

Abstract
CD47 is known to play an important role in CD4+ T cell homeostasis. We recently reported a reduction in mice deficient in the Cd47 gene (Cd47−/−) CD4+ T cell adhesion and transendothelial migration (TEM) in vivo and in vitro as a result of impaired expression of high‐affinity forms of LFA‐1 and VLA‐4 integrins. A prior study concluded that Cd47−/− mice were resistant to experimental autoimmune encephalomyelitis (EAE) as a result of complete failure in CD4+ T cell activation after myelin oligodendrocyte glycoprotein peptide 35–55 aa (MOG35–55) immunization. As the prior EAE study was published before our report, authors could not have accounted for defects in T cell integrin function as a mechanism to protect Cd47−/− in EAE. Thus, we hypothesized that failure of T cell activation involved defects in LFA‐1 and VLA‐4 integrins. We confirmed that Cd47−/− mice were resistant to MOG35–55‐induced EAE. Our data, however, supported a different mechanism that was not a result of failure of CD4+ T cell activation. Instead, we found that CD4+ T cells in MOG35–55‐immunized Cd47−/− mice were activated, but clonal expansion contracted within 72 h after immunization. We used TCR crosslinking and mitogen activation in vitro to investigate the underlying mechanism. We found that naïve Cd47−/− CD4+ T cells exhibited a premature block in proliferation and survival because of impaired activation of LFA‐1, despite effective TCR‐induced activation. These results identify CD47 as an important regulator of LFA‐1 and VLA‐4 integrin‐adhesive functions in T cell proliferation, as well as recruitment, and clarify the roles played by CD47 in MOG35–55‐induced EAE.
Keywords: neuroinflammation, autoimmune, TCR activation, T lymphocytes
Abbreviations
- ATCC
American Type Culture Collection
- CD40L/CD62L
cluster of differentiation 40/62 ligand
- Cd47−/−
mice deficient in the Cd47 gene
- DC
dendritic cell
- dLN
draining lymph node
- DPBS
Dulbecco’s PBS
- DPI
day postimmunization
- EAE
experimental autoimmune encephalomyelitis
- i.p.
intraperitoneal(ly)
- LN
lymph node
- MOG35–55
myelin oligodendrocyte glycoprotein peptide 35–55 aa
- MS
multiple sclerosis
- PD‐1
programmed cell death protein‐1
- PFA
paraformaldehyde
- PI
propidium iodide
- PT
pertussis toxin
- r
recombinant
- SIRP
signal regulatory protein
- TCR‐XL
TCR crosslinking
- TEM
transendothelial migration
- Tg
transgenic
- TSP
thrombospondin
- WT
wild‐type
Introduction
CD47 is a ubiquitously expressed glycoprotein that interacts “in cis” with multiple integrins and “in trans” with SIRPα and SIRPγ and TSP‐1 and TSP‐2 (reviewed in refs. [1, 2]). Previous studies have shown that CD47 interactions with SIRPs and TSP‐1 play an important role in leukocyte recruitment in models of inflammation and platelet adhesion and activation, in immune cell homeostasis and apoptosis, and as a modulator of CD4+ T cell functions (reviewed in ref. [3]). CD47 also is a marker of “self” and has been proposed as a cancer therapy target in murine hematopoietic cancer models [4, 5, 6, 7, 8–9].
Recently, we reported that CD47 is in close physical contact with β2 integrins and that CD47 is required for expression of high‐affinity forms of LFA‐1 and VLA‐4 integrins in human T cells [10]. In addition, murine Cd47−/− CD4+ Th1 cells have reduced adhesive interactions with TNF‐α‐inflamed cremaster muscle microvessels and a 50% reduction of TEM in vitro [11]. Importantly, it is well documented that both LFA‐1 and VLA‐4 are involved in T cell antigen priming by APCs and homing to lymphoid tissues and to sites of immune reactions and inflammation.
Recruitment and reactivation of self‐reactive T effector cells in the CNS are considered central mechanisms in the pathogenesis of MS. EAE is an established murine model for MS [12]. EAE is triggered primarily by autoreactive CD4+ Th subsets [13]. Given the importance of CD47 in immune cell function and in expression of high‐affinity forms of VLA‐4 and LFA‐1 and that the phenotype of Cd47−/− animals has not been explored in detail in neurologic autoimmune disease models, we examined whether CD47 regulates antigen‐dependent T cell responses in a model of MOG35–55‐induced EAE. A previous study by Han and colleagues [14] reported that Cd47−/− mice were resistant to active induction of EAE by MOG35–55 immunization. They also reported that passive induction of EAE by transfer of in vivo MOG35–55‐activated Cd47−/− T cells failed to induce disease in WT or recombination‐activating gene‐deficient mice, whereas transferring WT T cells induced disease in WT and Cd47−/− recipients. The authors attributed protection in EAE to complete failure of Cd47−/− CD4+ T cell activation. Their conclusion that in vivo MOG35–55‐activated Cd47−/− T cells transferred to WT recipients did not cause disease is flawed and inconsistent with literature. This is because Cd47−/− T cells transferred into WT mice cannot bind SIRPα, expressed by splenic macrophages or DCs to deliver a negative “don’t eat me” signal and thus, are rapidly phagocytosed and removed from the circulation, as reported previously [4, 5, 15]. With the use of the same in vivo EAE model, we confirm that Cd47−/− mice are completely resistant to MOG35–55‐induced EAE, but in contrast to Han and colleagues’ study [14], we demonstrate that CD4+ T cell activation does occur in MOG‐immunized Cd47−/− animals. Surprisingly, activated CD4+ T cells failed to sustain proliferation or clonal expansion. Specifically, MOG35–55 immunization of Cd47−/− mice induced CD4+ T cell activation and T cell entry into cell cycle and cell division, but activated T cells exhibited a striking reduction in clonal expansion within 48 h. We found that defective integrin activation in Cd47−/− CD4+ T cells after TCR‐XL treatment or mitogen treatments in vitro was responsible for impaired homotypic aggregation, poor proliferation, and subsequent increased apoptosis. Our results indicate that reduced LFA‐1 integrin‐mediated adhesion and a defect in proliferation of Cd47−/− CD4+ T cells upon MOG35–55 immunization—not failure in CD4+ T cell activation, as reported previously [14]—are the major mechanisms of protection in MOG35–55‐induced EAE.
MATERIALS AND METHODS
Mice
C57BL/6 WT mice from Charles River Laboratories (Wilmington, MA, USA) were used to establish a WT breeding colony in the pathogen‐free animal facility at Harvard Medical School, New Research Building (Boston, MA, USA). Generation of Cd47−/− mice on the C57BL/6J background has been previously described [16]. Cd47−/− mice were obtained from Dr. Eric Brown, while he was a faculty member at University of California (San Francisco, CA, USA) and were used to establish our colony that was used to generate the results (see Figs. 1 and 2). Subsequent studies (see 3, 7) were carried out with Cd47 −/− mice, purchased from The Jackson Laboratory (Bar Harbor, ME, USA), which also were completely protected in the EAE model. Animals from both sources had been backcrossed >10 generations. MOG35–55‐specific TCR Tg mice (2D2 mice) were provided by Dr. Vijay Kuchroo (Brigham and Womenˈs Hospital, Boston, MA, USA) [17]. Mice were used at 6–10 wk old for EAE induction or euthanized at 8–12 wk of age for harvest of cells or as otherwise noted.
Figure 1.
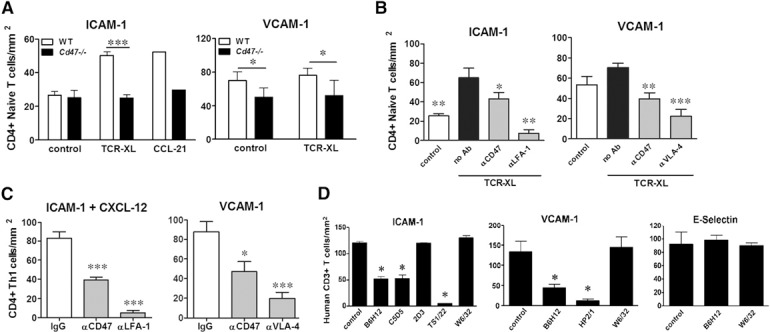
CD47 regulates TCR‐XL‐induced LFA‐1‐ and VLA‐4‐mediated arrest on ICAM‐1 and VCAM‐1 under shear flow conditions in vitro. (A) Naïve WT and Cd47−/− CD4+ T cells were stimulated by TCR‐XL in suspension, and their adhesion to immobilized ICAM‐1 or to VCAM‐1 was analyzed under flow conditions at an estimated shear rate of 0.5 (for ICAM‐1) and 0.75 (for VCAM‐1) dyne/cm2, as previously reported [21]. Data are means ± sem of 3 separate experiments, each done in duplicate coverslips. *P < 0.05; ***P < 0.001 by paired Student’s t test. Adhesion of T cells to ICAM‐1+ CCL21 (250 ng/ml) was the positive control (n = 2 duplicate coverslips). (B) WT naïve CD4+ T cell adhesion to ICAM‐1 and VCAM‐1 induced by TCR‐XL was reduced by anti‐CD47 mAb miap301 mAb (20 μg/ml, 30 min, 37°C). Likewise, blocking LFA‐1 and VLA‐4 mAb (20 μg/ml) inhibited cell adhesion. Data are means ± sem, n = 3 separate experiments performed in duplicate. *P < 0.05; **P < 0.01; ***P < 0.001 compared with WT T cells (black solid bars). (C) Adhesion of in vitro‐differentiated WT CD4+ Th1 cells to ICAM‐1 + CXCL12 (250 ng/ml) and to VCAM‐1 was reduced by blocking CD47 with miap301 mAb and by anti‐LFA‐1 and VLA‐4 mAb. Data are means ± sem, n = 3 separate experiments performed in duplicate. *P < 0.05; ***P < 0.001 vs. IgG control (all mAb used at 20 μg/ml). (D) Human peripheral blood CD3+ T cells (>95% purity) were pretreated with mAb to CD47 (B6H12 or C5D5), which blocks CD47 binding to SIRPα [42, 43] or VLA‐4 (HP2/1), class I (W6/32), nonblocking anti‐CD47 (2D3), or anti‐LFA‐1 mAb (TS1/22; all mAb are purified IgG, 20 μg/ml) for 30 min. T cell adhesion to human ICAM‐1‐Fc coimmobilized with CXCL12 or immobilized VCAM‐1‐Fc or E‐selectin‐Fc chimeras was measured under shear stress conditions, as we have published [21]. Data are means ± sem, performed in duplicate, n = 3–5 separate experiments. *P < 0.05 compared with no addition.
