

Abstract
Regulated proteolysis of signaling proteins under mechanical tension enables cells to communicate with their environment in a variety of developmental and physiologic contexts. The role of force in inducing proteolytic sensitivity has been explored using magnetic tweezers at the single-molecule level with bead-tethered assays, but such efforts have been limited by challenges in ensuring that beads are not restrained by multiple tethers. Here, we describe a multiplexed assay for single-molecule proteolysis that overcomes the multiple-tether problem using a flow extension (FLEX) strategy on a microscope equipped with magnetic tweezers. Particle tracking and computational sorting of flow-induced displacements allows assignment of tethered substrates into singly-captured and multiply-tethered bins, with the fraction of fully mobile, single-tethered substrates depending inversely on the concentration of substrate loaded on the coverslip. Computational exclusion of multiply-tethered beads enables robust assessment of on-target proteolysis by the highly specific tobacco etch virus protease and the more promiscuous metalloprotease ADAM17. This method should be generally applicable to a wide range of proteases and readily extensible to robust evaluation of proteolytic sensitivity as a function of applied magnetic force.
Graphical Abstract
INTRODUCTION
Force-dependent proteolysis of tension-sensing domains is a fundamental mechanism for signal transduction in biology 1–4. For example, in response to shear forces in the vasculature, von Willebrand Factor undergoes proteolysis in its force-sensing domain to regulate blood clotting5,6. At sites of cell-cell contact, a mechanosensing domain in the Notch receptor undergoes regulated proteolysis in response to tension applied by bound ligand, influencing cell fate decisions during development 7–9. In the cellular response to the extracellular matrix, force-induced cleavage of talin is essential for mechanosensation and the adhesion response 10,11.
Given the importance of force-dependent proteolysis in biological signaling, we sought to develop a robust, single-molecule assay for proteolytic cleavage that can be readily adapted for probing proteolytic sensitivity in response to force. In this regard, single-molecule magnetic tweezers methods enable direct, multiplexed observations of tension-dependent biochemical processes, in cell-based assays or in purified systems in vitro 12. These approaches typically rely on the capture of a substrate between a probe and anchor surface, normally a colloidal bead and glass coverslip, respectively. One problem that can confound these assays is the simultaneous capture of one bead by more than one immobilized substrate molecule, the so called “multiple-tether problem,” which has not been directly accounted for in previous multiplexed single-molecule proteolysis studies 7,13,14.
To address the multiple tethering problem, Dekker and coworkers used micropatterning to separate anchor points based on the contour length of a simple DNA substrate. Although the approach enriches for single tethers, it does not eliminate multiply tethered particles 15. In studying unzipping of DNA nanoswitches, Wong and colleagues used tethered particle motion and a length change signature to perform sorting for centrifugal force spectroscopy 16. This approach is highly specialized, however, and requires complex custom instrumentation that is not yet commercially available.
We present here a simple, readily accessible approach to identify monovalently tethered substrates for single-molecule enzymology and force spectroscopy, and apply our method to visualize single-molecule proteolysis in real time using a standard, inverted microscope equipped with magnetic tweezers (Figure 1). We capture beads onto chip-bound protease substrates in a microfluidic chamber, and computationally sort tracked substrate-tethered beads that are flow-stretched, showing that characteristic displacements under flow, or flow extension (FLEX) signatures, are a reliable method to identify beads tethered to a single substrate molecule. We then demonstrate specific proteolysis of bead-tethered substrates for two different classes of proteases. This technique should be a universal approach to sort putative substrates for a wide range of proteases, or other hydrolytic enzymes, and should be applicable to studies investigating proteolytic sensitivity as a function of force as well as to classic force-clamp spectroscopy experiments measuring the strength of adhesion bonds.
Figure 1.
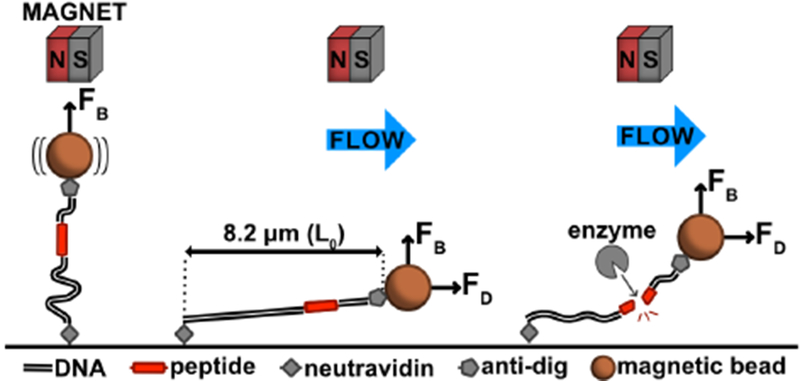
Schematic of the flow extension (FLEX) cleavage assay. Details of the FLEX approach are described in the text. FB, magnetic force; FD, drag force; L0, contour length.
MATERIALS AND METHODS
Chemicals
Sulfo-SMCC (sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate; 22322), and APMA (4-aminophenylmercurial acetate; A9563) were from Sigma. BB-94 or batimastat was from Calbiochem (196440). NeutrAvidin (31000) was from Thermo Fisher. The anti-digoxigenein Fab was purchased from Roche (11214667001). Nano-strip was from KMG (210034). Key reagents for the single-molecule working buffer were bovine serum albumin, heat shock fraction (A7906) and Pluronic F-127 (P2443) from Sigma. M-280 Tosyl activated Dynabeads (14203) were from Invitrogen. SDS-PAGE analysis was performed with either Bolt 4-12% Bis-Tris Plus gels (Life Technologies, NW04125BOX) or 4-20% tris-glycine gels (Bio-Rad, 456-1096). Denaturing PAGE of oligonucleotides and their conjugates was performed with Novex 15% TBE-Urea gels (Life Technologies, EC6885BOX). PD-10 desalting columns were from GE (95017-001). P-6 (7326221) and P-30 (7326223) Micro Bio-Spin columns were obtained from BioRad.
Lambda (λ) DNA dam(-) was purchased from New England Biolabs (N3013L). 3-aminopropyltrimethoxysilane was purchased from Sigma (281778-100ML) and stored under Argon at 4°C. Methyl-PEG5000-succinimidyl valerate (mPEG5000-SVA) and biotin-PEG5000-SVA (bioPEG5000SVA) were acquired from Laysan Biosciences and stored dessicated at −20 C.
