

Abstract
Heart failure (HF) is responsible for substantial morbidity and mortality and is increasing in prevalence. Although there has been remarkable progress in the treatment of HF with reduced ejection fraction (HFrEF), morbidity and mortality are still substantial. Cardiac contractility modulation (CCM) signals, consisting of biphasic high‐voltage bipolar signals delivered to the right ventricular septum during the absolute refractory period, have been shown to improve symptoms, exercise tolerance and quality of life and reduce the rate of HF hospitalizations in patients with ejection fractions (EF) between 25% and 45%. CCM therapy is currently approved in the European Union, China, India, Australia and Brazil for use in symptomatic HFrEF patients with normal or slightly prolonged QRS duration. CCM is particularly beneficial in patients with baseline EF between 35% and 45%, which includes half the range of HF patients with mid‐range EFs (HFmrEF). At the cellular level, CCM has been shown in HFrEF patients to improve calcium handling, to reverse the foetal myocyte gene programme associated with HF, and to facilitate reverse remodelling. This review highlights the preclinical and clinical literature related to CCM in HFrEF and HFmrEF and outlines the potential of CCM for HF with preserved EF, concluding that CCM may fill an important unmet need in the therapeutic approach to HF across the range of EFs.
Keywords: Cardiac contractility modulation, Heart failure, Pathophysiology, Treatment, Preserved ejection fraction
Introduction
Heart failure (HF) is the cardiovascular epidemic of the 21st century.1 Worldwide the prevalence of HF is estimated to exceed 25 million2 and its prevalence is rising. The HF epidemic can be explained by the paradox of clinical success, including more effective treatment of acute coronary syndromes, leading to a decrease in mortality following acute myocardial infarction. Unfortunately, this is accompanied by a greater incidence in cardiac dysfunction among survivors. An aging population also contributes to the development of HF,3 while the growing problems of obesity and diabetes are co‐morbidities that contribute to HF with preserved ejection fraction (HFpEF).4
Chronic HF patients stratified by categories of left ventricular ejection fraction (EF) represent different phenotypes in terms of demographics, clinical presentation, aetiology, mechanical and electrical remodelling, and pharmacotherapies. HF patients are currently classified as HF with reduced EF (HFrEF; EF < 40%), HF with mid‐range EF (HFmrEF; EF 40–49%) and HFpEF (EF ≥ 50%).5 Despite major improvements in pharmacological and device therapies for HFrEF treatment during the last several decades, the 5‐year survival rate has remained unchanged at 50%.6, 7, 8 Particularly problematic is that virtually all therapeutic innovations for HF have focused on HFrEF.9 Results of registry studies imply similar benefits of beta‐blockers and renin–angiotensin–aldosterone system inhibitors in HFmrEF and HFrEF, but such therapies have been unsuccessful in improving long‐term outcomes in HFmrEF and HFpEF patients,6 highlighting a critical gap in therapeutic options (Table 1).5, 10 The differential impact of therapies on HFrEF, HFmrEF and HFpEF suggests fundamental differences in the underlying pathophysiology and an incomplete understanding of the mechanisms involved.11, 12
Table 1.
Data from registries or subgroup analyses showing that treatments able to improve clinical outcome in heart failure with reduced ejection fraction seem to be beneficial in heart failure with mid‐range ejection fraction, but not in heart failure with preserved ejection fraction too
| HFrEF | HFmrEF | HFpEF | |
|---|---|---|---|
| ACEI | + | NA | − |
| ARB | + | (+) | − |
| BB | + | (+) | − |
| Ivabradine | + | NA | − |
| MRA | + | (+) | − |
| Digitalis | + | NA | − |
| ARNI | + | NA | NA |
| Diuretics | +c | +c | +c |
| Defibrillator | + | +* | +* |
| CRT | + | +c | NA |
| CCM | +c | +c | Case reports |
ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; ARNI, angiotensin receptor‐neprilysin inhibitor; BB, beta‐blocker; CRT, cardiac resynchronization therapy; CCM, cardiac contractility modulation; HFmrEF, heart failure with mid‐range ejection fraction; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; MRA, mineralocorticoid receptor blocker; NA, not available/not analysed.
+: positive results for mortality and/or morbidity in prospective randomized controlled trials.
(+): positive results from registries, subgroup analysis or retrospective analyses. No data from randomized controlled trials available.
−: negatively investigated for mortality and/or morbidity in prospective randomized controlled trials.
+*: secondary prevention.
+c: recommended to relieve symptoms and/or signs of congestion.
Specifically related to HFrEF, beta‐blockers, angiotensin‐converting enzyme inhibitors or angiotensin receptor blockers, and aldosterone antagonists have served as the mainstays of guideline‐directed medical treatment for nearly two decades. Each of these agents provide a mortality benefit in most patients with HFrEF.13 Since their introduction to treat HF in the ‘90s, the funny channel inhibitor, ivabradine, has been approved in 2010,14 and recently the combination of an angiotensin receptor blocker with a neprilysin inhibitor was launched.15 With respect to medical devices, implantable cardioverter defibrillator (ICD), atrio‐biventricular cardiac resynchronization therapy (CRT) and ventricular assist devices have been introduced to treat HF over the last decades. ICDs are indicated to prolong survival in HF subjects with an EF < 35% despite optimal medical therapy (OMT),16 but do not improve functional capacity or symptoms. The majority of HF patients are not candidates for CRT or a ventricular assist device since they lack a prolonged QRS or have insufficiently severe symptoms, respectively. Thus, despite all the progress, there remains the need for new, effective therapies across the entire EF spectrum.