Figure 2.
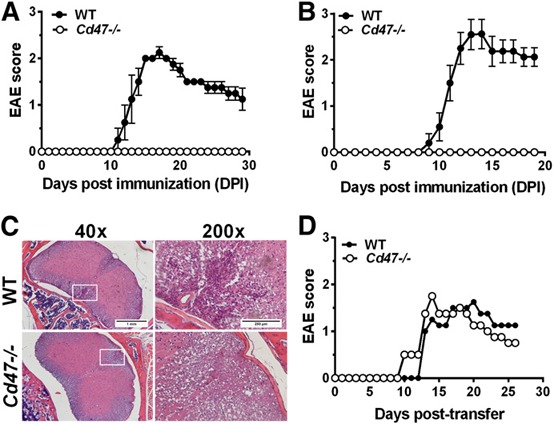
Cd47−/− mice are not susceptible to EAE. (A) Mice were immunized with MOG35–55, and the clinical score of male (n = 4 mice/group) and female (B; n = 10 mice/group) animals is reported (4 of 4 WT males and 10 of 10 WT female mice developed EAE; solid circles). No Cd47−/− mice developed EAE (open circles). Data are means ± sem of 3 separate experiments. (C) Histopathology of the spinal cord of WT and Cd47−/− mice, 15 d after MOG35–55 immunization. Original scale bars, 1 mm (40×, left) and 250 μm (200×, right). (D) Both Cd47−/− and WT mice are susceptible to passive transfer‐induced EAE after i.p. injection with 2 × 106 of MOG35–55 TCR‐specific T cells. Data are means ± sem from 4 mice of each group.
Figure 3.
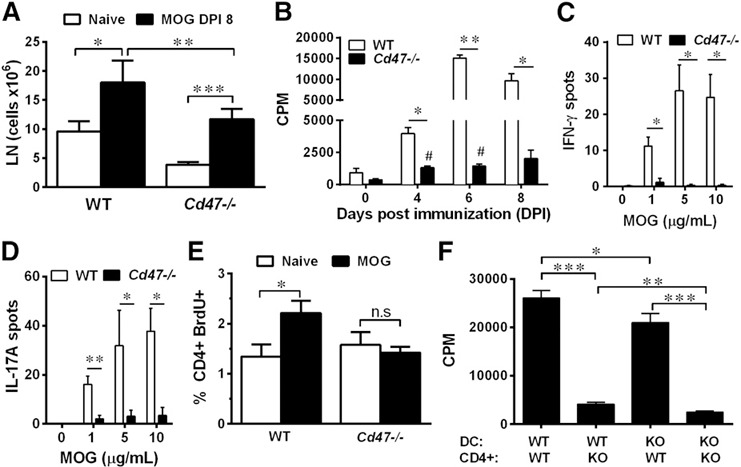
Reduced proliferation of Cd47−/− CD4+ T cells in vivo and in vitro upon antigen presentation. (A) Cervical and axillary LNs were harvested from naïve mice and mice, DPI 8. Total leukocyte number was determined by hemocytometer counting. Data are means ± sem from 5–10 mice of each group. (B) Total splenocytes were harvested from mice on DPI 4, 6, and 8 and assayed for recall proliferation to MOG35–55 (5 μg/ml). Data are means ± sem of triplicate determinations from 2 separate experiments. (C and D) ELISPOT data represent the number of cells producing IFN‐γ or IL‐17A, respectively, 48 h after rechallenge of 2 × 105 splenocytes with MOG35–55 (5 μg/ml). Data are means ± sem of triplicate determinations from 3 separate experiments. (E) dLNs were collected from naïve and immunized WT and Cd47−/− mice at DPI 3. Cells were stained with anti‐CD4+ mAb and for BrdU incorporation, and fluorescence in CD4+ T cells was quantified by flow cytometry. Data are means ± sem from 7 mice of each genotype performed in 3 independent experiments. (F) CD4+ T cell proliferation was determined in a rechallenge assay using CD11c+ DCs and CD4+ T cells purified from spleens of WT and Cd47−/− mice, 8 DPI, with MOG35–55 (5 μg/ml). KO, Knockout. Data are means ± sem, triplicate determinations, and are representative of 3 separate experiments. *P < 0.05; **P < 0.01; ***P < 0.001 vs. WT cells (A–F); #P < 0.05 vs. Cd47−/− cells at d 0 (B).
Figure 7.
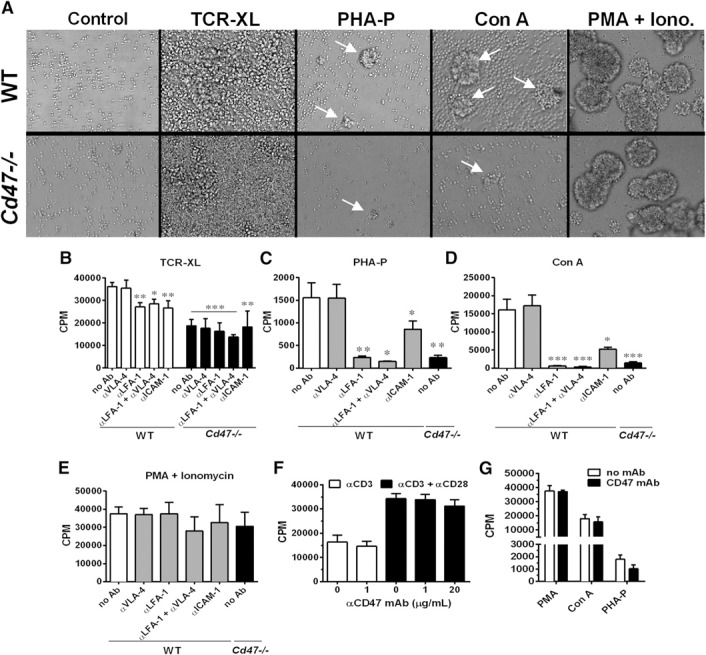
CD47 is necessary for integrin‐mediated splenic CD4+ T cell clustering and proliferation in vitro. (A) Representative images of naïve WT splenic CD4+ T cell (upper) and Cd47−/− (lower) homotypic cluster formation (white arrows), 48 h after activation induced by TCR‐XL (plate‐bound anti‐CD3ɛ with soluble anti‐CD28 mAb), PHA‐P, Con A, or PMA + ionomycin (Iono.). Proliferation of naïve CD4+ WT and Cd47−/− T cells induced by TCR‐XL (B), PHA‐P (C), Con A (D), or PMA + ionomycin (E). Note that only WT T cell proliferation was measured in the presence of blocking mAb to LFA‐1 and VLA‐4 and their combination and ICAM‐1. Data are means ± sem of triplicate samples pooled from 5 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 vs. WT T cells stimulated by TCR‐XL, PHA‐P, Con A, or PMA + ionomycin. (F) Proliferation of naïve WT splenic CD4+ T cells upon activation by plate‐bound anti‐CD3ɛ mAb (open bars) or plate‐bound anti‐CD3ɛ mAb plus soluble anti‐CD28 mAb (solid bars), with or without anti‐CD47 mAb miap301 at 1 or 20 µg/ml. Means ± sem of triplicate samples, n = 3 independent experiments. (G) Naïve WT splenic CD4+ T cells were activated with PMA + ionomycin, Con A, or PHA‐P in the absence (open bars) or presence (solid bars) of 20 μg/ml anti‐CD47 mAb miap301. Data are means ± sem of triplicate samples, n = 3 independent experiments.
Study approval
Animal research was performed in accordance with the guidelines of the Committee of Animal Research at the Harvard Medical School and the U.S. National Institutes of Health animal research guidelines, as set forth in the Guide for the Care and Use of Laboratory Animals. Blood was drawn and handled according to protocols for protection for human subjects, approved by the Brigham and Women’s Hospital Institutional Review Board. Informed consent was obtained from all volunteers in accordance with the Declaration of Helsinki.
Materials
RPMI 1640, cell tracer CFSE proliferation kit, ammonium‐chloride‐potassium lysing buffer, and nonessential amino acids were from Thermo Fisher Scientific (Waltham, MA, USA). DPBS+ and DPBS− Ca2+ and Mg2+ were purchased from Lonza (Walkersville, MD, USA). Con A and PHA‐P were obtained from Sigma‐Aldrich (St. Louis, MO, USA). PMA, ionomycin, PI, and FCS were purchased from Sigma‐Aldrich. MOG35–55 (M‐E‐V‐G‐W‐Y‐R‐S‐P‐F‐S‐R‐O‐V‐H‐L‐Y‐R‐N‐G‐K), corresponding to the mouse sequence, was synthesized by Quality Controlled Biochemicals (Hopkinton, MA, USA) and was purified to >99% by HPLC. The following rat mAb were purified, azide‐free IgG and purchased from BioLegend (San Diego, CA, USA): rat anti‐mouse mAb to IL‐4 (clone 11B11), CD3ɛ (clone 145‐2C11), CD28 (clone E18), CD47 (miap301), LFA‐1 (clone M17/4), and VLA‐4 (clones R1‐2 and 9C10). Other mAb were used as purified IgG: rat anti‐mouse ICAM‐1 (clone YN1.1), anti‐human β2 integrin (TS1/22), anti‐human VLA‐4 (HP2/1), and anti‐human MHC class I (W6/32) mAb, purchased from ATCC (Manassas, VA, USA); B6H12 and C5D5, function‐blocking mAb to human CD47, obtained from ATCC and from Dr. Parkos [18], respectively; 2D3, a nonblocking anti‐CD47 mAb, obtained from Dr. Eric Brown [19]; and anti‐Bcl‐xL (clone 2H12) from Southern Biotech (Birmingham, AL, USA). Mouse rIL‐12 and rIL‐2 were from BioLegend. Murine rIL‐7, rIL‐18, and rIL‐23 and murine and human VCAM‐1‐Fc and ICAM‐1‐Fc were from R&D Systems (Minneapolis, MN, USA). Murine CXCL12 and CCL21 were from PeproTech (Rocky Hill, NJ, USA). Fluorescent PE‐Annexin V apoptosis detection and FITC‐BrdU flow kits and the following fluorescent dye‐conjugated primary mAb were purchased from BD PharMingen (San Jose, CA, USA): anti‐mouse PE‐IL‐2, allophycocyanin‐IL‐4, PE‐IL‐9, allophycocyanin‐IL‐10, PE‐ and allophycocyanin‐IL‐17A, PE‐IFN‐γ, allophycocyanin‐forkhead box P3, PE‐CD25, FITC‐CD62L, PE‐Cy7‐CD69, PE‐ and allophycocyanin‐CD4, FITC‐ and Pacific Blue‐CD8, FITC‐ and PE‐CD11c, allophycocyanin‐CD11b, FITC‐ and allophycocyanin‐CD47, PE‐Cy7‐B220, FITC‐ and PE‐CD44, PE‐PD‐1, PE‐CD28, PE‐ICOS, PE‐CTLA‐4, and PE‐CD40L.
