

Abstract
Fibrosis is a pivotal player in heart failure development and progression. Measurements of (markers of) fibrosis in tissue and blood may help to diagnose and risk stratify patients with heart failure, and its treatment may be effective in preventing heart failure and its progression. A lack of pathophysiological insights and uniform definitions has hampered the research in fibrosis and heart failure. The Translational Research Committee of the Heart Failure Association discussed several aspects of fibrosis in their workshop. Early insidious perturbations such as subclinical hypertension or inflammation may trigger first fibrotic events, while more dramatic triggers such as myocardial infarction
and myocarditis give rise to full blown scar formation and ongoing fibrosis in diseased hearts. Aging itself is also associated with a cardiac phenotype that includes fibrosis. Fibrosis is an extremely heterogeneous phenomenon, as several stages of the fibrotic process exist, each with different fibrosis subtypes and a different composition of various cells and proteins — resulting in a very complex pathophysiology. As a result, detection of fibrosis, e.g. using current cardiac imaging modalities or plasma biomarkers, will detect only specific subforms of fibrosis, but cannot capture all aspects of the complex fibrotic process. Furthermore, several anti‐fibrotic therapies are under investigation, but such therapies generally target aspecific aspects of the fibrotic process and suffer from a lack of precision. This review discusses the mechanisms and the caveats and proposes a roadmap for future research.
Keywords: Fibrosis, Heart failure, Biomarkers, Fibroblast, Matrix, Prognosis, Imaging
Introduction
Fibrosis is a fundamental process observed in cardiac remodelling and considered to be a key contributor to heart failure and its progression. Importantly, the presence and extent of myocardial fibrosis has also prognostic implications, as it causes contractile dysfunction and arrhythmias in structural heart disease of various aetiologies.1, 2, 3, 4, 5, 6 Fibrosis is a direct and indirect target in the treatment of heart failure, either by established drug therapies (e.g. angiotensin‐converting enzyme inhibitors or mineralocorticoid receptor antagonists) or specific anti‐fibrotic drugs (e.g. pirfenidone). However, its resilience to therapy requires additional major efforts to control (and ideally prevent or reverse) fibrotic remodelling, being identified as a major contributor to heart failure progression.1, 2, 3, 4, 5, 6
While fibrosis is a widely used term, the exact definition is less precisely defined. Fibrosis in the broadest sense is defined as excessive accumulation of extracellular matrix (ECM). In simplified terms, fibrosis can be divided into (i) ‘reparative fibrosis’ and (ii) ‘reactive fibrosis’. The development of an organized scar after myocardial infarction (MI) can be best described as reparative or replacement fibrosis, which is necessary to mechanically stabilize the evolving (necrotic) tissue defect. In contrast, the fine interstitial ‘reactive fibrosis’ encountered in non‐ischaemic cardiomyopathies or in the surviving myocardium after MI appears to result from different pathological processes resulting in unique structural quality, ECM composition, and metabolic properties. Additionally, there is also a time component to scar development that has to be considered. For example, reactive fibrosis in the setting of pressure overload is initially characterized by perivascular fibrosis that later progresses to interstitial fibrosis. Fibrosis is also highly dynamic as it typically entails recruitment of fibroblasts and their conversion into myofibroblasts, excessive synthesis and secretion of ECM and ECM‐associated modulatory glycoproteins, posttranslational modification and cross‐linking of ECM proteins, and dysregulation of ECM production and breakdown by matrix metalloproteinases (MMPs) and their endogenous inhibitors (TIMPs). These different manifestations of fibrosis suggest that multiple targets or therapeutic opportunities may exist and that therapy may have to be personalized according to the diagnosis of specific remodelling processes and finally specific types of fibrosis.
Because todays' ‘one size fits all’ guideline approaches and broad heart failure patient classifications do not properly consider the different pathological processes that underlie fibrosis formation, it may not be a surprise that the outcomes of various studies are often contradictory and can only be rarely translated from one clinical setting to another. Also, as a consequence, fibrosis has not yet emerged as a primary target for heart failure therapies.
The Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC) organized a workshop on myocardial fibrosis with the aim to discuss and recommend strategies to address knowledge gaps in this field. This scientific roadmap paper summarizes the principal knowledge gaps that were identified, including the need for (i) more specific definitions of processes underlying the formation of fibrosis in heart failure under different pathological conditions, (ii) improved methods to detect fibrosis using imaging techniques and biomarkers associated with specific entities of fibrosis; and (iii) new therapies to directly target specific processes underlying cardiac fibrosis. Here, we provide a framework to better define fibrosis during various stages, aetiologies, and severities of heart failure. We propose a structured experimental scheme to assess fibrosis quality as well as quantity, and to provide a work‐up template that can be used in both translational and clinical research. Our goal is to direct future research to the identification of individual mechanisms of fibrosis formation, anticipating that this will provide insight into novel therapeutic targets and diagnostic tools for cardiac fibrosis stratification during heart failure progression.
More specific definitions are needed to describe the formation of myocardial fibrosis in heart failure
Myocardial fibrosis in heart failure is not a uniformly initiated process
Myocardial fibrosis is an endogenous, albeit suboptimal, repair response of the failing heart that can offer structural support while cardiomyocyte loss is occurring in the absence of appropriate cardiomyocyte replacement. Several key events characterize the fibrotic response to cardiac injury, which have been excellently reviewed elsewhere.1, 2, 3, 4, 5 Activation and conversion of fibroblasts into myofibroblasts are central events, critical also to non‐cardiac wound healing.1, 2, 3, 4, 5 Myofibroblasts produce and deposit ECM proteins, such as collagens, glycoproteins and proteoglycans (e.g. fibronectin, galectins, and periostin among many others), to offer local mechanic support to the failing heart. The formation of organized fibrotic structures and fibrils requires a multi‐step process that involves the degradation and processing of existing ECM to remove damaged tissue, and the production, secretion, cross‐linking, and maturation of new ECM. In addition to cardiac fibroblasts, which are the major source of ECM, monocytes and macrophages home to sites of injury and contribute to remodelling by secretion of pro‐fibrotic growth factors. Finally, cardiomyocytes also contribute to secretion of pro‐fibrotic growth factors into the ECM via paracrine mechanisms. In addition to fibrillary ECM constituents, non‐structural glycoproteins and proteoglycans are important accessory mediators of fibrosis. Glycosylation is a highly prominent post‐translational modification in ECM (reviewed by Rienks et al.6) and glycoproteomics is a novel tool with promise in the study of myocardial fibrosis.7, 8
Timely detection of fibrosis and determination of its state could potentially help to diagnose and stop heart failure progression early on. Capturing where fibrosis lies along the time continuum in a specific patient may inform physicians if fibrosis is developing as an early manifestation of the disease, and what type of targeted therapy could be employed. Early post‐MI therapy will most certainly differ from the therapy of non‐ischaemic diastolic dysfunction with a stiff left ventricle and even more from the treatment of end‐stage heart failure with an often severely fibrotic myocardium. The exact differences in disease states, including the identification of disease modulators, must be identified to improve and ideally establish an individualized therapy of heart failure‐related fibrosis.
Current classification of myocardial fibrosis
Traditionally, the form and stage of fibrosis have been denoted in line with the specific physiological phenomena that provoked the fibrotic response. Classically, the fibrotic process occurring after MI has been called ‘reparative’ or ‘replacement’ fibrosis. While scar formation is characterized by excessive accumulation of ECM, fibrosis is generally regarded as inevitable, as its absence would extend ventricular dilatation and could even result in ventricular rupture. Therefore, the infarct scar is a mandatory, albeit not perfect replacement, structural support. The post‐MI scar is the result of dramatic cardiomyocyte loss and the subsequent deposition of collagen fibrils that are cross‐linked to provide a strong ECM network. Reactive fibrosis, which is typically observed as perivascular or interstitial fibrosis, is stimulated by ongoing long‐run maladaptive signalling (e.g. by inflammatory cells, paracrine signals, and oxidative stress) that is part of progressive pathological cardiac remodelling. Figure 1 summarizes the different types of fibrosis which may all be observed in parallel in the same heart, making it a challenging exercise to target distinct fibrotic processes. It depicts an example of histology from a virtual cardiac tissue biopsy — it becomes instantly apparent that even within the tissue same sample, different forms of fibrosis may be present, and thus, the chances for sampling error in real life are very real. Some fibrotic manifestations will be ‘reactive’, some will have been ‘reparative’, some will be (or have become) static, and others dynamic. It is quite often not feasible to distinguish the various forms, as many features are shared and transition into one another is possible. Thus, identification of the specific and probably dominant type of fibrosis will be important for individualized anti‐fibrotic approaches.
