

Abstract
Aims
Despite recent advances in the treatment of chronic heart failure (HF), mortality and hospitalizations still remain high. Additional therapies to improve mortality and morbidity are urgently needed. The efficacy of cardiac glycosides – although regularly used for HF treatment – remains unclear. DIGIT‐HF was designed to demonstrate that digitoxin on top of standard of care treatment improves mortality and morbidity in patients with HF and a reduced ejection fraction (HFrEF).
Methods
Patients with chronic HF, New York Heart Association (NYHA) functional class III–IV and left ventricular ejection fraction (LVEF) ≤ 40%, or patients in NYHA functional class II and LVEF ≤ 30% are randomized 1:1 in a double‐blind fashion to treatment with digitoxin (target serum concentration 8–18 ng/mL) or matching placebo. Randomization is stratified by centre, sex, NYHA functional class (II, III, or IV), atrial fibrillation, and treatment with cardiac glycosides at baseline. A total of 2190 eligible patients will be included in this clinical trial (1095 per group). All patients receive standard of care treatment recommended by expert guidelines upon discretion of the treating physician. The primary outcome is a composite of all‐cause mortality or hospital admission for worsening HF (whatever occurs first). Key secondary endpoints are all‐cause mortality, hospital admission for worsening HF, and recurrent hospital admission for worsening HF.
Conclusion
The DIGIT‐HF trial will provide important evidence, whether the cardiac glycoside digitoxin reduces the risk for all‐cause mortality and/or hospital admission for worsening HF in patients with advanced chronic HFrEF on top of standard of care treatment.
Keywords: Heart failure, Cardiac glycosides, Digitalis, Digitoxin, Clinical trial
Introduction
Heart failure (HF) remains a major cause of mortality and morbidity in industrial countries representing the most frequent reason for hospitalization.1, 2 Due to demographical changes and promoted by current medical progress – i.e. lower mortality rates from ischaemic events – HF incidence and prevalence are expected to increase further.2, 3, 4 Despite advances in HF treatment, HF remains a disabling disorder that severely affects patients' prognosis and quality of life, and is associated with a high economic burden.5 Therefore, it is important to identify additional therapeutic strategies to improve the outcome of HF patients.
Cardiac glycosides have been used for the treatment of chronic HF for centuries. However, prospective trials investigating the impact of randomized initiation or withdrawal of cardiac glycoside therapy on all‐cause mortality or morbidity6, 7, 8, 9 in patients with HF and a reduced ejection fraction (HFrEF), respectively, failed to provide sufficient evidence and were exclusively performed using digoxin. The only randomized controlled and adequately powered trial investigating the impact of cardiac glycosides on mortality risk in HFrEF is the DIG trial. There, treatment with digoxin had no impact on mortality in the overall trial population,6 but significantly reduced hospitalizations for worsening HF by 28%. Baseline treatment of patients in the DIG trial included diuretics (81%) and angiotensin‐converting enzyme inhibitors (ACEi, 94%) according to guidelines at that time, but not beta‐blocker. A post‐hoc analysis indicated that HF patients [New York Heart Association (NYHA) class II–IV] treated with digoxin at serum concentrations of 0.5–0.9 ng/L (i.e. lower spectrum of the formerly used therapeutic range of 0.5–2.0 ng/mL) experienced a statistically significant mortality risk reduction (−19% to −23%) compared to placebo. In contrast, patients treated with digoxin at serum concentrations > 1.2 ng/mL had a significant increase in overall mortality.10, 11, 12
Current European Society of Cardiology (ESC) HF guidelines suggest treatment with cardiac glycosides as an option in patients with persistent symptoms despite optimal pharmacological therapy including ACEi/angiotensin receptor blocker (ARB), beta‐blocker, mineralocorticoid receptor antagonist (MRA), angiotension receptor–neprilysin inhibitor (ARNI), as well as cardiac resynchronization therapy (CRT) and ivabradine, if indicated.1 Furthermore, treatment with cardiac glycosides is recommended for rate control in HF patients with atrial fibrillation according to both ESC HF and atrial fibrillation guidelines.1, 13 Nevertheless, prescription rates of cardiac glycosides of currently 20–25% in European and ≈10% in U.S. HFrEF patient populations are steadily declining,14, 15, 16 possibly related to concerns about cardiac glycoside toxicity and because prospective randomized evidence in HF patients treated according to current standards is lacking.
Digitoxin shares the pharmacodynamic effects as digoxin, but has a more stable pharmacokinetic profile. Compared to digoxin, stable digitoxin serum levels are better maintained during treatment and the drug does not accumulate in patients with impaired renal function. This applies even in the elderly, in whom digitoxin dosage often has to be reduced because of a reduced skeletal muscle mass taking into account the high protein binding and main distribution of digitoxin in this compartment. Clinical drug–drug interactions extensively researched for in cardiac glycosides also have to be accounted for digitoxin.17 However, despite its similar pharmacodynamic but favourable pharmakokinetic profile compared to digoxin, adequately powered trials reporting on the effect of treatment with digitoxin on hard clinical endpoints in patients with HF are lacking.