Cell isolation
Human CD3+ T cells were isolated to >95% purity by negative selection using RosetteSep negative depletion kits (Stemcell Technologies, Vancouver, BC, Canada) from anti‐coagulated whole blood obtained from healthy volunteers [20]. Murine splenic CD11c+ DCs and CD4+ T cells were purified by magnetic bead positive separation kits (Miltenyi Biotec, Cambridge, MA, USA). Naïve CD4+ T cells were polarized to Th1 cells by culture with plate‐bound anti‐CD3ɛ mAb (5 μg/ml), and RPMI‐1640 medium containing anti‐CD28 mAb (1 μg/ml), anti‐IL‐4 mAb (1 μg/ml), and cytokines IL‐12 (10 ng/ml) and IL‐2 (25 U/ml), as previously detailed [21]. Polarization to Th1 cells was confirmed by measurement of intracellular IFN‐γ production by flow cytometry.
T cell adhesion to immobilized Fc chimera adhesion molecules under defined laminar shear flow conditions in vitro
T cell arrest on immobilized adhesion molecules in a parallel flow chamber has been described previously [21]. In brief, 5 × 105/ml T cells in DPBS with 0.1% BSA and 20 mM HEPES were drawn across immobilized ligands ICAM‐1‐Fc (10 μg/ml) or VCAM‐1‐Fc (5 μg/ml) under flow at 37°C. T cell arrest was monitored by video microscopy [10], and cell adhesion/mm2 was measured from review of recorded videos.
Induction of EAE and spinal‐cord histologic analysis
EAE was induced as described [22]. In brief, 6‐ to 10‐wk‐old WT and Cd47−/− mice were immunized subcutaneously with 100 μg MOG35–55 emulsified in CFA (H37Ra; Difco Laboratories, Detroit, MI, USA). Animals received 200 ng by i.p. injection of PT (List Biologic, Campbell, CA, USA) on d 0 and 2 DPI. Mice were observed daily, and EAE clinical signs were scored as follows: grade 0, no disease; grade 1, limp tail or isolated weakness; grade 2, partial hind‐limb paralysis; grade 3, total hind‐limb paralysis; grade 4, total hind‐limb and partial fore‐limb paralysis; grade 5, moribund or dead animal. To induce EAE by adoptive transfer of activated T cells, CD4+ T cells were isolated from spleens of naïve 2D2 mice and activated in vitro for 48 h in medium containing MOG35–55 peptide (20 μg/ml), IL‐2 (20 ng/ml), and IL‐7 (5 ng/ml). Activation was repeated and was followed by a final activation with plate‐bound anti‐CD3ɛ and anti‐CD28 mAb (1 μg/ml each mAb) for 24 h in media with IL‐12 (20 ng/ml), IL‐18 (25 ng/ml), and IL‐23 (10 ng/ml). These activated 2D2 Tg T cells were harvested, and 2 × 106 cells were transferred to naïve WT or Cd47−/− mice by i.p. injection. Animals also received 69 ng i.p. of PT on d 0 and d 2 after cell transfer, and clinical signs were scored daily. For histologic studies, mice were euthanized on DPI 15 and perfused transcardially with 4% PFA in PBS. Spinal cords were removed and fixed in 1% of PFA for 24 h, embedded in paraffin, sectioned, and stained with H&E.
T cell proliferation in vivo by BrdU incorporation
Mice were immunized with MOG35–55 and 48 h later, were injected with BrdU (2.5 mg/mouse i.p.). CD4+ T cells were isolated 24 h later from dLNs, and BrdU incorporation in CD4+ T cells was measured following the manufacturer’s instructions (BD PharMingen). Cell fluorescence was determined by flow cytometry (FACSCalibur flow cytometer; BD Biosciences, San Jose, CA, USA) and data analyzed by FlowJo software (Tree Star, Ashland, OR, USA).
T cell proliferation assays
Spleens from immunized mice were harvested, and aliquots of total splenocytes (1 × 105) were cultured with MOG35–55 for 48 h. Cultures were pulsed with 1 μCi [3H]‐thymidine in the last 16 h of incubation and harvested by automated sample harvester (Perkin‐Elmer, Waltham, MA, USA) [22]. For coculture‐recall assays of DC priming of the CD4+ T cell, purified CD4+ T cells and CD11c+ DCs from immunized mice were cultured with 5 μg/ml MOG35–55 for 48 h. Proliferation was monitored by [3H]‐thymidine incorporation. For in vitro proliferation assays, purified CD4+ T cells from spleens of naïve WT and Cd47−/− mice were activated by polyclonal activation TCR‐XL using plate‐bound anti‐CD3ɛ mAb (1 μg/ml) and soluble anti‐CD28 mAb (1 μg/ml) or by coculture with mitogens Con A (5 μg/ml) or PHA‐P (20 μg/ml) or ionomycin (5 μM) plus PMA (50 ng/ml). Proliferation was monitored by [3H]‐thymidine incorporation. Blocking mAb to LFA‐1, CD47, ICAM‐1, and a combination of 2 different function‐blocking anti‐VLA‐4 mAb were used as purified and azide‐free IgG at 20 μg/ml. T cell proliferation was also determined by a flow cytometric assay of CFSE dilution. Purified CD4+ T cells were added to a solution of 5 μM CFSE in PBS at 37°C for 10 min, followed by extensive washing with warm PBS. Cells were cultured as described in text and harvested at different time points after TCR‐XL. CFSE fluorescence of gated CD4+ cells was analyzed with FlowJo software to obtain proliferation indices (see Table 2 and Fig. 6).
Table 2.
CD4+ T cell proliferation upon TCR‐XL
| Live CD4+ gated cell | % Proliferating | Division index | Proliferation index | |||||
|---|---|---|---|---|---|---|---|---|
| Day | WT | KO | WT | KO | WT | KO | WT | KO |
| 1 | 15.8 ± 0 | 13.4 ± 0 | 6.8 ± 0 | 5.3 ± 0 | 0.1 ± 0 | 0.1 ± 0 | 1.1 ± 0 | 1.1 ± 0 |
| 2 | 32.2 ± 8.8 | 25.6 ± 6.6 | 72.4 ± 11.1 | 72.7 ± 8.5 | 1.5 ± 0.3 | 1.3 ± 0.2 | 2.0 ± 0.2 | 1.8 ± 0.2 |
| 3 | 36.4 ± 7.2 | 19.2 ± 4.4 a | 76.4 ± 7.5 | 79.9 ± 7.7 | 1.8 ± 0.1 | 1.6 ± 0.1 | 2.3 ± 0.1 | 2.0 ± 0.0 |
| 4 | 24.0 ± 6.3 | 11.2 ± 3.5 a | 77.0 ± 8.6 | 73.7 ± 10.7 | 2.0 ± 0.3 | 1.7 ± 0.2 | 2.6 ± 0.1 | 2.3 ± 0.1 |
| 5 | 14.9 ± 0.3 | 9.0 ± 3.4 | 77.2 ± 4.0 | 81.5 ± 8.2 | 2.1 ± 0.3 | 1.9 ± 0.2 | 2.6 ± 0.1 | 2.3 ± 0.1 |
KO, Knockout. Values represent means ± sem for n = 3 separate experiments.
P < 0.05 vs. WT.
Figure 6.
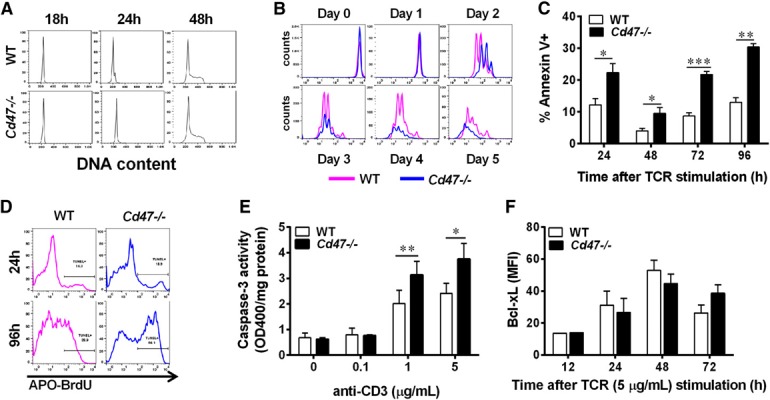
Cd47−/− CD4+ T cell proliferation after TCR‐XL using plate‐bound anti‐CD3ɛ and soluble anti‐CD28 mAb is impaired in vitro. (A) Cell cycle was analyzed in CD4+ T cells at 0 [overnight (o/n)], 24, and 48 h after TCR‐XL by the analysis of DNA content using flow cytometry and FlowJo cell‐cycle software. Data are representative of 3 independent experiments. (B) CD4+ T cell proliferation over a 5 d period was determined by CFSE dye dilution after TCR‐XL activation using FlowJo proliferation software. The histogram is representative of 3 independent experiments. FlowJo‐calculated indices are presented in Table 2. CD4+ T cell apoptosis was detected by Annexin V staining and flow cytometry. *P < 0.05; **P < 0.01; ***P < 0.001 compared with WT at each time point by paired Student’s t test. (D) DNA nicking in T cells was detected by the TUNEL assay at different times after TCR‐XL and analyzed by flow cytometry. Data are representative histograms of 2 separate experiments performed in duplicate. (E) Caspase‐3 activity in CD4+ T cells was quantified, 48 h after TCR‐XL, with anti‐CD3ɛ mAb plus 1 μg/ml anti‐CD28 mAb. *P < 0.05; **P < 0.01 compared with WT at each time point by paired Student’s t test. (F) Bcl‐xL expression in CD4+ T cells is shown after TCR‐XL. MFI, Mean fluorescence intensity.
