

Abstract
Aims
This study was designed to evaluate the safety, tolerability and haemodynamic effects of BMS‐986231, a novel second‐generation nitroxyl donor with potential inotropic, lusitropic and vasodilatory effects in patients hospitalized with decompensated heart failure and reduced ejection fraction (HFrEF).
Methods and results
Forty‐six patients hospitalized with decompensated HFrEF were enrolled into four sequential dose‐escalation cohorts in this double‐blind, randomized, placebo‐controlled Phase 2a study. Patients with baseline pulmonary capillary wedge pressure (PCWP) of ≥20 mmHg and a cardiac index of ≤2.5 L/min/m2 received one 6‐h i.v. infusion of BMS‐986231 (at 3, 5, 7 or 12 µg/kg/min) or placebo. BMS‐986231 produced rapid and sustained reductions in PCWP, as well as consistent reductions in time‐averaged pulmonary arterial systolic pressure, pulmonary arterial diastolic pressure and right atrial pressure. BMS‐986231 increased non‐invasively measured time‐averaged stroke volume index, cardiac index and cardiac power index values, and decreased total peripheral vascular resistance. There was no evidence of increased heart rate, drug‐related arrhythmia or symptomatic hypotension with BMS‐986231. Analyses of adverse events throughout the 30‐day follow‐up did not identify any toxicities specific to BMS‐986231, with the potential exception of infrequent mild‐to‐moderate headaches during infusion. There were no treatment‐related serious adverse events.
Conclusions
BMS‐986231 demonstrated a favourable safety and haemodynamic profile in patients hospitalized with advanced heart failure. Based on preclinical data and these study's findings, it is possible that the haemodynamic benefits may be mediated by inotropic and/or lusitropic as well as vasodilatory effects. The therapeutic potential of BMS‐986231 should be further assessed in patients with heart failure.
Keywords: BMS‐986231, CXL‐1427, Heart failure, Human, Nitroxyl
Introduction
Despite the substantial burden of heart failure (HF), few new pharmacological therapies have been introduced in decades. Diuretics, the current cornerstone of therapy for HF, act to reduce excess fluid that has been retained. A subset of patients with HF also receive vasodilators to reduce afterload, and an even smaller proportion receive inotropes to increase cardiac contractility.1, 2 However, current therapies are associated with a variety of side effects and limitations,1, 3 and thus there is a need for safe and effective therapy that reduces cardiac loads and enhances cardiac output (CO) by improving both diastolic and systolic function of the left ventricle.
Nitroxyl (HNO) is a reactive nitrogen molecule that exhibits many unique biological and pharmacological effects in the cardiovascular system.4 Nitroxyl improves cardiac function by a novel mechanism of action that includes direct augmentation of myocardial contractility and relaxation, together with combined arterial and venous dilation. The effect of HNO on cardiomyocytes is mediated through a unique cAMP‐independent mechanism that results in a reversible post‐translational modification of selective cysteine residues in proteins involved in excitation–contraction coupling machinery. Cysteine modifications of SERCA2a,5, 6 phospholamban7, 8 and the ryanodine receptor5 enhance calcium cycling without altering L‐type calcium channel activity or total sarcoplasmic calcium load.9 In addition, HNO modifies cardiac myofilaments and increases myofilament calcium sensitivity.10, 11 The vasodilatory effects of HNO are mediated via the activation of soluble guanylate cyclase.12 Consistent with this mechanism of action, earlier generations of HNO donors have shown beneficial effects on cardiac function by reducing cardiac preload and afterload, and increasing CO. Increase in CO is mediated in part by direct positive inotropic and lusitropic effects, as evidenced by preclinical mechanistic studies in isolated cardiomyocytes and pressure–volume loop studies in dogs with HF.13, 14, 15 CXL‐1020, a first‐generation HNO donor, was also associated with improvements in stroke volume index (SVI) and cardiac index (CI) in comparison with placebo in patients with decompensated HF, with no associated increase in heart rate (HR).13 Nitroxyl donors have not been associated with the adverse cardiac effects of currently available legacy inotropes, such as tachycardia and arrhythmias, and they possess many characteristics that distinguish them from nitric oxide‐releasing vasodilators.13, 15, 16, 17, 18
BMS‐986231, formerly CXL‐1427, is a novel second‐generation HNO donor with a half‐life of approximately 40–144 min in healthy individuals.19 Preliminary Phase 1 study data for BMS‐986231 in healthy volunteers demonstrated that it was well tolerated up to a dose of 10 µg/kg/min for 48 h and produced dose‐dependent changes in haemodynamic parameters without affecting HR.19 Studies with BMS‐986231 in canine models of HF have also reported reductions over time in Tau and the end‐diastolic pressure–volume relationship, and improvements in left ventricular (LV) ejection fraction (EF)/fractional area shortening, end‐systolic pressure volume and preload–recruitable stroke work relationship parameters.20, 21 This suggests BMS‐986231 has positive lusitropic and inotropic effects.
The primary objectives for this Phase 2a study included assessment of the safety, tolerability and haemodynamic effects of 6‐h i.v. infusions of BMS‐986231 or placebo in hospitalized patients with advanced HF [i.e. HF with reduced EF (HFrEF)], including patients hospitalized for acute decompensation.
Methods
Patient population and study protocol
This was a randomized, double‐blind, placebo‐controlled, multicentre, Phase 2a, dose–response study of BMS‐986231 in patients hospitalized for advanced HFrEF, including (but not exclusive to) those with acute decompensation (trial registration no. NCT02157506). Primary pharmacodynamic endpoints included changes from baseline during infusion in pulmonary capillary wedge pressure (PCWP), pulmonary arterial diastolic pressure (PADP), and CI using the Fick principle, as measured by an indwelling pulmonary artery catheter (PAC). Secondary endpoints included changes from baseline for additional haemodynamic parameters including right atrial pressure (RAP), pulmonary arterial systolic pressure (PASP), peripheral arterial systolic blood pressure (SBP), peripheral arterial diastolic blood pressure (DBP), calculated mean arterial pressure (MAP) and HR. Other secondary factors assessed included BMS‐986231 plasma concentrations at the end of infusion, as well as dose–pharmacodynamic (PD) effect and pharmacokinetic (PK)–PD effect relationships in central and peripheral haemodynamic parameters.
Patients were required to be aged 18–85 years and to have been admitted to hospital with HFrEF. The study population included, but was not limited to, patients admitted to hospital for acute decompensation. Patients were also required to have: an indwelling PAC in place for assessment of central haemodynamic parameters; an LVEF of ≤40%; a Fick and/or thermodilution determination of CI of ≤2.5 L/min/m2, and PCWP (or PADP, if a PCWP waveform could not be reliably obtained) of ≥20 mmHg if SBP was ≥100 mmHg or ≥22 mmHg if SBP was 95–99 mmHg.
Select exclusion criteria were: HR of <50 b.p.m. or >110 b.p.m. at baseline; screening or baseline SBP of >150 mmHg or <100 mmHg if PCWP was ≥20 mmHg but <22 mmHg, or <95 mmHg if PCWP was ≥22 mmHg; tricuspid or pulmonary valve prosthesis or endocarditis; right heart mass; history of pneumothorax or haemothorax, and a bleeding diathesis that would preclude the placement of a pulmonary artery line. Other criteria for exclusion were: a primary HF aetiology attributable to either restrictive or obstructive cardiomyopathy, idiopathic hypertrophic cardiomyopathy or uncorrected severe valvular disease; concomitant use of parenteral therapy with any antiarrhythmic drugs (oral therapy allowed), and atrial fibrillation/flutter with an uncontrolled rate (≥100 b.p.m.) at the time of randomization.
