

Abstract
The merger of open- and closed-shell elementary organometallic steps has enabled the selective intermolecular addition of nucleophilic radicals to unactivated alkynes. A range of carboxylic acids can be subjected to a CO2 extrusion, nickel capture, migratory insertion sequence with terminal and internal alkynes to generate stereodefined functionalized olefins. This platform has been further extended, via hydrogen atom transfer, to the direct vinylation of unactivated C–H bonds. Preliminary studies indicate that a Ni-alkyl migratory insertion is operative.
The direct addition of alkyl radicals to carbon–carbon π-bonds represents one of the most widely exploited transformations within the realm of open-shell chemistry. Indeed, this mechanism is central to a variety of bond-forming cascades in natural product total synthesis,1 the large-scale preparation of high-value polymers,2,3 and the synthesis of pharmaceutical agents.4 In the case of olefin SOMOphiles, radical additions often proceed rapidly and with predictable regioselectivity, a feature that has been broadly leveraged to effect carbon–carbon bond formations with simple alkenes, styrenes, and enones.5 In contrast, the direct addition of radicals to unactivated alkynes is often problematic due to (i) the diminished rate of C–C bond formation1,6 and (ii) the generation of high-energy vinyl radical intermediates that can readily participate in various undesirable open-shell pathways.
In recent years, metallaphotoredox has become a useful multicatalysis strategy, wherein traditionally inert functional groups are readily converted to carbon-centered radicals that subsequently engage in organometallic cross-couplings via a two-stage radical capture and reductive elimination cycle. Recently, we questioned if it might be possible to merge photoredox activation with an alternative elementary coupling step, namely migratory insertion. We recognized that such a merger of open-shell radical chemistry with a closed-shell migratory insertion pathway might provide a new strategy for the union of alkyl radicals and alkynes, an often-elusive coupling step. In this context, we hypothesized that a nickel catalyst might mediate the requisite coupling given that it readily participates in both 1 e− and 2 e− oxidation state changes,7 (i.e., it can readily function in both open- and closed-shell mechanisms). With respect to olefin geometry control, it is important to note that vinyl radicals undergo rapid inversion (kinv = 109 s−1, −133 °C)9 between (E)- and (Z)-isomers, whereas vinyl carbon–Ni bonds are configurationally stable and can be stereospecifically protonated.8 As such, the regio- and stereoselectivity of the alkyne addition process would be strictly enforced by the migratory insertion step.8
On the basis of recent studies in photocatalytic decarbox-ylative radical generation,10 we were optimistic that a photo-redox/nickel dual-catalysis platform would provide a mild route to the requisite alkyl-nickel species using simple yet abundant alkyl carboxylic acids (Figure 1). While decarboxylative vinylation with nickel has been demonstrated by our lab,11 and thereafter by Reisman12 and Baran,13 we recognized that such strategies require vinyl bromides or vinyl zinc systems, substrates that are typically prepared in one or two steps, respectively. As such, direct use of the parent alkyne14 in a cross-coupling vinylation would clearly be advantageous, a strategy recently employed by Wu in the Ni–H catalyzed hydroalkylation of phenylacetylenes and enynes.15
Figure 1.

Dual-catalytic alkyne hydroalkylation.
From a design perspective, we envisioned that this radical capture/migratory insertion mechanism would proceed as shown in Scheme 1. Upon irradiation with visible light, the photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) is known to access the highly oxidizing excited state (ES) species *Ir(III) (ES-2) (E½red [*IrIII/IrII] = +1.21 V vs the saturated calomelelectrode (SCE) in MeCN).16 Deprotonation of the carboxylic acid substrate (e.g., N-Boc-proline (3)) and subsequent single-electron oxidation of the resulting carboxylate functionality by ES-2 should generate alkyl radical 5 (upon CO2 extrusion)17 and reduced Ir(II) species 4. Open-shell alkyl species 5 is expected to rapidly engage in an oxidative radical capture with low-valent nickel species 6, generating alkyl-Ni(II) complex 7. At this stage, we hoped that this nucleophilic Ni(II) species would undergo the critical migratory insertion coupling step with alkyne 8 to generate vinyl-nickel complex 9 with a high degree of stereo- and regioselectivity based on the established mechanistic bias for cis-carbometalation and the studies by Bergman using stoichio-metric nickel complexes.8 Subsequent protodemetalation by either protonated base or carboxylic acid would then provide C(sp3)–C(sp2) coupled product 11. A single electron transfer event from Ir(II) species 4 (E½red [IrIII/IrII] = −1.37 V vs SCE in MeCN)16 to Ni(II) species 10 would regenerate both the Ir(III) species and low-valent nickel 6 (E½red[NiII/Ni0] = −1.2 V vs SCE in DMF), simultaneously completing both the nickel and photocatalyst cycles.18
Scheme 1.