Intracellular cytokine staining and cytokine production assays
For intracellular cytokine staining, cells were activated with 100 ng/ml PMA and 5 μg/ml ionomycin for 4 h in the presence of GolgiStop (BD PharMingen) and stained by anti‐cytokine mAb and appropriate isotype‐matched control antibodies [21]. Cell fluorescence was measured in the CD4+ population by flow cytometry. For analysis of IL‐4‐, IL‐6‐, IL‐17A‐, and IFN‐γ‐producing cells, splenocytes (2 × 105) from immunized mice were activated with MOG35–55 for 24 h in antibody‐coated ELISPOT plates, and spots were counted by an ELISPOT image analyzer (Cellular Technology, Cleveland, OH), as reported previously [22]. Leukocyte subtypes and expression of surface activation markers and costimulatory proteins were measured by appropriate mAb and flow cytometry [21, 22]. Intracellular staining of anti‐apoptotic Bcl‐xL protein was performed exactly as reported [23]. In brief, T cells were harvested, washed in DPBS, fixed in 2% PFA‐PBS for 20 min at room temperature, washed twice with PBS, and permeabilized with 0.5% saponin‐PBS containing 20% FBS (perm buffer). Cells were incubated with mouse anti‐Bcl‐xL (1 μg/ml) or no primary mAb in 50 μl perm buffer for 30 min at room temperature, washed twice with perm buffer, and incubated at room temperature for 20 min with goat anti‐mouse Alexa 647‐labeled secondary mAb in perm buffer. Cells were then washed and analyzed by flow cytometry.
Naïve T cell stimulation by TCR‐XL in suspension
Naïve CD4+ T cells (2 × 106 cells/ml) in RPMI 1640 + 0.2% FCS were treated with anti‐CD3ɛ (2.5 μg/ml) and anti‐CD28 (2.5 μg/ml) mAb for 15 min on ice. After washing to remove the first mAb, secondary antibodies (goat anti‐Armenian hamster and goat anti‐mouse; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were added (2.5 μg/ml) using the same medium and incubated for 15 min on ice. T cells were washed once and resuspended to 2 × 106 cells/ml in RPMI 1640 and kept on ice until stimulation was initiated at 37°C. Incubations were halted by addition of formaldehyde (diluted to 4% final concentration) and incubation at 37°C for 15 min. After chilling for 1 min on ice, T cells were centrifuged, resuspended in ice‐cold 90% methanol, and incubated on ice for 30 min. Then, cells were stained with primary‐labeled phospho‐specific mAb for 1 h at room temperature, washed, and resuspended in 1% formaldehyde in PBS for flow cytometry.
T cell‐cycle analysis
Naïve T cells were activated by TCR‐XL with plate‐bound mAb and incubated for different time points, harvested, fixed by drop‐wise addition of 100% ice‐cold ethanol, while vortexing to a final concentration of 75% ethanol, and incubated on ice for 1 h. Cells were washed in PBS and incubated in PI solution (50 μg/ml, 100 μg/ml RNase A, and 3.8 mM sodium citrate) for 3 h on ice. Cell‐cycle profiles were assessed by flow cytometry and analyzed by FlowJo software.
Analysis of CD4+ T cell apoptosis
Naïve, splenic WT and Cd47−/− CD4+ T cells were activated by TCR‐XL with plate‐bound anti‐CD3ɛ (1 μg/ml unless stated otherwise) and addition of 1 μg/ml anti‐CD28 mAb to the media, cultured for various times, and harvested to assess cell apoptosis. Annexin V staining and detection of DNA fragmentation using an APO‐BrdU TUNEL assay kit (Thermo Fisher Scientific) were performed by following the manufacturer’s instructions and analyzed by standard flow cytometry. T cell apoptosis was also assessed by caspase‐3 activity in cell lysates by quantification of light emission at 400 nm chromophore p‐nitroaniline after cleavage by caspase‐3 from the labeled DEVD‐p‐NA (Kit #ab39401; Abcam, Cambridge, MA, USA).
Statistics
Statistical analyses used Prism 5 software (GraphPad Software, La Jolla, CA, USA). Data are expressed as means ± sem. Data were analyzed by unpaired 2‐tailed Student’s t test between 2 groups and 1‐way ANOVA for >2 groups, with P < 0.05 considered significant.
RESULTS
CD47 regulates TCR‐XL and chemokine‐induced LFA‐1‐ and VLA‐4‐mediated arrest on ICAM‐1 and VCAM‐1 in vitro
We previously reported that CD47 is necessary for expression of high‐affinity forms of LFA‐1 and VLA‐4 integrins in CD4+ Th1 cells that bind to their endothelial cell‐expressed ligands ICAM‐1 and VCAM‐1, respectively, after CXCL12 stimulation [10]. To extend our observations, we investigated whether CD47 is necessary for TCR‐XL‐induced, naïve CD4+ T cell arrest on ICAM‐1 and VCAM‐1 in an in vitro flow‐chamber assay. We found that TCR‐XL or coimmobilized CCL21 chemokine increased adhesion of WT but not Cd47−/− naïve CD4+ T cells to immobilized ICAM‐1 and VCAM‐1 ( Fig. 1A ). Fewer Cd47−/− T cells bound to VCAM‐1 without a stimulus compared with WT T cells. These results show that CD47 is necessary for TCR‐XL and chemokine‐induced LFA‐1 and VLA‐4 activation and adhesion to ICAM‐1 and VCAM‐1 under fluid shear flow conditions.
Anti‐CD47 mAb miap301 reduces LFA‐1 and VLA‐4 integrin adhesion to ICAM‐1 and VCAM‐1
Although the amino acid(s) in CD47 that regulate integrin activation have not been identified, we tested whether mAb miap301, which blocks CD47 binding to its in trans ligand SIRPα [24], interferes with naïve CD4+ T cell adhesion. Pretreatment of WT CD4+ naïve T cells with miap301 mAb reduced TCR‐XL‐induced adhesion to ICAM‐1 and VCAM‐1 (Fig. 1B) and also reduced in vitro‐generated WT Th1 effector cell adhesion to ICAM‐1 coimmobilized with CXCL12 or to VCAM‐1 (Fig. 1C). Blocking mAb to LFA‐1 and VLA‐4, but not IgG, blocked T cell adhesion and demonstrated the specificity of this assay. Likewise, purified human CD3+ T cells pretreated with different anti‐CD47 mAb that block in trans CD47‐SIRPα adhesion (B6H12 and C5D5) also reduced adhesion (Fig. 1D). In contrast, a nonfunction‐blocking anti‐CD47 mAb (2D3) had no effect. These data demonstrate that CD47 is necessary for LFA‐1 and VLA‐4 integrin‐dependent adhesion of T cells to their endothelial cell‐expressed ligands, ICAM‐1 and VCAM‐1, respectively.
Cd47−/− mice are resistant to MOG35–55‐induced EAE but are susceptible to passive transfer of EAE
Based on our premise, Cd47−/− mice are not susceptible to EAE, as loss of CD47 impairs LFA‐1 and VLA‐4 activation and T cell recruitment (Fig. 1 and refs. [10, 11]), and as an anti‐VLA‐4 integrin mAb (natalizumab) is used to treat patients with relapsing and remitting MS, we examined the susceptibility of Cd47−/− mice to MOG35–55‐induced EAE. WT male ( Fig. 2A ) and female mice (Fig. 2B) immunized with MOG35–55 in CFA developed typical EAE disease scores. In striking contrast, Cd47−/− mice were completely resistant. Histologic analysis of spinal‐cord sections from DPI 15 (Fig. 2C) revealed abundant mononuclear infiltrates in WT mice but not in Cd47−/− animals. These findings corroborate the report by Han and colleagues [14] using this EAE model.
Based on our prior reports that both endothelial cell and T cell CD47 play important roles in T cell recruitment in in vivo and in vitro models [10, 11, 25, 26], we hypothesized that multiple defects converge to confer resistance in Cd47−/− mice to EAE. To address whether CD47 expressed in the host is required for susceptibility to EAE, we adoptively transferred in vitro‐polarized Thy1.1+ MOG35–55‐specific CD4+ T cells from 2D2 Tg mice into Thy1.2+ WT and Cd47−/− recipient female animals (passive EAE induction model). The TCR gene expressed in CD4+ T cells in 2D2 Tg mice recognizes MOG35–55 [17]. WT and Cd47−/− mice experienced comparable clinical signs of EAE (Fig. 2D) by DPI 13, indicating the defect(s) reside in the immune cell compartment. We note that transfer of Cd47−/− T cells into WT recipient animals leads to their rapid clearance from WT animals, as reported previously [15]; hence, this arm of the study was not performed. The mechanism involves phagocytosis of intravenous‐transferred Cd47−/− bone marrow or blood cells by splenic DCs and macrophages [4, 5].
CD4+ T cells do not proliferate in response to MOG35–55 in Cd47−/− mice
We first determined the number of total live cells in axillary and cervical dLNs from WT and Cd47−/− animals before and 8 d after MOG35–55 immunization. Although unimmunized, naïve Cd47−/− mice ( Fig. 3A ; open bars) showed a significant reduction (60 ± 5%) in cell number in LNs compared with age‐matched WT mice, the frequency of leukocyte types in WT and Cd47−/− remained similar ( Table 1 ). Prior studies also have reported reduced cell counts in the spleen and peripheral blood of Cd47−/− mice; however, this was because of a specific loss of CD4+ T cells [11, 16, 27].
Table 1.