Figure 1.
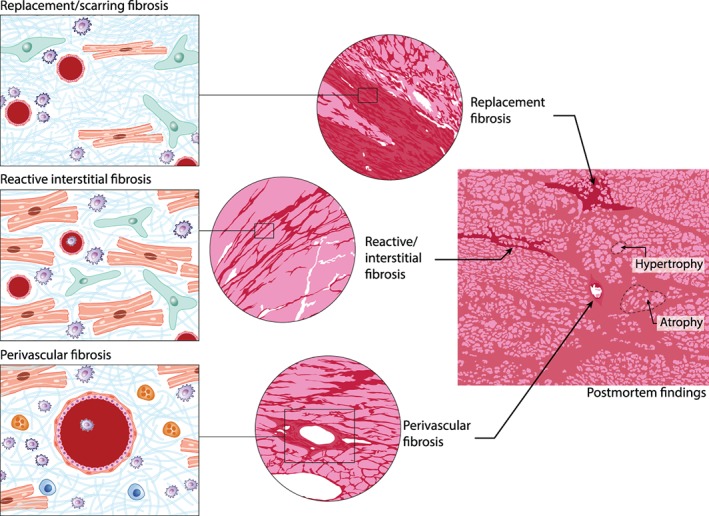
Different forms of fibrosis are not mutually exclusive. The left panels show replacement (upper panel), reactive interstitial (middle), and perivascular (lower) fibrosis, with different cells playing the major role: fibroblasts (green), inflammatory cells (blue), and myocytes (red), with fibrillar debris interpositioned. In reality, in a typical failing heart, all forms may occur (middle panel and right histology panels). (Illustration: Maartje Kunen, Medical Visuals.)
Understanding myocardial fibrosis in different phenotypes of heart failure
Fibrosis is frequently described in experimental heart failure in preclinical animal models. Given its complex pathophysiology, it is crucial in such studies to provide a minimum amount of information to allow the reader understanding what fibrosis is referred to. Further, the triggers, dynamics, and characteristics of the fibrotic process are very different among various aetiologies of heart failure. Below, we discuss cardiac fibrosis in the setting of MI, pressure overload, and aging (Figure 2), as well as genetic cardiomyopathies and heart failure with preserved ejection fraction (HFpEF).
Figure 2.
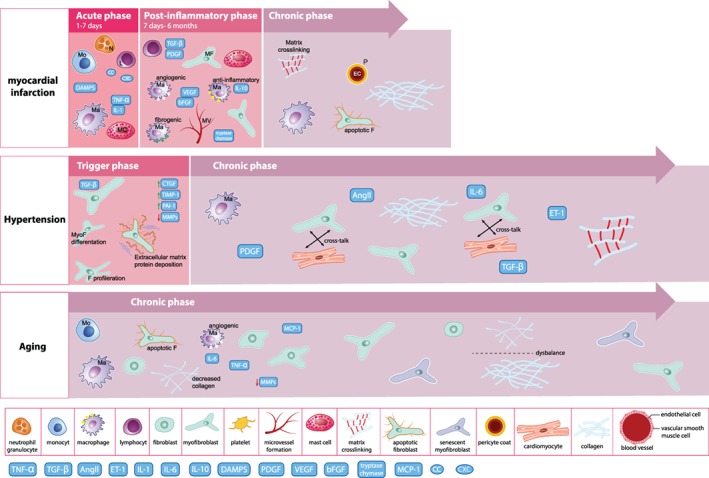
Graphical depiction of time‐dependent fibrosis formation in the heart after acute injury such as myocardial infarction, longstanding injury such as hypertension, and intrinsic tissue changes during aging and senescence. The aetiological factors underpinning fibrosis, as well as the (physiological) need for a fibrotic reparative response will dictate the extent and timing of the fibrotic process. (Illustration: Maartje Kunen, Medical Visuals.) AngII, angiotensin II; CTGF, connective tissue growth factor; DAMPS, danger‐associated molecular patterns; ET‐1, endothelin‐1; IL, interleukin; L, lymphocyte; Ma, macrophage; MC, mast cell; MCP‐1, monocyte chemoattractant protein‐1; MF/MyoF, myofibroblast; MMP, matrix metalloproteinase; MV, microvessel; N, neutrophil; PAI, plasminogen activator inhibitor; PDGF, platelet‐derived growth factor; TGF, transforming growth factor; TIMP, tissue inhibitor of metalloproteinase; TNF, tumour necrosis factor.
Myocardial fibrosis in post‐myocardial infarction
Myocardial infarction is one of the most common causes of heart failure. Several animal models have been developed to model human MI. Permanent ligation of a coronary artery induces a large transmural MI (rat, mouse, dog, sheep, pig), while transient ligation causes ischaemia–reperfusion damage (mouse, rat, dog, pig), with variable degrees of damage depending on duration of ischaemia, selection of coronary artery, location of the ligature, and pre‐treatment. MI causes a distinct tissue wound healing response with an initial strong inflammatory response, starting immediately after MI and peaking 3–7 days (depending on the species studied and model used) after MI. Neutrophils, monocytes, macrophages, but also fibroblasts themselves release factors that act on fibroblasts and trigger a pro‐fibrotic response to form the infarct scar. The controlled invasion of inflammatory cells is a prerequisite for proper infarct healing and prevention of myocardial rupture.9, 10, 11, 12, 13 After the initial phase, inflammation subsides and the proliferative phase starts, where fibroblasts convert into myofibroblasts, migrate and proliferate, resulting in an increased capacity for wound contraction and repair. Additionally, fibroblast progenitors as well as endothelial to mesenchymal transition are considered important post‐MI fibroblast sources. Collagen content begins to rise measurably 4–7 days after MI and peaks after 3–6 weeks, depending on the animal model used. Beside the amount of collagen, the type of collagen fibres formed and the degree of collagen cross‐linking affect the mechanical properties of the tissue. Finally, a maturation phase is reached, where a stable scar is formed. During this phase, it is unclear whether the reduction in ECM turnover is due to reduced matrix synthesis, increased ECM breakdown, or both.
The precise role of fibroblasts and the fibroblast cell sources in the post‐MI setting is incompletely understood, in part because of difficulties in labelling and identifying this cell type in vivo. Recently, fibroblast activating protein (FAP) was identified as a rather specific marker of activated, collagen‐synthesizing fibroblasts, whereas inactive fibroblasts, or fully differentiated myofibroblasts and non‐fibroblast cells in the infarct do not express FAP.11 Periostin is suggested as another marker of activated fibroblasts, suggestive for early fibroblast activation.14 Isolating post‐MI cardiac fibroblasts from an in vivo‐stimulated environment and evaluating these cells ex vivo has provided insight into their functional responses.12, 13
Myocardial fibrosis in models of pressure overload (e.g. hypertension)
The importance of fibrogenesis in pressure overload has been reviewed by Creemers and Pinto.3 Excessive myocardial ECM formation and collagen production take place in both human and experimental heart failure resulting from pressure overload, and collagen formation becomes disproportionate to left ventricular mass when the stress becomes chronic and sustained.3 Transforming growth factor (TGF)‐β is a central protein in the formation of pressure overload‐related fibrosis. It is activated by various circulating hormones such angiotensin II and endothelin‐1, but also by cellular stretch and strain. The TGF‐β pathway leads to activation of Smad2/3 and Rho/ROCK signalling, and activation of stress‐related kinases and proteins such as p38, ERK1/2 and elevated expression of connective tissue growth factor (CTGF). Fibroblasts in models of pressure overload have been identified as epicardial and endothelial cell‐derived and Pax3‐expressing cells (a major source under normal conditions and following pressure overload).15, 16 Premature senescence of myofibroblasts was identified as an essential anti‐fibrotic mechanism and potential therapeutic target in myocardial fibrosis in response to pressure overload.17
Aging
Aging is one of the key drivers of myocardial fibrosis (reviewed in18, 19, 20, 21, 22, 23, 24, 25). Animal models and human biopsy studies have demonstrated that collagen content of the heart progressively increase with advanced age, and collagen deposition is associated with increased wall stress, and with diastolic and systolic ventricular dysfunction. With aging, not only the production of collagen increases, but also the degradation becomes less effective.18, 20, 21 Also collagen processing and maturation is different, and cross‐linking seems to increase.18, 20, 21 The triggers for fibrosis in the aging heart are manifold, and, as a result, fibrosis may present in multiple forms. In response to cardiomyocyte injury and cell loss, replacement fibrosis may be seen. At the same time, with ongoing inflammation and age‐dependent increases in oxidative stress, interstitial fibrosis may occur. We must realize that age‐dependent fibrosis will usually develop alongside, so in concert with fibrosis that develops in response to cardiac injury, which complicates the understanding of what causes and then supports sustained fibrotic processes.