Background standard‐of‐care medications were allowed during the study. However, patients treated with or potentially requiring dopamine, dobutamine (within 2 h at US sites), enoximone, nesiritide, nitroglycerine or nitroprusside within 4 h, or levosimendan, amrinone or milrinone within 8 h of baseline haemodynamic assessment were excluded. Vasoactive medications, including i.v. or oral diuretics, were prohibited for ≥4 h prior to baseline haemodynamic assessment until completion of the 6‐h infusion, and the need to administer a diuretic (i.v. or oral) or any parenteral inotrope or vasodilator before completion of the infusion required discontinuation of the study drug infusion. The study was conducted in accordance with Good Clinical Practice [International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)] and the protocol approved by the relevant ethics committee for each institution. All participating patients provided written informed consent.
This study was designed to evaluate the safety, PD and PK effects of up to four ascending doses of BMS‐986231 in up to four cohorts of eight patients each (dose‐escalation cohorts, randomized six to active drug and two to placebo, with an initial sentinel 2:1 group), and subsequently to evaluate up to three of the initial dose levels in additional expansion cohorts (randomized three to active drug and one to placebo in blocks of four). Initially, patients were randomized in a double‐blind fashion to receive BMS‐986231 at a dose of 3 µg/kg/min or placebo continuously infused i.v. for 6 h. The pharmacist(s) at the study site who prepared the study drug infusion solutions and who was unblinded was responsible for randomizing patients using an interactive voice‐ or web‐based response system [IxRS (INC Research, Raleigh, NC, USA; S–Clinica, Inc., Iselin, NJ, USA)]. Study patients, the principal investigator and all other clinical staff members at the study site (in addition to the sponsor's clinical research associate monitors) were blinded to study drug assignments until after study completion. The dosing solutions for the placebo and the active study drug were indistinguishable in appearance. Subsequent BMS‐986231 dose levels of 5, 7 and 12 µg/kg/min were scheduled to be evaluated in a sequential fashion. After the first three patients in each dose‐escalation cohort had been randomized, dosed and evaluated, the safety and haemodynamic data were reviewed by a dose‐escalation review committee (DERC) which then decided whether the dose cohort should continue. After the eighth patient in each cohort had completed inpatient follow‐up, all safety and haemodynamic data were reviewed by the DERC before the study proceeded to the next dose.
The PAC was used to assess PCWP, RAP, PASP, PADP and Fick‐estimated CI at screening (≤4 h prior to initiation of the study drug infusion), baseline (≤30 min prior to initiation of the study drug infusion), during the 6‐h study drug infusion at 2 h, 4 h and 6 h, and at 2 h after the study drug infusion was completed (i.e. at 8 h). For the Fick estimation of CI, an assumed value for oxygen consumption (VO2) of 125 mg/mL/m2 was used. In a subset of 32 patients, CI was also assessed by thermodilution. Non‐invasive haemodynamic monitoring was performed using a non‐invasive continuous cardiac system (NICaS; NI Medical USA, Chapel Hill, NC, USA) device in all patients at baseline, during the 6‐h study drug infusion at 2 h, 4 h and 6 h, and at 2 h after the study drug infusion was completed. The NICaS device recorded SVI, which allowed for calculation of CI and cardiac power index (CPI). Measurements of blood pressure (BP) and HR were performed at baseline, every 30 min during infusion, and every 30 min for up to 2 h after the cessation of infusion.
Blood samples for PK analyses were collected at baseline, at the conclusion of the 6‐h infusion, and at 2 h after the cessation of infusion. Routine haematology, serum chemistry, coagulation and urinalysis parameters were assessed during screening, at baseline, at 6 h (serum chemistry only), at 24 h and at the first follow‐up visit on study day 8–12 (haematology and serum chemistry only). Plasma B‐type natriuretic peptide (BNP) was assessed at baseline and upon completion of the 6‐h infusion. Adverse events were recorded from the time a patient signed the consent form to 30 days after the patient's infusion of the study drug had been completed.
Statistical analyses
The safety, dose–PD and PK–PD relationship analyses datasets consisted of all randomized patients who received all or part of the infusion of the study drug; patient data were analysed according to the treatment the patient had received. The modified intent‐to‐treat (mITT) population was used for haemodynamic analyses and consisted of all randomized patients who received part or all of the infusion and underwent at least one baseline and post‐baseline invasive haemodynamic assessment. Comparisons between BMS‐986231 and placebo groups used a mixed‐effects model for repeated measures, with treatment, time (2 h, 4 h, 6 h and 8 h), time*treatment and baseline value as fixed effects. Time was a repeated measure factor and baseline value was a covariate. Time‐averaged models included results at 2 h, 4 h and 6 h. Statistical significance was defined by a P‐value of <0.05 and was tested separately for each treatment group. No adjustments were made for multiplicity. All data analyses were performed using SAS Version 9.3 or higher (SAS Institute, Inc., Cary, NC, USA).
Analyses of safety data were generally descriptive. Adverse events were listed individually and tabulated using standardized Medical Dictionary for Regulatory Activities (MedDRA) terminology. Quantitative 12‐lead electrocardiogram (ECG) data, BP, HR, body weight and clinical laboratory data were summarized with descriptive statistics. Clinical laboratory results were also evaluated on the basis of laboratory‐specified reference ranges.
Results
Patient randomization and disposition
A total of 46 hospitalized patients at 16 sites across five countries were randomized to treatment with either BMS‐986231 (n = 34) or placebo (n = 12) in four sequential dose‐escalating cohorts and the further expansion to two dose levels (5 and 7 µg/kg/min) between July 2014 and February 2015 (Figure 1). An additional 21 patients were considered as screening failures. The most common reason for screening failure was a screening PCWP below that required for inclusion (seven patients). An additional three patients were randomized but subsequently found to be ineligible for study inclusion prior to dosing and were therefore excluded from the analysis. One patient who was randomized and treated was excluded from the mITT analysis set because PCWP was below the minimum required for inclusion. Another patient was randomized to placebo but received 12 µg/kg/min of BMS‐986231 instead, and was therefore analysed according to the actual treatment received.
Figure 1.
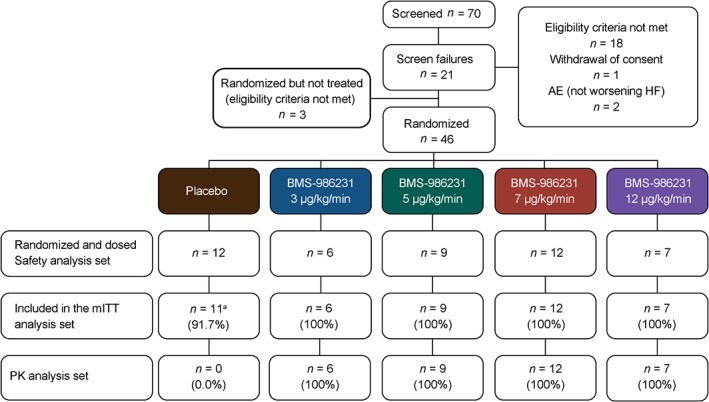
CONSORT diagram. aOne patient in the placebo group was randomized and dosed, but was excluded from the modified intent‐to‐treat (mITT) analysis set because screening and baseline pulmonary capillary wedge pressure (<20 mmHg) was below the minimum required for inclusion, and thus was not eligible for study entry as acknowledged by the investigator and confirmed by the study medical monitor and dose‐escalation review committee. AE, adverse event; HF, heart failure; PK, pharmacokinetic.
Baseline demographics and medical history were generally similar across the groups (Table 1). Most patients were male. Group mean ages ranged from 48.3 years to 63.5 years. The majority of patients had ischaemic (46%) or dilated (37%) cardiomyopathy as a primary aetiology, and all patients were in New York Heart Association (NYHA) class III or IV HF. Medication use was as expected for this patient population and was broadly similar across groups (Table 2). Evaluation of baseline haemodynamic parameters revealed that group mean SBP values ranged from 107 mmHg to 115 mmHg, group mean PCWP from 24 mmHg to 28 mmHg, and group mean Fick‐estimated CI from 1.77 L/min/m2 to 2.20 L/min/m2 (Table 3).