Frequency of leukocyte types in LNs from naïve or MOG‐immunized mice
| Naïve | MOG‐immunized | |||
|---|---|---|---|---|
| % | WT | Cd47−/− | WT | Cd47−/− |
| CD4+ | 32.9 ± 2.2 | 31.0 ± 2.9 | 31.8 ± 3.2 | 27.4 ± 3.9 |
| CD8+ | 26.2 ± 1.9 | 26.2 ± 2.6 | 26.3 ± 1.5 | 24.2 ± 2.6 |
| CD11c+ | 3.8 ± 0.6 | 5.3 ± 1.9 | 1.5 ± 1.2 | 1.6 ± 1.1 |
| B220+ | 32.4 ± 2.3 | 36.3 ± 3.6 | 41.0 ± 1.8 | 34.0 ± 4.7 |
LNs were harvested from naive and MOG‐immunized WT and Cd47−/− mice, DPI 8, and total leukocyte number was determined by hemocytometer counting. Data are means ± sem. The data are pooled from 5–10 mice in each cohort, from 3 independent studies. Analysis was performed with mAb that recognize CD4 and CD8 T cells, CD11c myeloid, and B220 B cells. Other cell types not identified represented <1–5% of the total LN populations.
On DPI 8, despite an increase in total cell number in immunized Cd47−/− mice, the cell number in dLNs remained significantly less than that in immunized WT mice (Fig. 3A, solid bars), and the populations were not skewed by loss of any 1 specific cell type (Table 1). Thus, the calculated absolute number of CD4+ T cells in Cd47 −/− mice was well below that of WT mice (WT, 5.73 × 106 vs. Cd47 −/−, 3.21 × 106). Given this significant reduction in total CD4+ T cells, we probed the animals for antigen‐specific, responsive CD4+ T cells in splenocytes at different DPI in an in vitro recall response to MOG35–55. Interestingly, proliferation of Cd47−/− cells at each time point was dramatically impaired compared with WT cells (Fig. 3B). Cd47−/− mice also produced significantly fewer IFN‐γ‐ and IL‐17A‐producing cells compared with WT mice (Fig. 3C and D), as well as IL‐4‐ and IL‐6‐producing cells (data not shown), as determined by ELISPOT assay. This general reduction in cytokine profile suggested a defect in proliferation rather than a skewed T cell effector differentiation. To investigate this proliferation defect in vivo, we monitored proliferation specifically in CD4+ T cells by BrdU incorporation and flow cytometric analysis. CD4+ T cells from dLNs of immunized WT mice proliferated by DPI 3, whereas no significant increase over baseline (1.6 ± 0.25%) was detected in immunized Cd47−/− mice (Fig. 3E). To distinguish whether lack of priming was a result of a defect in the Cd47−/− T cell or DC, we performed a “crisscross” MOG35–55‐driven in vitro proliferation assay. Cd47−/− CD4+ T cells showed a dramatic defect in proliferation when cultured with WT or Cd47−/− CD11c+ DCs (Fig. 3F). WT T cells showed less proliferation when cultured with Cd47−/− vs. WT DCs. Overall, these data demonstrate that Cd47−/− CD4+ T cells exhibit a significantly reduced proliferative response to MOG35–55 immunization compared with WT, and the defect resides primarily in the CD4+ T cell.
CD4+ T cells in Cd47−/− mice are activated after MOG35–55 immunization
We next investigated whether the observed reduction in CD4+ T cell proliferation in Cd47−/− mice was caused by failure of immune cells to activate, as reported by Han and colleagues [14], or to an inability of activated T cells to proliferate and expand in response to antigen. Intracellular IL‐2 production was the same in naïve Cd47−/− and WT CD4+ T cells (∼7% positive in WT and Cd47 −/− cells) and was elevated to similar levels in WT and Cd47−/− T cells isolated from spleen and from dLN on DPI 8. Figure 4A shows the gating scheme, and Fig. 4B shows the results. WT and Cd47−/− CD4+ T cells produced comparable levels of intracellular IL‐17A and IL‐4 cytokines, whereas Cd47 −/− produced greater amounts of IL‐9, IL‐10, and IFN‐γ. (Fig. 4C). Surprisingly, the percent of isolated, naïve CD4+ T cells that expressed surface activation markers CD44hi, CD69, and CD25 was significantly greater in Cd47−/− compared with WT animals (Fig. 4D). This finding is consistent with CD47 acting as a negative regulator of Th1 effector‐type responses, as reported previously [15]. On DPI 8, the percentage of Cd47−/− T cells expressing CD44hi, CD69, and CD25 increased significantly compared with naïve mice and was the same as or greater than WT cells. Taken together, these data demonstrate that CD4+ T cells in MOG35–55‐immunized Cd47−/− mice are activated to an equal or greater level than WT T cells, produce intracellular cytokines, yet proliferate poorly and have limited clonal expansion compared with WT CD4+ T effector cells.
Figure 4.
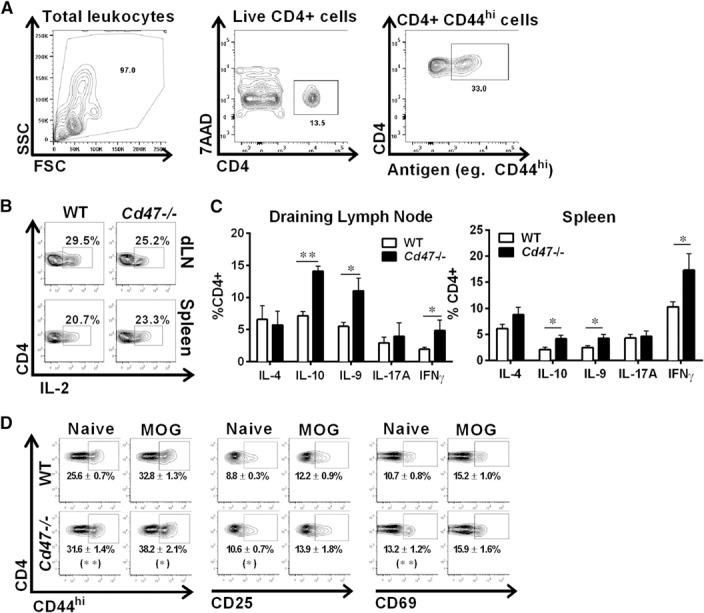
Cd47−/− CD4+ T cells are activated upon antigen stimulation in vivo. (A) The gating scheme to identify CD4+ T cells and expression of surface activation markers, for example, CD44hi, is shown. SSC, Side‐scatter; FSC, forward‐scatter; 7AAD, 7‐aminoactinomycin D. (B) dLNs and spleens were harvested from mice, DPI 8, and the production of intracellular IL‐2 by CD4+ T cells was determined by flow cytometry after 4 h of PMA‐ionomycin stimulation. (C) Intracellular production of cytokines in CD4+ T cells from dLN (left) and spleen (right) from MOG35–55‐immunized mice, DPI 8, was determined by flow cytometry after PMA‐ionomycin stimulation. *P < 0.05; **P < 0.01 for Cd47−/− values compared with WT. (D) Surface expression of CD44hi, CD25, and CD69 on CD4+ T cells was determined by flow cytometry. Representative contour plots of CD4+ T cells prepared from 5–10 mice of each genotype. Data in parentheses are means ± sem from 5–10 mice of each genotype. *P < 0.05; **P < 0.01 for Cd47−/− values compared with WT counterpart surface antigen in naïve and MOG35–55‐immunized mice.
Cd47−/− T cells are activated by TCR‐XL with CD28 costimulation
We next investigated whether Cd47−/− CD4+ T cells have a defect in antigen‐independent activation by TCR‐XL, a potent stimulus that bypasses APC priming and uniformly activates and induces robust proliferation of CD4+ T cells in vitro. As expected, based on the in vivo findings, TCR proximal signaling, measured by increased phosphorylation of Lck, ZAP70, ERK1/2, and AKT kinases, was essentially identical in WT and Cd47−/− CD4+ T cells ( Fig. 5A ). Moreover, distal activation markers, including increases in surface‐expressed CD44hi, CD25, CD69, and PD‐1; down‐regulation of CD62L surface expression; and elevated intracellular IL‐2 production were nearly identical in WT and Cd47−/− T cells (Fig. 5B and C). However, despite the fact that CD4+ Cd47−/− and WT T cells were activated to a similar level by TCR‐XL and express similar levels of CD28, ICOS, CTLA‐4, and CD40L costimulatory molecules (data not shown), Cd47−/− T cell proliferation was 32–48% less than WT T cells (Fig. 5D). These findings demonstrate that Cd47−/− CD4+ T cells do not exhibit a global defect in TCR proximal or distal signal transduction required for IL‐2 autocrine production and increased expression of surface markers of activation or intracellular cytokine production.
Figure 5.
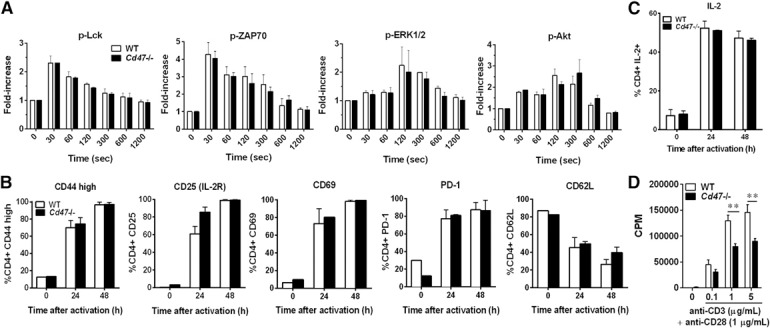
Cd47−/− CD4+ T cell activation is intact in vitro. CD4+ T cells were isolated from spleens of naïve WT or Cd47−/− mice, and their response to TCR‐XL stimulation was assessed. (A) Detection of phosphorylated (p) kinases in WT (open bars) and Cd47−/− T cells (solid bars) upon TCR‐XL in suspension by intracellular staining with mAb. Data represent the fold‐increase over time 0 s. Means ± sem, n = 3 independent studies. (B) Surface expression of CD44hi, CD25, CD69, PD‐1, and CD62L on naïve CD4+ T cells was determined by flow cytometry at 0, 24, or 48 h after TCR‐XL (plate‐bound anti‐CD3ɛ and soluble anti‐CD28 mAb). Data are means ± sem from n = 5 independent experiments. (C)_Intracellular IL‐2 production in CD4+ T cells after 0, 24, or 48 h after TCR‐XL was determined by standard flow cytometry after PMA‐ionomycin stimulation. Data are means ± sem, n = 3 independent experiments. (D) CD4+ T cell proliferation was measured by [3H]‐thymidine incorporation, 48 h after TCR‐XL stimulation. Data are means ± sem of duplicate samples from 3 independent experiments. **P < 0.01 for Cd47−/− values compared with WT cells.