Enzymes
T4 polynucleotide kinase (M0201S), T4 DNA ligase (M0202S), XbaI (R0145L), and XhoI (R0146S) were obtained from New England Biolabs. The enhanced activity SrtA pentamutant was expressed and purified from bacteria using the pET29-eSrtA vector (Addgene, #75144) as previously reported 17. TEV NIa protease was also expressed recombinantly in bacteria using the pTrc-7H-PRO plasmid and purified as described 18. The catalytic domain of ADAM17 (residues 215-477) was isolated after recombinant expression of its precursor form (1-477) with a C-terminal hexahistidine tag using a baculovirus expression system in insect cells. Purification was carried out in the presence of APMA as described 19. Enzyme activity was confirmed in bulk solution using a fluorogenic substrate (Mca-PLAQAV-Dpa-RSSSR-NH2; R&D systems, ES003) 20. Enzymes were snap frozen and stored at −80 C in aliquots before use.
Custom synthesized biopolymers
Oligonucleotides listed in Supplementary Table S2 were custom synthesized from Integrated DNA Technologies (IDT). Peptides listed in Supplementary Table S2 were custom synthesized from Genscript and supplied at 95% minimum purity. SMCC-activated amine-oligonucleotides (activated oligonucleotides, Table S2) were produced by mixing sulfo-SMCC (7.5 mM, dissolved in dimethylformamide) with 300 μM aminooligonucleotide at 25° C in 10 mM phosphate buffer, pH 7.2, containing 150 mM NaCl (PBS). After a 3-5 h reaction time, the activated oligonucleotide was purified away from residual free crosslinker on a PD-10 desalting column(s) pre-equilibrated in PBS, concentrated on a 3kDa MWCO centrifugal filter, and used immediately for peptide conjugation.
Other materials
Tubing was PE60 (Stoelting, 51162), coverslips were No. 1.5 VistaVision (VWR, 16004-312), double sided tape (Grace Bio-Labs, 620001), and 1 mm quartz tops were from Technical Glass Products.
Preparation of coverslips and flow chambers
Coverslips were prepared by first etching with stabilized piranha solution for 2 h, then silanizing with pre-hydrolyzed 3-(aminopropyl)trimethoxysilane in acidic methanol, and passivating by alkylation with PEG5000-succinimide containing 2-6 mol% biotinylated PEG 21,22. Coverslips were stored under vacuum before use. To generate flow cells, 15 × 5 mm channels were cut into double-sided tape, the tape was placed between the coverslip and a glass slide, and inlet and outlet ports were attached to the glass with epoxy as described21, 23.
Synthesis of double oligonucleotide peptide conjugates
The oligo-peptide conjugate (1) (Figure 2; oligo-peptide conjugate 1, Table S2) was synthesized by conjugation of the H2N-GGGK*GC-COOH (where the K* residue is ε-[5-amino-fluorescein (5-FAM)]-Lys) sortase donor peptide to a 5’-maleimide activated 17-base pair oligonucleotide (activated oligonucleotide 1 and peptideoligo conjugate 1, Table S2) 24. The conjugation reaction was allowed to proceed for 3 h at 22-25°C in PBS buffer pH 7.2 (10 mM phosphate, 150 mM NaCl) with 1 mM TCEP, containing 2 mM (~15 eq.) peptide 1 (Table S2) and 130 μM (1 eq) activated oligonucleotide 1 (Table S2). To purify the conjugate (1) (oligo-peptide conjugate 1, Table S2) from free peptide, the reaction was exchanged into in HBS buffer (25 mM HEPES pH 7.5, 150 mM NaCl) with a gravity desalting column and purified by size-exclusion chromatography to apparent homogeneity as assessed by TBE-Urea PAGE. The conjugate was stored at −80°C until use. The yield of oligo-peptide conjugate (1) from the oligonucleotide 1 starting material in this step is typically ~10 percent.
Figure 2.
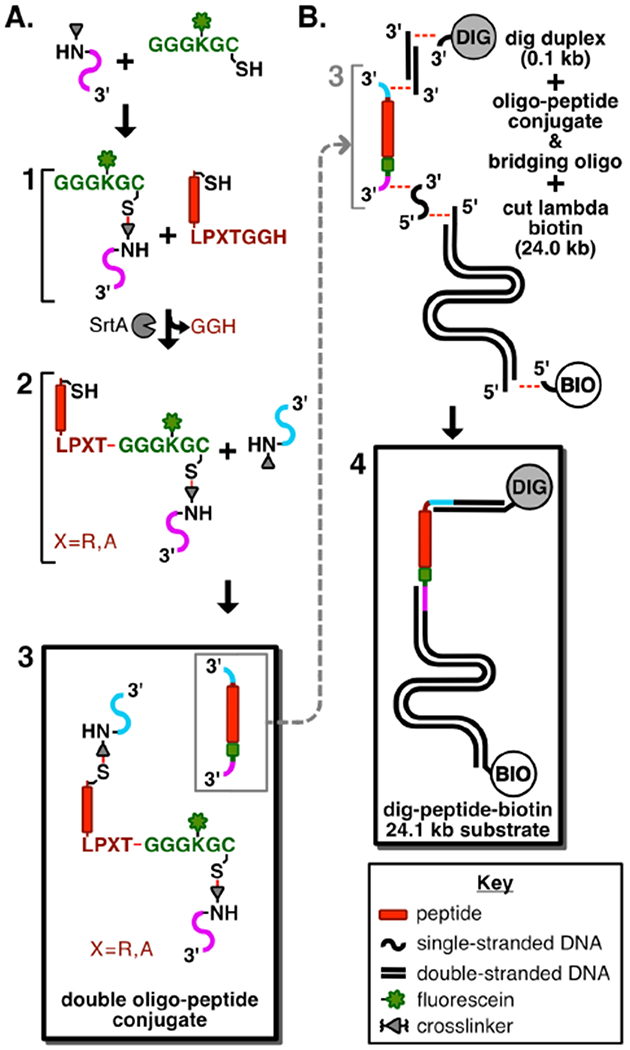
Scheme showing the route used to prepare DNA-conjugated peptide substrates for single-molecule proteolysis. A. Synthetic route to the double oligonucleotide-coupled peptide conjugate. Product 1 is a polyglycine conjugate made with a fluoresceinated lysine residue and a C-terminal thiol (green), coupled to a 5’-maleimide-oligo (magenta). Product 2 is the Sortase A-catalyzed conjugation of the substrate peptide containing a C-terminal LPXTG sortase acceptor motif (red) to the polyglycine-oligo (green-magenta). Product 3 (boxed) is the double oligonucleotide peptide conjugate made from linking the N-terminal cysteine of 2 to the 5’-maleimide-oligo (cyan). B. Synthesis of the digoxigenin-peptide-biotin substrate. Product 4 (boxed) is produced by an annealing and ligation reaction of a 5’ digoxigenin-containing oligonucleotide, a short (100 bp) DNA duplex and the N-terminal oligonucleotide end of the double oligo-peptide conjugate, along with simultaneous ligation of the C-terminal-end oligonucleotide to the XbaI-cleaved 24.0 kbp fragment of phage λ, and a terminal biotinylated oligonucleotide.