Proposed Dual-Catalytic Cycle
This new decarboxylation/metal capture/migratory insertion coupling protocol was first examined by exposure of N-Boc-proline and 1-heptyne to visible light irradiation (40 W blue LEDs) and catalytic quantities of NiCl2·dtbbpy, Ir[dF(CF3)-ppy]2(dtbbpy)PF6, and 1,1,3,3-tetramethylguanidine (TMG). To our delight, this dual-catalytic combination provided the desired hydroalkylation product 11 in modest yield, yet with excellent selectivity for the branched regioisomer (Table 1, entry 1). By lowering the base loading, the yield of the branched product could be increased to 80% (entry 2) with no erosion in regioselectivity. Given that direct radical-alkyne addition furnishes the linear olefin product, the marked selectivity for the branched isomer indicates that the nickel-mediated pathway is exclusively operative. Indeed, control experiments revealed that Ni, photocatalyst, and light are all critical for C–C bond formation (entries 3–5).
Table 1.
Control Experiments for the Alkyne Hydroalkylationa
 | |||
|---|---|---|---|
| entry | conditions | yield | r.r. |
| 1 | 1.0 equiv TMG | 17% | >20:1 |
| 2 | 0.1 equiv TMG | 80%(80%)b | >20:1 |
| 3 | no light | 0% | – |
| 4 | no photocatalyst | 0% | – |
| 5 | no Ni catalyst | <5% | – |
| 6 | 4-CzIPN catalystc | 70% | >20:1 |
Performed with H2O (20 equiv), carboxylic acid (1.0 equiv), and 1-heptyne (1.3 equiv). Yields by 1H NMR.
Yields in parentheses are isolated.
4-CzIPN (2 mol %) used in place of Ir catalyst.
With optimized conditions in hand, we next evaluated the scope of the alkyne component, utilizing N-Boc-proline as the common carboxylic acid coupling partner. As shown in Table 2, excellent regioselectivity and yield are maintained across a range of alkynes of varying steric demand at the propargylic position (12 and 13, 77 and 58% yield, respectively). Importantly, a large range of functional groups are readily tolerated, including alkyl chlorides, esters, nitriles, phthalimides, and perhaps most notable, unprotected alcohols (14–20, 72–85% yield). It should be noted that nearly all of the alkynes employed in Table 2 are commercially available, whereas only two of the vinyl halide or boronate analogs can be purchased, further demonstrating the advantages of directly employing alkynes in this new C(sp3)-vinylation reaction.
Table 2.
Alkyne and Carboxylic Acid Scope in the Ni/Photoredox-Mediated Decarboxylative Hydroalkylation of Alkynesa
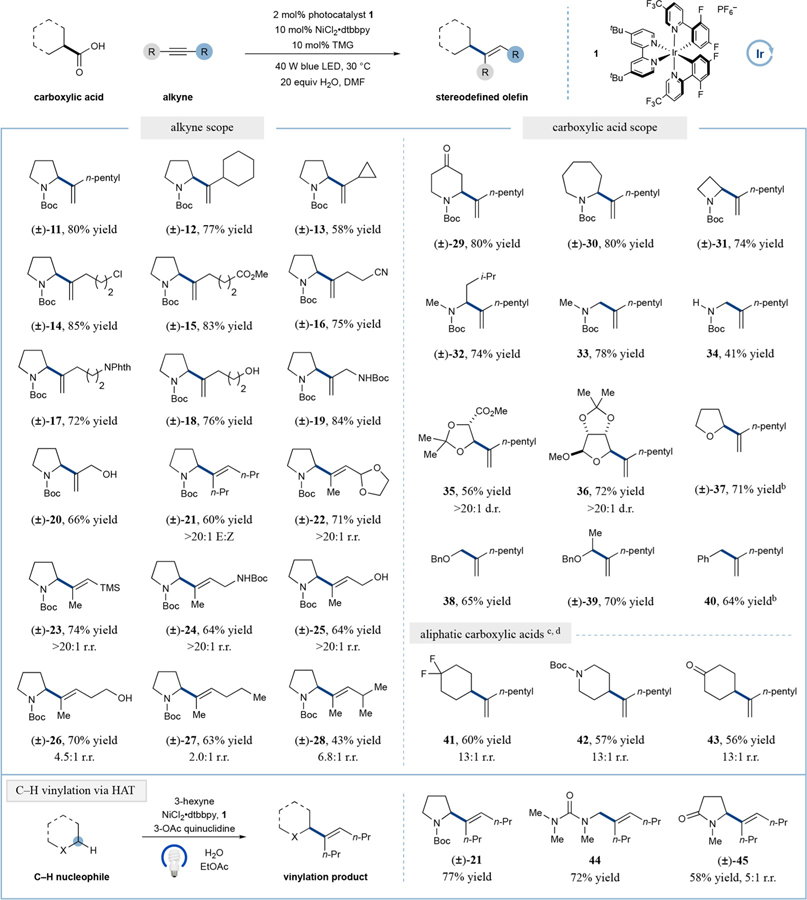 |
All yields are isolated. Performed with photocatalyst 1 (2 mol %), NiCl2·dtbbpy (10 mol %), TMG (10 mol %), carboxylic acid (1.0 equiv), alkyne (1.3 equiv). All products obtained in >20:1 r.r. and >20:1 E:Z, unless otherwise noted.