Based on these considerations, DIGIT‐HF (DIGitoxin to Improve ouTcomes in patients with advanced chronic Heart Failure) aims to determine the therapeutic potential and safety of digitoxin in a randomized, blinded and controlled clinical trial design with adequate sample size in patients with HFrEF on top of optimized HF treatment.
Study design
The DIGIT‐HF trial is a pragmatic, multicentre, randomized, double‐blind, parallel‐group, placebo‐controlled, phase IV trial. The objective is to demonstrate that digitoxin preferably ranging at serum concentrations of 8–18 ng/mL (10.5–23.6 nmol/L) on top of standard of care (SOC) reduces the composite endpoint of all‐cause mortality or first hospital admission for worsening HF (whatever occurs first) in patients with advanced chronic HFrEF.
Trial population
Adult patients may enter the screening phase if they present with advanced chronic HFrEF of NYHA functional class III–IV and left ventricular ejection fraction ≤ 40%, or NYHA functional class II and left ventricular ejection fraction ≤ 30%. After the screening visit, patients suitable for study enrolment by meeting all inclusion (Table 1) and not violating any exclusion criteria (Table 2) are randomized in a 1:1 fashion to either digitoxin or placebo treatment. A total number of 2190 patients in approximately 50 study centres in Germany and Austria will be included (1095 per group). All patients receive SOC as recommended by ESC guidelines,1 building on treatment with beta‐blocker, ACEi/ARB, MRA, as well as, if indicated, ARNI, ivabradine, CRT, and implantable cardioverter‐defibrillators upon the discretion of the treating physician. If present at baseline, cardiac glycoside pre‐treatment is stopped and immediately switched to double‐blind study medication. Inclusion of patients previously treated with cardiac glycosides was considered important because this is a relevant subpopulation with more advanced HF18, 19 and exclusion of these patients may lead to selection of a healthier patient population with lower event rates. However, because of potential withdrawal effects in patients with cardiac glycoside pre‐treatment, we re‐analysed data from the DIG trial. In the DIG trial, about 44% of patients received digoxin before randomization and were treated in a randomized withdrawal design, whereas 56% of patients were not treated with digoxin before randomization and were treated de novo with digoxin or placebo. In the subgroup analysis reported in the DIG trial, results on the composite of death or hospitalization for worsening HF of patients who received and did not receive digoxin before randomization were consistent with the overall trial population.6 In addition, the original data from the DIG trial requested and received from the National Heart, Lung, and Blood Institute (NHLBI) were analysed to exclude differential effects of withdrawal and onset of digoxin on endpoints and no inconsistencies were found. Based on these results, withdrawal effects by inclusion of patients previously treated with cardiac glycosides are not expected. Nonetheless, randomization is stratified for previous cardiac glycoside use and a subgroup analysis is planned to detect potential withdrawal effects.
Table 1.
Inclusion criteria
| 1. | Signed written informed consent and willingness to comply with treatment and follow‐up |
| 2. | Male and female patients aged ≥ 18 years at the day of inclusion |
| 3. | Capable to understand the investigational nature, potential risks and benefits of the clinical trial |
| 4. | Chronic heart failure with symptoms compatible with New York Heart Association functional class III–IV and a left ventricular ejection |
| fraction ≤ 40%, or New York Heart Association functional class II with left ventricular ejection fraction ≤ 30% (determined at screening by | |
| echocardiography or cardiac magnetic resonance tomography or within 8 weeks prior to study inclusion by left ventricular angiography, | |
| echocardiography, radionuclide ventriculography, cardiac magnetic resonance tomography) | |
| AND | |
| a heart failure therapy based on current ESC guideline recommendations for a duration of at least 6 months upon | |
| discretion of the treating physician | |
| 5. | Women without childbearing potential defined as one or more of the following: |
| a. Women at least 6 weeks after surgical sterilization by bilateral tubal ligation or bilateral oophorectomy with or without hysterectomy at theday of inclusion | |
| b. Women ≥ 50 years of age at the day of inclusion who have been postmenopausal since at least 1 year | |
| c. Women < 50 years and in postmenopausal state ≥ 1 year with serum FSH > 40 IU/L (ascertained by a second laboratory assessment after 4 weeks) | |
| OR | |
| Women of childbearing potential who have a negative hCG pregnancy test and agree to meet one or more of the following | |
| criteria from the time of screening/baseline, during the study and for a period of 40 days following the last administration of study medication: | |
| a. Correct use of reliable contraception methods. This includes hormonal contraceptive (oral contraceptives, implants, transdermal patches,hormonal vaginal devices or injections with prolonged release) or an intrauterine device/system or a barrier method of contraception suchas condom or occlusive cap (diaphragm or cervical/vault caps) with spermicide (foam/gel/film/cream/suppository) | |
| b. True abstinence (periodic abstinence and withdrawal are not acceptable methods of contraception) | |
| c. Sexual relationship only with female partners and/or sterile male partnersORMen |
Table 2.