To produce oligo-peptide conjugate (2) (Figure 2; oligo-peptide conjugates 2-N and 2-T, Table S2), conjugate (1) was coupled to the C-terminus of either the Notch S2 peptide (peptide 2, Table S2) or the TEV substrate peptide (peptide 3, Table S2) in a transpeptidation reaction using SrtA. The SrtA transpeptidation was performed over 4 h at 42°C in HBS pH 7.5 with 10 mM MgCl2 and 4 μM NiCl2 reaction buffer using 15 μM (1 eq.) of oligo-peptide conjugate 1 (1), 150 μM (10 eq.) peptide 2 or 3, and 15 μM (1 eq.) eSrtA, similar to previously reported methods 25, 26. After coupling, the conjugate (2) was further purified using a desalting column, which also was used for buffer exchange into HBS [25 mM HEPES pH 7.5, 150 mM NaCl]. The formation of this product was monitored and its purity was assessed by appearance of a new band corresponding to this material on TBE-Urea PAGE. Conjugate (2) was stored at −80°C until use.
The double oligo-peptide conjugate (3) was formed by attaching the N-terminal cysteine of conjugate (2) to a 5’-maleimide functionalized 20 base-pair oligonucleotide (activated oligonucleotide 2, Table S2), essentially as described above for synthesis of conjugate (1) except that activated oligonucleotide 2 was used in vast molar excess during this step. The Notch S2 and TEV double oligo-peptide conjugates (3) (Figure 2; oligo-peptide conjugates 3-N and 3-T, respectively, Table S2) were purified using size-exclusion chromatography to apparent homogeneity, as assessed by denaturing TBE-Urea PAGE, and were stored at −80°C. We estimate the yield of the double oligo-peptide conjugate (3) at ~5% from conjugate (2) as starting material.
Production of duplex DNA single-molecule substrates
Each double-oligonucleotide peptide conjugate (3) was annealed to a bridging oligonucleotide (oligonucleotide 3, Table S2), a 100 base-pair 5’-digoxigenin containing sequence from the pUC19 ori region (oligonucleotides 4, 5, and 6; Table S2), an XbaI digested fragment from the phage λ genome (λXbaI, 23,998 base-pairs, 3.75 nM, at 250 μg scale), and a 3’-biotin cohesive site cosR sequence (oligonucleotide 7, Table S2) in 20 mM HEPES buffer, pH 7.5 (annealing temperatures and times were similar to previously published methods 27). Prior to annealing, all annealed oligonucleotides with free 5’ ends were treated with T4 polynucleotide kinase in 1x PNK buffer for 1 hr at 37°C and purified with a Qiagen nucleotide cleanup kit. The annealed assembly was ligated with 0.16 U/μL T4 DNA ligase for 2 hr at room temperature. The fully ligated substrate was purified away from lower molecular weight contaminants (removal of the free biotinylated oligonucleotide is critical) by buffer exchange into HEPES-buffered saline (HBS), pH 7.5, using a centrifugal filter with a molecular weight cutoff of 100 kDa. The all-DNA control substrate (5) (Figure S2) was produced using the same assembly and ligation procedure, using a DNA insert (oligonucleotide 8, Table S2) in place of the double oligonucleotide conjugate. A synthesis at 500 μg scale of λXbaI yields a concentrated substrate stock sufficient to perform several hundred FLEX experiments at a working concentration of 0.2 pM.
Substrate capture and bead tethering procedure
Flow chambers were mounted on the stage of an Olympus IX51 inverted microscope on an air table (Technical Manufacturing Corporation), with off-axis LED gooseneck illuminator (Fisher), and custom-built magnetic tweezers, as previously described with minor modifications 7. The chambers were also equipped with a syringe pump (Harvard Apparatus) controlled using a stopcock valve and a 50 mL air spring to regulate fluid flow through the flow-cell. All images were acquired with a QIClick-R-F-M-12 Mono CCD camera (QImaging) controlled with Micro-Manager software 28.
Prior to capture of substrates on the flow-cell surface, the flow-cell chamber was first treated for a minimum of 10 min with 500 μL blocking buffer (HBS containing 0.5% (w/v) BSA and 1% (w/v) Pluronic F127) drawn manually into the flow cell. Then, the chamber was incubated for 3-5 min with 80 μL of NeutrAvidin (0.1 mg/mL in HBS) drawn manually into the chamber, and washed with 500 μL working buffer (HBS containing 0.1% (w/v) BSA and 1% (w/v) Pluronic F127) at a flow-rate of 100 μL/min. Substrate was diluted into working buffer (0.15 to 15 pM final concentration), and a total volume of 100 μL of substrate was drawn into the chamber first at 20 μL/min for one min to clear the inlet tubing, and at 5 μL/min thereafter before incubating in the chamber for an additional 2-5 min. The chamber was then washed with working buffer for 10 min at a flow rate of 50 μL/min.
To prepare beads for capture, tosyl-activated 2.8 μm Dynabeads were conjugated to anti-digoxigenin Fab, and stored in PBS with 0.01% (w/v) BSA at 4°C, as described (according to manufacturer’s Protocol and 21). Immediately before use, beads were diluted to 10 μg/mL in working buffer, vortexed, and sonicated to minimize clumping. The suspended beads (~250 μL) were then drawn into the chamber at 20 μL/min. The chamber was aligned in the direction of flow to make sure that a bead in motion across the chamber did not move more than half its diameter across the entire field of view (fov). The chamber was then washed with 400 μL ofworking buffer at 30 μL/min. After the washing step, the flow was halted to allow beads to relax from their extended position (typically for 5-10 min). After the syringe pump was turned off, a single 5mm NdFeB cube magnet on a hinged mount was positioned at 9 mm above the coverglass with a micrometer (equivalent to approximately 0.6 pN of applied force normal to the coverslip) in order to keep the beads above the surface. The magnet was positioned with its dipole parallel to plane of the flow cell as depicted in Figure 1 (note that orienting the dipole normal to the coverslip results in precession of the bead around its centered axial position). Force was estimated with a digoxigenin-λ-biotin (48,502 bp, full length prepared as described in 27) tether based on the variance in bead position (〈σy2〉) in the direction transverse to applied force, according to the method published by Strick and colleagues 29.
Single-molecule flow-extension procedure
For proof-of-concept flow extension experiments (Figure 3, supplementary movie V1), additional buffer was added to the inlet after substrate loading and bead capture, and the valve was opened and closed to allow pressure equilibration of the flow cell. Data acquisition was then initiated at 2 frames per second, initially in the absence of flow, but with the magnet in position (Figure 1), to allow determination of the initial position of each tracked bead. At the time of valve opening (at t = 200 sec), the buffer was drawn into the chamber at 5 μL/min using a syringe pump to induce flow-extension. The extension under flow reaches equilibrium at approximately 4 min. At the 5 min time point, the position of the bead at extension was recorded.