Cardiac contractility modulation overview
Cardiac contractility modulation (CCM) signals are biphasic relatively high‐voltage signals (7.5 V/22 ms duration) delivered to the right ventricular septum during the absolute refractory period.17, 18 Clinically, CCM is currently suggested for consideration by the European Society of Cardiology guidelines in patients with symptomatic HF on OMT and with normal or mildly prolonged QRS duration and reduced EF.19 Detailed descriptions of the device and the implantation procedure (essentially identical to implantation of a standard implanted pulse generator and pacing leads) have been provided previously.20, 21 CCM has been shown to improve quality of life [Minnesota Living with Heart Failure Questionnaire (MLHFQ)], left ventricular EF,22, 23 indexes of diastolic function,23 New York Heart Association (NYHA) classification, 6 min walk test,24 and peak oxygen consumption during cardiopulmonary stress testing25, 26 in patients with symptomatic HF on OMT (including ICD when indicated), with QRS duration < 130 ms and EF < 45%. These findings have recently been confirmed in the randomized FIX‐HF‐5C study, which also showed a reduction in the 6‐month composite rate of cardiac mortality and HF hospitalizations.27 Furthermore, this latter study also confirmed findings that patients with EF between 35% and 45% derive clinical benefits greater than those experienced by patients with EF < 35%. Most recently, all of these findings were also confirmed in a real‐world registry study (the CCM‐REG study, Personal communication of Prof. Gerd Hasenfuss.) that showed CCM decreased overall 3‐year mortality in patients with EF between 35% and 45% significantly below that predicted by both the Seattle Heart Failure Model28 and the Meta‐Analysis Global Group in Chronic Heart Failure (MAGGIC) score.29 These and other studies26, 30 have all shown that CCM is safe and non‐arrhythmogenic.
The main purpose of this review is to provide a summary of current knowledge of the mechanistic effects of CCM in the setting of HFrEF and explain how many of those mechanisms might also serve to improve cardiovascular function and clinical outcomes for HF patients with higher EFs.
Cardiac contractility modulation mechanisms of action
Cardiac contractility modulation signals increase contractile strength of isolated rabbit papillary muscle strips31 and trabeculae obtained from human hearts explanted from patients. With optimal parameter settings, contractile strength increased in these settings by an average of ∼30%.32 In early clinical studies, CCM signals were shown to increase left ventricular dP/dtmax more modestly by 5–10%.26, 30 In one study of dogs with intracoronary microembolization‐induced HF, active CCM monotherapy for 3 months induced an increase in left ventricular EF (27 ± 1% vs. 33 ± 1%, P < 0.0001) compared with a decrease in sham‐operated control animals (27 ± 1% vs. 23 ± 1%, P < 0.001).33 This increase was accompanied by reduced left ventricular volumes and improved myocardial structure. Importantly, the effects of CCM on function are not associated with increases in myocardial oxygen consumption as measured in patients with severe chronic HF under resting or stress conditions and independent of HF aetiology.34, 35
Concurrent with the mechanical effects of CCM, CCM exerts multiple effects at cellular and molecular levels (Table 2). As will be detailed below, CCM improves calcium (Ca2+) handling in cardiomyocytes, initiates molecular reverse remodelling of the foetal gene programme observed in HF back towards that of a normal adult33, 36 and a host of other pathways involved in myocyte and interstitial fibrosis. Furthermore, these effects are not only observed locally at the site of signal delivery but, over months, benefits extend remotely through global adaptive cardiac reverse remodelling.23, 37
Table 2.
Summary of the impact of cardiac contractility modulation in patients with heart failure with reduced ejection fraction
| CCM effect in HFrEF | |
|---|---|
|
Intracellular Ca2+ metabolism ↑ Improvement in diastolic Ca2+ levels (SERCA2a; phosphorylation of phospholamban) |
↑ |
|
Phosphorylation of myofilaments (troponin I, myosin light chain 2, myosin binding protein) |
↑ |
| Titin phosphorylation | ↑ |
| Titin distensibility | ↑ |
| Small heat shock protein (e.g. αB‐crystallin) | ↑ |
| Oxidative stress | ↓ |
| Cardiac fibrosis | ↓ |
| Sympathetic nerve activity | ↓ |
| Neutral metabolic activity | + |
| Improvement in LV systolic reserve | + |
| Improvement in LV diastolic filling (E/E´) | + |
| Improvement in QoL | + |
HFrEF, heart failure with reduced ejection fraction; LV, left ventricular; QoL, quality of life; SERCA2, sarcoplasmic reticulum Ca2+‐ATPase 2a.
Intracellular calcium metabolism
Disorders of intracellular Ca2+ homeostasis associated with HF have been shown to contribute to both systolic and diastolic dysfunction38 by interfering with the ryanodine receptor (RyR2),39 the sarcoplasmic reticulum Ca2+‐ATPase 2a (SERCA2a) pathway and the sodium–potassium pump.40 In the intracoronary microembolization‐induced HF study noted above,33 CCM also led to rapid normalization of phospholamban phosphorylation, which increased the ability of the sarcoplasmic reticulum to sequester calcium.33 In another study in dogs with HF, chronic therapy with CCM normalized the downregulated left ventricular expression of the Ca2+ binding protein S100A1.41 S100A1 interacts in a Ca2+‐dependent manner with the RyR2, the SERCA2a–phospholamban complex, cardiac titin, and mitochondrial F1‐ATPase. Its protein expression is reduced in cardiomyocytes from patients with end‐stage HFrEF.42 Its relevance in contractile function follows from experimental findings showing that downregulation of S100A1 protein contributes to contractile dysfunction of the diseased heart in mice,43 whereas S100A1 gene transfer restores contractile function of failing myocardium in rat models of HF.44
Butter et al.36 analysed endomyocardial biopsies at baseline and 3 and 6 months after CCM implantation from 11 HFrEF patients with an EF < 35% and NYHA functional class II/III despite OMT. CCM therapy was delivered in random order to be switched off and on for 3 months. The findings showed that 3 months of CCM therapy resulted in increased expression of SERCA2a, phospholamban, and RyR2, suggesting that CCM therapy in HFrEF patients normalized the expression of the key sarcoplasmic reticulum Ca2+ cycling and stretch response genes. In summary, CCM is able to induce beneficial molecular remodelling of intracellular Ca2+ regulatory proteins in HFrEF.