Yield determined by 1H NMR.
Reactions performed in the integrated photoreactor in DMSO, utilizing CsF (0.5 equiv) as the base.
Regioisomeric ratio refers to the branched and linear olefins.
We next turned our attention to the capacity of internal alkynes to participate in this decarboxylative hydroalkylation protocol. We first examined the symmetrical 4-octyne system, and were delighted to find that our optimized reaction conditions achieved useful yield and excellent selectively for production of the (E)-isomer (21, 60% yield, E:Z > 20:1), consistent with the proposed migratory insertion pathway. Next, we faced the challenge of unsymmetrical internal alkynes, a notoriously difficult substrate class with respect to regioselective hydrofunctionalization.19 Remarkably, we observed excellent regiocontrol in the hydroalkylation of a variety of nonsymmetrical acetylide systems (22–25, 64–74% yield, >20:1 r.r.), wherein the observed selectivity apparently arises from preferential alkyl-migratory insertion to position the nickel center at the C(sp) position of greatest electron density, and the incoming alkyl group at the site of highest electrophilicity based on the latent polarity of the alkyne. Indeed, examination of the products from reactions with three sterically similar alkynes, namely Me—C≡C—R, where R = CH2OH, (25), R = CH2CH2OH (26), and R = CH2CH2CH3 (27), reveals a consistent trend (>20:1 r.r., 4.5:1 r.r., 2.0:1 r.r., respectively), demonstrating that alkyne polarization plays a determining role in the selectivity of the migratory insertion step. Furthermore, comparing the products from reactions with sterically unsymmetrical alkynes, the incoming alkyl group is preferentially placed farthest from the bulky alkyne substituent (27 and 28 2.0:1 and 6.8:1 r.r., respectively), consistent with studies on the stoichiometric insertion of alkyl-nickel species into alkynes.8 We next focused on the scope of the carboxylic acid component. A number of cyclic and acyclic α-amino acids undergo efficient coupling with 1-heptyne (11, 29–34, 41–80% yield). Moreover, α-oxy carboxylic acids, in both cyclic and acyclic formats, undergo proficient alkyne coupling to accessallylic ethers in an analogous fashion (35–39, 56–72% yield). Alkyl carboxylic acids also participate in this Ni-mediated hydrofunctionalization pathway, albeit with a slight erosion in regioselectivity (41–43, 13:1 r.r.), wherein the minor regioisomer most likely forms via direct, nonmetal mediated radical addition to the alkyne substrate. Given the established nucleophilicity of nonstabilized aliphatic radicals, it is remarkable to consider that the traditional open-shell alkyne addition pathway is effectively bypassed using this radical capture/migratory insertion sequence.
From the outset, we envisioned that this combined nickel/photoredox catalysis reaction could be modified with respect to mode of radical generation while retaining the coupling efficiency of the nickel migratory insertion step. With this in mind, we have recently shown that the combination of photoredox and quinuclidine catalysts can selectively perform H· abstraction with hydridic C–H bonds.20 As shown in Table 2, the combination of light, 3-acetoxyquinuclidine, photocatalyst 1, and catalytic nickel allows for the efficient and site-selective vinylation of C–H bonds adjacent to carbamate, amide, and urea functionalities (21, 44, 45, 60–77% yield).15
To further probe our mechanistic design plan (Scheme 1), we studied the dependence of regioselectivity on the size of the carboxylic acid coupling partner with a sterically biased internal alkyne, 4-methylpent-2-yne (Scheme 2). Given that the possibility exists for an alternative Ni-hydride addition/oxidative radical capture pathway (Scheme 2B), we sought to distinguish that hypothesis from the migratory insertion sequence proposed herein. Under the nickel hydride mechanism, the regioselectivity-determining Ni–H insertion occurs prior to engaging the alkyl coupling partner.15 For this reason, the Ni–H mechanism predicts that the regioselectivity will be independent of the steric demand of carboxylic acid component. In contrast, the Ni-alkyl insertion step will involve nonbonding interactions between the alkyne substituents and incoming Ni-alkyl group, a feature that would lead to regioselectivity being a function of the steric demand of the carboxylic acid substrate (Scheme 2A). As such, we examined carboxylic acids of electronic natures similar to that of our model substrate (Boc-Pro), but with sterically smaller (Boc-Me-Gly) and sterically larger (Boc-Me-Leu) profiles. Consistent with a regioselectivity-determining Ni-alkyl insertion event, a positive correlation was observed between alkyl partner size and regioselectivity of hydroalkylation (46, 28, and 47, 4.4:1, 6.8:1, and 16:1 r.r., respectively), in complete accord with Scheme 1.