Figure 3.
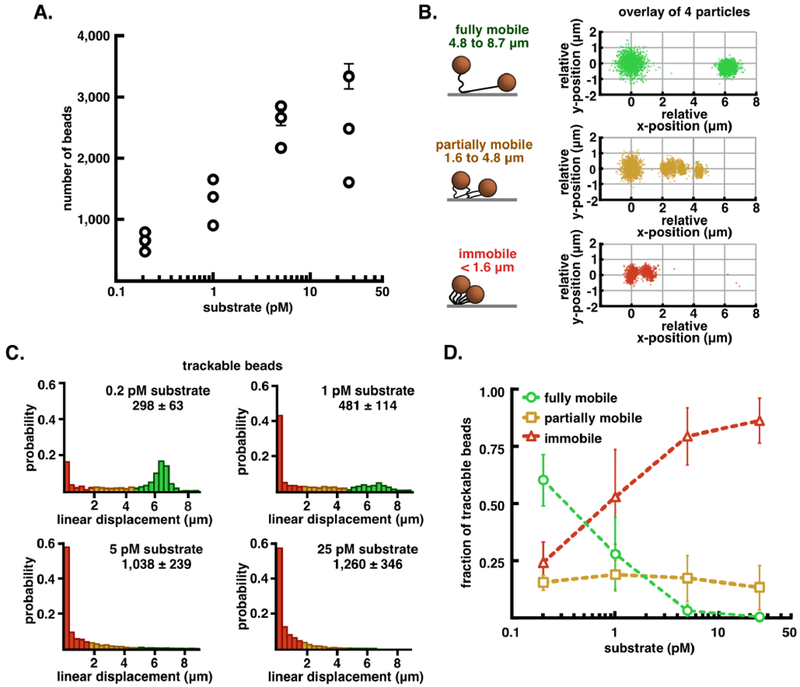
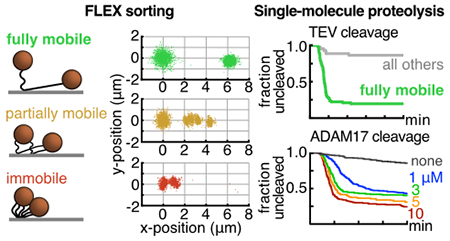
Tethered bead movement under flow extension as a function of tether density. A. Semi-log plot of the average number of captured beads in six fields-of-view from flow-chambers incubated with increasing amounts of dig-λXbaI-biotin (0.2, 1, 5, 25 pM), reported as mean ± S.E.M. B. Graphic representation of the three classes of beads that are observed in every experiment alongside an overlay of relative initial (t = 0-200 sec) and extended (t = 300-350 sec) position data from four trajectories from each class of displacement: fully mobile (4.8 - 8.7 μm, green), partially mobile (1.6 – 4.8 μm, orange), and immobile (< 1.6 μm, red). C. Probability normalized histograms of the linear displacement of bead trajectories at increasing amounts of substrate. The number of particle trajectories tracked in 3 replicates (mean ± S.E.M.) are listed above each histogram. D. Quantification of the fraction of the total trajectories that were sorted into each class of displacement, reported as mean ± S.E.M. See also Table S1.
Analysis and sorting of flow extension bead profiles
Time-lapse data sets were analyzed with custom MATLAB scripts written for this work, available upon request. Detected particles were identified and analyzed with colloidal particle tracking algorithms developed by Cocker and Grier 30, adapted for MATLAB by Kilfoil 31. The trajectories of monodisperse particles were inspected manually to ensure high-quality tracking output, using the msdanalyzer suite created by Tinevez 32.
The flow-extension trajectory of each monodisperse bead was analyzed to determine its linear displacement. Specifically, only bead trajectories that started at the first frame (t = 0 sec) of acquired data and that persisted through the time required for bead extension (t = 200 sec) were scored. The mean initial (0 to 200 sec) and extended (300 to 350 sec) positions were determined by fitting the bead positions to a Gaussian distribution. Linear flow-displacements were calculated by measuring the distance between these mean positions. The displacements were measured to sub-pixel accuracy (σσRMSE) and precision ().33.
Based on their mean displacements, the analyzed beads were sorted into accepted (fully mobile [8.7 to 4.8 μm]) and rejected (partially mobile [4.8 to 1.6 μm] and immobile [1.6 to 0 μm]) populations. The histogram of displacements for a population of tethered substrates was always inspected to qualitatively assess the character of the distribution relative to the fraction in each population. The mean extension for a high-quality fully mobile population was typically between 6.0 – 6.5 μm (L/L0 = 0.73 – 0.79) assuming L0 = 8.20.
Single-molecule proteolysis with TEV
Immediately before use, TEV protease was buffer-exchanged into TEV cutting buffer (25 mM HEPES pH 7.5, 25 mM NaCl, and 0.01%(w/v) Pluronic F-127) with a P-6 spin column to remove residual TCEP present during storage, and then diluted to a concentration of 20 μM before introduction into the flow cell. After substrate loading and bead capture, TEV was introduced into the flow cell at 5 μL/min and bead trajectories were monitored for assignment into mobile, partially mobile, or immobile categories. The field of view was then monitored for bead loss as a function of time to produce the cleavage plots in Figure 4 (see also supplementary movie V2).
Figure 4.
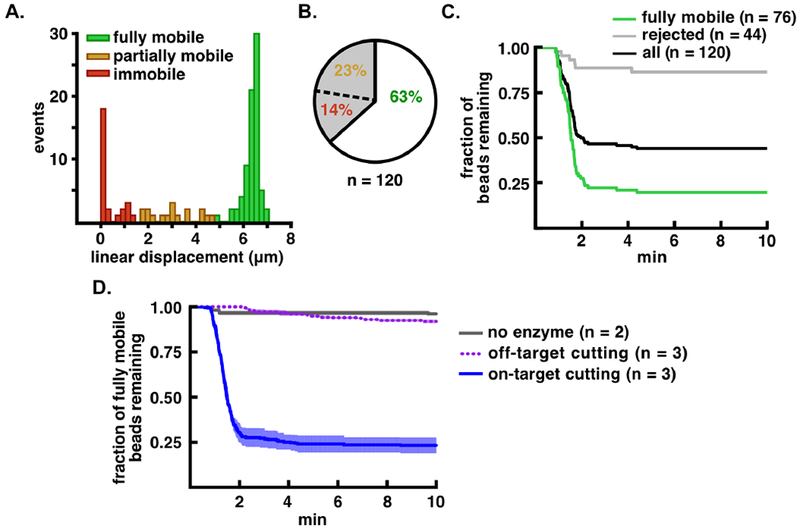
Improved analysis of TEV-catalyzed proteolysis using computational sorting of bead mobility. A. Distribution of flow extension distances for bead-tethered, TEV substrate molecules. The fully mobile population is shown in green, partially mobile in orange, and immobile in red. B. Pie chart showing the percentage of accepted, partially mobile, and immobile beads in the tethered population. C. Proteolysis of bead-tethered substrates upon addition of 20 μM TEV protease. The fraction of beads lost is plotted as a function of time for the total population (black), the accepted (fully mobile) population (green), and the rejected population (gray). D. Cleavage specificity analysis. Bead release from cleavage of the tethered peptide substrate is indicated with a blue solid line, release of the tethered all-DNA substrate is indicated with a purple dotted line, and bead release in the absence of enzyme is indicated in gray. Kinetic traces are normalized such that t = 0 min is set to 1 min before the start of bead loss, and are shown as mean ± S.E.M. for the number of trials indicated (n = 2 or n = 3).