Table 1.
Baseline patient characteristics
| BMS‐986231 treatment dosage | |||||
|---|---|---|---|---|---|
| Placebo group (n = 12) | 3 µg/kg/min (n = 6) | 5 µg/kg/min (n = 9) | 7 µg/kg/min (n = 12) | 12 µg/kg/min (n = 7) | |
| Age, years, mean ± SD | 62.7 ± 9.3 | 63.5 ± 3.5 | 61.1 ± 11.4 | 61.6 ± 10.3 | 48.3 ± 17.5 |
| Male, n (%) | 10 (83.3%) | 4 (66.7%) | 8 (88.9%) | 12 (100%) | 5 (71.4%) |
| Race, n (%) | |||||
| White | 8 (66.7%) | 5 (83.3%) | 7 (77.8%) | 9 (75.0%) | 7 (100%) |
| Black or African American | 4 (33.3%) | 1 (16.7%) | 1 (11.1%) | 3 (25.0%) | 0 |
| Primary cardiomyopathy aetiology, n (%) | |||||
| Ischaemic | 6 (50.0%) | 3 (50.0%) | 5 (55.6%) | 6 (50.0%) | 1 (14.3%) |
| Dilated | 4 (33.3%) | 2 (33.3%) | 1 (11.1%) | 4 (33.3%) | 6 (85.7%) |
| Other | 2 (16.7%) | 1 (16.7%) | 3 (33.3%) | 2 (16.7%) | 0 |
| NYHA class at study entry, n (%) | |||||
| III | 9 (75.0%) | 2 (33.3%) | 9 (100%) | 8 (66.7%) | 7 (100%) |
| IV | 3 (25.0%) | 4 (66.7%) | 0 | 4 (33.3%) | 0 |
| Coronary artery disease, n (%) | 7 (58.3%) | 3 (50.0%) | 5 (55.6%) | 8 (66.7%) | 1 (14.3%) |
| Hypertension, n (%) | 8 (66.7%) | 5 (83.3%) | 9 (100%) | 8 (66.7%) | 2 (28.6%) |
| History of arrhythmias, n (%) | 10 (83.3%) | 5 (83.3%) | 7 (77.8%) | 10 (83.3%) | 5 (71.4%) |
| Chronic renal failure, n (%) | 4 (33.3%) | 3 (50.0%) | 2 (22.2%) | 4 (33.3%) | 0 |
| Diabetes mellitus, n (%) | 3 (25.0%) | 4 (66.7%) | 4 (44.4%) | 5 (41.7%) | 1 (14.3%) |
NYHA, New York Heart Association; SD, standard deviation.
Table 2.
Baseline medications
| Medication | BMS‐986231 treatment dosage | ||||
|---|---|---|---|---|---|
|
Placebo group
(n = 12) |
3 µg/kg/min (n = 6) | 5 µg/kg/min (n = 9) | 7 µg/kg/min (n = 12) | 12 µg/kg/min (n = 7) | |
| Renin–angiotensin system blockers, n (%) | 9 (75.0%) | 3 (50.0%) | 9 (100%) | 11 (91.7%) | 6 (85.7%) |
| Beta‐blockers, n (%) | 12 (100%) | 4 (66.7%) | 8 (88.9%) | 12 (100%) | 7 (100%) |
| Calcium channel blockers, n (%) | 1 (8.3%) | 0 | 0 | 1 (8.3%) | 1 (14.3%) |
| Cardiac therapy, n (%) | 9 (75.0%) | 6 (100%) | 7 (77.8%) | 9 (75.0%) | 3 (42.9%) |
| Amiodarone | 3 (25.0%) | 1 (16.7%) | 3 (33.3%) | 3 (25.0%) | 1 (14.3%) |
| Digoxin | 4 (33.3%) | 2 (33.3%) | 2 (22.2%) | 3 (25.0%) | 0 |
| Dobutamine | 0 | 1 (16.7%) | 1 (11.1%) | 0 | 0 |
| Glyceryl trinitrate | 0 | 2 (33.3%) | 1 (11.1%) | 1 (8.3%) | 0 |
| Isosorbide dinitrate | 5 (41.7%) | 2 (33.3%) | 1 (11.1%) | 3 (25.0%) | 0 |
| Isosorbide dinitrate/hydralazine | 0 | 0 | 1 (11.1%) | 0 | 0 |
| Isosorbide mononitrate | 0 | 1 (16.7%) | 1 (11.1%) | 2 (16.7%) | 0 |
| Ivabradine | 0 | 0 | 0 | 1 (8.3%) | 1 (14.3%) |
| Milrinone | 0 | 0 | 0 | 0 | 1 (14.3%) |
| Diuretics, n (%) | 11 (91.7%) | 6 (100%) | 9 (100%) | 12 (100%) | 7 (100%) |
| Antithrombotic agents, n (%) | 11 (91.7%) | 6 (100%) | 8 (88.9%) | 11 (91.7%) | 6 (85.7%) |
| Lipid‐modifying agents, n (%) | 9 (75.0%) | 4 (66.7%) | 4 (44.4%) | 6 (50.0%) | 1 (14.3%) |
| Other antihypertensives, n (%) | 3 (25.0%) | 3 (50.0%) | 2 (22.2%) | 4 (33.3%) | 1 (14.3%) |
Table 3.
Baseline haemodynamic characteristics
| Parameter | BMS‐986231 treatment dosage | ||||
|---|---|---|---|---|---|
| Placebo group (n = 11 a ) | 3 µg/kg/min (n = 6) | 5 µg/kg/min (n = 9 b ) | 7 µg/kg/min (n = 12 c ) | 12 µg/kg/min (n = 7) | |
| SBP, mmHg, mean ± SD | 106.9 ± 9.5 | 112.5 ± 9.2 | 107.7 ± 12.0 | 114.0 ± 11.7 | 114.6 ± 17.8 |
| Most recent LVEF, %, mean ± SD | 23.6 ± 8.7 | 24.5 ± 6.8 | 21.8 ± 8.8 | 22.5 ± 10.4 | 27.6 ± 6.0 |
| BNP, pg/mL, mean ± SD | 365.5 ± 396.9 | 235.3 ± 178.9 | 174.2 ± 97.5 | 247.5 ± 195.3 | 288.6 ± 208.0 |
| RAP, mmHg, mean ± SD | 12.1 ± 4.4 | 12.4 ± 4.9 | 10.3 ± 3.7 | 12.1 ± 4.4 | 18.8 ± 3.8 |
| PASP, mmHg, mean ± SD | 57.8 ± 10.9 | 54.3 ± 12.2 | 52.4 ± 8.3 | 60.4 ± 11.4 | 48.3 ± 5.9 |
| PADP, mmHg, mean ± SD | 26.3 ± 5.4 | 24.8 ± 3.5 | 26.9 ± 4.5 | 27.7 ± 5.7 | 27.9 ± 5.0 |
| PCWP, or PADP if PCWP not available, mmHg, mean ± SD | 26.2 ± 4.9 | 24.3 ± 4.3 | 24.1 ± 5.4 | 26.9 ± 5.5 | 28.4 ± 5.4 |
| CI, L/min/m2 by Fick, mean ± SD | 1.8 ± 0.4 | 1.8 ± 0.4 | 1.9 ± 0.3 | 1.8 ± 0.3 | 2.2 ± 0.7 |
| CI, L/min/m2 by NICaS, mean ± SD | 2.0 ± 0.3 | 2.1 ± 0.7 | 2.2 ± 1.1 | 1.8 ± 0.5 | 2.4 ± 1.4 |
| CI, L/min/m2 by thermodilutiond, mean ± SD | 1.8 ± 0.4 | 1.6 ± 0.3 | 1.7 ± 0.3 | 1.5 ± 0.6 | 1.7 ± 0.5 |
| SVI, mL/m2 by NICaS, mean ± SD | 28.3 ± 5.7 | 29.0 ± 11.2 | 27.6 ± 9.3 | 24.8 ± 6.1 | 28.0 ± 15.1 |
BNP, B‐type natriuretic peptide; CI, cardiac index; LVEF, left ventricular ejection fraction; NICaS, non‐invasive continuous cardiac system (NI Medical); PADP, pulmonary arterial diastolic pressure; PASP, pulmonary arterial systolic pressure; PCWP, pulmonary capillary wedge pressure; RAP, right atrial pressure; SBP, systolic blood pressure; SD, standard deviation; SVI, stroke volume index.