Exclusion criteria
| 1. | Recent (< 2 months ago): myocardial infarction, coronary revascularization, surgery or catheter intervention for valvular heart disease, acute coronary syndrome, stroke or cerebral ischaemia, start of heart failure device therapy potentially improving left ventricular ejection fraction or heart failure symptoms (e.g. cardiac resynchronization therapy, cardiac contractility modulation, baroreflex activation therapy) |
| 2. | Scheduled surgery or catheter intervention for valvular heart disease or scheduled coronary revascularization |
| 3. | Active myocarditis |
| 4. | Complex congenital heart disease; this does not include: mild‐moderate valve disease, uncomplicated shunts (isolated patent foramen ovale, small atrial or ventricular septum defects without associated lesions), repaired secundum or sinus venosus atrial septal defects or ventricular septal defects without residua, previously ligated or occluded ductus arteriosus |
| 5. | High‐urgency listing for heart transplantation or scheduled therapy with left ventricular assist device |
| 6. | Heart rate < 60 b.p.m. (except if functional cardiac resynchronization therapy in place) |
| 7. | Sinoatrial/atrioventricular block >I degree, sick sinus syndrome or carotid sinus syndrome (except if pacemaker protected) |
| 8. | Proven or suspected accessory, atrioventricular pathways (e.g. Wolff–Parkinson–White syndrome) |
| 9. | History of symptomatic or sustained (≥ 30 s) ventricular arrhythmia unless a cardioverter‐defibrillator implanted |
| 10. | Current ventricular tachycardia or fibrillation (this means patients presenting with a running ventricular tachycardia or fibrillation. If ventricular arrhythmias are terminated and a cardioverter‐defibrillator is implanted, inclusion is allowed according to point 9) |
| 11. | Hypertrophic obstructive cardiomyopathy (idiopathic subaortic stenosis) |
| 12. | Cor pulmonale |
| 13. | Constrictive pericarditis |
| 14. | Thoracic aortic aneurysm (defined as diameter ≥ 45 mm) |
| 15. | Concomitant severe liver and renal disease |
| 16. | Persistent hypokalaemia (< 3.2 mM) |
| 17. | Hypercalcaemia or hypomagnesaemia, if clinically suspected and verified by laboratory testing (e.g. hyperparathyroidism, neoplasia‐induced hypercalcaemia, signs of neuromuscular hyperexcitability) |
| 18. | Present (within 6 weeks before baseline/day 0 visit) and continuous treatment with amiodarone [single or short‐term (up to 3 days), not continuous administration of amiodarone immediately before or during study treatment are acceptable] |
| 19. | Scheduled DCC in the next 24 h (e.g. patients not on cardiac glycosides with new‐onset atrial fibrillation. Patients already included and on treatment with IMP can continue IMP and study when scheduled for DCC) |
| 20. | Presence of both treatment with cardiac glycosides and atrial fibrillation |
| 21. | Simultaneous intravenous treatment with calcium salts |
| 22. | Evidence of cardiac glycoside intolerance or known hypersensitivity to any component of IMP |
| 23. | Suspected intoxication with cardiac glycosides |
| 24. | Unlikely compliance with protocol requirements |
| 25. | Pregnant and lactating women |
| 26. | Use of other investigational drugs or devices at the time of enrolment, or within 30 days prior to enrolment or 5 half‐lives for investigational drugs, whichever is longer |
| 27. | Life expectancy < 12 months (e.g. due to active cancer) |
DCC, direct current cardioversion; IMP, investigational medicinal product.
Patients with atrial fibrillation represent a substantial subgroup of patients with HFrEF. DIGIT‐HF aims to represent a common HFrEF population with a substantial proportion of atrial fibrillation, which is confirmed by current baseline characteristics with about 25% of patients having atrial fibrillation recruited to DIGIT‐HF. There is no proportion of patients with atrial fibrillation specified within the overall population of the trial. In order to balance prognostic differences, randomization is stratified for atrial fibrillation and respective subgroup analyses for all stratification factors will be carried out. Patients with atrial fibrillation are not excluded unless they are already treated with cardiac glycosides because randomization to placebo with termination of pre‐existing cardiac glycoside therapy could cause significant tachyarrhythmia. As atrial fibrillation is frequent in patients with HF and cardiac glycosides are commonly used for heart rate control, in the study protocol clear procedures were defined for treatment of atrial fibrillation during the course of the trial based on ESC guidelines for the treatment of HF and atrial fibrillation1, 13 (e.g. rhythm/rate control, direct current cardioversion, amiodarone use) (online supplementary material, DIGIT‐HF study protocol, Chapter 5.4, Treatment of atrial fibrillation). In particular, termination of study medication is defined if sufficient rate control requires treatment with cardiac glycosides or amiodarone, which are also exclusion criteria at baseline (exclusion criteria no. 18 and 20). Minimal or maximal heart rates are not specified at presence of atrial fibrillation at baseline. However, a heart rate ≤ 60 b.p.m. excludes patients with sinus rhythm as well as atrial fibrillation except if functional CRT is in place (exclusion criteria no. 6).