Single-molecule proteolysis with ADAM17
ADAM17 was diluted to the appropriate concentration into metalloprotease cutting buffer (HBS with 4 μM ZnCl2 and 1%(w/v) Pluronic F-127) immediately before use, and flowed into the chamber to generate the cleavage plots in Figure 5. Flow of enzyme into the chamber was halted after ~25 min, but trajectories were tracked for a total of 45 min to extract the rate constant for the slow exponential phase. To confirm that cleavage was due to ADAM17, the metalloprotease inhibitor BB-94 was used at 20 μM.
Figure 5.
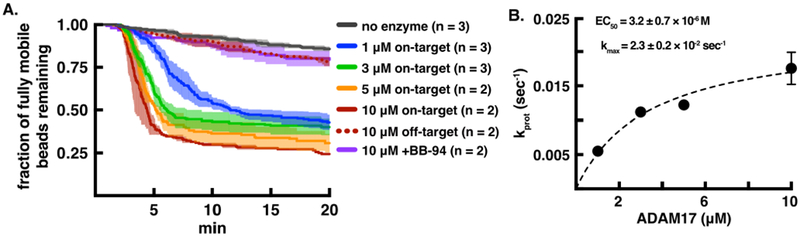
Single-molecule proteolysis of a Notch1 substrate by the ADAM17 catalytic domain. A. Plot of beads released as a function of time using different concentrations of enzyme and DNA control (dashed line) or Notch1 peptide substrate (solid lines). A cleavage reaction (at an ADAM17 concentration of 10 μM) performed in the presence of the metalloprotease inhibitor BB94 is shown in purple. B. Fit of the single-molecule cleavage rates from (A) as a function of ADAM17 concentration to the equation kprot = kmax × [ADAM17]/(EC50 + [ADAM17]). Kinetic traces are shown as mean ± S.E.M. for the number of trials indicated (n = 2 or n = 3).
Curve-fitting for cleavage data
Bead loss curves were fit to a double exponential using non-linear least squares fitting, where the fast rate constant corresponds to kprot. The start of bead loss was determined for each curve by manual inspection. For bead loss curves measured in the absence of enzyme or with inhibitor present, fits were to a single exponential over a time course of 15 min. For ADAM17 experiments, a plot of kprot as a function of enzyme concentration was fit to:
| (1) |
to extract kmax and EC50, which are analogous to kcat and KD, as described previously13.
RESULTS
In order to develop a reliable bead-tethered assay for single-molecule proteolysis, we developed an approach that relies on the use of flow extension (FLEX) to distinguish beads tethered to single substrates from beads with multiple tethers or beads that adhere to the flow-cell surface after capture. There are two independent measurements made in this experiment: first, we measure bead displacement in a flow extension assay to distinguish among different tether configurations, and then we measure bead release in a cleavage assay performed with protease (Figure 1). Our substrate contains a long DNA tether with an embedded polypeptide sequence between digoxigenin- and biotin-labeled DNA handles. The substrate is captured on a neutravidin surface using the biotinylated DNA end, and α-digoxigenin Fab coated magnetic beads are then tethered to the beads at the digoxigenin-labeled end. This bead-tethered substrate is then used for multiplexed, single-molecule proteolysis (Figure 1).
The assembly of the DNA-conjugated peptide substrate is schematically illustrated in Figure 2. First, we linked a single-stranded oligonucleotide to the C-terminal cysteine residue of a fluorescently labeled acceptor peptide using a maleimide-modified nucleotide (1 in Figure 2A and Figure S1). Second, we coupled a substrate peptide containing an N-terminal cysteine to this acceptor-DNA conjugate using evolved Sortase A (SrtA) (2 in Figure 2A and Figure S1). Third, using maleimide coupling, we attached a second single-stranded oligonucleotide to the N-terminal cysteine of this protein-DNA conjugate to generate a peptide substrate with single stranded oligonucleotides at each end (3 in Figure 2A and Figure S1). After purification on a size exclusion column, this molecule was then ligated to a short DNA duplex at the N-terminal end, and to an XbaI-cleaved fragment of λ phage (23,998 bp) at the C-terminal end. Ligation of a digoxigenin-modified oligonucleotide to the short duplex at the N terminus, and a biotinylated oligonucleotide to the λ phage fragment, resulted in production of a substrate with a biotin handle for surface attachment and a digoxigenin handle for capture of anti-digoxigenin beads (4 in Figure 2B).
After assembly as above, the final substrate for proteolysis contains a custom peptide embedded between DNA duplexes that together total 24,098 base-pairs and have a maximal extension length (L0) of 8.2 μm, assuming 0.34 nm per base pair and 0.35 nm per amino acid (Figures 1 and 2B). The modular nature of both N- and C-terminal DNA fragments enables the use of numerous surface attachment strategies and choice of DNA handles ofvariable lengths on either side of the substrate.
To establish proof-of-concept for the FLEX sorting method and to optimize conditions for single-bead capture, we first investigated a control all-DNA tether of comparable length, assembled similarly (Figure S2). We investigated beads captured onto these control DNA tethers loaded onto the flow cell at four different concentrations, ranging from 0.2 – 25 pM (Figure 3A). At each concentration, we performed a FLEX experiment by measuring the position of individual beads in the absence and presence of flow to classify bead trajectories into immobile (extension <1.6 μm), partially mobile (extension between 1.6 – 4.8 μm), or fully mobile (extension between 4.8 – 8.7 μm), eliminating clumped beads or trajectories that were incomplete over the flow-extension period (Table S1). The trajectory data show that loading substrates at a concentration of 0.2 pM results in a majority of single beads with full trajectories falling into the fully mobile category, and that a concentration of 5 pM or greater results in multiple tethering or immobility of virtually all loaded beads (Figure 3C, D; see also Table S1). For the trajectories that are fully mobile, displacements fit to a normal distribution (Figure 3C and 3D, green population) of 6.37 ± 0.54 μm (mean ± S.D., n = 518). It is also clear that there is an inverse correlation between the fraction of fully mobile and immobile beads as the concentration of tethered beads increases, indicating that as more trajectories are observed there are sufficient bound substrates to cause multiple tethering for nearly every bead (Figure 3C and 3D). Overall, the bead mobility analysis indicates that the FLEX signature is a robust method to determine whether a bead is tethered to a single substrate molecule.
