, Wang R, Yang Y, Liu Y (2013) Identification of circulating microRNAs as potential biomarkers for detecting acute myeloid leukemia. PLoS ONE 8: e56718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo Q, Luan J, Li N, Zhang Z, Zhu X, Zhao L, Wei R, Sun L, Shi Y, Yin X et al (2017) MicroRNA‐181 as a prognostic biomarker for survival in acute myeloid leukemia: a meta‐analysis. Oncotarget 8: 89130–89141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Leeuw DC, van den Ancker W, Denkers F, de Menezes RX, Westers TM, Ossenkoppele GJ, van de Loosdrecht AA, Smit L (2013) MicroRNA profiling can classify acute leukemias of ambiguous lineage as either acute myeloid leukemia or acute lymphoid leukemia. Clin Cancer Res 19: 2187–2196 [DOI] [PubMed] [Google Scholar]
- 44. Tanoue A, Nakamura T, Endo F, Nishiyama S, Sakiyama H, Matsuda I (1992) Sex‐determining region Y (SRY) in a patient with 46, XX true hermaphroditism. Jpn J Hum Genet 37: 311–320 [DOI] [PubMed] [Google Scholar]
- 45. Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ (2014) Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15: 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC et al (2014) Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512: 198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ogawa LM, Baserga SJ (2017) Crosstalk between the nucleolus and the DNA damage response. Mol BioSyst 13: 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kumar B, Garcia M, Murakami JL, Chen CC (2016) Exosome‐mediated microenvironment dysregulation in leukemia. Biochem Biophys Acta 1863: 464–470 [DOI] [PubMed] [Google Scholar]
- 49. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, Wang Y, Battula VL, Mak PY, Schallmoser K et al (2014) Reciprocal leukemia‐stroma VCAM‐1/VLA‐4‐dependent activation of NF‐kappaB mediates chemoresistance. Blood 123: 2691–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Doron B, Abdelhamed S, Butler JT, Hashmi SK, Horton TM, Kurre P (2019) Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia 33: 918–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boyd AL, Campbell CJ, Hopkins CI, Fiebig‐Comyn A, Russell J, Ulemek J, Foley R, Leber B, Xenocostas A, Collins TJ et al (2014) Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J Exp Med 211: 1925–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW et al (2014) Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature 506: 328–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Corces‐Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R (2014) Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA 111: 2548–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boyiadzis M, Whiteside TL (2018) Exosomes in acute myeloid leukemia inhibit hematopoiesis. Curr Opin Hematol 25: 279–284 [DOI] [PubMed] [Google Scholar]
- 55. Lai CP, Kim EY, Badr CE, Weissleder R, Mempel TR, Tannous BA, Breakefield XO (2015) Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun 6: 7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fumagalli S, Thomas G (2011) The role of p53 in ribosomopathies. Semin Hematol 48: 97–105 [DOI] [PubMed] [Google Scholar]
- 57. Galili N, Qasim SA, Raza A (2009) Defective ribosome biogenesis in myelodysplastic syndromes. Haematologica 94: 1336–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D (2007) Ribosomal protein S17 gene (RPS17) is mutated in Diamond‐Blackfan anemia. Hum Mutat 28: 1178–1182 [DOI] [PubMed] [Google Scholar]
- 59. Gazda HT, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE et al (2008) Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond‐Blackfan anemia patients. Am J Hum Genet 83: 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al‐Shahrour F, Hasserjian RP, Scadden EO et al (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464: 852–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Atsushi Hirao TH (2013) Mechanistic/mammalian target protein of rapamycin signaling in hematopoietic stem cells and leukemia. Cancer Sci 104: 977–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang J, Nguyen‐McCarty M, Hexner EO, Danet‐Desnoyers G, Klein PS (2012) Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med 18: 1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C, Sinha AU, Lane SW, Souza AL, Clish CB et al (2012) mTOR complex 1 plays critical roles in hematopoiesis and Pten‐loss‐evoked leukemogenesis. Cell Stem Cell 11: 429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fabian MR, Sonenberg N, Filipowicz W (2010) Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 79: 351–379 [DOI] [PubMed] [Google Scholar]
- 65. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63 [DOI] [PubMed] [Google Scholar]
- 66. Pillai RS, Bhattacharyya SN, Filipowicz W (2007) Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol 17: 118–126 [DOI] [PubMed] [Google Scholar]
- 67. Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA, Nucera S, Ciceri F, Kaufmann KB, Takayama N et al (2016) miR‐126 regulates distinct self‐renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell 29: 214–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qian P, He Xi C, Paulson A, Li Z, Tao F, Perry John M, Guo F, Zhao M, Zhi L, Venkatraman A et al (2016) The Dlk1‐Gtl2 locus preserves LT‐HSC function by inhibiting the PI3K‐mTOR pathway to restrict mitochondrial metabolism. Cell Stem Cell 18: 214–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bianchi N, Finotti A, Ferracin M, Lampronti I, Zuccato C, Breveglieri G, Brognara E, Fabbri E, Borgatti M, Negrini M et al (2015) Increase of microRNA‐210, decrease of raptor gene expression and alteration of mammalian target of rapamycin regulated proteins following mithramycin treatment of human erythroid cells. PLoS ONE 10: e0121567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim G, An H‐J, Lee M‐J, Song J‐Y, Jeong J‐Y, Lee J‐H, Jeong H‐C (2016) Hsa‐miR‐1246 and hsa‐miR‐1290 are associated with stemness and invasiveness of non‐small cell lung cancer. Lung Cancer 91: 15–22 [DOI] [PubMed] [Google Scholar]
- 71. Li XJ, Ren ZJ, Tang JH, Yu Q (2017) Exosomal microRNA MiR‐1246 promotes cell proliferation, invasion and drug resistance by targeting CCNG2 in breast cancer. Cell Physiol Biochem 44: 1741–1748 [DOI] [PubMed] [Google Scholar]
- 72. Moshiri F, Salvi A, Gramantieri L, Sangiovanni A, Guerriero P, De Petro G, Bassi C, Lupini L, Sattari A, Cheung D et al (2018) Circulating miR‐106b‐3p, miR‐101‐3p and miR‐1246 as diagnostic biomarkers of hepatocellular carcinoma. Oncotarget 9: 15350–15364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mohrin M, Bourke E, Alexander D, Warr MR, Barry‐Holson K, Le Beau MM, Morrison CG, Passegue E (2010) Hematopoietic stem cell quiescence promotes error‐prone DNA repair and mutagenesis. Cell Stem Cell 7: 174–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yahata T, Takanashi T, Muguruma Y, Ibrahim AA, Matsuzawa H, Uno T, Sheng Y, Onizuka M, Ito M, Kato S et al (2011) Accumulation of oxidative DNA damage restricts the self‐renewal capacity of human hematopoietic stem cells. Blood 118: 2941–2950 [DOI] [PubMed] [Google Scholar]
- 75. Jan M, Snyder TM, Corces‐Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R (2012) Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 4: 149ra118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Oguro H, Ding L, Morrison SJ (2013) SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13: 102–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75: 1895–1904 [DOI] [PubMed] [Google Scholar]
- 78. da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- 79. Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–d450 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Figure 6
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 79 partner repository with the dataset identifier PXD013688 (http://www.ebi.ac.uk/pride/archive/projects/PXD013688).