Study flow and dose adjustment
Digitoxin or matching placebo is given orally as continuous treatment until the end of the study on top of SOC. The target range of digitoxin serum concentration is 8–18 ng/mL (10.5–23.6 nmol/L) throughout the study. The starting dose is 0.07 mg digitoxin once daily (o.d.) or placebo. After 6 weeks of treatment, serum concentrations of digitoxin are determined in a core laboratory and dose adjustments are initiated centrally to avoid unblinding. In the placebo group, dose adjustment is randomly assigned. In the digitoxin group, dose adjustment employs a pre‐defined algorithm. If digitoxin serum levels are outside the target range, doses are reduced or increased to 0.05 or 0.1 mg digitoxin o.d., accordingly. Otherwise, the dose of digitoxin is maintained. In patients requiring up‐titration, a second measurement of digitoxin serum concentrations is performed 6 weeks later (i.e. 12 weeks after randomization) to confirm digitoxin serum concentrations within the target range. Again, in the placebo group, dose adjustment is randomly assigned. In the digitoxin group, dose is reduced to 0.07 mg o.d. if serum concentration is above target range. Otherwise, the present dose of 0.1 mg digitoxin o.d. is maintained. An overview of the study flow is depicted in Figure 1.
Figure 1.
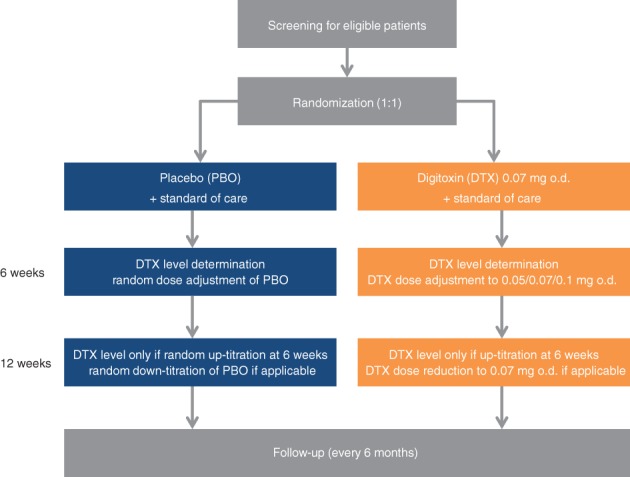
Flowchart of the DIGIT‐HF trial.
If digitoxin serum levels after 6 weeks exceed a concentration of 25 ng/mL (33 nmol/L), i.e. the upper limit of the therapeutic range formerly used in clinical practice, it is considered highly unlikely that the target range can be achieved with the above described dose adjustment. For safety reasons (as the study hypothesis is based on association between mortality and serum concentrations retrospectively observed in the DIG trial), patients with digitoxin serum levels > 25 ng/mL (> 33 nmol/L) should not continue digitoxin treatment with the proposed doses. In these patients, an adapted dose adjustment will be performed. If digitoxin serum concentrations are > 25 ng/mL (> 33 nmol/L) after 6 weeks, treatment will be paused for 6 weeks and re‐started with a dose of 0.05 mg taken every second day. If digitoxin serum levels determined 12 weeks or 12 months after randomization (the latter time‐point applicable only for scientific and safety reasons, see below) exceed 25 ng/mL (33 nmol/L), patients should not continue active treatment for safety reasons. To keep the trial pragmatic and to avoid unblinding of these patients, treatment is then switched to placebo. Digitoxin serum concentrations are also determined in all patients after 12 months for scientific and safety reasons only.
Outpatient follow‐up is scheduled every 6 months. The course of the disease including severity, signs and symptoms of HF, and medication are documented in detail. Quality of life is assessed at baseline and after 12 months by means of the 12‐item Short‐Form Health Survey. All patients will be followed until the end of the study and will have an end of treatment visit.
Outcomes
The primary endpoint is a composite of all‐cause mortality and hospital admission for worsening HF (whatever occurs first). The latter is defined by the presence of all of the following: (i) worsening of HF based on clinical judgement (presence of HF symptoms) by the treating physician; (ii) hospital stay overnight; and (iii) intravenous treatment with diuretics or vasoactive substances (e.g. nitroglycerine) or inotropes (e.g. dobutamine).
Key secondary endpoints are components of the composite and recurrent hospital admission for worsening HF. Further secondary endpoints are cardiovascular death, death from HF, any non‐cardiovascular death, fatal or non‐fatal myocardial infarction, fatal or non‐fatal stroke, any cardiovascular hospitalization, hospital admission for any cause, implantation of a cardioverter‐defibrillator, implantation of a CRT device, implantation of a pacemaker, sudden cardiac death, and change in NYHA functional class.
Safety and tolerability of treatment are documented and adverse events, serious adverse events and laboratory abnormalities will be compared between the two treatment groups.
Blinding and randomization
The study is double‐blind. Randomization is stratified by centre, sex, NYHA functional class (II, III, or IV), atrial fibrillation, and pre‐treatment with cardiac glycosides at baseline. Randomization, assessment of digitoxin serum levels and blinded dose adjustment are performed centrally via a web portal, which is also used for emergency unblinding if necessary. Placebo patients receive random dose adjustments. Investigators can stop or pause study medication without unblinding the patient, if deemed necessary.
Study duration
Patients are followed until 734 adjudicated primary events have been recorded. At 367 (50%) events an interim analysis is planned, and the study may be terminated for proven efficacy, but not for futility. This interim analysis is performed by an independent statistician and termination for proven efficacy is judged by the independent Data Monitoring Committee (see below) to maintain blinding. All patients will be followed until the end of the study. A final visit will be conducted for each patient 40 days after finishing treatment with the study medication; patients who withdraw study participation are encouraged to take part in this final visit and in follow‐up via telephone visits. At the end of the study, patient treatment including cardiac glycosides is left at the discretion of the treating physician, who may request unblinding of the patient at this time‐point.