We next used our FLEX sorting procedure to assess the single-molecule proteolysis of a bead-tethered peptide substrate. For proof-of-concept studies, we assembled a double-DNA conjugated 19-residue peptide substrate (see Figure 2) containing a consensus Tobacco Etch Virus (TEV) protease cleavage site 34–36 (ENLYFQ/G, with the scissile bond indicated by a backslash), and subjected this substrate to cleavage with TEV (Figure 4). Beads captured on the DNA-peptide substrate show a distribution of FLEX behavior (Figure 4A, B), with 63% ofthe tethered beads (76/120) being fully mobile. Upon TEV treatment (Figure 4C), the population of tethered substrates (black) shows two kinetic phases and a residual population of approximately 45% of the beads that remain uncleaved after 10 min. Computational sorting of the beads into accepted and rejected beads based on their FLEX trajectories shows that the computationally rejected population (gray, inclusive of both partially mobile and immobile tracks) resists specific cleavage by TEV, whereas the fully mobile population (green) shows more complete cleavage with a residual bead population of approximately 20% uncleaved beads after 10 min. The accepted bead population exhibits a rate of decay that is much faster than that of the rejected bead population (Figure 4C, compare gray and green decay curves). Whereas the accepted population of molecules for the specific peptide substrate shows rapid decay with a kloss of 3.09 ± 0.03 × 10−2 sec−1 (n = 3, mean ± 95% CI), the accepted molecules for control bead-tethered substrates that lack peptide or tethered substrates treated with buffer under flow without added TEV exhibit only slow phases of bead loss that are nearly equivalent (kloss= 3.3 ± 0.2 × 10−5 sec−1 for the DNA-only control substrate, n = 3 mean ± 95% CI) likely attributable to dissociation of the anti-digoxigenin/digoxigenin (or neutravidin/biotin) interaction, for which the off rates in the literature range from 0.015 sec−1 37 to 1.5 × 10−5 sec−1 38 (Figure 4D).
We next tested whether our assay system could be used to investigate proteolysis at the single-molecule level for a less selective protease, ADAM17. ADAM17 catalyzes the cleavage of a number of different proteins involved in intracellular signaling, including the epidermal growth factor precursor, tumor necrosis factor α, and, under certain pathophysiologic contexts, the force-sensitive substrate human Notch1 39, 40. Unlike TEV, the catalytic domain of ADAM17 does not recognize a highly specific consensus sequence, and is instead capable of cleaving a wide range of substrates with preference only for a bulky aliphatic residue in the P1’ position (i.e. Val or Ile) 41–43.
The ADAM 17 substrate contains a ten-residue equence from human Notch1 (NIPYKIEA/VQS, with the scissile bond indicated by a backslash) that spans the metalloprotease cleavage site (called S2 40, 44) with additional short flanking sequences embedded between the N- and C-terminal DNA handles (Figure 2). Cleavage of the Notch1 substrate by ADAM17 shows both a fast phase of bead release, dependent on the concentration of ADAM17 in the cleavage buffer, followed by a slow phase (Figure 5A). Controls testing for release of tethered beads in the absence of enzyme for bead release in the presence of both ADAM17 and the metalloprotease inhibitor BB-94, or for ADAM17 dependent release of beads tethered to a DNA-only substrate analog show a slow phase of non-specific bead detachment, confirming that the fast exponential phase for ADAM17-catalyzed cleavage of the Notch1 substrate is due to specific single-molecule proteolysis. This result also strongly suggests that ADAM17 cleavage of Notch occurs when the target sequence is in an extended conformation. The rate of enzyme-catalyzed bead release plotted as a function of ADAM17 concentration shows saturable kinetics (Figure 5B), with an estimated kmax of 2.3 ± 0.2 × 10−2 sec−1 and EC50 for [ADAM17] of 3.2 ± 0.7 × 10−6 M (reported as mean ± S.E.M., analogous to values for kcat and KD, as proposed previously 13). These values for kmax and EC50 compare favorably to reported values of kcat (0.07 sec−1) and KM (4.0 μM) for ADAM17 cleavage of a TNFα substrate 45.
DISCUSSION
Here, we report a method that uses flow extension sorting of tethered beads to enable stringent analysis of single-molecule proteolysis for two different enzymes and their substrates. The cleavage rates we observe are consistent with published studies of these enzymes in bulk proteolysis experiments 20, 45. Our approach relies on computational sorting of substrate-tethered beads loaded at low density, using their mobility under flow to exclude immobile or multiply tethered beads and thereby only analyze beads tethered to a single substrate molecule.
In previous studies measuring proteolysis of bead-tethered substrates, approaches to classify different beads within the field of view were not used, and therefore it was not possible to make strong claims about monovalency7, 13. Our data show that tether valency is highly sensitive to the concentration of substrate used for capture in the flow cell. Even when DNA-only molecules are used for the flow-extension analysis, a pronounced increase in fully mobile beads is readily evident as the decrease in grafting density is reduced to the lowest loading concentration (0.2 pM). The FLEX procedure enables empirical optimization ofsubstrate concentration to maintain enough beads for multiplexing statistics while minimizing the population of beads rejected because of surface adsorption or multiple tethering. In the studies reported here, we were able to load all-DNA molecules into the chamber so that as many as 202 fully mobile (of 355 total) DNA-tethered beads were present in a single field of view. Even the double-oligonucleotide conjugate peptide substrates, which are likely to be more adsorption prone because of their embedded peptide sequences, can be delivered to the flow cell at concentrations sufficient to produce 120 fully mobile (of 165 total) substrate-tethered beads in a single field of view. We also note that our empirical data are consistent with statistical predictions about multiple tethering made using an ideal surface model dependent upon the grafting density of substrates and the sum of tether contour length and bead diameter 33.
The use of the flow-extension signature to perform computational exclusion of beads that are not held by a single tether results in a demonstrable improvement in the measurement of substrate cleavage kinetics (Figure 4). The approach should be particularly valuable in identifying reaction conditions suitable for investigation of protein or peptide substrates that are prone to adsorption on surfaces. Optimization of reaction conditions can be carried out by selecting blocking conditions and substrate variants that minimize the number of transiently or permanently adsorbed beads, enriching for the fully mobile beads that report faithfully on cleavage of a single substrate molecule during the enzyme-catalyzed reaction.