Myocardial fibrosis in (genetic) cardiomyopathies
Fibrosis in (mono‐) genetic cardiomyopathies can occur as fine interstitial fibrosis or replacement fibrosis, both due to structural changes in response to the gene defect. Therefore, the observation of fibrosis for instance on cardiac magnetic resonance imaging (MRI) is generally regarded as an early sign of the disease, even when systolic function is still normal.26, 27 Early fibrosis in cardiomyopathies is regarded as a malicious event as the need for cardiac repair usually is minimal. Clearly, the events triggering fibrosis in cardiomyopathies are very heterogeneous, and encompass events such as cell death, metabolic derangements, neurohormonal activation, and direct toxic effects of mutated proteins.28
Myocardial fibrosis in heart failure with preserved ejection fraction
Heart failure with preserved ejection fraction accounts for almost half of the cases of heart failure. Co‐morbidities, including aging, obesity, hypertension, and diabetes, are key factors for HFpEF progression into overt heart failure. Recent evidence suggests that in HFpEF the extent of myocardial fibrosis (as measured by T1‐MRI, see below) is related to the degree of diastolic dysfunction.29, 30 Clearly, pro‐fibrotic signals are diverse and differ from classical, systolic, heart failure signals. Fibrosis in HFpEF usually presents as perivascular and fine interstitial fibrosis and is associated with systemic inflammation.31 As a consequence, fibrosis in HFpEF will likely be multifaceted, with fibrosis due to aging, due to hypertension, and in response to inflammatory and metabolic (obesity) triggers, with occasional superimposed reparative fibrosis, in case of (small) MI or myocarditis. Clearly, the current call to better phenotype HFpEF resonates particularly for the understanding of fibrosis in this complex disease.31
The ‘chronic fibrotic response’ in heart failure
From the discussion above it becomes clear that in heart failure (and in fact in more chronic diseases), a sustained fibrotic response is observed, that initially may be reparative, but at some point, rather contributes to organ damage and failure. So, it seems that in certain forms and stages of heart failure the fibrotic response cannot be switched off, and that a certain degree of fibrogenesis remains persistent. This is different from physiological healing, where the termination of the reparative phase is identified by the disappearance of activated myofibroblasts from the tissue.32, 33
It is currently unknown how to differentiate the endogenous, necessary and beneficial fibrotic response or matrix turnover from the excessive, ongoing and harmful chronic fibrotic response that leads to matrix deposition and tissue stiffening.34 We postulate that these triggers that cause this chronic fibrotic response are multifold, including sustained fibroblast proliferation via feedback loops, cardiomyocyte‐mediated fibroblast activation, inhibition of myofibroblast apoptosis, and the presence of sustained low‐grade systemic and local inflammation.
Although mechanistically this remains largely a black box, several players have been recognized. As described above, TGF‐β plays a central role in fibroblast proliferation and fibroblast‐to‐myofibroblast conversion. TGF‐β is produced in high numbers by myofibroblasts to create a vicious cycle of myofibroblast activation. TGF‐β stimulates several growth factors (epidermal growth factor, insulin‐like growth factor‐1, growth differentiation factor‐11), which mediate proliferation of fibroblasts, involving autocrine signalling via fibroblast growth factor‐2 and/or CTGF.35, 36 TGF‐β also prevents myofibroblast apoptosis, via stimulation of PI3K/AKT pro‐survival signalling pathway.37 ‘Myofibroblast persistence’ may lead to non‐resolving and progressive fibrosis, as exemplified by human idiopathic pulmonary fibrosis.38 Experimental drugs targeting the TGF‐β and MAPK pathways indicate that the myofibroblast phenotype can be reversed, but whether this also can be achieved in vivo remains unclear.39, 40
The low‐grade, persistent systemic inflammation that is observed in heart failure41, 42, 43, 44 is a major driver of fibrosis. TGF‐β has pleiotropic effects on the immune system and has both immunosuppressive and pro‐inflammatory functions,44 and may polarize macrophages and neutrophils towards a M2 phenotype, which produces large quantities of inflammatory cytokines.
Experimental studies suggest that regulating the inflammatory and immunomodulatory response may be effective in reducing MI‐related remodelling, fibrosis and outcomes. For example, in a recent mouse study, neuregulin‐1, an epidermal growth factor family member released by cardiac endothelial cells, attenuated myocardial interstitial fibrosis by inhibiting activation of myocardial macrophages.45 Most compelling evidence form the preclinical field was generated for tumour necrosis factor (TNF)‐α inhibition43, 46 but surprisingly, clinical studies e.g. with steroids and TNF‐α blockers showed no or even detrimental effects, so that initial translation of experimental observations to clinical medicine has failed,44 suggesting that broad targeting of the immune system will not be a useful therapeutic strategy. Of interest, the recent CANTOS trial showed that targeted inhibition of inteleukin‐1α did reduce cardiovascular (and cancer) outcomes, albeit to a small degree.47
Proposal for a minimum assessment profile to screen cardiac fibrosis
The term ‘myocardial fibrosis’ needs to be specified with more precision and potentially individualized to offer effective therapeutics to patients with heart failure and cardiac fibrosis. For experimental studies, we propose a minimum set of parameters that should be provided to allow readers to appreciate the nuances of the fibrosis phenotype present. These items are listed in Table 1. In Table 2, we summarize the key functions, disease conditions, analytical methods and biomarkers in different forms of fibrosis. These insights are crucial to identify therapeutic opportunities for various subtypes of fibrosis‐induced heart failure.
Table 1.
Proposed minimal dataset to describe fibrosis in animal studies
| Parameter | Example(s) |
|---|---|
| Species |
Mouse, rat, sheep, pig Precise reporting of strain, genetic background, age |
| Perturbation |
Pressure overload, MI by permanent LAD ligation or ischaemia/reperfusion injury, diet, salt loading Precise reporting of duration of intervention and period of ischaemia and pressure overload |
| Time course | Reporting the time course of disease progression, with samples taken before and at several time points [acute, subacute (days) and chronic (months)] post‐disease induction |
| Assessments | |
| Histology |
For instance: Masson, Picrosirius red Percentage of LV tissue affected, sampled ROIs; Use of validated antibodies for immune histology
|
| Inflammation glycoproteins‐proteoglycans in the heart at RNA and protein level |
‐ Acute vs. chronic process (duration of disease)? Glycoproteins/proteoglycans:
‐ Quantification of inflammation (myeloperoxidase, CD45‐ and CD68‐staining leucocytes). ‐ Collagen crosslinking enzymes (LOX's) ‐ MMP/TIMPs at transcript level and zymography |
| Blood biomarkers |
Galectin‐3 CITP PIIINP ST2 |
| Imaging | MRI (T1 mapping, late enhancement fibrosis) |
| Functional analyses | Echocardiography and invasive haemodynamics for determining load‐dependent diastolic and systolic function |
CITP, C‐terminal propeptide of procollagen type I; CTGF, connective tissue growth factor; LAD, left anterior descending artery; LOX, lysyl oxidase; LV, left ventricular; MI, myocardial infarction; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; PIIINP, procollagen type III N‐terminal propeptide; ROI, region of interest; TGF, transforming growth factor; TIMP, tissue inhibitor of metalloproteinases.
Table 2.