Impaired proliferation in Cd47−/− T cells upon TCR‐XL activation in vitro
To identify the underlying defects in Cd47−/− T cell proliferation farther downstream of TCR‐XL, we investigated the cell‐cycle and cell proliferation. Cd47−/− T cells displayed no significant differences from WT in the cell‐cycle progression, up to 48 h, as illustrated in Fig. 6A ; however, Cd47 −/− T cells exhibited a striking reduction in proliferation, as demonstrated by the differences in the kinetics of TCR‐XL‐induced CFSE dye dilution (Fig. 6B). The calculated indices using FlowJo software are summarized in Table 2 . These data demonstrate that the number of live Cd47 −/− T cells dropped by 50% beyond d 2 compared with WT, even though the Cd47 −/− and WT T cells exhibited similar division and proliferation indexes. Consistent with this dramatic reduction in cell number, which is consistent with results in Figs. 3A, D, and E and 6A and B, a significant increase in apoptosis of Cd47−/− T cells occurred after TCR‐XL, as detected by Annexin V binding (Fig. 6C). This increase in apoptosis of Cd47 −/− compared with WT CD4+ T cells was corroborated by increased TUNEL staining of fragmented DNA and by elevated caspase‐3 activity (Fig. 6D and E). The anti‐apoptotic proteins of the Bcl‐2 family are actively regulated in T cells (reviewed in ref. [28]). As previous studies showed that the Bcl‐xL gene was an important survival factor induced in CD4+ T cells by TCR‐dependent activation, we examined whether its expression was impaired in Cd47 −/− T cells [23, 29, 30–31]. As shown in Fig. 6F, this was not the case, as Bcl‐xL protein was elevated, to a similar level, in both cell types by TCR‐XL activation. This result suggests other family members, including anti‐ and proapoptotic, are involved, and their identity requires additional detailed studies.
CD47 and LFA‐1 are essential for CD4+ T cell proliferation in vitro
As lack of CD47 impaired the expression of high‐affinity LFA‐1 and VLA‐4 conformations in the human Jurkat CD4+ T cell line [10], we examined TCR‐XL‐induced homotypic aggregation of naïve WT and Cd47−/− CD4+ T cells after 48 h of in vitro culture. From the literature, LFA‐1 and VLA‐4 integrin‐dependent homotypic T cell aggregation is an established hallmark of efficient TCR‐ and mitogen‐mediated activation and proliferation in vivo and in vitro [32, 33, 34, 35–36]. Cd47−/− T cells exhibited smaller homotypic aggregates ( Fig. 7A ) and a 48% reduction in proliferation of Cd47−/− CD4+ T cells compared with WT T cells after TCR‐XL (Fig. 7B; “no Ab”). Inclusion of function‐blocking mAb to either LFA‐1 or ICAM‐1 significantly reduced proliferation of WT T cells but not to the level of Cd47−/− cells. Anti‐LFA‐1 or ICAM‐1 mAb did not further block proliferation by Cd47−/− T cells. WT and Cd47−/− T cell proliferation was not reduced by 2 different function‐blocking mAb to VLA‐4 that we have previously shown to block VLA‐4–VCAM‐1 interactions [10] (Fig. 7B). A more striking observation was the near‐complete lack of proliferation by Cd47−/− T cells incubated with PHA‐P or Con A lectins, which initiate proliferation by capping surface proteins, including the TCR, CD2, signaling lymphocyte activating molecule family (SLAMs), and other costimulatory molecules (Fig. 7C and D). We next validated the role of LFA‐1 and ICAM‐1 in proliferation of WT T cells induced by PHA‐P or Con A. Proliferation was totally abolished by anti‐LFA‐1 to the level of Cd47−/− T cells and significantly reduced by ICAM‐1 mAb. In contrast, a combination of 2 function‐blocking VLA‐4 mAb again had no inhibitory effect (Fig. 7C and D). Given these results, we next asked if failure of Cd47−/− T cells to proliferate was an intrinsic T cell defect by culturing these cells with PMA plus ionomycin, a stimulus that bypasses surface receptors and activates key intracellular pathways of proliferation. PMA plus ionomycin induced robust clustering and proliferation in both WT and Cd47−/− CD4+ T cells. This indicates that Cd47−/− T cells do not have intrinsic defects in proliferation or cytokine production pathways (Figs. 4 and 5). As our prior studies revealed that CD47 is in close physical contact with and regulates β2 integrin activation [10], these results further underscore the critical requirement for CD47 in TCR‐XL and mitogen‐stimulated T cell clustering and clonal expansion in this model and in vitro proliferation assays.
In the final set of experiments, we tested whether targeting CD47 altered T cell proliferation. A rat anti‐mouse CD47 mAb (clone miap301) was shown to inhibit completely CD47‐dependent thymocyte and erythrocyte binding to immobilized SIRPα, its “trans” ligand [24]. Whereas this mAb reduced T cell arrest on ICAM‐1 and VCAM‐1 (Fig. 1), surprisingly, miap301 mAb did not reduce TCR‐XL‐, PHA‐P‐, and Con A‐stimulated proliferation (Fig. 7F, filled bars, and G). Thus, this mAb does not reproduce the effect on T cell proliferation observed in Cd47−/− CD4+ T cells. Whereas a prior study in human T cells reported that some but not all mAb to CD47 were capable of costimulation with anti‐CD3ɛ mAb [37], we did not detect any effect on proliferation (Fig. 7F, open bars).
DISCUSSION
Our observation that mice lacking CD47 are protected in EAE is consistent with defects in T cell LFA‐1 and VLA‐4 integrin‐adhesive functions and emphasizes the importance of CD47 in adaptive immune cell responses in a self‐antigen‐driven murine disease model. The data herein demonstrate that CD4+ T cells become activated in MOG35–55‐immunized Cd47−/− animals and produce normal or enhanced levels of intracellular cytokines. Remarkably, CD4+ T cell proliferation was severely blunted starting 48 h after MOG35–55 immunization in Cd47−/− mice and resulted in increased apoptosis and a very low frequency of antigen‐primed T cells that was insufficient to initiate clinical symptoms or evidence of histologic CNS disease (Figs. 2 and 3). We do not exclude, however, the contribution of impaired T cell homing and recruitment of T effectors cells, including regulatory T cells, Th1 or Th17, as well as myeloid cells to the CNS during initiation and/or effector phases of EAE. Future studies are needed to address this possibility.
Based on our recent report that CD47 associates β2 integrin in T cells and that CD47 is necessary to induce high‐affinity conformations of LFA‐1 and VLA‐4 integrins required for adhesion and TEM [10], we initially postulated that defects in VLA‐4 and LFA‐1 function were responsible for the blockade in proliferation and increased apoptosis. Indeed, integrins are well documented to participate in homotypic aggregation and proliferation of T cells and in resistance to apoptotic stimuli, especially signals that activate the intrinsic (mitochondrial) death pathway (reviewed in ref. [38]). Our investigation shows both MOG35–55 immunization and TCR‐XL stimulation in vitro failed to induce proliferation and clonal expansion comparable with their WT counterpart. Whereas our early results revealed that MOG immunization of Cd47 −/− mice led to an increase in cell numbers in the dLN (Fig. 3A), further results from in vivo and in vitro experiments showed that Cd47−/− CD4+ T cells were not able to cause disease, mostly likely as a result of failure to proliferate (Fig. 3B–E) or failure to migrate from dLNs to the CNS or both. In fact, activation of Cd47 −/− CD4+ T cells resulted in elevated apoptosis, which was not related to impaired expression of the anti‐apoptotic protein Bcl‐xL (Fig. 6). There is significant literature for CD47‐mediated cell death in a variety leukocytes and other cell types, as reviewed by Oldenborg [3], and therefore, additional experiments are necessary to identify the molecular mechanisms underlying apoptosis of activated Cd47 −/− T cells that we report here. Lastly, our data demonstrate that CD4+ T cell proliferation relies, in part, on LFA‐1 activation and binding to ICAM‐1, especially in response to mitogens. We infer that MOG35–55‐activated Cd47−/− T effector cells that do proliferate and survive in immunized Cd47−/− mice are likely to have defects in homing to the CNS, and additional studies are required to assess this possibility. These findings highlight the important and multiple roles of Cd47 in T cell activation and the cellular mechanisms that lead to protection of Cd47−/− mice in the MOG35–55‐induced EAE model.
Although our data identify defects in T cell integrins as the dominant mechanism for failure of clonal cell expansion with resulting increased apoptosis, we suggest that defects in integrin functions in DCs and monocytes also contribute to protection in EAE. Prior studies found impaired DC migration to LNs upon antigen immunization in vivo in Cd47 −/− animals [39, 40]. Other investigators found that anti‐CD47 mAb miap301 blocked monocyte transmigration across brain endothelial monolayers in a murine in vitro model [41]. Although the authors of these publications did not identify a role for CD47 regulation of leukocyte integrins, we propose such a defect would have contributed to impaired myeloid cell migration. The latter report also found that blocking mAb to monocyte SIRPα or to endothelial CD47 also attenuated transmigration, further implicating the CD47‐SIRPα pathway in monocyte recruitment.