The assay tools reported here should be readily adaptable for the investigation of any tension dependent hydrolytic reaction, simply by using the magnetic tweezers to vary the magnitude of applied force, as long as the enzyme can be supplied at concentrations on the order of its Km for substrate. Apart from testing mechanosensing domains, our assay can be used to study the stability of peptide and foldamer secondary and tertiary structural elements by investigating the force-dependence of denaturation of such structural elements using proteolytic cleavage as a proxy. Two classes of molecules with changes in cleavage sensitivity can be envisioned: those substrates that have cryptic cleavage sites that are exposed upon denaturation or conformational change 46, 47, or those substrates that only present a productive cleavage site when they fold 48–51.
The FLEX sorting technique should also be applicable to in vitro single-molecule force-spectroscopy 52–54. For bond rupture experiments, it is equally important to ensure that only monovalent interactions are scored to evaluate catch-or flex-bond versus slip-bond behavior 55–57.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to Tom C. M. Seegar for providing the baculovirus used for ADAM17 production, Andrew Moreno for help with coding, and both Thomas G. W. Graham and Dan Song for help with flow cell preparation. This work is supported by NIH R35 CA220340 (to S.C.B.) and R01 GM114065 (to J.J.L.).
Funding Sources
Supported by grants for the NIH (R35 CA200340 to S.C.B. and R01 GM114065 to J.J.L). S.C.B. receives research funding for an unrelated project from Novartis, and is a consultant on unrelated projects for IFM and Ayala Therapeutics.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website, and includes the following:
1) Supporting Figures S1 and S2, Supporting Tables S1 and S2, Legends for Supporting Movies V1 and V2, combined into a single PDF file.
2) Supporting Movies V1 and V2 (file type AVI)
REFERENCES
- [1].Henrique D, and Schweisguth F (2019) Mechanisms of Notch signaling: a simple logic deployed in time and space, Development 146, dev172148. [DOI] [PubMed] [Google Scholar]
- [2].Sadler JE (2005) New concepts in von Willebrand disease, Annu. Rev. Med 56, 173–191. [DOI] [PubMed] [Google Scholar]
- [3].Haining AW, Lieberthal TJ, and Del Rio Hernandez A (2016) Talin: a mechanosensitive molecule in health and disease, FASEB J. 30, 2073–2085. [DOI] [PubMed] [Google Scholar]
- [4].Wirtz D, Konstantopoulos K, and Searson PC (2011) The physics of cancer: the role of physical interactions and mechanical forces in metastasis, Nat. Rev. Cancer 11, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tsai H (1996) Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion, Blood 87, 4235–4244. [PubMed] [Google Scholar]
- [6].Zhang X, Halvorsen K, Zhang CZ, Wong WP, and Springer TA (2009) Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor, Science 324, 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, Tiyanont K, McArthur DG, Aster JC, Perrimon N, Loparo JJ, and Blacklow SC (2015) Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch, Dev. Cell 33, 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Langridge PD, and Struhl G (2017) Epsin-Dependent Ligand Endocytosis Activates Notch by Force, Cell 171, 1383–1396 e1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Parks AL, Klueg KM, Stout JR, and Muskavitch MA (2000) Ligand endocytosis drives receptor dissociation and activation in the Notch pathway, Development 127, 1373–1385. [DOI] [PubMed] [Google Scholar]
- [10].Saxena M, Changede R, Hone J, Wolfenson H, and Sheetz MP (2017) Force-Induced Calpain Cleavage of Talin Is Critical for Growth, Adhesion Development, and Rigidity Sensing, Nano Lett. 17, 7242–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, and Huttenlocher A (2004) Calpain-mediated proteolysis of talin regulates adhesion dynamics, Nat. Cell Biol 6, 977–983. [DOI] [PubMed] [Google Scholar]
- [12].Vilfan ID, Lipfert J, Koster DA, Lemay SG, and Dekker NH (2009) Magnetic Tweezers for Single-Molecule Experiments, In Handbook of Single-Molecule Biophysics, pp 371–395. [Google Scholar]
- [13].Adhikari AS, Chai J, and Dunn AR (2011) Mechanical load induces a 100-fold increase in the rate of collagen proteolysis by MMP-1, J. Am. Chem. Soc 133, 1686–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kirkness MWH, and Forde NR (2018) Single-Molecule Assay for Proteolytic Susceptibility: Force-Induced Collagen Destabilization, Biophys. J 114, 570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].De Vlaminck I, Henighan T, van Loenhout MT, Pfeiffer I, Huijts J, Kerssemakers JW, Katan AJ, van Langen-Suurling A, van der Drift E, Wyman C, and Dekker C (2011) Highly parallel magnetic tweezers by targeted DNA tethering, Nano Lett. 11, 5489–5493. [DOI] [PubMed] [Google Scholar]
- [16].Yang D, Ward A, Halvorsen K, and Wong WP (2016) Multiplexed single-molecule force spectroscopy using a centrifuge, Nat. Commun 7, 11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dorr BM, Ham HO, An C, Chaikof EL, and Liu DR (2014) Reprogramming the specificity of sortase enzymes, Proc. Natl. Acad. Sci. U. S. A 111, 13343–13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Parks TD, Howard ED, Wolpert TJ, Arp DJ, and Dougherty WG (1995) Expression and purification of a recombinant tobacco etch virus Nla proteinase: biochemical analyses of the full-length and a naturally occurring truncated proteinase form, Virology 210, 194–194. [DOI] [PubMed] [Google Scholar]
- [19].Milla ME, Leesnitzer MA, Moss ML, Clay WC, Carter HL, Miller AB, Su J-L, Lambert MH, Willard DH, and Sheeley DM (1999) Specific sequence elements are required for the expression of functional tumor necrosis factor-αconverting enzyme (TACE), J. Biol. Chem 274, 30563–30570. [DOI] [PubMed] [Google Scholar]
- [20].Minond D, Cudic M, Bionda N, Giulianotti M, Maida L, Houghten RA, and Fields GB (2012) Discovery of novel inhibitors of adisintegrin and metalloprotease 17 (ADAM17) using glycosylated and non-glycosylated substrates, J. Biol. Chem 287, 36473–36487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tanner NA, and van Oijen AM (2010) Visualizing DNA Replication at the Single-Molecule Level, In Single Molecule Tools, Part B:Super-Resolution, Particle Tracking, Multiparameter, and Force Based Methods, pp 259–278. [DOI] [PubMed] [Google Scholar]
- [22].Chandradoss SD, Haagsma AC, Lee YK, Hwang JH, Nam JM, and Joo C (2014) Surface passivation for single-molecule protein studies, J. Vis. Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gambino S, Mousley B, Cathcart L, Winship J, Loparo JJ, and Price AC (2016) A single molecule assay for measuring site-specific DNA cleavage, Anal. Biochem 495, 3–5. [DOI] [PubMed] [Google Scholar]