Overview of key functions, conditions, analytical methods and biomarkers in different forms of fibrosis
| Type of fibrosis | Function | Disease conditions | Methods to detect in tissue | Blood biomarkers |
|---|---|---|---|---|
| Reparative fibrosis | Replacing necrotic cells | Myocyte necrosis (ischaemia, infection, autoimmunity, toxicity, gene mutations) |
Histochemistry (Sirius red and Azan blue) Cardiac MRI, late enhancement |
CITP, PICP PINP, PIIINP |
| Reactive fibrosis | New matrix production in between cells | Matrix production in response to an acquired or genetic trigger, such as stretch |
Histochemistry Cardiac MRI, T1 mapping of extracellular volume |
CITP, PICP, Galectin‐3, sST2, and periostin |
| Perivascular fibrosis | New matrix produced around vessels | Perivascular fibrosis upon pressure overload or vascular stress | Histochemistry | Unknown |
| Cells involved | ||||
| (Myo)fibroblasts | Reparative and reactive | Most diseased hearts | Staining of periostin, FAP, SMA | Periostin, mimecan, SPARC |
| Smooth muscle cells | Perivascular and reactive | Pressure overload, vasculitis | Staining of SMA | |
| Inflammatory cells | Reparative and reactive | Acute and chronic cardiac disease | Staining of macrophages, monocytes, neutrophils (CD45) | sST2, CRP, galectin‐3 |
| Content/mechanisms | ||||
| Collagen production | Structural proteins | Healthy and diseased heart | Immunostaining, rtPCR | CITP, PICP, PINP, PIIINP |
| Glycoproteins/proteoglycans | Inter‐cellular communication | Most diseased hearts | Immunostaining, rtPCR | Galectin‐3, sST2, periostin, mimecan, SPARC |
| MMP and their inhibitors (TIMPs) | Collagen degradation/production balance | Healthy and diseased heart | Immunostaining and activity assays, rtPCR | MMP‐1 and ‐9, TIMP‐1 and ‐2 |
| Growth factors: TGF, CTGF, FGF, Wnt pathways | Stimulate collagen production, affects inflammation | Most diseased heart | Immunoblots, signalling pathways | ND |
CITP, C‐terminal propeptide of procollagen type I; CRP, C‐reactive protein; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; PINP, amino‐terminal propeptide of type I collagen; PIIINP, procollagen type III N‐terminal propeptide; PINP, procollagen type I N‐terminal propeptide; rtPCR, reverse transcriptase‐polymerase chain reaction; sST2, soluble ST2; TIMP, tissue inhibitor of metalloproteinases.
Improved methods are needed to detect fibrosis using biomarkers and imaging techniques
Detection of myocardial fibrosis is not straightforward in animal models and is even less so in the clinical setting where myocardial tissue sampling is not readily available. The gold standard for human studies is to assess the quality (i.e. focal, interstitial or perivascular distribution) and to quantify fibrosis in myocardial tissue biopsies using histological techniques (i.e. Masson's Trichrome or Sirius Red histochemical staining). Although endomyocardial biopsies (EMBs) are limited by sampling error and small tissue fragments, the study of explanted hearts for heart transplantation offers unique possibilities with regard to spatiotemporal histological analyses and modern ‐omics techniques, not hampered by lack of tissue. We aware of several (national) initiatives, where all explanted hearts will be archived centrally to ensure proper sample size (so‐called heart banks), and we foresee these will generate valuable information on fibrosis as well. But to date, tissue studies are mostly still carried out on EMBs, requiring an invasive procedure with the associated risk for complications and sampling errors.48 Thus, alternative, non‐invasive and ideally equally or even more reliable methods should be developed and broadly applied.
Circulating biomarkers of myocardial fibrosis
Extracellular matrix proteins or cleaved processing products are often released into the systemic circulation and therefore measurable in serum or plasma using reliable and approved methods (e.g. ELISAs). Commonly used fibrosis biomarkers give insight into collagen production [e.g. procollagen type I N‐terminal propeptide, procollagen type III N‐terminal propeptide (PIIINP)] or the secretion of non‐structural (glyco)proteins that modulate the collagen production itself or its maturation (e.g. periostin, mimecan, monocyte chemoattractant protein‐1, or galectin‐3). In addition, several MMPs (e.g. MMP‐3, MMP‐9, MMP‐11, and MMP‐12) and their tissue inhibitors (e.g. TIMP‐1 and TIMP‐3) involved in the balance of collagen degradation, are released into the blood stream and can be measured reliably. In fact, there is a wide body of literature on the potential utility of these markers alone or in concert.49 In general, many of these markers are valuable for clinical risk prediction. Interestingly, several factors have been shown to predict the response to treatment with anti‐fibrotic properties, such as mineralocorticoid receptor antagonists.50 However, the use of these markers has not become a clinical standard because of limited power in fully adjusted models with clinical variables, and because of technical difficulties in measuring the proteins, often requiring laborious and expensive radioimmunoassays. Recently, several new emerging fibrotic markers have been studied, including galectin‐3, sST2, and periostin.51 These markers can generally be measured with Food and Drug Administration‐cleared ELISA assays which allow fast turnaround times.
Circulating levels of fibrosis markers may not parallel findings in histologically proven cardiac fibrosis and therefore caution is required in the interpretation of systemic venous circulating biomarker levels in relation to myocardial disease. López et al.52 measured circulating levels of many fibrotic markers and correlated these in the same patients to local cardiac tissue fibrosis volumes measured with histology. The results show that out of 28 potential biomarkers associated with build‐up or breakdown of myocardial fibrosis, only C‐terminal propeptide of procollagen type I (CITP) and PIIINP correlated with histological findings. This may be explained because in heart failure patients, several other organs undergo fibrotic changes as well: liver, lungs, kidneys, and vessels. A recent article by Du and colleagues showed that galectin‐3, growth differentiation factor‐15, and TIMP‐1 plasma levels do not reflect myocardial fibrosis in mouse models of post‐MI heart failure, hypertensive heart failure, and HFpEF.53 Instead, production in extracardiac tissues such as fatty and lung tissue had much greater impact on plasma levels of these markers. So ideally, we would need a marker that is specific for cardiac fibrosis, or at the very least, adequately reflects changes in myocardial fibrosis. However, since most fibrotic pathways are shared amongst organs, such a marker may not exist and it is therefore simply impossible to use circulating fibrosis markers as a perfect surrogate for myocardial fibrosis (Figure 3). Further, the markers generally do no clearly distinguish between various forms of fibrosis, or between the trigger that causes fibrosis. Therefore, the use of these factors for precision diagnostics is questionable, but excess local production however may be used to target specific treatment. Further, the potential of fibrotic markers as surrogate outcomes in phase I/II trials is likely to be limited at this point, in view of the limited specificity of available fibrotic (bio‐)markers.
Figure 3.
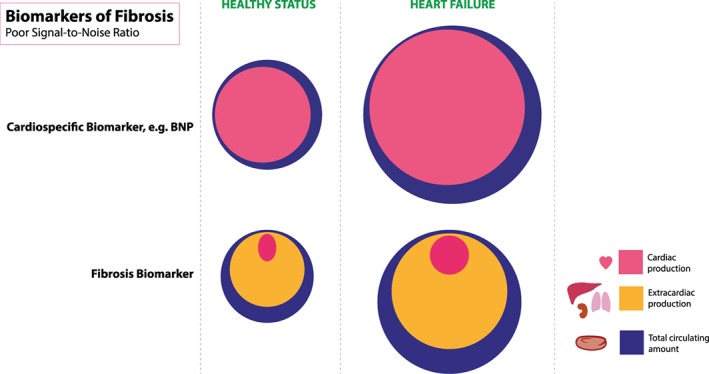
Systemic biomarkers, measured in the plasma of patients with heart failure, ideally reflect changes in the heart muscle. For cardio‐specific biomarkers, such as natriuretic peptides and troponins, this is very accurate. However, for many (more novel) markers that are expressed by many organs outside the heart as well, the systemic levels only marginally reflects cardiac production. BNP, B‐type natriuretic peptide. (Illustration: Maartje Kunen, Medical Visuals.)