n = 10 for RAP and BNP.
n = 8 for RAP.
n = 11 for CI by Fick and SVI by NICaS.
n‐values for CI by thermodilution were 9 in the placebo group and 3, 7, 9 and 4 in the 3‐, 5‐, 7‐ and 12‐µg/kg/min groups, respectively.
Haemodynamic assessments
Infusion of BMS‐986231 rapidly reduced PCWP, which was one of the primary endpoints of the study. At the first time‐point assessed (i.e. at 2 h), the mean change from baseline in PCWP with BMS‐986231 was −4.7 mmHg across all dose groups (range: −5.1 mmHg to −4.0 mmHg) vs. 0.2 mmHg with placebo (Figure 2). These changes equated to an 18% mean reduction with BMS‐986231 from baseline PCWP values that ranged from 24 mmHg to 28 mmHg. The reduction in PCWP with BMS‐986231 was sustained throughout the duration of infusion and returned towards baseline at the 2‐h post‐infusion assessment time‐point without a rebound increase above baseline (Figure 2 A). A time‐averaged analysis of mean change in PCWP over the entire duration of infusion demonstrated a statistically significant mean reduction in PCWP throughout the duration of infusion in all dose groups (Figure 2 B). Similarly, greater mean maximal reductions in PCWP during study drug infusion were noted with BMS‐986231 (range: 4.8–6.9 mmHg) than with placebo (2.0 mmHg), with significant differences noted in all but the 12‐µg/kg/min group (supplementary material online, Figure S1).
Figure 2.
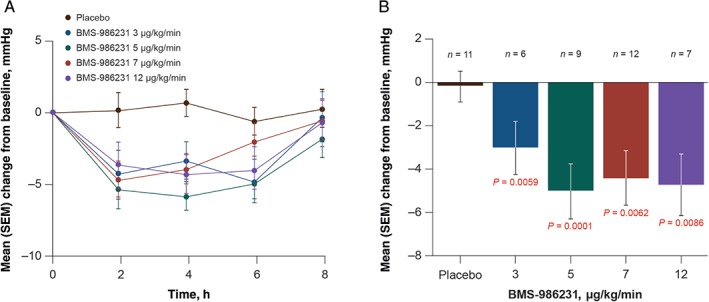
(A) Time course of mean change and (B) mean time‐averaged change (to 6 h) from baseline over the course of infusion of BMS‐986231 or placebo in adjudicated pulmonary capillary wedge pressure (PCWP) on a modified intent‐to‐treat basis. SEM, standard error of the mean.
Consistent with the observed reductions in PCWP, time‐averaged reductions in PADP (also a primary endpoint) were observed in all BMS‐986231 dose groups, and significant reductions in comparison with placebo were demonstrated in the 3‐µg/kg/min and 5‐µg/kg/min groups (Figure 3A). In comparison with placebo, reductions in time‐averaged PASP from baseline were significant in all BMS‐986231 dose groups (Figure 3 B), and in RAP with the highest dose (Figure 3 C).
Figure 3.
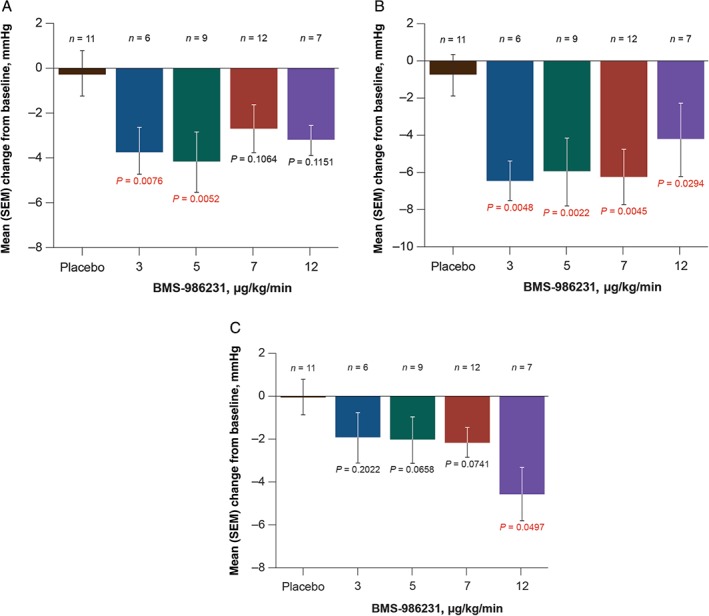
Mean time‐averaged change from baseline in adjudicated (A) pulmonary arterial diastolic pressure, (B) pulmonary arterial systolic pressure, and (C) right atrial pressure during infusion of BMS‐986231 or placebo on a modified intent‐to‐treat basis. SEM, standard error of the mean.
NICaS‐assessed SVI increased in a dose‐dependent manner with BMS‐986231 (supplementary material online, Figure S2). The difference from placebo was significant in the 12‐µg/kg/min group (84% vs. 4%). Similarly, NICaS‐assessed CI was increased with all doses of BMS‐986231, with significance shown at the two highest doses, and CI obtained in a subset of patients using thermodilution showed numerical increases in all BMS‐986231 dose groups, although none reached significance (Table 4). Cardiac index estimated using Fick (also a primary endpoint) did not show a clear pattern of effect, and no significant differences between BMS‐986231 and placebo were reported (Table 4). BMS‐986231 increased NICaS‐assessed CPI, with significance noted in the highest dose group (Figure S2), and calculated total peripheral resistance was significantly reduced in all BMS‐986231 groups in comparison with placebo (Figure S2).
Table 4.
Time‐averaged percentage change from baseline in cardiac index (CI)
| CI method | Change from baseline, mean ± SEM, % | ||||
|---|---|---|---|---|---|
| Placebo group | BMS‐986231 treatment dosage | ||||
| 3 µg/kg/min | 5 µg/kg/min | 7 µg/kg/min | 12 µg/kg/min | ||
| Fick (estimated), n = 44 | 7.95 ± 4.87% | 0.53 ± 7.40% | 13.41 ± 7.84% | 9.59 ± 3.59% | −9.58 ± 6.81% |
| Thermodilution, n = 32 | 9.86 ± 5.15% | 25.87 ± 5.40% | 18.36 ± 10.69% | 17.73 ± 4.48% | 21.20 ± 8.66% |
| NICaS, n = 44 | 1.99 ± 7.46% | 23.91 ± 8.11% | 17.87 ± 6.36% | 31.48 ± 6.10%a | 62.06 ± 27.15%b |
NICaS, non‐invasive continuous cardiac system (NI Medical); SEM, standard error of the mean.
P < 0.05 vs. placebo.
P < 0.0001 vs. placebo.