A surprising result was that although the anti‐CD47 mAb miap301 was found to block CD47 binding to murine SIRPα [24] and reduced Th1 T cell arrest on ICAM‐1 and VCAM‐1 (Fig. 1D), the mAb did not block naïve CD4+ T cell proliferation in vitro induced by Con A or PHA‐P lectins or TCR‐XL, as shown in Fig. 7G. The blocking effect of this mAb on adhesion of murine T cells to ICAM‐1 and VCAM‐1 was proportional to the level of inhibition of human T cell arrest on ICAM‐1 and VCAM‐1 by anti‐human CD47 mAb (Fig. 1D) that also blocked CD47 binding to SIRPα [42, 43]. We did not, however, predict the lack of inhibition on T cell proliferation by miap301 mAb, and differences in results between T cells deficient in CD47 and the anti‐CD47 antibody treatment remain unexplained. A likely explanation is that mAb miap301 does not fully block CD47 in cis and in trans association with other proteins required in proliferation of T cells and potentially other immune cells. Alternatively, perhaps CD47 performs actions that promote proliferation in the absence of binding ligand. We previously reported that miap301 mAb stained WT T cells, neutrophils, monocytes, and endothelial cells but not these same cell types from Cd47−/− animals, indicating specificity for murine CD47 [26]. We speculate that miap301 mAb interferes with in cis CD47‐integrin association as a result of steric hindrance, but this clearly requires future study. Our results with the miap301 mAb provide some insight into the previous report by Han and colleagues [14] that coadministration of MOG35–55 with anti‐CD47 mAb reduced paralysis, whereas mAb, given at peak of paralysis, worsened disease (a Janus‐like, opposing effect). We speculate that worsening paralysis by mAb miap301 infusions is a result of impaired recruitment and/or proliferation of T regulatory cells or other cell types that normally dampen the immune response to MOG35–55 [44, 45, 46, 47–48]. On the other hand, it is difficult to predict the outcome when using rat anti‐mouse blocking mAb to CD47 interactions in vivo as a result of short half‐life of a rat mAb in mice and of CD47 multiple in cis and in trans partners in immune and nonimmune (i.e., endothelium and epithelium) cell types. Given the emerging role of antibodies that target CD47 or SIRPα as an immunotherapy in cancer models [49, 50], a better understanding of the effect of current or newly generated mAb on CD47 binding to SIRPs or impairing leukocyte or endothelial cell integrin functions is important.
Han and colleagues [14] recently reported that Cd47−/− mice were protected in the MOG35–55‐induced EAE model as a result of complete failure in T cell activation. Our much more in‐depth in vivo and in vitro studies do not support this conclusion. It is likely the differences are related to their not appreciating that CD47 is necessary for LFA‐1 integrin function in adhesion and antigen‐ and lectin‐dependent proliferation. These authors also did not appreciate prior reports demonstrating that Cd47−/− cells injected into WT animals do not survive [5, 15]. Thus, their conclusion that Cd47−/− T cells isolated from MOG35–55‐immunized Cd47−/− mice and then transferred into WT did not cause disease is not interpretable.
Prior studies supported an important role for CD47 and its ligands SIRPα and TSP‐1 in immune‐mediated disease and as a potential therapeutic target in such diseases. Knock‐in Tg mice expressing a nonsignaling, truncated SIRPα molecule are also protected in MOG35–55‐induced EAE as a result of failure of DCs to prime T cells and to proliferate [51] and in a collagen‐induced arthritis model [52, 53]. Tsp1−/− mice also show milder disease in EAE [54]. We suggest the Janus‐like, opposing effect of CD47 does not negate its value as a therapeutic target based on the literature. Notably, initial studies that targeted other therapeutically important immune molecules, including CTLA‐4, VLA‐4, and CD28, have also reported Janus‐like opposing effects before their functions in immune cells were fully understood [55, 56, 57–58]. We conclude that lack of sustained LFA‐1 integrin‐dependent proliferation in Cd47−/− CD4+ T cells upon MOG35–55 immunization is the major mechanism for protection in the MOG35–55‐induced EAE model, although we do not discount the potential contribution of impaired T cell homing and/or recruitment of T effector cells (regulatory T cells, Th1, or Th17) or myeloid to the CNS.
AUTHORSHIP
V.A., W.E., and F.W.L. share senior authorship. R.B., J.M.H., G.N., D.E., A.A., and W.E. designed and conducted experiments and acquired and analyzed data. C.A.P. provided key reagents and wrote the manuscript. S.J.K., T.M., and A.H.L. provided reagents and wrote the manuscript. V.A. and F.W.L. designed experiments, analyzed data, and wrote and organized the manuscript.
DISCLOSURES
Content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
ACKNOWLEDGMENTS
The research was supported by an American Heart Association Postdoctoral Award (Postdoctoral Award 11POST7730055; to V.A.) and awards from the U.S. National Institutes of Health (NIH; R01 HL124780) and an American Heart Association Grant‐in‐Aid (15GRNT25080164; both to F.W.L.). Support was also given with awards from the NIH (AI071448, to S.J.K.; AI093838, to S.J.K. and W.E.; and DK79392, DK72564, and DK61379, to C.A.P.) and the National Multiple Sclerosis Society (RG3945, to S.J.K., and PP1734, to W.E.). The authors acknowledge the contribution of Ms. Kay Case to making hybridoma culture supernatants used in this study, Thomas Buttrick for technical assistance with T cell proliferation assays, and Drs. Anthony Letai (Dana‐Farber Cancer Institute, Boston, MA, USA) and Vicki A. Boussiotis (Beth Israel Deaconess Medical Center, Boston, MA, USA) for helpful discussions on T cell apoptosis.
Contributor Information
Wassim Elyaman, Email: elyaman@rics.bwh.harvard.edu.
Francis W. Luscinskas, Email: fluscinskas@partners.org
REFERENCES
- 1. Brown, E. J. , Frazier, W. A. (2001) Integrin‐associated protein (CD47) and its ligands. Trends Cell Biol. 11, 130–135. [DOI] [PubMed] [Google Scholar]
- 2. Barclay, A. N. , Van den Berg, T. K. (2014) The interaction between signal regulatory protein alpha (SIRPa) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol. 32, 25–50. [DOI] [PubMed] [Google Scholar]
- 3. Oldenborg, P. A. (2013) CD47: a cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol. 2013, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oldenborg, P. A. , Zheleznyak, A. , Fang, Y. F. , Lagenaur, C. F. , Gresham, H. D. , Lindberg, F. P. (2000) Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054. [DOI] [PubMed] [Google Scholar]
- 5. Blazar, B. R. , Lindberg, F. P. , Ingulli, E. , Panoskaltsis‐Mortari, A. , Oldenborg, P. A. , Iizuka, K. , Yokoyama, W. M. , Taylor, P. A. (2001) CD47 (integrin‐associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J. Exp. Med. 194, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Majeti, R. , Chao, M. P. , Alizadeh, A. A. , Pang, W. W. , Jaiswal, S. , Gibbs, K. D., Jr. , van Rooijen, N. , Weissman, I. L. (2009) CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chao, M. P. , Tang, C. , Pachynski, R. K. , Chin, R. , Majeti, R. , Weissman, I. L. (2011) Extranodal dissemination of non‐Hodgkin lymphoma requires CD47 and is inhibited by anti‐CD47 antibody therapy. Blood 118, 4890–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chao, M. P. , Weissman, I. L. , Majeti, R. (2012) The CD47‐SIRPa pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 24, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ho, C. C. , Guo, N. , Sockolosky, J. T. , Ring, A. M. , Weiskopf, K. , Özkan, E. , Mori, Y. , Weissman, I. L. , Garcia, K. C. (2015) “Velcro” engineering of high affinity CD47 ectodomain as signal regulatory protein a (SIRPa) antagonists that enhance antibody‐dependent cellular phagocytosis. J. Biol. Chem. 290, 12650–12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Azcutia, V. , Routledge, M. , Williams, M. R. , Newton, G. , Frazier, W. A. , Manica, A. , Croce, K. J. , Parkos, C. A. , Schmider, A. B. , Turman, M. V. , Soberman, R. J. , Luscinskas, F. W. (2013) CD47 plays a critical role in T‐cell recruitment by regulation of LFA‐1 and VLA‐4 integrin adhesive functions. Mol. Biol. Cell 24, 3358–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Azcutia, V. , Stefanidakis, M. , Tsuboi, N. , Mayadas, T. , Croce, K. J. , Fukuda, D. , Aikawa, M. , Newton, G. , Luscinskas, F. W. (2012) Endothelial CD47 promotes vascular endothelial‐cadherin tyrosine phosphorylation and participates in T cell recruitment at sites of inflammation in vivo. J. Immunol. 189, 2553–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuchroo, V. K. , Anderson, A. C. , Waldner, H. , Munder, M. , Bettelli, E. , Nicholson, L. B. (2002) T Cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross‐reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu. Rev. Immunol. 20, 101–123. [DOI] [PubMed] [Google Scholar]
- 13. Jäger, A. , Dardalhon, V. , Sobel, R. A. , Bettelli, E. , Kuchroo, V. K. (2009) Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 183, 7169–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han, M. H. , Lundgren, D. H. , Jaiswal, S. , Chao, M. , Graham, K. L. , Garris, C. S. , Axtell, R. C. , Ho, P. P. , Lock, C. B. , Woodard, J. I. , Brownell, S. E. , Zoudilova, M. , Hunt, J. F. , Baranzini, S. E. , Butcher, E. C. , Raine, C. S. , Sobel, R. A. , Han, D. K. , Weissman, I. , Steinman, L. (2012) Janus‐like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J. Exp. Med. 209, 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bouguermouh, S. , Van, V. Q. , Martel, J. , Gautier, P. , Rubio, M. , Sarfati, M. (2008) CD47 expression on T cell is a self‐control negative regulator of type 1 immune response. J. Immunol. 180, 8073–8082. [DOI] [PubMed] [Google Scholar]