Imaging techniques to visualize and quantify fibrosis
The most widely applied imaging technique in contemporary heart failure management is echocardiography. Classical two‐dimensional echocardiography, however, provides little information about the presence or extent of fibrosis. More modern techniques such as tissue velocity imaging and global longitudinal systolic strain provide mechanical tissue details that associate with myocardial fibrosis by biopsy examination.54, 55, 56 In addition to echocardiography, cardiac MRI is a technique that is replacing EMB as gold standard for human cardiac fibrosis identification and quantification. Clearly, the use of cardiac MRI cannot fully replace echocardiography as first‐choice imaging modality, given the high costs. But in order to perform in deep phenotyping, with the aim to choose patients with specific pathophysiological characteristics, or to monitor these during (drug) treatment, cardiac MRI has distinct benefits. Using delayed gadolinium enhancement, it is possible to visualize scar tissue, for instance after transmural myocardial infarction. While delayed gadolinium enhancement mainly identifies focal reparative fibrosis, modern techniques in cardiac MRI are developing that may be able to provide more granularity in imaging fibrosis. Most interestingly, T1 mapping is such an emerging cardiac MRI technique, measuring the longitudinal relaxation time of individual protons, which is depicted as a pixelated map. T1 mapping allows the quantification of extracellular volume (ECV) fraction of the myocardium. ECV is not a pure measure of fibrosis, although it has been evaluated to this aim,57, 58 but ECV rather mirrors diffuse changes including fibrosis, but also interstitial oedema, protein degradation and aggregation, lipid accumulation, and deposition of iron or amyloid. A recent HFA position paper discusses in detail the (fast) developments in imaging techniques.59 Furthermore, the presence and the extent of MRI‐proven fibrosis have been related to poor clinical outcomes.5 Collectively, different imaging (MRI) techniques may be applied as surrogate measures for the presence and extent of myocardial fibrosis. The next challenge is to develop a therapeutic plan to reverse or prevent further development of fibrosis based on the cardiac magnetic resonance findings. A promising approach would be to label specific molecules with established relation to myocardial fibrosis, and image those, e.g. by nuclear techniques, with the ultimate aim to target this specifically.60
New therapies are needed to directly target myocardial fibrosis with specific drugs
It has been proposed for several years that direct targeting fibrosis might be useful in heart failure.1, 61, 62 Angiotensin‐converting enzyme inhibitors and mineralocorticoid receptor antagonists reduce fibrosis formation, but clearly a residual fibrotic burden remains and with that, a potential need to target it in order to improve outcomes. It is crucial to ascertain where the fibrotic process is, at a given time point, what the triggers are, and which cells and proteoglycans play a role. The complexity of targeting fibrosis is illustrated by a recent article by Clarke and colleagues,62 providing an example for why MMP inhibition may not be as effective as previously hypothesized. They postulate that early during the remodelling phase, MMP inhibition might be less effective because there is little collagen to degrade, while at later fibrotic phases it is less effective because MMP levels have fallen to low levels. Therefore, the setting, the aetiology and the timing, all appear very important in MMP inhibition and this could in part explain the disappointing results thus far. Next, we will discuss a few novel options that are on the horizon.
Connective tissue growth factor and pirfenidone
Inhibition of fibrosis formation in pressure overloaded heart is best achieved by alleviating the primary stressor,3 i.e. the elevated pressure. TGF‐β (e.g. pirfenidone), angiotensin or endothelin receptor blockers, ERK inhibitors or inhibition of CTGF are being tested as specifically fibrosis targeted treatments. Different from post‐MI fibrosis, it appears that inhibition of the excess fibrosis in pressure overload is generally safe and well‐tolerated.
Connective tissue growth factor (or CCN2) is a matricellular protein and modulates the signalling of many cytokines and ECM signals including those of TGF‐β, bone morphogenetic protein, Wnt, vascular endothelial growth factor, and integrins. Because it modulates multiple pathways simultaneously, and via several different mechanisms, the effects of CTGF are combinatorial and context‐dependent (i.e. dependent on the environment and mediators present), and therefore, the biology of CTGF is very complex.63 CTGF expression is induced by many different pathophysiological insults, and when it becomes overexpressed, it helps promote differentiation of cells to become activated myofibroblasts that deposit and remodel the ECM. CTGF is involved in multiple positive feedback loops that can propagate tissue remodelling and fibrosis, and therefore it should be considered a central mediator of fibrosis. Consequently, the goal of inhibiting CTGF is to disrupt these positive feedback loops and arrest the progressive nature of fibrosis (and possibly reverse it). A human monoclonal antibody, FG‐3019, that binds to CTGF and interferes with its activity has proven beneficial in rodent models of MI (unpublished), neonatal (rat) bronchopulmonary dysplasia,64 and thoracic aorta constriction model.65 Clinical testing of FG‐3019 indicates an excellent safety profile and has been tested in approximately 400 patients with diabetic kidney disease, pancreatic cancer, idiopathic pulmonary fibrosis, or liver fibrosis.
Pirfenidone is a synthetic molecule that has been reported to decrease the expression of various pro‐fibrotic factors, including TGF‐β1, TNF‐α, platelet derived growth factor and collagen.66 Results from experimental models provided evidence for a therapeutic utility of pirfenidone in pressure overload67, 68, 69 and hypertension,68 leading to a reduction in arrhythmogenic substrate.70
Matrix metalloproteinases
The effects of regulating MMP activity on cardiac fibrosis and left ventricular remodelling outcomes have primarily been assessed in MI models. Targeted deletion and transgenic mice or MMP inhibitors (MMPi) reveal both beneficial and detrimental consequences.71 While early promises of MMPi in animal models were encouraging, these findings have not translated to humans. This has been due to selectivity and specificity issues, as well as our lack of understanding of the full range of MMP functions.71 For example, the MMP‐9 substrate list includes hundreds of substrates ranging from collagen and fibronectin, interleukin‐1β, pro‐enzymes and citrate synthase.72, 73 Further, not all MMPi have been beneficial, as MMP‐12i given at 3 h post‐MI suppressed neutrophil apoptosis to prolong inflammation, resulting in exacerbated left ventricular dilatation.74 MMP‐28 deletion inhibited M2 anti‐inflammatory macrophage activation to stimulate left ventricular dysfunction and increase cardiac rupture rates.75 If anything, these results indicate how complex the fibrotic process in heart failure is, and the specific role of MMPs herein. There is a need, therefore, to delineate individual MMP roles under specific conditions and times.
Galectin‐3 inhibitors
Galectin‐3 is a lectin binding galactoside, and has been shown to be upregulated in heart failure by myofibroblasts, monocytes and macrophages that are recruited towards sites of injury and fibrosis.76, 77 Studies in mice deficient for galectin‐3 have suggested that galectin‐3 is not a bystander but rather a culprit for myocardial fibrosis.78, 79 Inhibition of galectin‐3, either achieved with large carbohydrates,78, 79 or antisense RNA,80 or small designer molecules,81 effectively reduces organ fibrosis.82
Non‐coding RNAs
Recently, non‐coding RNA (both microRNA and long non‐coding RNAs‐based)‐based treatment strategies for fibrosis have been put forward.83, 84 Specific non‐coding RNAs seem to play crucial roles in the regulation of the cardiac fibroblast phenotype and their modulation seem to be effective both in animal as well as clinical studies; indeed there is currently a phase II trial in patients with kidney fibrosis using an inhibitor of microRNA‐21 (http://regulusrx.com/programs/pipeline/). Thus, non‐coding RNA‐based treatment approaches might provide an opportunity also for the treatment of cardiac fibrosis and remodelling, if those microRNA inhibitors could be selectively delivered in the heart — miRNA‐21 is ubiquitously expressed. Other microRNAs, such as miRNA‐29b, appear to have a more cardio‐specific effect.85
Future directions and conclusions
In this article we discuss several challenges and requirements in the study of fibrosis (Table 3). For future applications, strategies that allow for differentiation of fibrosis subtype are needed. Preclinical studies in relevant animal models to better understand the dynamics of fibrosis formation, degradation, and their importance for the functional phenotype are therefore required. More advanced in vitro models might allow a better functional control and mechanistic understanding. Whereas further optimizations are needed to implement human cardiac disease parameters,86 these well controlled environments allow longitudinal evaluations and screening of anti‐fibrotic strategies. Accumulating data linking fibrosis to a stronger inflammatory response are at the initial phases. At later time points, fibrosis‐specific mediators and pathways (predominantly fibroblast‐specific factors such as TGF‐β, osteopontin, and galectins) contribute to the progression of fibrosis, and are distinct from the mechanisms driving inflammation. The presence of co‐morbidities should be considered — e.g. it has recently been discussed that co‐morbidities such as cancer may obscure the biomarkers' signals.87 Thus, to design effective therapeutics for fibrotic disease, inflammation triggering fibrosis is to be considered, and the challenge ahead is to target specific molecules and pathways that act on fibrosis (specific interleukins) while leaving the (often beneficial) effects of the inflammatory response uninhibited. A vast diversity of inflammatory, immunological, and molecular mechanisms collectively contribute to cardiac fibrosis: the complex interplay between adaptive immune system activation, fibroblasts‐to‐myofibroblast conversion and proliferation, mast cell activation, neutrophil influx, and production, modulation, maturation and apposition of collagens, the embedding in the extracellular milieu. These should all be considered and taken into account during the design and testing of new anti‐fibrotic therapies.