Myofilaments
Cardiac force generation results from protein interactions between the thin filaments [α‐actin, α‐tropomyosin and the troponin complex, comprised of troponin I (TnI), troponin T and troponin C] and the thick filaments [the myosin complex, comprised of a pair of myosin heavy chains (MHC) and two pairs each of myosin light chain 1 and 2 (MLC1, MLC2), and associated proteins such as myosin binding protein C].45 TnI and MLC2 are important myofibrillar proteins involved in the regulation of myofilament Ca2+ sensitivity and cardiac inotropy. The sensitivity of the cardiac myofilaments to Ca2+ is primarily regulated by the phosphorylation state of TnI and MLC2.46 Butter et al.36 demonstrated that 3 months of CCM therapy reverses the downregulated expression of the α isoform of MHC in HFrEF patients. In HFrEF patients, CCM has shown an increase in the phosphorylation state of TnI and of myosin‐binding protein C in the left and right ventricle, which occurred as soon as 30 min after signal delivery and which remained after 3 months of therapy (Figure 1).
Figure 1.
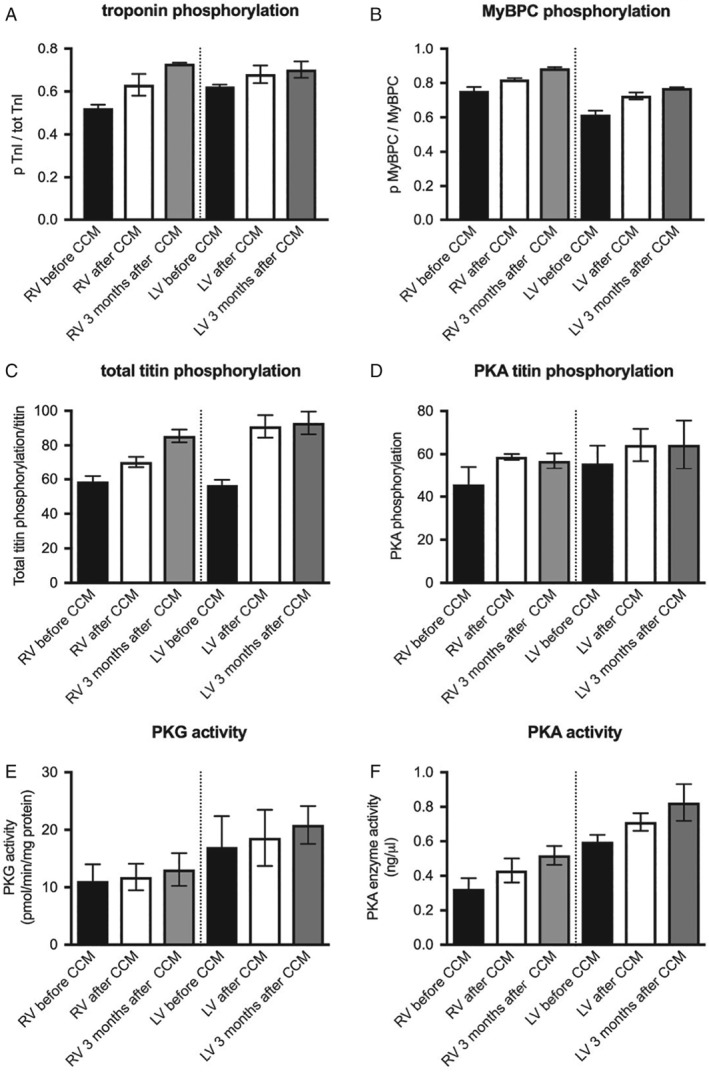
Impact of cardiac contractility modulation (CCM) on phosphorylation of troponin I (A), myosin‐binding protein C (MyBPC) (B), titin (C), and protein kinase G (PKG) and A (PKA) activity (D–F) in patients with heart failure with reduced ejection fraction (HFrEF). (A–D) Mean ± standard error of the mean of the ratio of phosphorylated to total MyBPC, of phosphorylated to total troponin I (TnI), of phosphorylated to total titin, and of PKA‐induced phosphorylation of titin to total titin, respectively, in endomyocardial biopsies from the right ventricle (RV) or left ventricle (LV) before (black bars), 30 min after (white bars), and 3 months after initiating CCM (grey bars) of two HFrEF patients, as indicated. (E, F) Mean ± standard error of the mean of PKG activity (pmol/min/mg protein) and PKA activity (ng/µL), respectively, in the RV and LV at the same time points after initiating CCM in two HFrEF patients.
The large cytoskeletal protein titin acts as a bidirectional spring and is involved in early diastolic recoil and late diastolic distensibility of cardiomyocytes.46 Its characteristics are modified through isoform shifts and through phosphorylation by several kinases including protein kinase A (PKA),47 G (PKG),48 and C (PKC).49 PKA and PKG promote titin compliance, but PKC reduces compliance.11 In a dog model of HF, CCM therapy reversed the downregulated expression of titin.37 Large‐scale analyses are not yet available from human HF myocardium. However, these findings were also confirmed in two HFrEF patients (Figure 1).