Scheme 2.

Alkylation Selectivity as a Function of Acid Size
Supplementary Material
ACKNOWLEDGMENTS
Financial support provided by the NIHGMS (RO1 GM103558–05) and kind gifts from Merck, BMS, Firmenich, Pfizer, Janssen, and Eli Lilly.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02834.
Experimental details and characterization data (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Wille U Chem. Rev 2013, 113, 813–853. [DOI] [PubMed] [Google Scholar]
- (2).Carothers WH Chem. Rev 1931, 8, 353–426. [Google Scholar]
- (3).Chen M; Zhong M; Johnson JA Chem. Rev 2016, 116, 10167. [DOI] [PubMed] [Google Scholar]
- (4).Douglas JJ; Sevrin MJ; Stephenson CR J. Org. Process Res. Dev 2016, 20, 1134. [Google Scholar]
- (5) (a).Chu L; Ohta C; Zuo Z; MacMillan DW C. J. Am. Chem. Soc 2014, 136, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Capacci AG; Malinowski JT; McAlpine NJ; Kuhne J; MacMillan DW C. Nat. Chem 2017, 9, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Gόmez-Balderas R; Coote ML; Henry DJ; Fischer H; Radom LJ Phys. Chem. A 2003, 107, 6082. [Google Scholar]; (b) Giese B; Lachhein S Angew. Chem., Int. Ed. Engl 1982, 21, 768. [Google Scholar]
- (7).Tasker SZ; Standley EA; Jamison TF Nature 2014, 509, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Huggins JM; Bergman RG J. Am. Chem. Soc 1981, 103, 3002. [Google Scholar]
- (9).Fessenden RW; Schuler RH J. Chem. Phys 1963, 39, 2147. [Google Scholar]
- (10).Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DW C. Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Noble A; McCarver SJ; MacMillan DW C. J. Am. Chem. Soc 2015, 137, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Suzuki N; Hofstra JL; Poremba KE; Reisman SE Org. Lett 2017, 19, 2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Edwards JT; Merchant RR; McClymont KS; Knouse KW; Qin T; Malins LR; Vokits B; Shaw SA; Bao D-H; Wei F-L; Zhou T; Eastgate MD; Baran PS Nature 2017, 545, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) For examples of Cu-, Fe-, and Ni-catalyzed alkyne hydroalkylations, see: Uehling MR; Suess AM; Lalic GJ J. Am. Chem. Soc 2015, 137, 1424. [DOI] [PubMed] [Google Scholar]; (b) Lu X-Y; Liu J-H; Lu X; Zhang Z-Q; Gong T-J; Xiao B; Fu Y Chem. Commun 2016, 52, 5324. [DOI] [PubMed] [Google Scholar]; (c) Cheung CW; Zhurkin FE; Hu X J. Am. Chem. Soc 2015, 137, 4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Deng H-P; Fan X-Z; Chen Z-H; Xu Q-H; Wu JJ Am. Chem. Soc 2017, 139, 13579. [DOI] [PubMed] [Google Scholar]
- (16).Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA; Malliaras GG; Bernhard S Chem. Mater 2005, 17, 5712. [Google Scholar]
- (17).Bockman TM; Hubig SM; Kochi JK J. Org. Chem 1997, 62, 2210–2221. [DOI] [PubMed] [Google Scholar]
- (18).Durandetti M; Devaud M; Périchon J New J. Chem 1996, 20, 659. [Google Scholar]
- (19).Liu Z; Derosa J; Engle KM J. Am. Chem. Soc 2016, 138, 13076. [DOI] [PubMed] [Google Scholar]
- (20).Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DW C. Science 2016, 352, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.