Table 3.
Key recommendations
| Challenges | Requirements |
|---|---|
| Improvement and refinement in describing fibrosis |
|
| Improvement in detecting fibrosis |
|
| Targeting fibrosis |
|
Clinically, disease‐specific (bio‐)markers and imaging modalities — acting as surrogate parameters of specific temporal stages of fibrosis — will help to identify patients who might benefit from a specific therapy. Until recently, attempts to inhibit fibrosis have been mostly focusing on single pro‐fibrotic factors. Since fibrosis is driven and sustained by the activation of multiple interconnecting and intercommunicating pro‐fibrotic pathways, a multi‐target approach will likely help to slow down the progression of fibrosis. Using systems biology approaches and multi‐omics technologies to understand network signalling will aid in these efforts. Ultimately, a concerted anti‐fibrotic strategy that collectively targets important inflammatory signalling molecules, pro‐fibrotic cytokines, and cellular functions should be considered in developing therapies to adequately treat fibrosis.
In conclusion, in this position paper we have summarized the current knowledge gaps in the myocardial fibrosis field and provided templates for assessing fibrosis in both translational and clinical studies.
Funding
R.A.d.B. is supported by the Netherlands Heart Foundation (CVON DOSIS, grant 2014‐40, CVON SHE‐PREDICTS‐HF, grant 2017‐21, and CVON RED‐CVD, grant 2017‐11); and the Innovational Research Incentives Scheme program of the Netherlands Organization for Scientific Research (NWO VIDI, grant 917.13.350). J.B. is supported is supported by the Deutsche Forschungsgemeinschaft (DFG), Clinical Research Group 311 (KFO 311) ‘(Pre)terminal heart and lung failure: unloading and repair’ (DFG; TP1, BA 1742/9‐1) and ‘MR‐Focus’‘ (DFG BA 1742/8‐1). J.S. has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (consolidator grant Evicare #725229) and by the Netherlands Heart Foundation (CVON‐HUSTCARE). M.M. is a BHF Chair Holder (CH/16/3/32406), with BHF program grant support (RG/16/14/32397), and was awarded a BHF Special Project grant to participate in the ERA‐CVD Translational Grant MacroERA. M.M. and T.T. are members of a network funded by the Foundation Leducq. C.G.T. is supported by a Federico II University/Ricerca di Ateneo grant. S.H. has received funding from the European Union Commission's Seventh Framework programme under grant agreement n. 305507 (HOMAGE), n. 602904 (FIBROTARGETS) and FP7‐Health‐2013‐Innovations‐1 n. 602156 (HECATOS), CVON2016‐Early HFPEF, 2015‐10, and CVON SHE‐PREDICTS‐HF, grant 2017‐21. C.M. is supported by the DFG (Ma 2528/7‐1, SFB 894, TRR‐219) and the Federal Ministry of Education and Science (BMBF; 01EO150, CF.3, RC2).
Conflict of interest: The UMCG, which employs R.A.d.B., has received research grants and/or fees from AstraZeneca, Abbott, Bristol‐Myers Squibb, Novartis, Roche, Trevena, and ThermoFisher GmbH. R.A.d.B. is a minority shareholder of scPharmaceuticals, Inc.; received personal fees from MandalMed Inc, Novartis, and Servier. J.B. received honoraria for lectures and/or consulting from Novartis, Pfizer, Vifor, Bayer, Servier, Orion, CVRx, Abiomed, Abbott, Medtronic, and research support from Zoll, CVRx, Bayer, Vifor, Abiomed, Medtronic. T.T. has filed and licensed patents in the field of noncoding RNAs; is founder of Cardior Pharmaceuticals GmbH. A.R.L. reports grants from Servier and Pfizer, and speaker fees, advisory board fees and/or consultancy fees from Servier, Pfizer, Novartis, Roche, Takeda, Boehringer Ingelheim, Amgen, Clinigen Group, Ferring Pharmaceuticals, Bristol Myers Squibb, Eli Lily and Janssens‐Cilag Ltd. C.G.T. has a patent issued in the field of heart failure, and reports speaker fees from Alere. C.M. is a consultant to Servier and has received speaker honoraria from Bayer, Bristol‐Myers Squibb, Boehringer Ingelheim, Berlin Chemie, Daiichi Sankyo, Novartis, Pfizer and Servier. The other authors report no conflicts of interest.
References
- 1. Heymans S, González A, Pizard A, Papageorgiou AP, López‐Andrés N, Jaisser F, Thum T, Zannad F, Díez J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur J Heart Fail 2015;17:764–771. [DOI] [PubMed] [Google Scholar]
- 2. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014;71:549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure‐overloaded heart. Cardiovasc Res 2011;89:265–272. [DOI] [PubMed] [Google Scholar]
- 4. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 2012;18:1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piek A, de Boer RA, Silljé HH. The fibrosis‐cell death axis in heart failure. Heart Fail Rev 2016;21:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S. Myocardial extracellular matrix: an ever‐changing and diverse entity. Circ Res 2014;114:872–888. [DOI] [PubMed] [Google Scholar]
- 7. Lindsey ML, Mayr M, Gomes AV, Delles C, Arrell DK, Murphy AM, Lange RA, Costello CE, Jin YF, Laskowitz DT, Sam F, Terzic A, Van Eyk J, Srinivas PR. American Heart Association Council on Functional Genomics and Translational Biology, Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular and Stroke Nursing, Council on Hypertension, and Stroke Council. Transformative impact of proteomics on cardiovascular health and disease: a scientific statement from the American Heart Association. Circulation 2015;132:852–872. [DOI] [PubMed] [Google Scholar]
- 8. Barallobre‐Barreiro J, Gupta SK, Zoccarato A, Kitazume‐Taneike R, Fava M, Yin X, Werner T, Hirt MN, Zampetaki A, Viviano A, Chong M, Bern M, Kourliouros A, Domenech N, Willeit P, Shah AM, Jahangiri M, Schaefer L, Fischer JW, Iozzo RV, Viner R, Thum T, Heineke J, Kichler A, Otsu K, Mayr M. Glycoproteomics reveals decorin peptides with anti‐myostatin activity in human atrial fibrillation. Circulation 2016;134:817–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frantz S, Hofmann U, Fraccarollo D, Schäfer A, Kranepuhl S, Hagedorn I, Nieswandt B, Nahrendorf M, Wagner H, Bayer B, Pachel C, Schön MP, Kneitz S, Bobinger T, Weidemann F, Ertl G, Bauersachs J. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J 2013;27:871–881. [DOI] [PubMed] [Google Scholar]
- 10. Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S. Neutrophils orchestrate post‐myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 2017;38:187–197. [DOI] [PubMed] [Google Scholar]
- 11. Tillmanns J, Hoffmann D, Habbaba Y, Schmitto JD, Sedding D, Fraccarollo D, Galuppo P, Bauersachs J. Fibroblast activation protein alpha expression identifies activated fibroblasts after myocardial infarction. J Mol Cell Cardiol 2015;87:194–203. [DOI] [PubMed] [Google Scholar]
- 12. McCurdy SM, Dai Q, Zhang J, Zamilpa R, Ramirez TA, Dayah T, Nguyen N, Jin YF, Bradshaw AD, Lindsey ML. SPARC mediates early extracellular matrix remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol 2011;301:H497–H505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol 2014;70:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SH. Origin of cardiac fibroblasts and the role of periostin. Circ Res 2009;105:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Müller AM, Volz KS, Tang Z, Red‐Horse K, Ardehali R. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res 2014;115:625–635. [DOI] [PubMed] [Google Scholar]
- 16. Moore‐Morris T, Tallquist MD, Evans SM. Sorting out where fibroblasts come from. Circ Res 2014;115:602–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J Am Coll Cardiol 2016;67:2018–2028. [DOI] [PubMed] [Google Scholar]
- 18. Burlew BS. Diastolic dysfunction in the elderly‐‐the interstitial issue. Am J Geriatr Cardiol 2004;13:29–38. [DOI] [PubMed] [Google Scholar]
- 19. Singh M, Foster CR, Dalal S, Singh K. Role of osteopontin in heart failure associated with aging. Heart Fail Rev 2010;15:487–494. [DOI] [PubMed] [Google Scholar]