Statistical analysis
The primary analysis is based on the intention‐to‐treat population, which comprises all randomized patients who took at least one dose of the study drug. The primary null hypothesis of this study is that both treatment groups have equal hazard rates. This null hypothesis will be evaluated with a multivariable Cox proportional hazards regression model including the time to death or first hospitalization for worsening HF as a dependent variable, and study treatment allocation (digitoxin or placebo), centre, sex, NYHA functional class (II, III, or IV), atrial fibrillation, and treatment with cardiac glycosides at baseline as independent factors. Superiority of digitoxin over placebo regarding the composite endpoint can be concluded if the median time to event is longer in the digitoxin‐treated group and the corresponding P‐value of the treatment effect estimated from the Cox regression model is less than the pre‐specified two‐sided significance level of 1% at interim or 4.5% in the final analysis.20
As soon as the primary hypothesis is rejected, non‐inferiority of digitoxin against placebo concerning all‐cause mortality will be assessed as the first secondary analysis to exclude detrimental effects. Non‐inferiority will be concluded if the upper bound of the one‐sided 97.5% confidence interval of the digitoxin/placebo hazard ratio is below 1.303, whereby the hazard ratio and the confidence interval are derived from a Cox regression model as specified for the primary analysis. Superiority will be concluded if there is a significant difference between digitoxin and placebo regarding overall survival in the respective Cox regression model as specified for the primary analysis (i.e. the upper bound of the one‐sided 97.5% confidence interval of the digitoxin/placebo hazard ratio is below 1). If superiority of digitoxin over placebo regarding overall mortality is demonstrated, substudies will be tested at the significance level of 5%. Otherwise, substudy analysis will be conducted non‐confirmatory. There are no multiplicity issues as the test procedure is hierarchical. All secondary analyses will be explorative. Adverse events, serious adverse events and laboratory abnormalities will be compared between treatment groups with a chi‐square test and other appropriate tests.
Determination of sample size
The required sample size is estimated for a log‐rank test. Assumptions are based on the data from the SHIFT trial (ISRCTN70429960).21 Since eligible patients are expected to have a more severe heart disease than patients studied in SHIFT, it is assumed that the treatment effect of digitoxin is in the same order as the effect of ivabradine observed in the SHIFT trial, although some of the patients are treated with ivabradine as part of SOC. Based on the SHIFT trial, event rates of 26% (digitoxin) and 31% (placebo) after 24 months are expected for the primary outcome (2% were added to the rates of 24% vs. 29% for the composite of hospitalization for HF or cardiovascular death reported in SHIFT, because all‐cause mortality was 2% higher than cardiovascular mortality). Exponential survival is assumed. Additionally, it is assumed that adjustment for stratification variables – as planned for the primary analysis – will not reduce power. The two‐sided family‐wise type‐I error rate is set to 0.05 (0.01 at interim and 0.045 at the final analysis20) and the power is set to 0.8. The required sample size is estimated for an accrual period of 36 months and a maximum length of follow‐up of 48 months. Under these assumptions, a total of 2190 patients (1095 patients per group) with 734 events (total) is required to prove superiority of digitoxin over placebo. Should recruitment of patients require more time than anticipated, the follow‐up period will be extended and because this is an event‐driven trial, a smaller sample size will then be needed to observe the required number of events.
Substudies
Pre‐specified substudies will investigate the effect of digitoxin on endothelial function, arrhythmic burden, left ventricular remodelling and function, and exercise capacity. Substudies will be included in the hierarchical test procedure of the trial after primary and key secondary analyses have been performed. The hierarchical order of testing will be determined before unblinding.
Trial organization
The principal investigators and the Hannover Clinical Trail Center (HCTC) representing the trial sponsor (Hannover Medical School) are responsible for all aspects of the study protocol and amendments. The trial Steering Committee ensures scientific quality in all study‐related aspects. An independent Data Monitoring Committee is responsible for reviewing unblinded safety data at regular intervals during the study and may recommend stopping the trial because of harmful effects. An independent Clinical Event Adjudication Committee (CEAC) will rate all causes and circumstances of hospitalizations and death according to pre‐specified criteria in the CEAC charter.
This study is conducted in compliance with the German Drug Law (AMG), the German Good Clinical Practice (GCP) ordinance, ICH GCP guidelines, and other applicable ethical and regulatory requirements. The study is supported by the Federal Ministry of Education and Research (BMBF) under grant number 01KG1303. The DIGIT‐HF trial is registered at EudraCT (2013–005326‐38). Further information can be obtained from the trial protocol (online supplementary material, DIGIT‐HF study protocol).