- 16. Lindberg, F. P. , Bullard, D. C. , Caver, T. E. , Gresham, H. D. , Beaudet, A. L. , Brown, E. J. (1996) Decreased resistance to bacterial infection and granulocyte defects in IAP‐deficient mice. Science 274, 795–798. [DOI] [PubMed] [Google Scholar]
- 17. Bettelli, E. , Pagany, M. , Weiner, H. L. , Linington, C. , Sobel, R. A. , Kuchroo, V. K. (2003) Myelin oligodendrocyte glycoprotein‐specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197, 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parkos, C. A. , Colgan, S. P. , Liang, T. W. , Nusrat, A. , Bacarra, A. E. , Carnes, D. K. , Madara, J. L. (1996) CD47 mediates post‐adhesive events required for neutrophil migration across polarized intestinal epithelia. J. Cell Biol. 132, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lindberg, F. P. , Gresham, H. D. , Schwarz, E. , Brown, E. J. (1993) Molecular cloning of integrin‐associated protein: an immunoglobulin family member with multiple membrane‐spanning domains implicated in alpha v beta 3‐dependent ligand binding. J. Cell Biol. 123, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rao, R. M. , Betz, T. V. , Lamont, D. J. , Kim, M. B. , Shaw, S. K. , Froio, R. M. , Baleux, F. , Arenzana‐Seisdedos, F. , Alon, R. , Luscinskas, F. W. (2004) Elastase release by transmigrating neutrophils deactivates endothelial‐bound SDF‐1alpha and attenuates subsequent T lymphocyte transendothelial migration. J. Exp. Med. 200, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alcaide, P. , Maganto‐Garcia, E. , Newton, G. , Travers, R. , Croce, K. J. , Bu, D. X. , Luscinskas, F. W. , Lichtman, A. H. (2012) Difference in Th1 and Th17 lymphocyte adhesion to endothelium. J. Immunol. 188, 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elyaman, W. , Bradshaw, E. M. , Wang, Y. , Oukka, M. , Kivisäkk, P. , Chiba, S. , Yagita, H. , Khoury, S. J. (2007) JAGGED1 and delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J. Immunol. 179, 5990–5998. [DOI] [PubMed] [Google Scholar]
- 23. Mueller, K. L. , Thomas, M. S. , Burbach, B. J. , Peterson, E. J. , Shimizu, Y. (2007) Adhesion and degranulation‐promoting adapter protein (ADAP) positively regulates T cell sensitivity to antigen and T cell survival. J. Immunol. 179, 3559–3569. [DOI] [PubMed] [Google Scholar]
- 24. Jiang, P. , Lagenaur, C. F. , Narayanan, V. (1999) Integrin‐associated protein is a ligand for the P84 neural adhesion molecule. J. Biol. Chem. 274, 559–562. [DOI] [PubMed] [Google Scholar]
- 25. Martinelli, R. , Newton, G. , Carman, C. V. , Greenwood, J. , Luscinskas, F. W. (2013) Novel role of CD47 in rat microvascular endothelium: signaling and regulation of T‐cell transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 33, 2566–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stefanidakis, M. , Newton, G. , Lee, W. Y. , Parkos, C. A. , Luscinskas, F. W. (2008) Endothelial CD47 interaction with SIRPgamma is required for human T‐cell transendothelial migration under shear flow conditions in vitro. Blood 112, 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Legrand, N. , Huntington, N. D. , Nagasawa, M. , Bakker, A. Q. , Schotte, R. , Strick‐Marchand, H. , de Geus, S. J. , Pouw, S. M. , Böhne, M. , Voordouw, A. , Weijer, K. , Di Santo, J. P. , Spits, H. (2011) Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T‐ and natural killer‐ (NK) cell homeostasis in vivo. Proc. Natl. Acad. Sci. USA 108, 13224–13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dunkle, A. , He, Y. W. (2011) Apoptosis and autophagy in the regulation of T lymphocyte function. Immunol. Res. 49, 70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boise, L. H. , Minn, A. J. , Noel, P. J. , June, C. H. , Accavitti, M. A. , Lindsten, T. , Thompson, C. B. (1995) CD28 costimulation can promote T cell survival by enhancing the expression of Bcl‐xL. Immunity 3, 87–98. [DOI] [PubMed] [Google Scholar]
- 30. Kerstan, A. , Hünig, T. (2004) Cutting edge: distinct TCR‐ and CD28‐derived signals regulate CD95L, Bcl‐xL, and the survival of primary T cells. J. Immunol. 172, 1341–1345. [DOI] [PubMed] [Google Scholar]
- 31. Khoshnan, A. , Tindell, C. , Laux, I. , Bae, D. , Bennett, B. , Nel, A. E. (2000) The NF‐kappa B cascade is important in Bcl‐xL expression and for the anti‐apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J. Immunol. 165, 1743–1754. [DOI] [PubMed] [Google Scholar]
- 32. Rothlein, R. , Springer, T. A. (1986) The requirement for lymphocyte function‐associated antigen 1 in homotypic leukocyte adhesion stimulated by phorbol ester. J. Exp. Med. 163, 1132–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwartz, D. , Wong, R. C. , Chatila, T. , Arnaout, A. , Miller, R. , Geha, R. (1989) Proliferation of highly purified T cells in response to signaling via surface receptors requires cell‐cell contact. J. Clin. Immunol. 9, 151–158. [DOI] [PubMed] [Google Scholar]
- 34. Hommel, M. , Kyewski, B. (2003) Dynamic changes during the immune response in T cell‐antigen‐presenting cell clusters isolated from lymph nodes. J. Exp. Med. 197, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kandula, S. , Abraham, C. (2004) LFA‐1 on CD4+ T cells is required for optimal antigen‐dependent activation in vivo. J. Immunol. 173, 4443–4451. [DOI] [PubMed] [Google Scholar]
- 36. Zumwalde, N. A. , Domae, E. , Mescher, M. F. , Shimizu, Y. (2013) ICAM‐1‐dependent homotypic aggregates regulate CD8 T cell effector function and differentiation during T cell activation. J. Immunol. 191, 3681–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reinhold, M. I. , Lindberg, F. P. , Kersh, G. J. , Allen, P. M. , Brown, E. J. (1997) Costimulation of T cell activation by integrin‐associated protein (CD47) is an adhesion‐dependent, CD28‐independent signaling pathway. J. Exp. Med. 185, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stupack, D. G. , Cheresh, D. A. (2002) Get a ligand, get a life: integrins, signaling and cell survival. J. Cell Sci. 115, 3729–3738. [DOI] [PubMed] [Google Scholar]
- 39. Van, V. Q. , Lesage, S. , Bouguermouh, S. , Gautier, P. , Rubio, M. , Levesque, M. , Nguyen, S. , Galibert, L. , Sarfati, M. (2006) Expression of the self‐marker CD47 on dendritic cells governs their trafficking to secondary lymphoid organs. EMBO J. 25, 5560–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hagnerud, S. , Manna, P. P. , Cella, M. , Stenberg, A. , Frazier, W. A. , Colonna, M. , Oldenborg, P. A. (2006) Deficit of CD47 results in a defect of marginal zone dendritic cells, blunted immune response to particulate antigen and impairment of skin dendritic cell migration. J. Immunol. 176, 5772–5778. [DOI] [PubMed] [Google Scholar]
- 41. De Vries, H. E. , Hendriks, J. J. , Honing, H. , De Lavalette, C. R. , van der Pol, S. M. , Hooijberg, E. , Dijkstra, C. D. , van den Berg, T. K. (2002) Signal‐regulatory protein alpha‐CD47 interactions are required for the transmigration of monocytes across cerebral endothelium. J. Immunol. 168, 5832–5839. [DOI] [PubMed] [Google Scholar]
- 42. Lee, W. Y. , Weber, D. A. , Laur, O. , Severson, E. A. , McCall, I. , Jen, R. P. , Chin, A. C. , Wu, T. , Gernert, K. M. , Parkos, C. A. (2007) Novel structural determinants on SIRP alpha that mediate binding to CD47. J. Immunol. 179, 7741–7750. [DOI] [PubMed] [Google Scholar]
- 43. Hatherley, D. , Graham, S. C. , Turner, J. , Harlos, K. , Stuart, D. I. , Barclay, A. N. (2008) Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell 31, 266–277. [DOI] [PubMed] [Google Scholar]
- 44. Liu, Y. , Carlsson, R. , Comabella, M. , Wang, J. , Kosicki, M. , Carrion, B. , Hasan, M. , Wu, X. , Montalban, X. , Dziegiel, M. H. , Sellebjerg, F. , Sørensen, P. S. , Helin, K. , Issazadeh‐Navikas, S. (2014) FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat. Med. 20, 272–282. [DOI] [PubMed] [Google Scholar]
- 45. Wohler, J. E. , Smith, S. S. , Zinn, K. R. , Bullard, D. C. , Barnum, S. R. (2009) Gammadelta T cells in EAE: early trafficking events and cytokine requirements. Eur. J. Immunol. 39, 1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bassil, R. , Zhu, B. , Lahoud, Y. , Riella, L. V. , Yagita, H. , Elyaman, W. , Khoury, S. J. (2011) Notch ligand delta‐like 4 blockade alleviates experimental autoimmune encephalomyelitis by promoting regulatory T cell development. J. Immunol. 187, 2322–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McGeachy, M. J. , Stephens, L. A. , Anderton, S. M. (2005) Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 175, 3025–3032. [DOI] [PubMed] [Google Scholar]
- 48. O'Connor, R. A. , Malpass, K. H. , Anderton, S. M. (2007) The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J. Immunol. 179, 958–966. [DOI] [PubMed] [Google Scholar]
- 49. Liu, X. , Pu, Y. , Cron, K. , Deng, L. , Kline, J. , Frazier, W. A. , Xu, H. , Peng, H. , Fu, Y. X. , Xu, M. M. (2015) CD47 blockade triggers T cell‐mediated destruction of immunogenic tumors. Nat. Med. 21, 1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vonderheide, R. H. (2015) CD47 blockade as another immune checkpoint therapy for cancer. Nat. Med. 21, 1122–1123. [DOI] [PubMed] [Google Scholar]
- 51. Tomizawa, T. , Kaneko, Y. , Kaneko, Y. , Saito, Y. , Ohnishi, H. , Okajo, J. , Okuzawa, C. , Ishikawa‐Sekigami, T. , Murata, Y. , Okazawa, H. , Okamoto, K. , Nojima, Y. , Matozaki, T. (2007) Resistance to experimental autoimmune encephalomyelitis and impaired T cell priming by dendritic cells in Src homology 2 domain‐containing protein tyrosine phosphatase substrate‐1 mutant mice. J. Immunol. 179, 869–877. [DOI] [PubMed] [Google Scholar]
- 52. Verdrengh, M. , Lindberg, F. P. , Ryden, C. , Tarkowski, A. (1999) Integrin‐associated protein (IAP)‐deficient mice are less susceptible to developing Staphylococcus aureus‐induced arthritis. