

Abstract
We report a Pd-catalyzed intermolecular hydrophosphinylation of 1,3-dienes to afford chiral allylic phosphine oxides. Commodity dienes and air stable phosphine oxides couple to generate organophosphorus building blocks with high enantio- and regiocontrol. This method constitutes the first asymmetric hydrophosphinylation of dienes.
Graphical Abstract
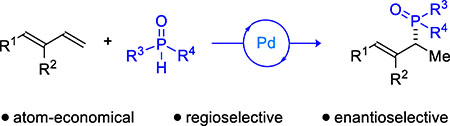
Conjugated dienes are versatile motifs for constructing molecules that range from natural products to synthetic polymers.1,2 In recent years, hydrofunctionalization has emerged as an attractive and atom-economical3 method to transform dienes into valuable building blocks.4 In comparison to other hydrofunctionalizations (e.g., hydroboration or hydroformylation), hydrophosphinylation remains in its infancy (Figure 1). Hirao first coupled isoprene and diethyl phosphonate to furnish an allylic phosphonate, albeit with low efficiency (10% yield) and at an elevated temperature (150 °C).5 Tanaka later improved the hydrophosphorylation of 1,3-dienes by using a more reactive pinacol-derived phosphonate to synthesize allylphosphonates.6 While promising, this strategy has been restricted to producing achiral regioisomers or racemic mixtures.7 Given the potential for chiral phosphines in catalysis,8 as well as the need for novel phosphine motifs in medicine9 and agrochemical space,10 we sought to develop an enantioselective hydrophosphinylation.11 Herein, we report the transformation of several petroleum feedstocks and readily available dienes into chiral phosphine oxide building blocks, with high region- and enantiocontrol.
Figure 1.

Inspiration for asymmetric hydrophosphinylation of 1,3-dienes.
Given previously reported asymmetric hydroamination12 and hydrothiolation13 of 1,3-dienes, we chose to focus on a phosphorus nucleophile that would possess intermediate nucleophilicity compared to amines and thiols. As part of our reaction design, we imagined using phosphine oxides (2) as P-based nucleophiles because they are air stable, commercially available, and readily reduced to the corresponding phosphine.14 In addition, the pKa of 2 (ca. 25)15 is between that of amines and thiols. Although the phosphine oxide reagent and its corresponding product could inhibit catalysis, hydrophosphinylation of alkenes16 and alkynes17 using transition-metal catalysis and photocatalysis has been reported. Encouraged by these examples, we set out to identify a catalyst that would overcome the established 1,4-addition pathway to furnish the desired chiral isomer.
We began our investigations with the coupling of 1-phenylbutadiene (1a) and commercially available 2a (Table 1). We examined a range of achiral bisphosphine ligands, with both Rh and Pd precatalysts. While Rh showed no reactivity, Pd was promising for the hydrophosphinylation of 1a. As highlighted in Table 1A, we observed that the ligand bite angle affected efficiency of the hydrophosphinylation.18 Combining Pd2(dba)3 and ferrocene-based dppf offered optimal results (90%, >20:1 rr). Catalytic amounts of acid provided an increase in the reaction rate; P(V)-based Bransted acids proved to be the most effective for hydrophosphinylation (Table 1B). In the absence of an acid co-catalyst, we observe 16% of product 3aa after 3 hours and 87% yield after 24 hours. Based on these results, we focused on the Josiphos ligand family with diphenylphosphinic acid as a co-catalyst.19 As seen in Table 1C, with Pd(L3) we could lower the catalyst loading to 0.50 mol% and synthesize 3aa on gram scale while retaining high reactivity (1.05 g, 91%) and selectivity (>20:1 rr, 95:5 er). The er in the presence of different acids shows little variation and ranges from 95:5 to 96:4.
Table 1.
Ligand and Acid Effects on Asymmetric Hydrophosphinylation of 1aa
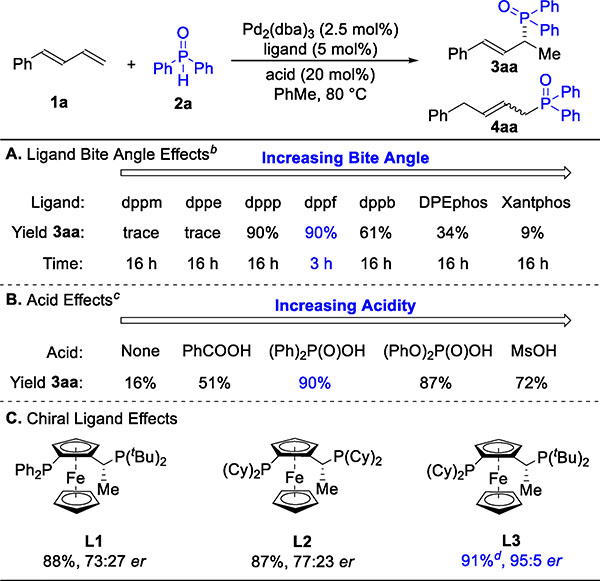 |
Reaction conditions: 1a (0.12 mmol), 2a (0.10 mmol), Pd2(dba)3 (2.5 mol%), ligand (5.0 mol%), acid (20 mol%), toluene (0.40 mL), 3 h (unless otherwise noted). Yield determined by GC-FID analysis of the reaction mixture, which was referenced to 1,3,5-trimethoxybenzene. Regioselectivity ratio (rr) is the ratio of 3aa to 4aa, which is determined by 31P NMR analysis of reaction mixture. Enantioselectivity ratio (er) determined by chiral SFC. See SI for full structure of abbreviations used. Unless otherwise noted, rr is >20:1.
Standard conditions with (Ph)2P(O)OH as acid.
Standard conditions with dppf as ligand.
Isolated yield of 3aa, 3.47 mmol scale, using Pd2(dba)3 (0.50 mol%) and L3 (1.0 mol%) with standard conditions, 18 h.
With these conditions in hand, we investigated the hydrophosphinylation of various 1,3-dienes with phosphine oxide 2a (Table 2). We found that a variety of 1-aryl substituted dienes could be transformed to chiral products 3ba-3ja with moderate to high reactivity (36–88%) and selectivity (>20:1 rr, 88:12–96:4 er). Dienes containing aryl chlorides (3ca, 3ha, 3ia) offer higher reactivity than aryl bromides (3da), potentially due to the mitigation of side pathways initiated by oxidative addition into the C–X bond. The petroleum feedstocks butadiene (1m) and isoprene (1n) can be coupled with 2a to furnish chiral building blocks 3ma and 3na, respectively. We observed product mixtures of 3ma and 3na that equally, or moderately, favor 3,4-addition over the established 1,4-addition previously reported for the hydrophosphorylation of butadiene6 (1m) and isoprene5,6 (1n). To examine if the allylic phosphine products (3ma and 3na) could racemize by a sigmatropic rearrangement,20 we resubjected 3ma to the standard reaction conditions. After 12 hours, we observed no change in the enantiomeric excess. The 1,2-disubstituted diene (1k) and 1-alkyl substituted diene (1l) transform to products 3ka and 3la, respectively, in the presence of (S)-DTBM-SegPhos. This result suggests that the diene substitution pattern must be matched with the appropriate ligand family, an observation in agreement with our previous studies on Rh-catalyzed hydrothiolation of 1,3-dienes.13
Table 2.
Hydrophosphinylation of Various 1,3-Dienesaa
 |
Reaction conditions: 1 (0.12 mmol), 2a (0.10 mmol), Pd2(dba)3 (2.5 mol%), ligand (5.0 mol%), (Ph)2P(O)OH (20 mol%), toluene (0.40 mL), 6 h. Isolated yield of 3. Regioselectivity ratio (rr) is the ratio of 3 to 4, which is determined by 31P NMR analysis of reaction mixture. Enantioselectivity determined by chiral SFC.
(S)-DTBM-SegPhos (5.0 mol%) instead of L3, see SI for structure, 24 h.
Next, we investigated the hydrophosphinylation of 1a with structurally and electronically different phosphine oxides (Table 3). We observed high reactivity (3ab-3am, 51–88%), regioselectivity (>20:1 rr), and enantioselectivity (74:26–98:2 er). This coupling tolerates aryl (3ab-3ai), heterocyclic (3aj), and alkyl (3ak) phosphine oxides. Mono- (2a-2g), di- (2h), and trisubstituted (2i) aryl groups on the phosphine oxide partner can be coupled with 1a to afford enantioenriched products (3aa-3ai). Fused ring motifs, which are the basis of a large class of ligand scaffolds, can also be incorporated in the phosphine oxide partner to generate products 3al and 3am.
Table 3.
Hydrophosphinylation of 1a with Various Phosphine Oxidesa
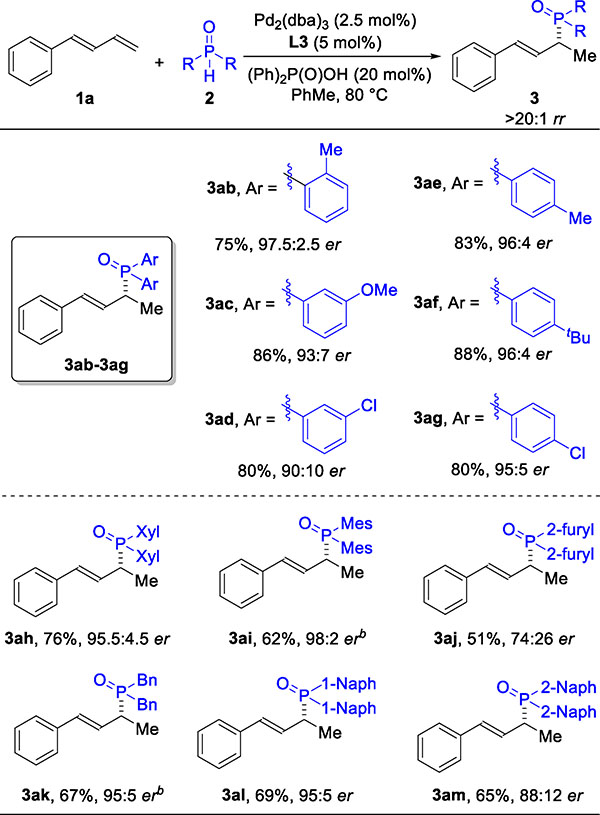 |
Reaction conditions: 1a (0.12 mmol), 2 (0.10 mmol), Pd2(dba)3 (2.5 mol%), ligand (5.0 mol%), (Ph)2P(O)OH (20 mol%), toluene (0.40 mL), 6 h. Isolated yield of 3. Regioselectivity ratio (rr) is the ratio of 3 to 4, which is determined by 31P NMR analysis of reaction mixture. Enantioselectivity determined by chiral SFC. See SI for full structure of abbreviations used.
Reaction time is 24 h.
Catalyst-controlled C–P bond formation would enable selective access to diastereomers. To test this idea, we prepared enantiopure phosphine oxide 2n bearing a tert-butyl and phenyl group, a popular motif in chiral ligand design (Figure 2).21 Depending on the enantiomer of the ligand L3 used, the (R,R)-diastereomer 3an22 or (R,S)-diastereomer 3an′ can be obtained with high diastereocontrol (95:5 and 91:9 dr, respectively). This result represents a diastereodivergent strategy for making phosphine oxides.
Figure 2.

Diastereodivergent hydrophosphinylation of 1a.
Based on literature precedence and our own observations, we propose the mechanism depicted in Figure 3A. The Pd(0) precatalyst undergoes ligand substitution with the bisphosphine ligand to form a chiral monomeric species I, subsequent oxidative addition to diphenylphosphinic acid (HX) forms Pd–H species II. A related oxidative addition has been implicated as a key step in the hydrophosphinylation of terminal alkynes.17e In the absence of acid additives, we observe a significant induction period.23 We reason that the addition of an acid co-catalyst (i.e., diphenylphosphinic acid) shortens the induction period and favors the Pd–H catalyst (e.g., II). At this point, two different modes of diene 1 coordination lead to the major product 3 (path a) and the minor product 4 (path b). In path a, species III undergoes hydropalladation to provide the key Pd-π-allyl intermediate IV. Species IV then undergoes a ligand exchange with phosphine oxide 2 to form species V. Subsequent reductive elimination of V furnishes the allylic phosphine oxide 3 and regenerates the Pd-catalyst I.
Figure 3.

Proposed mechanism and initial investigations of the Pd-catalyzed hydrophosphinylation of 1,3-dienes.
To probe the mechanism, we conducted the following experiments (Figure 3B–D). First, a deuterium-labeled phosphine oxide d-2a was subjected to the standard reaction conditions. In this experiment, we see deuterium incorporation at the C1 (10% D) and C4 (64% D) positions of d-3ea. If hydropalladation were irreversible, we should observe about a 6:1 mixture of regioisomers. In contrast, we observe >20:1 rr and thus conclude that hydropalladation is reversible. Second, (Z)-1-phenylbutadiene (Z-1a) was subjected to the hydrophosphinylation. We observed only the (E)-product 3aa (>95% E content) in similar yield (74%) and regioselectivity (>20:1 rr) compared to the model substrate (Table 1, 3aa, 90% yield, >20:1 rr). This result suggests that isomerization occurs faster than C–P bond formation. Furthermore, excess diene Z-1a is recovered with ca. 25% Z content, which is consistent with a reversible hydropalladation and reversible diene coordination. By subjecting toluoyl phosphine oxide 2e to product 3aa, under otherwise standard conditions, we confirmed that the allylic phosphine oxide 3aa cannot undergo further substitution to form 3ae. Our proposal is in line with a study on alkyne hydrophosphinylation where Pd–P bond cleavage requires elevated temperatures and reductive elimination is the turnover-limiting step.17e We observed that alkyl-substituted dienes (1l-1n) form products (3la-3na) with lower regioselectivity compared to the aryl-substituted dienes (3ba-3ka). Thus, reductive elimination to form the conjugated product appears to be favorable.
The direct construction of chiral phosphines and phosphine oxides has previously been achieved via additions to Michael acceptors or transition-metal catalyzed substitutions.24,25 Herein, we reported a complementary way to access chiral phosphine oxides. This study features the first enantioselective hydrophosphinylation of dienes. Phosphine oxides and 1,3-dienes can be coupled to furnish chiral allylic products in high yields, regioselectivity, and enantioselectivity. Mechanistic studies suggest that the coupling proceeds through a reversible hydropalladation of the 1,3-diene partner followed by irreversible reductive elimination to afford the chiral phosphine oxide building blocks.
Supplementary Material
ACKNOWLEDGMENT
Funding provided by UC Irvine, the National Institutes of Health (R35GM127071) and the National Science Foundation (CHE-1465263). Shao-Zhen Nie thanks Liaocheng University for a scholarship. We thank Dr. Joseph Ziller and Austin Ryan for X-ray crystallographic analysis and the Jarvo Lab for use of their SFC.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectral data for all new compounds (PDF)
Crystallographic Data (CIF)
The authors declare no competing financial interests.
REFERENCES
- (1).For selected reviews on 1,3-dienes as building blocks, see:Nicolaou KC; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels-Alder Reaction in Total Synthesis. Angew. Chem., Int. Ed 2002, 41, 1668–1698.Reymond S; Cossy J Copper-Catalyzed Diels-Alder Reactions. Chem. Rev 2008, 108, 5359–5406.Chen J-R; Hu X-Q; Lu L-Q; Xiao W-J Formal [4+1] Annulation Reactions in the Synthesis of Carbocyclic and Heterocyclic Systems. Chem. Rev 2015, 115, 5301–5365.Büschleb M; Dorich S; Hanessian S; Tao D; Schenthal KB; Overman LE Synthetic Strategies toward Natural Products Containing Contiguous Stereogenic Quaternary Carbon Atoms. Angew. Chem., Int. Ed 2016, 55, 4156–4186.
- (2).For reviews on polymerization of 1,3-dienes, see:Monakov YB; Mullagaliev IR Ionic and coordination diene polymerization and organic derivatives of main group metals. Russ. Chem. Bull.; Int. Ed 2004, 53, 1–9.Friebe L; Nuyken O; Obrecht W Neodymium Based Ziegler/Natta Catalysts and their Application in Diene Polymerization. Adv. Polym. Sci 2006, 204, 1–154.Zhang Z; Cui D; Wang B; Liu B; Yang Y Polymerization of 1,3-Conjugated Dienes with Rare-Earth Metal Precursors. Struct. Bonding (Berlin) 2010, 137, 49–108.Valente A; Mortreux A; Visseaux M; Zinck P Coordinative Chain Transfer Polymerization. Chem. Rev 2013, 113, 3836–3857.Takeuchi D Stereoselective Polymerization of Conjugated Dienes In Encyclopedia of Polymer Science and Technology; Wiley: New York, 2013; pp 1–25.
- (3).Trost BM The atom economy—a search for synthetic efficiency. Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- (4).For select hydrofunctionalizations of dienes, see:Löber O; Kawatsura M; Hartwig JF Palladium-Catalyzed Hydroamination of 1,3-Dienes: A Colorimetric Assay and Enantioselective Additions. J. Am. Chem. Soc 2001, 123, 4366–4367.Page JP; RajanBabu TV Asymmetric Hydrovinylation of 1-Vinylcycloalkenes. Reagent Control of Regio- and Stereoselectivity. J. Am. Chem. Soc 2012, 134, 6556–6559.Zbieg JR; Yamaguchi E; McInturff EL; Krische MJ Enantioselective C–H Crotylation of Primary Alcohols via Hydrohydroxyalkylation of Butadiene. Science 2012, 336, 324–327.Park BY; Montgomery TP; Garza VJ; Krische MJ Ruthenium Catalyzed Hydrohydroxyalkylation of Isoprene with Heteroaromatic Secondary Alcohols: Isolation and Reversible Formation of the Putative Metallacycle Intermediate. J. Am. Chem. Soc 2013, 135, 16320–16323.Chen Q-A; Kim DK; Dong VM Regioselective Hydroacylation of 1.3-Dienes by Cobalt Catalysis. J. Am. Chem. Soc 2014, 136, 3772–3775.Saini V; O’Dair M; Sigman MS Synthesis of Highly Functionalized Tri- and Tetrasubstituted Alkenes via Pd-Catalyzed 1,2-Hydrovinylation of Terminal 1,3-Dienes. J. Am. Chem. Soc 2015, 137, 608–611.Marcum JS; Roberts CC; Manan RS; Cervarich TN; Meek SJ Chiral Pincer Carbodicarbene Ligands for Enantioselective Rhodium-Catalyzed Hydroarylation of Terminal and Internal 1,3-Dienes with Indoles. J. Am. Chem. Soc 2017, 139, 15580–15583.Yang X-H; Lu A; Dong VM Intermolecular Hydroamination of 1,3-Dienes To Generate Homoallylic Amines. J. Am. Chem. Soc 2017, 139, 14049–14052.Gui Y-Y; Hu N; Chen X-W; Liao L-L; Ju T; Ye J-H; Zhang Z; Li J; Yu D-G Highly Regio- and Enantioselective Copper-Catalyzed Reductive Hydroxymethylation of Styrenes and 1,3-Dienes with CO2. J. Am. Chem. Soc 2017, 139, 17011–17014.Adamson NJ; Hull E; Malcolmson SJ Enantioselective Intermolecular Addition of Aliphatic Amines to Acyclic Dienes with a Pd–PHOX Catalyst. J. Am. Chem. Soc 2017, 139, 7180–7183.Adamson NJ; Wilbur KCE; Malcolmson SJ Enantioselective Intermolecular Pd-Catalyzed Hydroalkylation of Acyclic 1,3-Dienes with Activated Pronucleophiles. J. Am. Chem. Soc 2018, 140, 2761–2764.Schmidt VA; Kennedy CR; Bezdek MJ; Chirik PJ Selective [1,4]-Hydrovinylation of 1.3-Dienes with Unactivated Olefins Enabled by Iron Diimine Catalysts. J. Am. Chem. Soc 2018, 140, 3443–3453. For reviews, see: Hydrofunctionalization In Topics in Organometallic Chemistry; Ananikov VP, Tanaka M Eds.; Springer: Berlin, 2014; Vol. 343.McNeill E; Ritter T 1,4-Functionalization of 1,3-Dienes With Low-Valent Iron Catalysts. Acc. Chem. Res 2015, 48, 2330–2343.Bezzenine-Lafollée S; Gil R; Prim D; Hannedouche J First-Row Late Transition Metals for Catalytic Alkene Hydrofunctionalisation: Recent Advances in C–N, C–O, and C–P Bond Formation. Molecules 2017, 22, 1901–1929.
- (5).Hirao T; Masunaga T; Yamada N; Ohshiro Y; Agawa T Palladium-catalyzed New Carbon-Phosphorous Bond Formation. Bull. Chem. Soc. Jpn 1982, 55, 909–913. [Google Scholar]
- (6).Mirzaei F; Han L-B; Tanaka M Palladium-catalyzed hydrophosphorylation of 1,3-dienes leading to allylphosphonates. Tet. Lett 2001, 42, 297–299. [Google Scholar]
- (7).Two additional examples have been reported for the coupling of hydrophosphorous acid (H3PO2) and a 1,3-diene that yield allylic phosphinic acids via Pd-catalyzed 1,4-addition, see:Bravo-Altamirano K; Abrunhosa-Thomas I; Montchamp J-L Palladium-Catalyzed Reactions of Hypophosphorous Compounds with Allenes, Dienes, and Allylic Electrophiles: Methodology for the Synthesis of H-Phosphinates. J. Org. Chem 2008, 73, 2292–2301.
- (8).For select reviews of phosphines in catalysis, see:Tolman CA Steric effects of phosphorus ligands in organometallic chemisTry and homogeneous catalysis. Chem. Rev 1977, 77, 313–348.van Leeuwen PWNM; Kamer PCJ; Reek JNH; Dierkes P Ligand Bite Angle Effects in Metal-catalyzed C–C Bond Formation. Chem. Rev 2000, 100, 2741–2770.Noyori R; Ohkuma T Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angew. Chem., Int. Ed 2001, 40, 40–73.Xiao Y; Guo H; Kwon O Nucleophilic Chiral Phosphines: Powerful and Versatile Catalysts for Asymmetric Annulations. Aldrichimica Acta. 2016, 49, 3–13.Ni H; Chan W-L; Lu Y Phosphine-Catalyzed Asymmetric Organic Reactions. Chem. Rev 2018, 118, 9344–9411.
- (9).For reviews of phosphines in medicine, see:Tiekink ERT Phosphinegold(I) thiolates—pharmacological use and potential. Bioinorg. Chem. Appl 2003, 1, 53–67.Phillips AD; Gonsalvi L; Romerosa A; Vizza F; Peruzzini M Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA): Transition metal complexes and related catalytic, medicinal, and photoluminescent applications. Coord. Chem. Rev 2004, 248, 955–993.Dominelli B; Correia JDG; Kühn FE Medicinal Applications of Gold(I/nI)-Based Complexes Bearing V-Heterocyclic Carbene and Phosphine Ligands. J. Organomet. Chem 2018, 866, 153–164.
- (10).For a select example of phosphines in agrochemicals, see:Schlipalius DI; Valmas N; Tuck AG; Jagadeesan R; Ma L; Kaur R; Goldinger A; Anderson C; Kuang J; Zuryn S; Mau YS; Cheng Q; Collins PJ; Nayak MK; Schirra HJ; Hilliard MA; Ebert PR A Core Metabolic Enzyme Mediates Resistance to Phosphine Gas. Science 2012, 338, 807–810. For a review, see: Nath NS; Bhattacharya I; Tuck AG; Schlipalius DI; Ebert PR Mechanisms of Phosphine Toxicity. J. Toxicol 2011, 2011, 1–9.
- (11).We are using the definition of hydrophosphinylation that refers to the addition of a phosphine oxide to a degree of unsaturation, see:Han L-B; Choi N; Tanaka M Oxidative Addition of HP(O)Ph2 to Platinum(0) and Palladium(0) Complexes and Palladium-Catalyzed Regio- and Stereoselective Hydrophosphinylation of Alkynes. Organometallics 1996, 15, 3259–3261.
- (12).Yang X-H; Dong VM Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes. J. Am. Chem. Soc 2017, 139, 1774–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Yang X-H; Davison RT; Dong VM Catalytic Hydrothiolation: Regio- and Enantioselective Coupling of Thiols and Dienes J. Am. Chem. Soc 2018, 140, 10443–10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For reviews on reducing phosphine oxides to phosphines, see:Hérault D; Nguyen DH; Nuel D; Buono G Reduction of secondary and tertiary phosphine oxides to phosphines. Chem. Soc. Rev 2015, 44, 2508–2528.Kovács T; Keglevich G The Reduction of Tertiary Phosphine Oxides by Silanes. Curr. Org. Res 2017, 21, 569–585.
- (15).Grayson M; Farley CE; Streuli CA Secondary phosphine oxides: The effect of structure on acid strength and rates of cleavage of disulfides. Tetrahedron 1967, 23, 1065–1078. [Google Scholar]
- (16).For select hydrophosphinylations of alkenes, see:Kawaguchi S-I; Nomoto A; Sonoda M; Ogawa A Photoinduced hydrophosphinylation of alkenes with diphenylphosphine oxide. Tet. Lett 2009, 50, 624–626.Yoo W-J; Kobayashi S Hydrophosphinylation of unactivated alkenes with secondary phosphine oxides under visible-light photocatalysis. Green. Chem 2013, 15, 1844–1848.Li Z; Fan F; Zhang Z; Xiao Y; Liu D; Liu Z-Q A silver-initiated free-radical intermolecular hydrophosphinylation of unactivated alkenes. RSC Adv. 2015, 5, 27853–27856.