Another recently identified protective mechanism for I‐band titin domains involves the small heat shock proteins (sHSPs) HSP27 (HSPB1) and αB‐crystallin (HSPB5). These are abundantly expressed in cardiac myocytes and are further induced by stress such as cardiac ischaemic injury or end‐stage HF. Their overexpression protects cells from oxidative stress, energy depletion, and other unfavourable conditions.50 HSP27 and αB‐crystallin are translocated under acidic stress preferentially to the sarcomeres; in particular to the I‐band region. Binding of HSP27/αB‐crystallin to unfolded titin domains prevents titin aggregation under stress, thereby maintaining normal myocyte stiffness. Contractility of myocytes is also strongly affected by intracellular acidosis, which increases the passive stiffness of the heart. These findings suggest that aggregation of unfolded titin‐immunoglobulin domains under mechanical and acidic stress stiffens cardiomyocytes, but that sHSPs translocate to these domains to prevent this aggregation and protect against stiffening, thus preserving diastolic function.50
Consistent with these findings, it has been shown that mouse hearts deficient in αB‐crystallin and HSPB2 display normal systolic function but exhibit increased passive stiffness and diastolic dysfunction when exposed to stress,51 suggesting a potential beneficial role of sHSPs on titin‐based passive stiffness in patients with HFpEF. This hypothesis has now been supported by Franssen et al.52 who recently demonstrated that αB‐crystallin reverses the pronounced diastolic stiffness of failing human cardiomoycytes probably through relief of titin aggregation. Interestingly, sHSPs are upregulated by exposure to magnetic fields as shown in cardiomyocytes for HSP7053 and HSP27, 70, and 90 in endothelial cells.54 Imaging mass spectrometry55 findings from a HFrEF patient who underwent CCM therapy illustrate an upregulation of αB‐crystallin 3 months post‐CCM (Figure 2).56 These insights lead to the hypothesis that heat shock activation by CCM may mimic the benefit observed during preconditioning.
Figure 2.
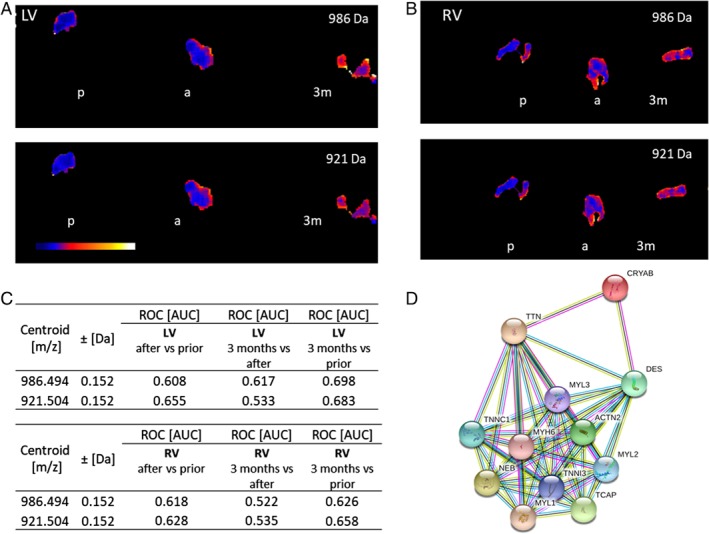
Imaging mass spectrometry of endomyocardial biopsies. Ion density distributions of m/z‐values 986 Da and 921 Da (α‐crystallin B chain) are significantly increased after 3 months (3m) in comparison to prior (p) cardiac contractility modulation intervention. (A) Left ventricle (LV) and (B) right ventricle (RV) endomyocardial biopsies (P < 0.001). (C) Receiver operating characteristic (ROC) values show the discrimination capability of m/z 956 Da/921 Da between shortly after/prior, 3 months/shortly after and 3 months/prior cardiac contractility modulation intervention in the LV (upper table) and RV (lower table) [area under the curve (AUC) > 0.6] of two heart failure with reduced ejection fraction patients. (D) String database analysis56 demonstrating the interaction between α‐crystallin B chain (CryAB) and titin. (a) stands for shortly after cardiac contractility modulation intervention.
Extracellular matrix: fibrosis
In dogs with chronic HF, chronic CCM monotherapy increases left ventricular EF and stroke volume, which is paralleled by a reduction in volume fraction of replacement fibrosis and interstitial fibrosis.33 Further evaluation of the impact of 3 months of CCM therapy on cardiac remodelling in dogs with HF showed upregulation and normalization of the matrix metalloproteinases 1, 2, and 9.37 In a chronic rabbit model of HF, CCM lasting 6 h per day for 4 weeks attenuated myocardial fibrosis and collagen deposition potentially by inhibiting transforming growth factor‐β1/Smad3 signalling.57
Autonomic nervous system
Since CCM initially increases septal contractility, this has been shown to activate vagal afferent fibres.58 Accordingly, a reduction of excess sympathetic activation associated with HF is expected with a resulting improvement in autonomic balance. Similarly, CRT improves cardiac haemodynamics in part by a reduction of excessive sympathetic activity. Normalization of sympathetic activity is also characteristic of clinical responders to atrio‐biventricular pacing. Our own findings in a patient with HFrEF illustrate that CCM also decreased muscle sympathetic nerve activity (MSNA) after several months of treatment (Figure 3). There was, however, no immediate effect of CCM stimulation on MSNA burst incidence (bursts/min and bursts/100 heart beats), which is in line with the CRT results.59
Figure 3.
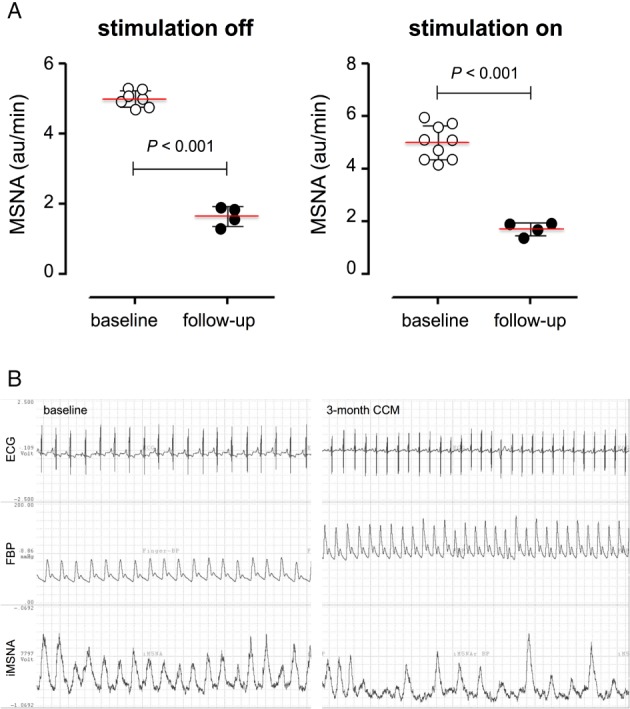
Impact of cardiac contractility modulation (CCM) on muscle sympathetic nerve activity (MSNA). (A) Bar graphs depict MSNA (au/min) at baseline and 3 months after (follow‐up), off stimulation (left panel), or on stimulation (right panel). MSNA did not acutely change during short on/off stimulations, either at baseline (white circles left panel vs. white circles right panel) nor 3 months later (black circles left panel vs. black circles right panel). After 3 months of treatment, MSNA basal levels (black circles) were significantly reduced compared to baseline (white circles), suggesting that CCM induces a remodelling process, which includes at least indirectly also the sympathetic nerve activity. (B) Representative sympathetic nerve recording of a heart failure with reduced ejection fraction patient with CCM stimulation one day after implantation (left) and after 3 months of intermittent therapy. Note the remarkable reduction in sympathetic burst incidence with chronic CCM stimulation (at baseline; 100%; after 3‐month CCM stimulation: ∼50%). ECG, electrocardiogram; FBP, phospholamban.
Cardiac contractility modulation in patients with higher ejection fractions: clinical effects and mechanistic implications
As noted above, while the focus of prior studies of CCM therapy has been on patients with HFrEF, significant amounts of data are available on the effects of CCM in patients with NYHA class III and IV symptoms with EFs between 35% and 45%. This includes half of the range of EFs of the HF population now designated as HFmrEF (defined as patients with EFs from 40% to 49%). It was initially observed in a small subset of 40 patients from the FIX‐HF‐5 study that CCM improved exercise tolerance (indexed by both peak oxygen consumption and 6 min walking test), and quality of life (indexed by both MLHFQ and NYHA classification) more in patients with EFs between 35% and 45% than in patients with EF < 35%.21 This finding was reconfirmed prospectively in the recently completed FIX‐HF‐5C study which showed, in this same subgroup, a 1.76 ml O2/min/kg increase of peak oxygen consumption, a 15 point improvement in MLHFQ and 59.3 m improvement in 6 min walk test with 71% of patients exhibiting at least one NYHA class improvement.27 In all parameters, these effects were larger than in the group with EF < 35%. These clinical findings motivated us to undertake a preliminary investigation into possible mechanisms by which CCM could impact cardiovascular properties in HFmrEF and HFpEF that will be summarized below.
Interestingly, the mechanisms of action of CCM impact certain processes that are also mechanistically implicated in the pathophysiology of HFpEF.60 Therefore, consistent with results discussed above concerning even greater clinical effects in patients with EF between 35% and 45%,21, 27, 61 CCM has potential to provide a new therapeutic approach for HF patients with higher EFs.
Recent data increasingly recognize the roles of non‐cardiac systemic processes such as vascular tone, renal dysfunction, metabolic disorders, pro‐inflammatory, pro‐fibrotic, immunological alterations11 and right heart dysfunction62 in the development of clinical HFpEF and HFmrEF. Such abnormalities, which are often linked with co‐morbidities commonly present in these populations (e.g. obesity, diabetes, renal dysfunction, hypertension), promote myocyte hypertrophy, increase resting myocyte tension, impaire calcium metabolism and increase interstitial fibrosis via many of the same mechanisms detailed above for HFrEF.
Accordingly, it is possible that mechanisms by which CCM improves myocyte function in HFrEF may also play a beneficial role in HFmrEF and HFpEF. Studies into this possibility have just begun with the publication summarizing the cellular and molecular effects of CCM in one HFpEF patient and one HFmrEF patient in whom functional class and exercise tolerance where improved. In these cases, CCM was shown to downregulate the expression of the foetal gene product myosin 7 and to increase phosphorylation of MLC2 and TnI in the left and right ventricle both early (30 min) and late (3 months) following the initiation of CCM therapy.60 This change was associated with an increase in contractile reserve induced by stress echocardiography. Furthermore, following both 30 min and 3 months of CCM therapy, PKA and PKG activity and the degree of phosphorylated titin in the right and left ventricles were higher when compared to values obtained prior to initiating CCM, which can contribute to improved relaxation.60 Similarly, the effects of CCM therapy on markers of cardiac fibrosis in the right ventricle of the patient with severe HFpEF showed that the expression of collagen I, collagen III and of the myofibroblast marker α‐smooth muscle actin were reduced by 29%, 22%, and 22%, respectively, after 3 months of CCM therapy.60 Consistent reductions in collagen I, collagen III and α‐smooth muscle actin expression 3 months following CCM were also observed in both ventricles of the HFmrEF patient.60 Blinded evaluation of the region‐dependent proteome signature via imaging mass spectrometry55 further revealed a reduction in the expression of collagen 2a (VI) chain in the right and left ventricles 3 months after initiating CCM in the HFmrEF patient, suggesting an impact of CCM on cardiac collagen regulation.
Summary
Cardiac contractility modulation improves a variety of myocardial and systemic cardiovascular properties that are involved in the pathophysiology of HFrEF (Table 2). The evidence from animal models and patients with HFrEF demonstrates that CCM therapy has the potential to have beneficial effects in HF via processes involved in Ca2+ handling, the cytoskeleton, the extracellular matrix, and potentially the autonomous nervous system. Clinical studies show trends for greater improvements in exercise tolerance, quality of life and functional status in patients with EF 35–45% vs. those with lower EFs, a finding that was reproduced in separate studies. It is noteworthy that this EF range spans half that used to define the HFmrEF population. Whether the mechanisms observed in HFrEF also apply to patients with HFpEF needs to be investigated. Therefore, a prospective, multicentre, single arm, open‐label 24‐week exploratory study evaluating CCM therapy in patients who are symptomatic despite OMT is planned: Cardiac Contractility Modulation (CCM™) Therapy in Subjects with Heart Failure with preserved Ejection Fraction, in brief CCM‐HFpEF (EUDAMED; number CIV1612017844).
Conflict of interest: C.T. and B.K. received lecturing fees from Impulse Dynamics. D.G. and D.B. are consultants for Impulse Dynamics. The other authors have no conflicts to declare.
References
- 1. Luscher TF. Heart failure: the cardiovascular epidemic of the 21st century. Eur Heart J 2015;36:395–397. [DOI] [PubMed] [Google Scholar]
- 2. Liu L, Eisen HJ. Epidemiology of heart failure and scope of the problem. Cardiol Clin 2014;32:1–8, vii. [DOI] [PubMed] [Google Scholar]
- 3. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, DK MG, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics‐‐2015 update: a report from the American Heart Association. Circulation 2015;131:e29–e322. [DOI] [PubMed] [Google Scholar]
- 4. Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 2011;32:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tschöpe C, Pieske B. Heart failure with mid‐range EF (HFmrEF): a mildly reduced EF does not imply a mild disease. Heart and Metabolism 2017;74:8–12. [Google Scholar]
- 6. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006;355:251–259. [DOI] [PubMed] [Google Scholar]
- 7. Zarrinkoub R, Wettermark B, Wandell P, Mejhert M, Szulkin R, Ljunggren G, Kahan T. The epidemiology of heart failure, based on data for 2.1 million inhabitants in Sweden. Eur J Heart Fail 2013;15:995–1002. [DOI] [PubMed] [Google Scholar]
- 8. Mamas MA, Sperrin M, Watson MC, Coutts A, Wilde K, Burton C, Kadam UT, Kwok CS, Clark AB, Murchie P, Buchan I, Hannaford PC, Myint PK. Do patients have worse outcomes in heart failure than in cancer? A primary care‐based cohort study with 10‐year follow‐up in Scotland. Eur J Heart Fail 2017;19:1095–1104. [DOI] [PubMed] [Google Scholar]
- 9. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62:e147–e239. [DOI] [PubMed] [Google Scholar]
- 10. Tschope C, Birner C, Bohm M, Bruder O, Frantz S, Luchner A, Maier L, Stork S, Kherad B, Laufs U. Heart failure with preserved ejection fraction: current management and future strategies: expert opinion on the behalf of the Nucleus of the “Heart Failure Working Group” of the German Society of Cardiology (DKG). Clin Res Cardiol 2017;107:1–19. [DOI] [PubMed] [Google Scholar]
- 11. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 12. Tschope C, Van Linthout S. New insights in (inter)cellular mechanisms by heart failure with preserved ejection fraction. Curr Heart Fail Rep 2014;11:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abi‐Samra F, Gutterman D. Cardiac contractility modulation: a novel approach for the treatment of heart failure. Heart Fail Rev 2016;21:645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost-Brama A, Lerebours G, Tavazzi L and Investigators S. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo‐controlled study. Lancet 2010;376:875–885. [DOI] [PubMed] [Google Scholar]
- 15. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR; PARADIGM‐HF Investigators and Committees . Angiotensin‐neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014;371:993‐1004. [DOI] [PubMed] [Google Scholar]
- 16. Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, Domanski M, Troutman C, Anderson J, Johnson G, SE MN, Clapp‐Channing N, Davidson‐Ray LD, Fraulo ES, Fishbein DP, Luceri RM, Ip JH; Sudden Cardiac Death in Heart Failure Trial (SCD‐HeFT) Investigators . Amiodarone or an implantable cardioverter‐defibrillator for congestive heart failure. N Engl J Med 2005;352:225–237. [DOI] [PubMed] [Google Scholar]
- 17. Kleemann T. Cardiac contractility modulation. A new form of therapy for patients with heart failure and narrow QRS complex? Herz 2015;40:945–951. [DOI] [PubMed] [Google Scholar]
- 18. Kuschyk J, Kloppe A, Schmidt‐Schweda S, Bonnemeier H, Rousso B, Roger S. Cardiac contractility modulation: a technical guide for device implantation. Rev Cardiovasc Med 2017;18:1–13. [DOI] [PubMed] [Google Scholar]
- 19. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 20. Abraham WT, Burkhoff D, Nademanee K, Carson P, Bourge R, Ellenbogen KA, Parides M, Kadish A; FIX‐HF‐5 Investigators and Coordinators . A randomized controlled trial to evaluate the safety and efficacy of cardiac contractility modulation in patients with systolic heart failure: rationale, design, and baseline patient characteristics. Am Heart J 2008;156:641–648.e1. [DOI] [PubMed] [Google Scholar]
- 21. Abraham WT, Lindenfeld J, Reddy VY, Hasenfuss G, Kuck KH, Boscardin J, Gibbons R, Burkhoff D; FIX‐HF‐5C Investigators and Coordinators. A randomized controlled trial to evaluate the safety and efficacy of cardiac contractility modulation in patients with moderately reduced left ventricular ejection fraction and a narrow QRS duration: study rationale and design. J Card Fail 2015;21:16‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Muller D, Remppis A, Schauerte P, Schmidt‐Schweda S, Burkhoff D, Rousso B, Gutterman D, Senges J, Hindricks G, Kuck KH. Clinical effects of long‐term cardiac contractility modulation (CCM) in subjects with heart failure caused by left ventricular systolic dysfunction. Clin Res Cardiol 2017;106:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu CM, Chan JY, Zhang Q, Yip GW, Lam YY, Chan A, Burkhoff D, Lee PW, Fung JW. Impact of cardiac contractility modulation on left ventricular global and regional function and remodeling. JACC Cardiovasc Imaging 2009;2:1341–1349. [DOI] [PubMed] [Google Scholar]