- 20. Susic D, Frohlich ED. The aging hypertensive heart: a brief update. Nat Clin Pract Cardiovasc Med 2008;5:104–110. [DOI] [PubMed] [Google Scholar]
- 21. Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev 2010;15:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dai DF, Chen T, Johnson SC, Szeto H, Rabinovitch PS. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal 2012;16:1492–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loffredo FS, Nikolova AP, Pancoast JR, Lee RT. Heart failure with preserved ejection fraction: molecular pathways of the aging myocardium. Circ Res 2014;115:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sorriento D, Franco A, Rusciano MR, Maione AS, Soprano M, Illario M, Iaccarino G, Ciccarelli M. Good at heart: preserving cardiac metabolism during aging. Curr Diabetes Rev. 2015;12:90–99. [DOI] [PubMed] [Google Scholar]
- 25. Horn MA, Trafford AW. Aging and the cardiac collagen matrix: novel mediators of fibrotic remodelling. J Mol Cell Cardiol 2016;93:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Almaas VM, Haugaa KH, Strøm EH, Scott H, Smith HJ, Dahl CP, Geiran OR, Endresen K, Aakhus S, Amlie JP, Edvardsen T. Noninvasive assessment of myocardial fibrosis in patients with obstructive hypertrophic cardiomyopathy. Heart 2014;100:631–638. [DOI] [PubMed] [Google Scholar]
- 27. Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TD, Ismail NA, Dweck MR, Di Pietro E, Roughton M, Wage R, Daryani Y, O'Hanlon R, Sheppard MN, Alpendurada F, Lyon AR, Cook SA, Cowie MR, Assomull RG, Pennell DJ, Prasad SK. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013;309:896–908. [DOI] [PubMed] [Google Scholar]
- 28. Bondue A, Arbustini E, Bianco AM, Ciccarelli M, Dawson D, De Rosa M, Hamdani N, Hilfiker‐Kleiner D, Meder B, Leite Moreira A, Thum T, Gabriele Tocchetti C, Varricchi G, Van der Velden J, Walsh R, Heymans S. Complex roads from genotype to phenotype in dilated cardiomyopathy: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res 2018;114:1287–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Su MY, Lin LY, Tseng YH, Chang CC, Wu CK, Lin JL, Tseng WY. CMR‐verified diffuse myocardial fibrosis is associated with diastolic dysfunction in HFpEF. JACC Cardiovasc Imaging 2014;7:991–997. [DOI] [PubMed] [Google Scholar]
- 30. Rommel KP, von Roeder M, Latuscynski K, Oberueck C, Blazek S, Fengler K, Besler C, Sandri M, Lücke C, Gutberlet M, Linke A, Schuler G, Lurz P. Extracellular volume fraction for characterization of patients with heart failure and preserved ejection fraction. J Am Coll Cardiol 2016;67:1815–1825. [DOI] [PubMed] [Google Scholar]
- 31. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype‐specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation 2016;134:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 2003;4:500–503. [DOI] [PubMed] [Google Scholar]
- 33. Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest 2018;128:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol 2018;71:1696–1706. [DOI] [PubMed] [Google Scholar]
- 35. Strutz F, Zeisberg M, Renziehausen A, Raschke B, Becker V, van Kooten C, Muller G. TGF‐beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF‐2). Kidney Int 2001;2:579–592. [DOI] [PubMed] [Google Scholar]
- 36. Grotendorst GR, Rahmanie H, Duncan MR. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J 2004;3:469–479. [DOI] [PubMed] [Google Scholar]
- 37. Kulasekaran P, Scavone CA, Rogers DS, Arenberg DA, Thannickal VJ, Horowitz JC. Endothelin‐1 and transforming growth factor‐beta1 independently induce fibroblast resistance to apoptosis via AKT activation. Am J Respir Cell Mol Biol 2009;4:484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding Q, Luckhardt T, Hecker L, Zhou Y, Liu G, Antony VB, de Andrade J, Thannickal VJ. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis. Drugs 2011;8:981–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Driesen RB, Nagaraju CK, Abi‐Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc Res 2014;3:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kosla J, Dvorakova M, Dvorak M, Cermak V. Effective myofibroblast dedifferentiation by concomitant inhibition of TGF‐beta signaling and perturbation of MAPK signaling. Eur J Cell Biol 2013;12:363–373. [DOI] [PubMed] [Google Scholar]
- 41. Weber KT, Sun Y, Díez J. Fibrosis: a living tissue and the infarcted heart. J Am Coll Cardiol 2008;52:2029–2031. [DOI] [PubMed] [Google Scholar]
- 42. Oikonomou E, Tousoulis D, Siasos G, Zaromitidou M, Papavassiliou AG, Stefanadis C. The role of inflammation in heart failure: new therapeutic approaches. Hellenic J Cardiol 2011;1:30–40. [PubMed] [Google Scholar]
- 43. Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail 1996;3:243–249. [DOI] [PubMed] [Google Scholar]
- 44. Suthahar N, Meijers WC, Silljé HHW, de Boer RA. From inflammation to fibrosis‐molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep 2017;14:235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vermeulen Z, Hervent AS, Dugaucquier L, Vandekerckhove L, Rombouts M, Beyens M, Schrijvers DM, De Meyer GRY, Maudsley S, De Keulenaer GW, Segers VFM. Inhibitory actions of the NRG‐1/ErbB4 pathway in macrophages during tissue fibrosis in the heart, skin, and lung. Am J Physiol Heart Circ Physiol 2017;313:H934–H945. [DOI] [PubMed] [Google Scholar]
- 46. Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically relevant concentrations of tumor necrosis factor‐alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation 1998;97:1382–1391. [DOI] [PubMed] [Google Scholar]
- 47. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJ, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida‐Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PR, Troquay RP, Libby P, Glynn RJ; CANTOS Trial GroupAntiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 48. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, Virmani R. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. Eur Heart J 2007;28:3076–3093. [DOI] [PubMed] [Google Scholar]
- 49. Zannad F, Rossignol P, Iraqi W. Extracellular matrix fibrotic markers in heart failure. Heart Fail Rev 2010;15:319–329. [DOI] [PubMed] [Google Scholar]
- 50. Iraqi W, Rossignol P, Angioi M, Fay R, Nuée J, Ketelslegers JM, Vincent J, Pitt B, Zannad F. Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: insights from the Eplerenone Post‐Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) study. Circulation 2009;119:2471–2479. [DOI] [PubMed] [Google Scholar]
- 51. de Boer RA, Daniels LB, Maisel AS, Januzzi JL Jr. State of the art: newer biomarkers in heart failure. Eur J Heart Fail 2015;17:559–669. [DOI] [PubMed] [Google Scholar]
- 52. López B, González A, Ravassa S, Beaumont J, Moreno MU, San José G, Querejeta R, Díez J. Circulating biomarkers of myocardial fibrosis: the need for a reappraisal. J Am Coll Cardiol 2015;65:2449–2456. [DOI] [PubMed] [Google Scholar]
- 53. Du W, Piek A, Schouten EM, van de Kolk CWA, Mueller C, Mebazaa A, Voors AA, de Boer RA, Silljé HH. Plasma levels of heart failure biomarkers are primarily a reflection of extracardiac production. Theranostics 2018;8:593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang SA, Lee SC, Choe YH, Hahn HJ, Jang SY, Park SJ, Choi JO, Park SW, Oh JK. Effects of hypertrophy and fibrosis on regional and global functional heterogeneity in hypertrophic cardiomyopathy. Int J Cardiovasc Imaging 2012;28 (Suppl. 2):133–140. [DOI] [PubMed] [Google Scholar]
- 55. Krämer J, Niemann M, Liu D, Hu K, Machann W, Beer M, Wanner C, Ertl G, Weidemann F. Two‐dimensional speckle tracking as a non‐invasive tool for identification of myocardial fibrosis in Fabry disease. Eur Heart J 2013;34:1587–1596. [DOI] [PubMed] [Google Scholar]
- 56. Carluccio E, Biagioli P, Zuchi C, Bardelli G, Murrone A, Lauciello R, D'Addario S, Mengoni A, Alunni G, Ambrosio G. Fibrosis assessment by integrated backscatter and its relationship with longitudinal deformation and diastolic function in heart failure with preserved ejection fraction. Int J Cardiovasc Imaging 2016;32:1071–1080. [DOI] [PubMed] [Google Scholar]
- 57. Iles L, Pfluger H, Phrommintikul A, Cherayath J, Aksit P, Gupta SN, Kaye DM, Taylor AJ. Evaluation of diffuse myocardial fibrosis in heart failure with cardiac magnetic resonance contrast‐enhanced T1 mapping. J Am Coll Cardiol 2008;52:1574–1580. [DOI] [PubMed] [Google Scholar]
- 58. Sibley CT, Noureldin RA, Gai N, Nacif MS, Liu S, Turkbey EB, Mudd JO, van der Geest RJ, Lima JA, Halushka MK, Bluemke DA. T1 mapping in cardiomyopathy at cardiac MR: comparison with endomyocardial biopsy. Radiology 2012;265:724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Čelutkienė J, Plymen CM, Flachskampf FA, de Boer RA, Grapsa J, Manka R, Anderson L, M G, Barberis V, Filardi PP, Gargiulo P, Zamorano JL, Lainscak M, Seferovic P, Ruschitzka F, Rosano GM, Nihoyannopoulos P. Innovative imaging methods in heart failure: a shifting paradigm in cardiac assessment. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2018;20:1615–1633. [DOI] [PubMed] [Google Scholar]
- 60. Arias T, Petrov A, Chen J, de Haas H, Pérez‐Medina C, Strijkers GJ, Hajjar RJ, Fayad ZA, Fuster V, Narula J. Labeling galectin‐3 for the assessment of myocardial infarction in rats. EJNMMI Res 2014;4:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, González A, Thum T, Díez J, Jaisser F, Pizard A, Zannad F. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail 2017;19:177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Clarke SA, Richardson WJ, Holmes JW. Modifying the mechanics of healing infarcts: is better the enemy of good? J Mol Cell Cardiol 2016;93:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012;5(Suppl. 1):S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alapati D, Rong M, Chen S, Hehre D, Rodriguez MM, Lipson KE, Wu S. Connective tissue growth factor antibody therapy attenuates hyperoxia‐induced lung injury in neonatal rats. Am J Respir Cell Mol Biol 2011;45:1169–1177. [DOI] [PubMed] [Google Scholar]
- 65. Szabó Z, Magga J, Alakoski T, Ulvila J, Piuhola J, Vainio L, Kivirikko KI, Vuolteenaho O, Ruskoaho H, Lipson KE, Signore P, Kerkelä R. Connective tissue growth factor inhibition attenuates left ventricular remodeling and dysfunction in pressure overload‐induced heart failure. Hypertension 2014;63:1235–1240. [DOI] [PubMed] [Google Scholar]
- 66. Lopez‐de la Mora DA, Sanchez‐Roque C, Montoya‐Buelna M, Sanchez‐Enriquez S, Lucano‐Landeros S, Macias‐Barragan J, Armendariz‐Borunda J. Role and new insights of pirfenidone in fibrotic diseases. Int J Med Sci 2015;12:840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yamagami K, Oka T, Wang Q, Ishizu T, Lee JK, Miwa K, Akazawa H, Naito AT, Sakata Y, Komuro I. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure‐overloaded hearts. Am J Physiol Heart Circ Physiol 2015;309:H512–H522. [DOI] [PubMed] [Google Scholar]
- 68. Yamazaki T, Yamashita N, Izumi Y, Nakamura Y, Shiota M, Hanatani A, Shimada K, Muro T, Iwao H, Yoshiyama M. The antifibrotic agent pirfenidone inhibits angiotensin II‐induced cardiac hypertrophy in mice. Hypertens Res 2012;35:34–40. [DOI] [PubMed] [Google Scholar]
- 69. Mirkovic S, Seymour AM, Fenning A, Strachan A, Margolin SB, Taylor SM, Brown L. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA‐salt hypertensive rats. Br J Pharmacol 2002;135:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm 2010;7:1438–1445. [DOI] [PubMed] [Google Scholar]
- 71. Phatharajaree WA, Phrommintikul A, Chattipakorn N. Matrix metalloproteinases and myocardial infarction. Can J Cardiol 2007;23:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yamamoto D, Takai S. Pharmacological implications of MMP‐9 inhibition by ACE inhibitors. Curr Med Chem 2009;16:1349–1354. [DOI] [PubMed] [Google Scholar]
- 73. Yamamoto D, Takai S, Jin D, Inagaki S, Tanaka K, Miyazaki M. Molecular mechanism of imidapril for cardiovascular protection via inhibition of MMP‐9. J Mol Cell Cardiol 2007;43:670–676. [DOI] [PubMed] [Google Scholar]
- 74. Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Brás LE, Lindsey ML. Early matrix metalloproteinase‐12 inhibition worsens post‐myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol 2015;185:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, Lindsey ML. Matrix metalloproteinase‐28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res 2013;112:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, André S, Crijns HJ, Gabius HJ, Maessen J, Pinto YM. Galectin‐3 marks activated macrophages in failure‐prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004;110:3121–3128. [DOI] [PubMed] [Google Scholar]
- 77. de Boer RA, Voors AA, Muntendam P, van Gilst WH, van Veldhuisen DJ. Galectin‐3: a novel mediator of heart failure development and progression. Eur J Heart Fail 2009;11:811–817. [DOI] [PubMed] [Google Scholar]
- 78. Yu L, Ruifrok WP, Meissner M, Bos EM, van Goor H, Sanjabi B, van der Harst P, Pitt B, Goldstein IJ, Koerts JA, van Veldhuisen DJ, Bank RA , van Gilst WH, Silljé HH, de Boer RA. Genetic and pharmacological inhibition of galectin‐3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail 2013;6:107–117. [DOI] [PubMed] [Google Scholar]
- 79. Calvier L, Miana M, Reboul P, Cachofeiro V, Martinez‐Martinez E, de Boer RA, Poirier F, Lacolley P, Zannad F, Rossignol P, López‐Andrés N. Galectin‐3 mediates aldosterone‐induced vascular fibrosis. Arterioscler Thromb Vasc Biol 2013;33:67–75. [DOI] [PubMed] [Google Scholar]
- 80. Li X, Tang X, Lu J, Yuan S. Therapeutic inhibition of galectin‐3 improves cardiomyocyte apoptosis and survival during heart failure. Mol Med Rep 2018;17:4106–4112. [DOI] [PubMed] [Google Scholar]
- 81. Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J, Sethi T. Regulation of transforming growth factor‐β1‐driven lung fibrosis by galectin‐3. Am J Respir Crit Care Med 2012;185:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Suthahar N, Meijers WC, Silljé HH, Ho JE, Liu FT, de Boer RA. Galectin‐3 activation and inhibition in heart failure and cardiovascular disease: an update. Theranostics 2018;8:593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA‐21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008;456:980–984. [DOI] [PubMed] [Google Scholar]
- 84. Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, Garg A, Remke J, Zimmer K, Batkai S, Thum T. Inhibition of the cardiac fibroblast‐enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res 2017;121:575–583. [DOI] [PubMed] [Google Scholar]
- 85. Abonnenc M, Nabeebaccus AA, Mayr U, Barallobre‐Barreiro J, Dong X, Cuello F, Sur S, Drozdov I, Langley SR, Lu R, Stathopoulou K, Didangelos A, Yin X, Zimmermann WH, Shah AM, Zampetaki A, Mayr M. Extracellular matrix secretion by cardiac fibroblasts: role of microRNA‐29b and microRNA‐30c. Circ Res 2013;113:1138–1147. [DOI] [PubMed] [Google Scholar]
- 86. Deddens JC, Sadeghi AH, Hjortnaes J, van Laake LW, Buijsrogge M, Doevendans PA, Khademhosseini A, Sluijter JP. Modeling the human scarred heart in vitro: toward new tissue engineered models. Adv Healthc Mater 2017;6:1600571. [DOI] [PubMed] [Google Scholar]
- 87. Ameri P, Canepa M, Anker MS, Belenkov Y, Bergler‐Klein J, Cohen‐Solal A, Farmakis D, López‐Fernández T, Lainscak M, Pudil R, Ruschitska F, Seferovic P, Filippatos G, Coats A, Suter T, Von Haehling S, Ciardiello F, de Boer RA, Lyon AR, Tocchetti CG. Heart Failure Association Cardio‐Oncology Study Group of the European Society of Cardiology. Cancer diagnosis in patients with heart failure: epidemiology, clinical implications and gaps in knowledge. Eur J Heart Fail 2018;20:879–887. [DOI] [PubMed] [Google Scholar]