Administration of BMS‐986231 was associated with numerical but not statistically significant reductions in SBP (2.4–8.7 mmHg vs. 3.2 mmHg with placebo) that were generally noted within 2 h of infusion initiation; SBP returned to baseline 2–4 h later (Figure 4A). Decreases in SBP of ≥20 mmHg from baseline during the infusion were noted in similar percentages of placebo‐ and BMS‐986231‐treated patients (16.7% with placebo vs. 20.6% with BMS‐986231); the incidence of SBP reductions of ≥20 mmHg in BMS‐986231 groups did not demonstrate dose‐dependency, with the highest incidence in the 7‐µg/kg/min cohort. Reductions in SBP with BMS‐986231 were somewhat less pronounced at higher doses. BMS‐986231 was associated with dose‐dependent reductions in mean DBP (4.4–7.7 mmHg vs. 0.1 mmHg for placebo) that were more sustained over the duration of infusion, although none of the reductions in mean time‐averaged change from baseline reached significance (Figure 4 B). Mean arterial pressure was reduced by 4.8–7.2 mmHg across the BMS‐986231 dose groups and by 1.1 mmHg with placebo; none of the reductions with BMS‐986231 were statistically significant in comparison with placebo (Figure 4 C). Pulse pressure (PP) was generally maintained or improved across all BMS‐986231 doses; changes were not statistically significant compared with placebo. There were no significant effects of BMS‐986231 vs. placebo on HR throughout the infusion period (supplementary material online, Figure S3).
Figure 4.
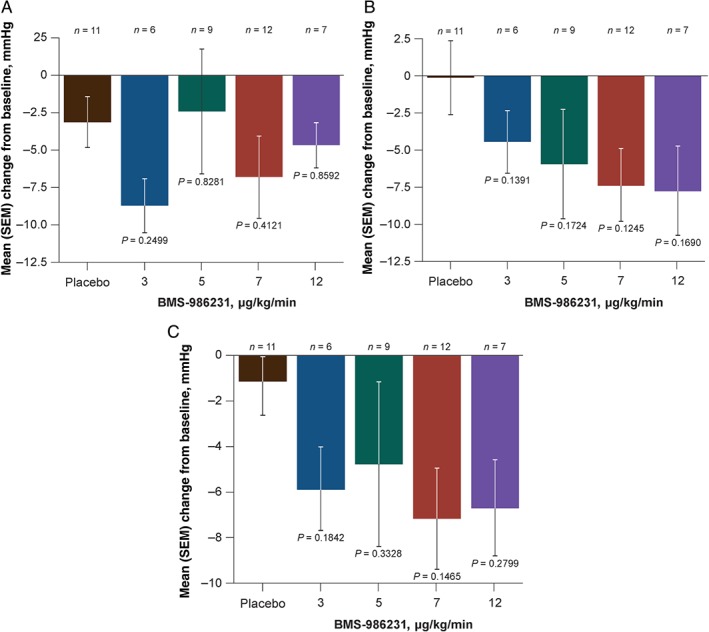
Mean time‐averaged change from baseline in (A) systolic blood pressure, (B) diastolic blood pressure, and (C) mean arterial pressure during infusion of BMS‐986231 or placebo on a modified intent‐to‐treat basis. SEM, standard error of the mean.
Pharmacokinetic and pharmacodynamic analyses
Plasma concentrations of BMS‐986231 at the end of the 6‐h infusion were dose‐dependent (supplementary material Table S1, online). Analysis of the relationship between BMS‐986231 plasma concentration at the end of infusion and time‐averaged reduction from baseline in PCWP across the infusion revealed a non‐significant trend. Similar analyses of NICaS‐derived SVI and CI data revealed significant correlations between BMS‐986231 plasma concentration and time‐averaged change in these variables (supplementary material online, Figure S4).
Adverse event reporting
Treatment‐emergent adverse events (TEAEs) were noted more frequently in patients treated with BMS‐986231 than in those given placebo (range: 42.9–83.3% vs. 25%), although no dose‐dependency was noted and the highest incidence occurred at the lowest BMS‐986231 dose (Table 5). Hypotension (four cases; 11.8% incidence) and headache (three cases; 8.8% incidence) were the most common TEAEs reported across the combined BMS‐986231 dose groups. One case of hypotension (8.3%) and no headaches were reported in the placebo group (Table 5). Cases of hypotension were considered mild and unrelated to the study drug, except in one patient receiving 12 µg/kg/min of BMS‐986231, in whom hypotension was considered moderate and related to the study drug. There were no cases of symptomatic hypotension. Study infusion was discontinued prior to completion in one patient with asymptomatic hypotension who was thought to be oliguric, but this was not subsequently confirmed by the independent safety review committee. Headaches were mild to moderate in severity and did not lead to interruption or early termination of infusion. Across the four BMS‐986231 dose groups, 11 TEAEs were classed as ‘cardiac disorders’ (32.4% incidence) compared with none in the placebo group. However, none of these 11 cardiac events were considered by the investigators to be related to treatment. In addition to hypotension and headache, two other preferred‐term TEAEs occurred in at least two patients receiving BMS‐986231. These were cardiac failure (in one patient in the 5‐µg/kg/min group and one patient in the 7‐µg/kg/min group) and congestive heart failure (in one patient in the 3‐µg/kg/min group and one patient in the 5‐µg/kg/min group) (Table 5). Again, none of these four events were considered treatment‐related.
Table 5.
Summary of treatment‐emergent adverse events (TEAEs)
| Patients with: | BMS‐986231 treatment dosage | |||||
|---|---|---|---|---|---|---|
|
Placebo group
(n = 12) |
3 µg/kg/min (n = 6) | 5 µg/kg/min (n = 9) | 7 µg/kg/min (n = 12) | 12 µg/kg/min (n = 7) | Combined (n = 34) | |
| ≥1 TEAE, n (%) | 3 (25.0%) | 5 (83.3%) | 5 (55.6%) | 7 (58.3%) | 3 (42.9%) | 20 (58.8%) |
| ≥2 TEAEs (for combined active treatment) by preferred term, n (%) | ||||||
| Cardiac disorders | ||||||
| Cardiac failure | 1 (11.1%) | 1 (8.3%) | 2 (5.9%) | |||
| Cardiac failure, congestive | 1 (16.7%) | 1 (11.1%) | 2 (5.9%) | |||
| Nervous system disorders | ||||||
| Headache | 2 (16.7%) | 1 (14.3%) | 3 (8.8%) | |||
| Vascular disorders | ||||||
| Hypotension | 1 (8.3%) | 3 (33.3%) | 1 (14.3%) | 4 (11.8%) | ||
| ≥1 serious TEAE, n (%) | 1 (8.3%) | 3 (50.0%) | 1 (11.1%) | 3 (25.0%) | 1 (14.3%) | 8 (23.5%) |
| Cardiac disorders | ||||||
| Atrial flutter | 1 (8.3%) | 1 (2.9%) | ||||
| Atrioventricular block, complete | 1 (14.3%) | 1 (2.9%) | ||||
| Cardiac failure | 1 (8.3%) | 1 (2.9%) | ||||
| Cardiac failure, congestive | 1 (16.7%) | 1 (11.1%) | 2 (5.9%) | |||
| Infections/infestations | ||||||
| Rhinovirus infection | 1 (16.7%)a | 1 (2.9%) | ||||
| Metabolism/nutrition disorders | ||||||
| Dehydration | 1 (8.3%) | 0 | ||||
| Renal and urinary disorders | ||||||
| Renal failure | 1 (8.3%) | 1 (2.9%) | ||||
| Respiratory, thoracic and mediastinal disorders | ||||||
| Respiratory failure | 1 (16.7%) | 1 (2.9%) | ||||
| Skin and subcutaneous disorders | ||||||
| Toxic epidermal necrolysis | 1 (16.7%) | 1 (2.9%) | ||||
| ≥1 severe TEAE, n (%) | 4 (66.7%) | 2 (16.7%) | 1 (14.3%) | 7 (20.6%) | ||
| ≥1 fatal TEAE | 1 (16.7%) | 1 (2.9%) | ||||
| ≥1 drug‐related TEAEb, n (%) | 1 (8.3%) | 1 (11.1%) | 2 (16.7%) | 1 (14.3%) | 4 (11.8%) | |
| TEAE leading to drug interruption, n (%) | 0 | |||||
| TEAE leading to drug discontinuation, n (%) | 1 (14.3%) | 1 (2.9%) | ||||
This patient received study infusion on 23 July 2014 and was diagnosed with Stevens–Johnson syndrome on 13 August 2014, which was empirically considered by the investigator to be iatrogenic and related to the use of the cephalosporin antibiotic for rhinovirus, and not to the study drug.