Discussion
Study rationale
The effect of cardiac glycosides on patient outcome remains unclear despite the long history of its clinical use and available data from prospective randomized trials6, 7, 8 in HF (see comparison of RADIANCE, PROVED, and DIG with DIGIT‐HF in the online supplementary Table S1). While the prescription rates of cardiac glycosides declined substantially in recent years, it is unclear whether this is justified against the benefit in HF hospitalizations seen with digoxin in the DIG trial (number needed to treat for 3 years to avoid one HF hospitalization: 10–12).6, 10, 11, 12 Moreover, the large randomized trials showing the benefits of ACEi, beta‐blockers, MRA, ivabradine, CRT, and recently also ARNI, have been performed with sizable proportions of background therapy including cardiac glycosides, i.e. in 25–90% of the study populations.21, 22, 23, 24, 25, 26, 27, 28 Currently, it is unclear whether the above listed interventions may have exerted their benefits independent of cardiac glycoside treatment. Importantly, the survival benefit of spironolactone on top of a background therapy with ACEi (95% of trial population) in the RALES trial was only significant in patients receiving digoxin; in this trial, background therapy with beta‐blocker was very low (10%), but was high with cardiac glycosides (75%).27 In addition, a recent retrospective analysis of the DIG trial using entry criteria (heart rate > 70 b.p.m.) and the primary composite endpoint used in SHIFT (i.e. cardiovascular death or hospital admission for worsening HF) found a similar risk reduction regarding both the composite outcome and its components among patients receiving digoxin or ivabradine.29 Yet, data demonstrating the impact of cardiac glycosides on mortality and morbidity in patients with HF and atrial fibrillation are scarce and only available from observational studies or retrospective analyses. Results were conflicting and contradictory, even if performed in the same data set. This was mainly due to limitations such as prescription bias and invisible confounders, which cannot be biometrically rectified, as well as missing information on cardiac glycoside serum concentrations for proper risk adjustment.18, 19, 30
Drug and dose rationale
In contrast to digoxin, no trial has yet investigated the effect of digitoxin on clinically relevant cardiovascular endpoints in HF patients, despite its more stable pharmacokinetics and higher pharmacological stability.17 In patients with chronic HF (NYHA class II–III), daily administration of 0.07 mg digitoxin decreased end‐systolic diameters and increased posterior wall motion amplitude, stroke volume, shortening fraction, and early diastolic left ventricular filling speed.31 When applied at mean doses of 0.06–0.07 mg o.d., digitoxin serum concentrations remained constantly low, ranging between 6.4–9.4 ng/mL (8.5–12.2 nM) – even in patients with impaired renal function.32 Concentrations of digitoxin in a range of 8–18 ng/mL increased parameters of systolic function in dose–response analyses in man equivalent to digoxin concentrations of 0.5–0.9 ng/mL,33 i.e. the concentration associated with a reduced overall mortality risk in patients with HFrEF studied in the DIG trial.6 Our own studies demonstrated that treatment of endothelial cells with digitoxin at concentrations of 10–30 nM (7.65–22.95 ng/mL) effectively inhibited cytokine‐induced pro‐inflammatory processes (e.g. expression of monocyte chemoattractant protein‐1, vascular cell adhesion molecule‐1, monocyte adhesion), production of reactive oxygen species and apoptosis, but increased the expression and activation of the endothelial nitric oxide synthase.34 Based on these studies, concentrations of digitoxin ≥ 8 ng/mL seem to be necessary for improving myocardial function, endothelial dysfunction35 and ultimately prognosis in patients with advanced chronic HFrEF. Therefore, digitoxin target serum concentrations of 8–18 ng/mL (10.5–23.6 nmol/L) were chosen for the DIGIT‐HF trial. Analysis of blinded dose adjustments to achieve target serum concentrations in the first 300 patients randomized into DIGIT‐HF verified the chosen dose adjustment protocol supporting simplicity of trial conduct.36 The verified dose adjustment protocol also supports safe use of digitoxin even in elderly patients with a reduced skeletal muscle mass and hence reduced volume of distribution due to the high protein binding of digitoxin. Possible pharmacodynamic and pharmacokinetic drug–drug interactions between digitoxin and a variety of drugs have to be taken into account. However, recommendations not to use certain concomitant medication affecting pharmacodynamics and pharmacokinetics (e.g. colestyramine, colestipol) in the study protocol (online supplementary material, DIGIT‐HF study protocol, Chapter 5.4, Concomitant therapy) and in particular the exclusion of amiodarone, which is known to reduce hepatic digitoxin metabolism by inducing enzyme inhibition resulting in significantly increased digitoxin levels,17 seem to be effective measures to avoid serious side effects so far.
Although direct comparison of digoxin with digitoxin on outcome in HFrEF in a prospective, placebo‐controlled, randomized clinical trial would be preferable, implementation of this trial most likely will not be feasible. However, if present data of the DIG trial are supported by the outcome data of DIGIT‐HF and because of the almost identical pharmacodynamic properties,17 analogous use of digoxin and digitoxin should be possible for evidence‐based HF therapy.
Summary
The DIGIT‐HF clinical outcome trial will provide important and urgently required evidence, whether digitoxin against a background of contemporary drug and device HF therapy may improve mortality and/or morbidity in patients with advanced chronic HFrEF presenting with sinus rhythm or atrial fibrillation.
Funding
The study is funded by the Federal Ministry of Education and Research (BMBF; 01KG1303).
Conflict of interest: none declared.
Supporting information
Supplementary material. DIGIT‐HF study protocol.
Table S1. Summary of prospective studies on cardiac glycoside use in patients with heart failure.
DIGIT‐HF Investigators and Committees
Principal Investigators: Udo Bavendiek, Johann Bauersachs.
Steering Committee: Udo Bavendiek, Johann Bauersachs, Armin Koch, Heiko von der Leyen, Christian Veltmann, Michael Böhm, Stefan Störk.
Clinical Event Adjudication Committee: Ulrich Tebbe, Stephan von Haehling, Markus Haass.
Data Safety Monitoring Committee: Stefan Anker, Paul Mohacsi, Gerhard Pölzl, Helmut Trampisch.
Biometry: Lukas Aguirre Dávila, Kristina Weber.
Project and Data Management: Silke Zimmermann, Barbara Neuhaus.
Sponsor: Hannover Medical School.
Study Coordinator: Udo Bavendiek.
Contributor Information
Udo Bavendiek, Email: bavendiek.udo@mh-hannover.de.
on behalf of the DIGIT‐HF Investigators and Committees (see Appendix):
Udo Bavendiek, Johann Bauersachs, Armin Koch, Heiko von der Leyen, Christian Veltmann, Michael Böhm, Stefan Störk, Ulrich Tebbe, Stephan von Haehling, Markus Haass, Stefan Anker, Paul Mohacsi, Gerhard Pölzl, Helmut Trampisch, Lukas Aguirre Dávila, Kristina Weber, Silke Zimmermann, and Barbara Neuhaus
References
- 1. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 2. Christ M, Stork S, Dorr M, Heppner HJ, Muller C, Wachter R, Riemer U; Trend HF Germany Project . Heart failure epidemiology 2000‐2013: insights from the German Federal Health Monitoring System. Eur J Heart Fail 2016;18:1009–1018. [DOI] [PubMed] [Google Scholar]
- 3. Neumann T, Biermann J, Erbel R, Neumann A, Wasem J, Ertl G, Dietz R. Heart failure: the commonest reason for hospital admission in Germany: medical and economic perspectives. Dtsch Arztebl Int 2009;106:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guha K, McDonagh T. Heart failure epidemiology: European perspective. Curr Cardiol Rev 2013;9:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jhund PS, Macintyre K, Simpson CR, Lewsey JD, Stewart S, Redpath A, Chalmers JW, Capewell S, McMurray JJ. Long‐term trends in first hospitalization for heart failure and subsequent survival between 1986 and 2003: a population study of 5.1 million people. Circulation 2009;119:515–523. [DOI] [PubMed] [Google Scholar]
- 6. Digitalis Investigation Group . The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 1997;336:525–533. [DOI] [PubMed] [Google Scholar]
- 7. Uretsky BF, Young JB, Shahidi FE, Yellen LG, Harrison MC, Jolly MK. Randomized study assessing the effect of digoxin withdrawal in patients with mild to moderate chronic congestive heart failure: results of the PROVED trial. PROVED Investigative Group. J Am Coll Cardiol 1993;22:955–962. [DOI] [PubMed] [Google Scholar]
- 8. Packer M, Gheorghiade M, Young JB, Costantini PJ, Adams KF, Cody RJ, Smith LK, Van Voorhees L, Gourley LA, Jolly MK. Withdrawal of digoxin from patients with chronic heart failure treated with angiotensin‐converting‐enzyme inhibitors. RADIANCE Study. N Engl J Med 1993;329:1–7. [DOI] [PubMed] [Google Scholar]
- 9. Young JB, Gheorghiade M, Uretsky BF, Patterson JH, Adams KF Jr. Superiority of "triple" drug therapy in heart failure: insights from the PROVED and RADIANCE trials. Prospective Randomized Study of Ventricular Function and Efficacy of Digoxin. Randomized Assessment of Digoxin and Inhibitors of Angiotensin‐Converting Enzyme. J Am Coll Cardiol 1998;32:686–692. [DOI] [PubMed] [Google Scholar]
- 10. Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 2003;289:871–878. [DOI] [PubMed] [Google Scholar]
- 11. Ahmed A, Rich MW, Love TE, Lloyd‐Jones DM, Aban IB, Colucci WS, Adams KF, Gheorghiade M. Digoxin and reduction in mortality and hospitalization in heart failure: a comprehensive post hoc analysis of the DIG trial. Eur Heart J 2006;27:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahmed A, Pitt B, Rahimtoola SH, Waagstein F, White M, Love TE, Braunwald E. Effects of digoxin at low serum concentrations on mortality and hospitalization in heart failure: a propensity‐matched study of the DIG trial. Int J Cardiol 2008;123:138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, Castella M, Diener HC, Heidbuchel H, Hendriks J, Hindricks G, Manolis AS, Oldgren J, Popescu BA, Schotten U, Van Putte B, Vardas P. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J 2016;37:2893–2962. [DOI] [PubMed] [Google Scholar]
- 14. Cox JL, Ramer SA, Lee DS, Humphries K, Pilote L, Svenson L, Tu JV. Pharmacological treatment of congestive heart failure in Canada: a description of care in five provinces. Can J Cardiol 2005;21:337–343. [PubMed] [Google Scholar]
- 15. Patel N, Ju C, Macon C, Thadani U, Schulte PJ, Hernandez AF, Bhatt DL, Butler J, Yancy CW, Fonarow GC. Temporal trends of digoxin use in patients hospitalized with heart failure: analysis from the American Heart Association Get With the Guidelines‐Heart Failure Registry. JACC Heart Fail 2016;4:348–356. [DOI] [PubMed] [Google Scholar]