- [24].Hermanson GT (2013) Bioconjugate Techniques, 3 ed., Academic Press. [Google Scholar]
- [25].David Row R, Roark TJ, Philip MC, Perkins LL, and Antos JM (2015) Enhancing the efficiency of sortase-mediated ligations through nickel-peptide complex formation, Chem. Commun. (Camb.) 51, 12548–12551. [DOI] [PubMed] [Google Scholar]
- [26].Levary DA, Parthasarathy R, Boder ET, and Ackerman ME (2011) Protein-protein fusion catalyzed by sortase A, PLoS ONE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim H, and Loparo JJ (2016) Multistep assembly of DNA condensation clusters by SMC, Nat. Commun 7, 10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014) Advanced methods of microscope control using muManager software, J. Biol. Methods 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Strick TR, Allemand JF, Bensimon D, Bensimon A, and Croquette V (1996) The elasticity of a single supercoiled DNA molecule, Science 271, 1835–1837. [DOI] [PubMed] [Google Scholar]
- [30].Crocker JC, and Grier DG (1996) Methods of digital video microscopy for colloidal studies, J Colloid Interface Sci. [Google Scholar]
- [31].Pelletier V, Gal N, Fournier P, and Kilfoil ML (2009) Microrheology of microtubule solutions and actin-microtubule composite networks, Phys. Rev. Lett 102, 188303. [DOI] [PubMed] [Google Scholar]
- [32].Tarantino N, Tinevez JY, Crowell EF, Boisson B, Henriques R, Mhlanga M, Agou F, Israel A, and Laplantine E (2014) TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO-IKK supramolecular structures, J. Cell. Biol 204, 231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yin H, Landick R, and Gelles J (1994) Tethered particle motion method for studying transcript elongation by a single RNA polymerase molecule, Biophys. J 67, 2468–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Carrington JC, and Dougherty WG (1988) A viral cleavage site cassette: identification of amino acid sequences required for tobacco etch virus polyprotein processing, Proc. Natl. Acad. Sci. U. S. A 85, 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Phan J, Zdanov A, Evdokimov AG, Tropea JE, Peters Iii HK, Kapust RB, Li M, Wlodawer A, and Waugh DS (2002) Structural Basis for the Substrate Specificity of Tobacco Etch Virus Protease, J. Biol. Chem 277, 50564–50572. [DOI] [PubMed] [Google Scholar]
- [36].Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, and Waugh DS (2001) Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency, Protein Eng. 14, 993–1000. [DOI] [PubMed] [Google Scholar]
- [37].Neuert G, Albrecht C, Pamir E, and Gaub HE (2006) Dynamic force spectroscopy of the digoxigenin-antibody complex, FEBS Lett. 580, 505–509. [DOI] [PubMed] [Google Scholar]
- [38].Koussa MA, Halvorsen K, Ward A, and Wong WP (2015) DNA nanoswitches: a quantitative platform for gel-based biomolecular interaction analysis, Nat. Meth 12, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, and et al. (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha, Nature 385, 733–736. [DOI] [PubMed] [Google Scholar]
- [40].Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, and Israel A (2000) A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE, Mol. Cell 5, 207–216. [DOI] [PubMed] [Google Scholar]
- [41].Caescu CI, Jeschke GR, and Turk BE (2009) Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10, Biochem. J 424, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mohan MJ, Seaton T, Mitchell J, Howe A, Blackburn K, Burkhart W, Moyer M, Patel I, Waitt GM, and Becherer JD (2002) The Tumor Necrosis Factor-α Converting Enzyme (TACE): A Unique Metalloproteinase with Highly Defined Substrate Selectivity, Biochemistry 41, 9462–9469. [DOI] [PubMed] [Google Scholar]
- [43].Tucher J, Linke D, Koudelka T, Cassidy L, Tredup C, Wichert R, Pietrzik C, Becker-Pauly C, and Tholey A (2014) LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries, J. Proteome Res 13, 2205–2214. [DOI] [PubMed] [Google Scholar]
- [44].Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan D, Ray WJ, and Kopan R (2000) A ligand-induced extracellular cleavage regulates γ-secretase-like proteolytic activation of Notch1, Mol. Cell 5, 197–206. [DOI] [PubMed] [Google Scholar]
- [45].Stawikowska R, Cudic M, Giulianotti M, Houghten RA, Fields GB, and Minond D (2013) Activity of ADAM17 (a disintegrin and metalloprotease 17) is regulated by its noncatalytic domains and secondary structure of its substrates, J. Biol. Chem 288, 22871–22879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stanger HE, and Gellman SH (1998) Rules for Antiparallel β-Sheet Design: D-Pro-Gly Is Superior to L-Asn-Gly for β-Hairpin Nucleation1, J. Amer Chem. Soc 120, 4236–4237. [Google Scholar]
- [47].Cline LL, and Waters ML (2009) The structure of well-folded beta-hairpin peptides promotes resistance to peptidase degradation, Biopolymers 92, 502–507. [DOI] [PubMed] [Google Scholar]
- [48].Zasloff M (1987) Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor, Proc. Natl. Acad. Sci. U. S. A 84, 5449–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Resnick NM, Maloy WL, Guy HR, and Zasloff M (1991) A novel endopeptidase from Xenopus that recognizes α-helical secondary structure, Cell 66, 541–554. [DOI] [PubMed] [Google Scholar]
- [50].Bayer P, Arndt A, Metzger S, Mahajan R, Melchior F, Jaenicke R, and Becker J (1998) Structure determination of the small ubiquitinrelated modifier SUMO-1, J. Mol. Biol 280, 275–286. [DOI] [PubMed] [Google Scholar]
- [51].Mossessova E, and Lima CD (2000) Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast, Mol. Cell 5, 865–876. [DOI] [PubMed] [Google Scholar]
- [52].Liu B, Chen W, and Zhu C (2015) Molecular Force Spectroscopy on Cells, Annu. Rev. Phys. Chem 66, 427–451. [DOI] [PubMed] [Google Scholar]
- [53].Thomas WE, Vogel V, and Sokurenko E (2008) Biophysics of Catch Bonds, Annu. Rev. Biophys 37, 399–416. [DOI] [PubMed] [Google Scholar]
- [54].Thomas WE (2009) Mechanochemistry of receptor–ligand bonds, Curr. Opin. Struct. Biol 19, 50–55. [DOI] [PubMed] [Google Scholar]
- [55].Thomas WE, Trintchina E, Forero M, Vogel V, and Sokurenko EV (2002) Bacterial adhesion to target cells enhanced by shear force, Cell 109, 913–923. [DOI] [PubMed] [Google Scholar]
- [56].Kim J, Zhang C-Z, Zhang X, and Springer TA (2010) A mechanically stabilized receptor–ligand flex-bond important in the vasculature, Nature 466, 992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Thomas W (2008) Catch Bonds in Adhesion, Annu. Rev. Biomed. Eng 10, 39–57 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.