No reported study drug‐related TEAEs were serious, severe or fatal.
A total of 10 serious TEAEs were recorded in nine patients across all groups within 30 days of infusion, none of which were considered to be drug‐related by the investigators or the independent safety review committee (Table 5). One patient treated with 3 µg/kg/min of BMS‐986231 experienced two serious TEAEs (rhinovirus infection and Stevens–Johnson syndrome), neither of which was considered by the investigator to be related to the study drug. One death occurred 5 days after the start of 3 µg/kg/min of BMS‐986231 in a 62‐year‐old man with ischaemic cardiomyopathy and severe pulmonary hypertension, who developed respiratory failure thought to have resulted from aspiration pneumonia/pneumonitis. This death was considered by the investigator and the DERC as unlikely to be related to the study drug. All serious TEAEs occurred at least 4 days after the study drug infusion period, and eight of the 10 events occurred at least 9 days after the infusion period.
Laboratory and ECG assessments
No clinically meaningful changes in haematology parameters were observed (supplementary material online, Table S2). Specifically, no clinically significant or drug‐related changes were observed in differential blood counts or in haemoglobin, haematocrit or platelet counts. Despite baseline abnormalities associated with advanced HF, no clinically relevant changes in chemistry parameters were noted in the 6‐ and 24‐h timeframes (Table S2). No drug‐related changes were observed in hepatocellular enzymes or in total and direct bilirubin levels; there was also no evidence of increased renal compromise, either from assessments of mean values of blood urea nitrogen or creatinine or from review of the shift tables for these parameters (Table S2). There were no clinically meaningful changes in coagulation parameters (e.g. activated partial thromboplastin time) associated with study drug treatment, and no treatment‐related changes in BNP, cystatin C or troponin I levels were observed (Table S2).
QT and QTc intervals did not change from baseline in either the placebo or BMS‐986231 groups, and no drug‐related findings were observed. No clinically meaningful changes in ECG‐derived RR, PR or QRS intervals were observed during treatment with BMS‐986231 at any dose in comparison with placebo.
Discussion
This randomized, double‐blind, placebo‐controlled study demonstrated that a 6‐h infusion of BMS‐986231, a novel second‐generation HNO donor, was associated with a favourable safety and haemodynamic profile in patients with advanced HF. The patients included in this study are representative of the advanced HF population according to their baseline vital signs and central haemodynamics. In these patients, BMS‐986231 was associated with rapid and sustained significant decreases in PCWP that returned towards baseline values at 2 h post‐infusion without rebound. As referred to in the introduction, recent preclinical studies in dogs with pacing‐ and microembolization‐induced HF have demonstrated findings that are consistent with the vasodilatory, inotropic and lusitropic properties attributed to BMS‐986231.20, 21 The current study also shows haemodynamic outcomes with BMS‐986231 that appear to support the findings of these preclinical studies.
Similar to the reductions observed in PCWP, BMS‐986231 reduced PADP and PASP, effects which were likely to have been mediated by pulmonary and systemic vasodilation. These effects would be likely to benefit patients with acute decompensated HF because they are accompanied by clinically significant improvements in dyspnoea.22, 23, 24 In a manner consistent with PCWP, PASP and PADP results, RAP was reduced numerically to a greater extent with all doses of BMS‐986231 than with placebo.
BMS‐986231 increased NICaS‐assessed SVI at all doses, with significance noted at the highest dose. Because there were no significant effects on HR, the effects of BMS‐986231 on NICaS‐assessed CI and CPI were similar and predominantly attributable to those on SVI. Thermodilution‐obtained CI showed numerical increases in all BMS‐986231 cohorts, but CI estimated with Fick did not show a clear difference between BMS‐986231 and placebo. Caution should be taken when interpreting Fick‐estimated CI results in the current study because Fick utilized an assumed estimate of VO2 rather than a direct measurement, and an inherent discrepancy between actual and Fick‐estimated VO2 has been demonstrated.25, 26, 27 A recent publication reported instances of both under‐ and overestimation of CO using the Fick technique, and suggested that thermodilution‐derived CO is more reliable, at least in the types of patient included in the current study.28 Although NICaS is a comparatively new method of assessing CI, a number of validation studies have previously been published. For example, Paredes and colleagues reported that NICaS‐derived CO measurements correlated well with thermodilution‐derived measurements in 35 cardiac patients at a single centre (two‐tailed Pearson correlation, r = 0.91).29 In another study, Leitman et al. compared CI measurement using NICaS with that of Doppler echocardiography in 60 patients undergoing dobutamine stress echocardiography.30 Again, NICaS‐derived CI results correlated well with Doppler‐derived findings (Spearman's rank correlation, r = 0.81).
Fick‐estimated CI results aside, therefore, the increases in SVI, CI and CPI obtained using NICaS and thermodilution without any effect on HR may have been mediated primarily by vasodilatory effects; however, inotropic and/or lusitropic effects of BMS‐986231 may also have contributed because all three mechanisms have been reported with HNO donation in preclinical studies using HF models.13, 15, 20, 21
The administration of BMS‐986231 was associated with mild reductions in SBP that occurred early, were not considered by the investigators to be clinically significant, and returned to baseline levels by the end of infusion without the need for intervention. In contrast to its effect on SBP, BMS‐986231 was associated with more sustained, albeit non‐significant, reductions in DBP throughout infusion. These DBP results are most probably attributable to the potentially greater impact of arterial vasodilation on DBP than on SBP. Importantly, no cases of symptomatic hypotension were noted, and despite the reductions in SBP and DBP, which amounted to an approximate 5‐mmHg decrease in calculated MAP, PP was predominantly maintained or increased, suggesting maintenance or potential improvement of end‐organ perfusion with BMS‐986231.
Generally, BMS‐986231 was well tolerated when administered for 6 h at doses up to 12 µg/kg/min. There were 11 reported cases of cardiac disorders across all BMS‐986231 dose groups and none in the placebo group. There were also eight cases of serious TEAEs in the active treatment groups (23.5% incidence) combined compared with one (8.3%) in the placebo cohort. It is important to note that patients were randomized to BMS‐986231 or placebo at a 3:1 ratio and hence this difference partially reflects the larger number of patients in the active treatment groups. The small sample size is also undoubtedly a factor. There was also no indication of dose‐dependency for TEAEs observed with BMS‐986231. The most common trigger for a serious TEAE report across the treatment and placebo groups was prolonged hospitalization or readmission following discharge. In the advanced HF patients in this study, high complication and readmission rates were expected as a function of their underlying disease state. Published rates of readmission for HF patients within 30 days of discharge from the index hospitalization are approximately 20–35%.31, 32, 33 In this context, the frequency of readmission as a cause of serious TEAEs in this study is consistent with the known natural history of patients with this condition. In addition, individual serious TEAEs, such as acute renal failure or atrial flutter requiring cardioversion, are not unexpected in this population. General complications related to prolonged hospitalization in these patients may also occur frequently and can be fatal. No serious TEAEs reported within 30 days after the completion of infusion were considered to have been possibly or probably related to BMS‐986231, and there was no evidence of a dose‐dependent incidence of serious TEAEs. All serious TEAEs occurred at least 4 days after the infusion of BMS‐986231 was terminated; given the short half‐life of BMS‐986231 (40–144 min), the absence of TEAEs classed as ‘serious’ during the first 3 days after infusion is encouraging. Furthermore, BMS‐986231 did not adversely impact laboratory parameters such as BNP, cystatin C, troponin I, creatinine, blood urea nitrogen, haemoglobin and platelet count. Similarly, there were no clinically meaningful changes in ECG parameters.
This study has some limitations that should be outlined. Firstly, the study population was broadly defined as having ‘advanced’ HF; although the study included patients with acute decompensated HF, we were unable to assess the effects of the study drug in this subpopulation alone. Secondly, although BMS‐986231 has shown positive inotropy and lusitropy in preclinical studies, it was not possible to fully assess for inotropy or lusitropy here. Conductance catheter technology is reliable for measuring the end‐systolic pressure–volume relationship, which is generally accepted as a load‐independent index of LV function. However, the use of such technology under clinical conditions is limited. In the current study, it is possible that a positive inotropic effect of BMS‐986231 may have been masked by a primarily vasodilatory effect, or that the primary mechanism of BMS‐986231 may be dose‐related (i.e. vasodilatory at lower doses and inotropic at higher doses). This can only be addressed through a new trial. Other limitations include the restriction of the infusion period to 6 h. Patients were required to be in a sufficiently stable condition in which they would not require vasoactive medications (including diuretics) for a period of at least 10 h (i.e. ≥4 h before the infusion and during the 6‐h infusion itself). The 6‐h infusion period was considered sufficiently long to allow observation of the effects of the study drug while patients were stable and not receiving vasoactive medication. However, research into the effects of BMS‐986231 beyond a 6‐h infusion is needed. The sequential panel design of the study also meant that patients in the different dosing arms were not randomized at the same time and in the same location, which can potentially lead to imbalances between study arms. The blinding was conducted in a way in which study personnel (i.e. the pharmacist) had knowledge of the treatment assignment. Although appropriate steps were taken to separate the pharmacist from the investigators, such blinding is less robust than that afforded by the use of pre‐prepared, completely blinded kits. Finally, the small sample size limits the interpretation of findings.
In summary, in this Phase 2a trial, BMS‐986231 was well tolerated and was shown to lower PCWP and increase CO as shown with SVI and CI assessment (by NICaS and thermodilution). Although positive inotropy and/or lusitropy cannot be extrapolated from these findings alone, the haemodynamic findings are consistent with the drug's purported mechanisms of action derived from preclinical studies, and are highly applicable therapeutic findings in this patient population. The clinical significance of these findings is currently unknown, however, and further studies to assess the safety and efficacy of BMS‐986231 in patients with HF are needed.
Disclosure: this work was previously presented in part at Heart Failure 2016, European Society of Cardiology Congress, 21–24 May 2016, Florence.
Supporting information
Figure S1. Mean maximal decrease from baseline in adjudicated pulmonary capillary wedge pressure during the infusion of BMS‐986231 or placebo on a modified intent‐to‐treat basis.
Figure S2. Change from baseline in stroke volume index, cardiac power index and total peripheral resistance assessed by NICaS (non‐invasive continuous cardiac system; NI Medical) over the course of infusion of BMS‐986231 or placebo (to 6 h).
Figure S3. Change from baseline in heart rate over the course of infusion of BMS‐986231 or placebo (to 6 h).
Figure S4. Time‐averaged change from baseline in adjudicated pulmonary capillary wedge pressure, stroke volume index and cardiac index vs. BMS‐986231 plasma concentrations at 6 h.
Table S1. Plasma concentrations of BMS‐986231 in patients hospitalized with heart failure with reduced ejection fraction.
Table S2. Laboratory parameters at baseline, 6 h and 24 h in patients hospitalized with heart failure with reduced ejection fraction and given BMS‐986231 or placebo.
Acknowledgements
Medical writing support was provided by Joshua Rodman of HCG Communications (New York, NY, USA) and Geraint Owens of Chameleon Communications International (Wilmslow, Cheshire, UK), and funded by Bristol‐Myers Squibb.
Funding
This work was supported by Cardioxyl Pharmaceuticals and Bristol‐Myers Squibb.
Conflict of interest: E.M.G., J.G., K.B.S. and T.Z. have received research grants from Cardioxyl Pharmaceuticals. A.B.V.B. has received research grants from Cardioxyl Pharmaceuticals, CVRx, Celladon and Novartis. S.S.G. has provided consultancy to Bristol‐Myers Squibb and received research grants from Cardioxyl Pharmaceuticals. T.S. has provided consultancy to AstraZeneca, Bayer, Abbott and Sanofi. R.P.V. was Senior Director of Clinical Development at Cardioxyl Pharmaceuticals at the time of this study. D.C. was Executive Vice President & Development and Regulatory Affairs at Cardioxyl Pharmaceuticals at the time of this study. S.Y.F. was Chief Medical Officer of Cardioxyl Pharmaceuticals at the time of this study. C.T., G.J.H., M.J., S.H.D., M.K., P.C.P., R.P., A.V. and V.M. report no conflicts of interest.
References
- 1. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62:e147–239. [DOI] [PubMed] [Google Scholar]
- 2. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 3. Greenberg B. Acute decompensated heart failure: treatments and challenges. Circ J 2012;76:532–543. [DOI] [PubMed] [Google Scholar]
- 4. Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, Hobbs AJ, Fukuto JM. The pharmacology of nitroxyl (HNO) and its therapeutic potential: not just the Janus face of NO. Pharmacol Ther 2007;113:442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tocchetti CG, Wang W, Froehlich JP, Huke S, Aon MA, Wilson GM, Di Benedetto G, O'Rourke B, Gao WD, Wink DA, Toscano JP, Zaccolo M, Bers DM, Valdivia HH, Cheng H, Kass DA, Paolocci N. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res 2007;100:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lancel S, Zhang J, Evangelista A, Trucillo MP, Tong X, Siwik DA, Cohen RA, Colucci WS. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ Res 2009;104:720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Froehlich JP, Mahaney JE, Keceli G, Pavlos CM, Goldstein R, Redwood AJ, Sumbilla C, Lee DI, Tocchetti CG, Kass DA, Paolocci N, Toscano JP. Phospholamban thiols play a central role in activation of the cardiac muscle sarcoplasmic reticulum calcium pump by nitroxyl. Biochemistry 2008;47:13150–13152. [DOI] [PubMed] [Google Scholar]
- 8. Sivakumaran V, Stanley BA, Tocchetti CG, Ballin JD, Caceres V, Zhou L, Keceli G, Rainer PP, Lee DI, Huke S, Ziolo MT, Kranias EG, Toscano JP, Wilson GM, O'Rourke B, Kass DA, Mahaney JE, Paolocci N. HNO enhances SERCA2a activity and cardiomyocyte function by promoting redox‐dependent phospholamban oligomerization. Antioxid Redox Signal 2013;19:1185–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kohr MJ, Kaludercic N, Tocchetti CG, Dong Gao W, Kass DA, Janssen PM, Paolocci N, Ziolo MT. Nitroxyl enhances myocyte Ca2+ transients by exclusively targeting SR Ca2+‐cycling. Front Biosci (Elite Ed) 2010;2:614–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai T, Tian Y, Tocchetti CG, Katori T, Murphy AM, Kass DA, Paolocci N, Gao WD. Nitroxyl increases force development in rat cardiac muscle. J Physiol 2007;580:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao WD, Murray CI, Tian Y, Zhong X, DuMond JF, Shen X, Stanley BA, Foster DB, Wink DA, King SB, Van Eyk JE, Paolocci N. Nitroxyl‐mediated disulfide bond formation between cardiac myofilament cysteines enhances contractile function. Circ Res 2012;111:1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu G, Groneberg D, Sikka G, Hori D, Ranek MJ, Nakamura T, Takimoto E, Paolocci N, Berkowitz DE, Friebe A, Kass DA. Soluble guanylate cyclase is required for systemic vasodilation but not positive inotropy induced by nitroxyl (HNO) in the mouse. Hypertension 2015;65:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sabbah HN, Tocchetti CG, Wang M, Daya S, Gupta RC, Tunin RS, Mazhari R, Takimoto E, Paolocci N, Cowart D, Colucci WS, Kass DA. Nitroxyl (HNO) a novel approach for the acute treatment of heart failure. Circ Heart Fail 2013;6:1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Nitroxyl anion exerts redox‐sensitive positive cardiac inotropy in vivo by calcitonin gene‐related peptide signaling. Proc Natl Acad Sci U S A 2001;98:10463–10468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paolocci N, Katori T, Champion HC, St John ME, Miranda KM, Fukuto JM, Wink DA, Kass DA. Positive inotropic and lusitropic effects of HNO/NO− in failing hearts: independence from beta‐adrenergic signaling. Proc Natl Acad Sci U S A 2003;100:5537–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arcaro A, Lembo G, Tocchetti CG. Nitroxyl (HNO) for treatment of acute heart failure. Curr Heart Fail Rep 2014;11:227–235. [DOI] [PubMed] [Google Scholar]
- 17. Chin KY, Qin C, Cao N, Kemp‐Harper BK, Woodman OL, Ritchie RH. The concomitant coronary vasodilator and positive inotropic actions of the nitroxyl donor Angeli's salt in the intact rat heart: contribution of soluble guanylyl cyclase‐dependent and ‐independent mechanisms. Br J Pharmacol 2014;171:1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp‐Harper BK. Nitroxyl (HNO): the Cinderella of the nitric oxide story. Trends Pharmacol Sci 2008;29:601–608. [DOI] [PubMed] [Google Scholar]
- 19. Cowart D, Venuti R, Guptill J, Noveck R, Foo S. A phase 1 study of the safety and pharmacokinetics of the intravenous nitroxyl prodrug, CXL‐1427. J Am Coll Cardiol 2015;65(S10):A876. [Google Scholar]
- 20. Del Rio CL, Hartman JC, Reardon J. Evaluation of the cardiovascular profile of a novel nitroxyl donor BMS‐986231 in chronically instrumented dogs with normal cardiac function and pacing‐induced cardiomyopathy. Eur J Heart Fail 2017;19(Suppl 1):515. [abstract P2025] [Google Scholar]
- 21. Sabbah HN, Hartman JC, Reardon J. Effects of acute intravenous infusion of nitroxyl donor BMS‐986231 on left ventricular function and cardiac rhythm in anaesthetised dogs with intracoronary microembolisation‐induced heart failure. Eur J Heart Fail 2017;19(Suppl 1):7. [abstract 37]28052544 [Google Scholar]
- 22. Butler J, Emerman C, Peacock WF, Mathur VS, Young JB; VMAC Study Investigators . The efficacy and safety of B‐type natriuretic peptide (nesiritide) in patients with renal insufficiency and acutely decompensated congestive heart failure. Nephrol Dial Transplant 2004;19:391–399. [DOI] [PubMed] [Google Scholar]
- 23. Mitrovic V, Seferovic PM, Simeunovic D, Ristic AD, Miric M, Moiseyev VS, Kobalava Z, Nitsche K, Forssmann WG, Lüss H, Meyer M. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J 2006;27:2823–2832. [DOI] [PubMed] [Google Scholar]
- 24. Solomonica A, Burger AJ, Aronson D. Hemodynamic determinants of dyspnea improvement in acute decompensated heart failure. Circ Heart Fail 2013;6:53–60. [DOI] [PubMed] [Google Scholar]
- 25. Thrush DN. Spirometric versus Fick‐derived oxygen consumption: which method is better? Crit Care Med 1996;24:91–95. [DOI] [PubMed] [Google Scholar]
- 26. Stock MC, Ryan ME. Oxygen consumption calculated from the Fick equation has limited utility. Crit Care Med 1996;24:86–90. [DOI] [PubMed] [Google Scholar]
- 27. Narang N, Thibodeau JT, Levine BD, Gore MO, Ayers CR, Lange RA, Cigarroa JE, Turer AT, de Lemos JA, McGuire DK. Inaccuracy of estimated resting oxygen uptake in the clinical setting. Circulation 2014;129:203–210. [DOI] [PubMed] [Google Scholar]
- 28. Fanari Z, Grove M, Rajamanickam A, Hammami S, Walls C, Kolm P, Saltzberg M, Weintraub WS, Doorey AJ. Cardiac output determination using a widely available direct continuous oxygen consumption measuring device: a practical way to get back to the gold standard. Cardiovasc Revasc Med 2016;17:256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paredes OL, Shite J, Shinke T, Watanabe S, Otake H, Matsumoto D, Imuro Y, Ogasawara D, Sawada T, Yokoyama M. Impedance cardiography for cardiac output estimation: reliability of wrist‐to‐ankle electrode configuration. Circ J 2006;70:1164–1168. [DOI] [PubMed] [Google Scholar]
- 30. Leitman M, Sucher E, Kaluski E, Wolf R, Peleg E, Moshkovitz Y, Milo‐Cotter O, Vered Z, Cotter G. Non‐invasive measurement of cardiac output by whole‐body bio‐impedance during dobutamine stress echocardiography: clinical implications in patients with left ventricular dysfunction and ischaemia. Eur J Heart Fail 2006;8:136–140. [DOI] [PubMed] [Google Scholar]
- 31. Krumholz HM, Merrill AR, Schone EM, Schreiner GC, Chen J, Bradley EH, Wang Y, Wang Y, Lin Z, Straube BM, Rapp MT, Normand SL, Drye EE. Patterns of hospital performance in acute myocardial infarction and heart failure 30‐day mortality and readmission. Circ Cardiovasc Qual Outcomes 2009;2:407–413. [DOI] [PubMed] [Google Scholar]
- 32. Dharmarajan K, Hsieh AF, Lin Z, Bueno H, Ross JS, Horwitz LI, Barreto‐Filho JA, Kim N, Bernheim SM, Suter LG, Drye EE, Krumholz HM. Diagnoses and timing of 30‐day readmissions after hospitalization for heart failure, acute myocardial infarction, or pneumonia. JAMA 2013;309:355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bergethon KE, Ju C, DeVore AD, Hardy NC, Fonarow GC, Yancy CW, Heidenreich PA, Bhatt DL, Peterson ED, Hernandez AF. Trends in 30‐day readmission rates for patients hospitalized with heart failure: findings from the Get With The Guidelines‐Heart Failure Registry. Circ Heart Fail 2016;9:e002594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mean maximal decrease from baseline in adjudicated pulmonary capillary wedge pressure during the infusion of BMS‐986231 or placebo on a modified intent‐to‐treat basis.
Figure S2. Change from baseline in stroke volume index, cardiac power index and total peripheral resistance assessed by NICaS (non‐invasive continuous cardiac system; NI Medical) over the course of infusion of BMS‐986231 or placebo (to 6 h).
Figure S3. Change from baseline in heart rate over the course of infusion of BMS‐986231 or placebo (to 6 h).
Figure S4. Time‐averaged change from baseline in adjudicated pulmonary capillary wedge pressure, stroke volume index and cardiac index vs. BMS‐986231 plasma concentrations at 6 h.
Table S1. Plasma concentrations of BMS‐986231 in patients hospitalized with heart failure with reduced ejection fraction.
Table S2. Laboratory parameters at baseline, 6 h and 24 h in patients hospitalized with heart failure with reduced ejection fraction and given BMS‐986231 or placebo.