- (17).For select hydrophosphinylations of alkynes, see:Han L-B; Hua R; Tanaka M Phosphinic Acid Induced Reversal of Regioselectivity in Pd-Catalyzed Hydrophosphinylation of Alkynes with Ph2P(O)H. Angew. Chem., Int. Ed 1998, 37, 94–96.Han L-B; Zhao C-Q; Tanaka M Rhodium-Catalyzed Regio- and Stereoselective Addition of Diphenylphosphine Oxide to Alkynes. J. Org. Chem 2001, 66, 5929–5932.Han L-B; Zhang C; Yazawa H; Shimada S Efficient and Selective Nickel-Catalyzed Addition of H–P(O) and H–S Bonds to Alkynes. J. Am. Chem. Soc 2004, 126, 5080–5081.Rooy SV; Cao C; Patrick BO; Lam A; Love JA Alkyne hydrophosphinylation catalyzed by rhodium pyrazolylborate complexes. Inorg. Chim. Acta 2006, 359, 2918–2923.Chen T; Zhao C-Q; Han L-B Hydrophosphorylation of Alkynes Catalyzed by Palladium: Generality and Mechanism. J. Am. Chem. Soc 2018, 140, 3139–3155. For a review, see: Xu Q; Han L-B Metal-catalyzed additions of H–P(O) bonds to carbon–carbon unsaturated bonds. J. Organomet. Chem 2011, 696, 130–140.
- (18).For a review on ligand bite angle effects, see: ref 8b.
- (19).We also tried the PHOX ligand scaffold, which has demonstrated high reactivity and enantioselectivity in Pd-catalyzed diene hydroamination (see: ref 4j) and hydroalkylation (see: ref 4k), but observed no reactivity.
- (20).Herriott AW; Mislow K Rearrangement of allyl phosphinites and optical stability of allyl phosphine oxides. Tet. Lett 1968, 9, 3013–3016. [Google Scholar]
- (21).Lühr S; Holz J; Börner A The Synthesis of Chiral Phosphorus Ligands for use in Homogeneous Metal Catalysis. Chem-CatChem 2011, 3, 1708–1730. [Google Scholar]
- (22).X-ray crystallography data confirmed the absolute configuration of 3an, CCDC: 1868886. The absolute configuration of compounds 3aa-3am, 3an′ and 3ba-3na were assigned by analogy.
- (23).See the Supporting Information for more details.
- (24).For select enantioselective additions to Michael acceptors, see:Carlone A; Bartoli G; Bosco M; Sambri L; Melchiorre P Organocatalytic Asymmetric Hydrophosphination of α,β-Unsaturated Aldehydes. Angew. Chem., Int. Ed 2007, 46, 4504–4506.Ibrahem I; Rios R; Vesely J; Hammar P; Eriksson L; Himo F; Córdova A Enantioselective Organocatalytic Hydrophosphination of α,β-Unsaturated Aldehydes. Angew. Chem., Int. Ed 2007, 46, 4507–4510.Feng J-J; Chen X-F; Shi M; Duan W-L Palladium-Catalyzed Asymmetric Addition of Diarylphosphines to Enones toward the Synthesis of Chiral Phosphines. J. Am. Chem. Soc 2010, 132, 5562–5563.Chew RJ; Teo KY; Huang Y; Li B-B; Li Y; Pullarkat SA; Leung P-H Enantioselective phospha-Michael addition of diarylphosphines to β,γ-unsaturated α-ketoesters and amides. Chem. Commun 2014, 50, 8768–8770. For a review, see: Pullarkat SA Recent Progress in Palladium-Catalyzed Asymmetric Hydrophosphination. Synthesis 2016, 48, 493–503.
- (25).For select enantioselective transition-metal catalyzed sub-stitutions, see:Butti P; Rochat R; Sadow AD; Togni A Palladium-Catalyzed Enantioselective Allylic Phosphination. Angew. Chem., Int. Ed 2008, 47, 4878–4881.Zhang L; Liu W; Zhao X Carbon-Phosphorous Bond Formation by Enantioselective Palladium-Catalyzed Allylation of Diphenylphosphine Oxide. Eur. J. Org. Chem 2014, 6846–6849.Liu C; Wang Q Alkenylation of C(sp3)–H Bonds by Zincation/Copper-Catalyzed Cross-Coupling with Iodonium Salts. Angew. Chem., Int. Ed 2018, 57, 4727–4731.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.