- 16. Chioncel O, Lainscak M, Seferovic PM, Anker SD, Crespo‐Leiro MG, Harjola VP, Parissis J, Laroche C, Piepoli MF, Fonseca C, Mebazaa A, Lund L, Ambrosio GA, Coats AJ, Ferrari R, Ruschitzka F, Maggioni AP, Filippatos G. Epidemiology and one‐year outcomes in patients with chronic heart failure and preserved, mid‐range and reduced ejection fraction: an analysis of the ESC Heart Failure Long‐Term Registry. Eur J Heart Fail 2017;19:1574–1585. [DOI] [PubMed] [Google Scholar]
- 17. Belz GG, Breithaupt‐Grogler K, Osowski U. Treatment of congestive heart failure – current status of use of digitoxin. Eur J Clin Invest 2001;31:10–17. [DOI] [PubMed] [Google Scholar]
- 18. Ziff OJ, Lane DA, Samra M, Griffith M, Kirchhof P, Lip GY, Steeds RP, Townend J, Kotecha D. Safety and efficacy of digoxin: systematic review and meta‐analysis of observational and controlled trial data. BMJ 2015;351:h4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bavendiek U, Aguirre Davila L, Koch A, Bauersachs J. Assumption versus evidence: the case of digoxin in atrial fibrillation and heart failure. Eur Heart J 2017;38:2095–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fleming TR, Harrington DP, O'Brien PC. Designs for group sequential tests. Control Clin Trials 1984;5:348–361. [DOI] [PubMed] [Google Scholar]
- 21. Swedberg K, Komajda M, Bohm M, Borer JS, Ford I, Dubost‐Brama A, Lerebours G, Tavazzi L; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo‐controlled study. Lancet 2010;376:875–885. [DOI] [PubMed] [Google Scholar]
- 22. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR; PARADIGM‐HF Investigators and Committees . Angiotensin‐neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014;371:993–1004. [DOI] [PubMed] [Google Scholar]
- 23. CONSENSUS Trial Study Group . Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med 1987;316:1429–1435. [DOI] [PubMed] [Google Scholar]
- 24. Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN; SOLV Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991;325:293–302. [DOI] [PubMed] [Google Scholar]
- 25. Effect of metoprolol CR/XL in chronic heart failure . Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT‐HF). Lancet 1999;353:2001–2007. [PubMed] [Google Scholar]
- 26. Moss AJ, Hall WJ, Cannom DS, Klein H, Brown MW, Daubert JP, Estes NA 3rd, Foster E, Greenberg H, Higgins SL, Pfeffer MA, Solomon SD, Wilber D, Zareba W; MADIT‐CRT Trial Investigators. Cardiac‐resynchronization therapy for the prevention of heart‐failure events. N Engl J Med 2009;361:1329–1338. [DOI] [PubMed] [Google Scholar]
- 27. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 28. Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B; EMPHASIS‐HF Study Group . Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011;364:11–21.21073363 [Google Scholar]
- 29. Castagno D, Petrie MC, Claggett B, McMurray J. Should we SHIFT our thinking about digoxin? Observations on ivabradine and heart rate reduction in heart failure. Eur Heart J 2012;33:1137–1141. [DOI] [PubMed] [Google Scholar]
- 30. Rush CJ, Campbell RT, Jhund PS, Petrie MC, McMurray JJ. Association is not causation: treatment effects cannot be estimated from observational data in heart failure. Eur Heart J 2018;39:3417–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Staiger J, Spath J, Dickhuth HH, Keul J. Ventricle function in low‐dose digitoxin in patients with chronic heart failure (stage II/III). Z Kardiol 1983;72:448–455. [PubMed] [Google Scholar]
- 32. Storstein L. Studies on digitalis. XI. Digitoxin metabolism in patients with impaired renal function. Clin Pharmacol Ther 1977;21:536–546. [DOI] [PubMed] [Google Scholar]
- 33. Belz GG, Erbel R, Schumann K, Gilfrich HJ. Dose‐response relationships and plasma concentrations of digitalis glycosides in man. Eur J Clin Pharmacol 1978;13:103–111. [DOI] [PubMed] [Google Scholar]
- 34. Jagielska J, Salguero G, Schieffer B, Bavendiek U. Digitoxin elicits anti‐inflammatory and vasoprotective properties in endothelial cells: therapeutic implications for the treatment of atherosclerosis? Atherosclerosis 2009;206:390–396. [DOI] [PubMed] [Google Scholar]
- 35. Bauersachs J, Widder JD. Endothelial dysfunction in heart failure. Pharmacol Rep 2008;60:119–126. [PubMed] [Google Scholar]
- 36. Bavendiek U, Aguirre Davila L, Schwab J, Phillip SA, Westenfeld R, Maier LS, Stoerk S, Weber K, Koch A, Bauersachs J; DIGIT‐HF Study Group . Digitoxin serum concentrations affecting patient safety and potential outcome in patients with HFrEF – analyses of the ongoing DIGIT‐HF‐trial. Eur Heart J 2017;38(Suppl 1): P6168 (abstr). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material. DIGIT‐HF study protocol.
Table S1. Summary of prospective studies on cardiac glycoside use in patients with heart failure.