- 24. Stix G, Borggrefe M, Wolpert C, Hindricks G, Kottkamp H, Bocker D, Wichter T, Mika Y, Ben‐Haim S, Burkhoff D, Wolzt M, Schmidinger H. Chronic electrical stimulation during the absolute refractory period of the myocardium improves severe heart failure. Eur Heart J 2004;25:650–655. [DOI] [PubMed] [Google Scholar]
- 25. Borggrefe MM, Lawo T, Butter C, Schmidinger H, Lunati M, Pieske B, Misier AR, Curnis A, Bocker D, Remppis A, Kautzner J, Stuhlinger M, Leclerq C, Taborsky M, Frigerio M, Parides M, Burkhoff D, Hindricks G. Randomized, double blind study of non‐excitatory, cardiac contractility modulation electrical impulses for symptomatic heart failure. Eur Heart J 2008;29:1019–1028. [DOI] [PubMed] [Google Scholar]
- 26. Kadish A, Nademanee K, Volosin K, Krueger S, Neelagaru S, Raval N, Obel O, Weiner S, Wish M, Carson P, Ellenbogen K, Bourge R, Parides M, Chiacchierini RP, Goldsmith R, Goldstein S, Mika Y, Burkhoff D, Abraham WT. A randomized controlled trial evaluating the safety and efficacy of cardiac contractility modulation in advanced heart failure. Am Heart J 2011;161:329–337.e1‐2. [DOI] [PubMed] [Google Scholar]
- 27. Abraham WT, Kuck KH, Goldsmith RL, Lindenfeld J, Reddy VY, Carson PE, Mann DL, Saville B, Parise H, Chan R, Wiegn P, Hastings JL, Kaplan AJ, Edelmann F, Luthje L, Kahwash R, Tomassoni GF, Gutterman DD, Stagg A, Burkhoff D, Hasenfuss G. A randomized controlled trial to evaluate the safety and efficacy of cardiac contractility modulation. JACC Heart Fail 2018;6:874–883. [DOI] [PubMed] [Google Scholar]
- 28. Levy WC, Mozaffarian D, Linker DT, Sutradhar SC, Anker SD, Cropp AB, Anand I, Maggioni A, Burton P, Sullivan MD, Pitt B, Poole‐Wilson PA, Mann DL, Packer M. The Seattle Heart Failure Model: prediction of survival in heart failure. Circulation 2006;113:1424–1433. [DOI] [PubMed] [Google Scholar]
- 29. Pocock SJ, Ariti CA, McMurray JJ, Maggioni A, Kober L, Squire IB, Swedberg K, Dobson J, Poppe KK, Whalley GA, Doughty RN, Meta‐Analysis Global Group in Chronic Heart Failure . Predicting survival in heart failure: a risk score based on 39 372 patients from 30 studies. Eur Heart J 2013;34:1404–1413. [DOI] [PubMed] [Google Scholar]
- 30. Pappone C, Rosanio S, Burkhoff D, Mika Y, Vicedomini G, Augello G, Shemer I, Prutchi D, Haddad W, Aviv R, Snir Y, Kronzon I, Alfieri O, Ben‐Haim SA. Cardiac contractility modulation by electric currents applied during the refractory period in patients with heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 2002;90:1307–1313. [DOI] [PubMed] [Google Scholar]
- 31. Brunckhorst CB, Shemer I, Mika Y, Ben‐Haim SA, Burkhoff D. Cardiac contractility modulation by non‐excitatory currents: studies in isolated cardiac muscle. Eur J Heart Fail 2006;8:7–15. [DOI] [PubMed] [Google Scholar]
- 32. Burkhoff D, Shemer I, Felzen B, Shimizu J, Mika Y, Dickstein M, Prutchi D, Darvish N, Ben‐Haim SA. Electric currents applied during the refractory period can modulate cardiac contractility in vitro and in vivo. Heart Fail Rev 2001;6:27–34. [DOI] [PubMed] [Google Scholar]
- 33. Imai M, Rastogi S, Gupta RC, Mishra S, Sharov VG, Stanley WC, Mika Y, Rousso B, Burkhoff D, Ben‐Haim S, Sabbah HN. Therapy with cardiac contractility modulation electrical signals improves left ventricular function and remodeling in dogs with chronic heart failure. J Am Coll Cardiol 2007;49:2120–2128. [DOI] [PubMed] [Google Scholar]
- 34. Goliasch G, Khorsand A, Schutz M, Karanikas G, Khazen C, Sochor H, Schmidinger H, Wolzt M, Graf S. The effect of device‐based cardiac contractility modulation therapy on myocardial efficiency and oxidative metabolism in patients with heart failure. Eur J Nucl Med Mol Imaging 2012;39:408–415. [DOI] [PubMed] [Google Scholar]
- 35. Butter C, Wellnhofer E, Schlegl M, Winbeck G, Burkhoff D, Fleck E. Enhanced inotropic state by cardiac contractility modulation signals is not associated with changes in myocardial oxygen consumption [abstract]. Heart Rhythm 2004;1:S278. [DOI] [PubMed] [Google Scholar]
- 36. Butter C, Rastogi S, Minden HH, Meyhofer J, Burkhoff D, Sabbah HN. Cardiac contractility modulation electrical signals improve myocardial gene expression in patients with heart failure. J Am Coll Cardiol 2008;51:1784–1789. [DOI] [PubMed] [Google Scholar]
- 37. Rastogi S, Mishra S, Zaca V, Mika Y, Rousso B, Sabbah HN. Effects of chronic therapy with cardiac contractility modulation electrical signals on cytoskeletal proteins and matrix metalloproteinases in dogs with heart failure. Cardiology 2008;110:230–237. [DOI] [PubMed] [Google Scholar]
- 38. Lompre AM, Hajjar RJ, Harding SE, Kranias EG, Lohse MJ, Marks AR. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation 2010;121:822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Currie S, Elliott EB, Smith GL, Loughrey CM. Two candidates at the heart of dysfunction: the ryanodine receptor and calcium/calmodulin protein kinase II as potential targets for therapeutic intervention‐An in vivo perspective. Pharmacol Ther 2011;131:204–220. [DOI] [PubMed] [Google Scholar]
- 40. Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol 2002;34:951–969. [DOI] [PubMed] [Google Scholar]
- 41. Gupta RC, Mishra S, Rastogi S, Wang M, Rousso B, Mika Y, Remppis A, Sabbah HN. Ca2+‐binding proteins in dogs with heart failure: effects of cardiac contractility modulation electrical signals. Clin Transl Sci 2009;2:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, Heizmann CW. Altered expression of the Ca2+‐binding protein S100A1 in human cardiomyopathy. Biochim Biophys Acta 1996;1313:253–257. [DOI] [PubMed] [Google Scholar]
- 43. Kelder JC, Cramer MJ, van Wijngaarden J, van Tooren R, Mosterd A, Moons KG, Lammers JW, Cowie MR, Grobbee DE, Hoes AW. The diagnostic value of physical examination and additional testing in primary care patients with suspected heart failure. Circulation 2011;124:2865–2873. [DOI] [PubMed] [Google Scholar]
- 44. Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, Loffler E, Janssen PM, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A, Koch WJ. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest 2004;114:1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Tombe PP. Cardiac myofilaments: mechanics and regulation. J Biomech 2003;36:721–730. [DOI] [PubMed] [Google Scholar]
- 46. LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation 2010;121:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fukuda N, Wu Y, Nair P, Granzier HL. Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform‐dependent manner. J Gen Physiol 2005;125:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res 2009;104:87–94. [DOI] [PubMed] [Google Scholar]
- 49. Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res 2009;105:631–638 17 p following 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kotter S, Unger A, Hamdani N, Lang P, Vorgerd M, Nagel‐Steger L, Linke WA. Human myocytes are protected from titin aggregation‐induced stiffening by small heat shock proteins. J Cell Biol 2014;204:187–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Golenhofen N, Redel A, Wawrousek EF, Drenckhahn D. Ischemia‐induced increase of stiffness of alphaB‐crystallin/HSPB2‐deficient myocardium. Pflugers Arch 2006;451:518–525. [DOI] [PubMed] [Google Scholar]
- 52. Franssen C, Kole J, Musters R, Hamdani N, Paulus WJ. Alpha‐B crystallin reverses high diastolic stiffness of failing human cardiomyocytes. Circ Heart Fail 2017;10:e003626. [DOI] [PubMed] [Google Scholar]
- 53. Goodman R, Blank M. Insights into electromagnetic interaction mechanisms. J Cell Physiol 2002;192:16–22. [DOI] [PubMed] [Google Scholar]
- 54. Bernardini C, Zannoni A, Turba ME, Bacci ML, Forni M, Mesirca P, Remondini D, Castellani G, Bersani F. Effects of 50 Hz sinusoidal magnetic fields on Hsp27, Hsp70, Hsp90 expression in porcine aortic endothelial cells (PAEC). Bioelectromagnetics 2007;28:231–237. [DOI] [PubMed] [Google Scholar]
- 55. Klein O, Strohschein K, Nebrich G, Oetjen J, Trede D, Thiele H, Alexandrov T, Giavalisco P, Duda GN, von Roth P, Geissler S, Klose J, Winkler T. MALDI imaging mass spectrometry: discrimination of pathophysiological regions in traumatized skeletal muscle by characteristic peptide signatures. Proteomics 2014;14:2249–2260. [DOI] [PubMed] [Google Scholar]
- 56. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. STRING 8 – a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009;37:D412–D416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang F, Dang Y, Li Y, Hao Q, Li R, Qi X. Cardiac contractility modulation attenuate myocardial fibrosis by inhibiting TGF‐beta1/Smad3 signaling pathway in a rabbit model of chronic heart failure. Cell Physiol Biochem 2016;39:294–302. [DOI] [PubMed] [Google Scholar]
- 58. Sengupta J, Kannampalli P, Belligoli A, Rousso B, Ben‐Haim S, Gutterman D. Cardiac vagal afferent response in rats during cardiac contractility modulation (CCM) [abstract]. FASEB J 2015;29:651.6. [Google Scholar]
- 59. Najem B, Unger P, Preumont N, Jansens JL, Houssiere A, Pathak A, Xhaet O, Gabriel L, Friart A, De Roy L, Vandenbossche JL, van de Borne P. Sympathetic control after cardiac resynchronization therapy: responders versus nonresponders. Am J Physiol Heart Circ Physiol 2006;291:H2647–H2652. [DOI] [PubMed] [Google Scholar]
- 60. Tschope C, Van Linthout S, Spillmann F, Klein O, Biewener S, Remppis A, Gutterman D, Linke WA, Pieske B, Hamdani N, Roser M. Cardiac contractility modulation signals improve exercise intolerance and maladaptive regulation of cardiac key proteins for systolic and diastolic function in HFpEF. Int J Cardiol 2016;203:1061–1066. [DOI] [PubMed] [Google Scholar]
- 61. Borggrefe M, Burkhoff D. Clinical effects of cardiac contractility modulation (CCM) as a treatment for chronic heart failure. Eur J Heart Fail 2012;14:703–712. [DOI] [PubMed] [Google Scholar]
- 62. Gorter TM, van Veldhuisen DJ, Bauersachs J, Borlaug BA, Celutkiene J, AJS C, Crespo‐Leiro MG, Guazzi M, Harjola VP, Heymans S, Hill L, Lainscak M, CS L, Lund LH, Lyon AR, Mebazaa A, Mueller C, Paulus WJ, Pieske B, Piepoli MF, Ruschitzka F, Rutten FH, Seferovic PM, Solomon SD, Shah SJ, Triposkiadis F, Wachter R, Tschope C, de Boer RA. Right heart dysfunction and failure in heart failure with preserved ejection fraction: mechanisms and management. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2018;20:16–37. [DOI] [PubMed] [Google Scholar]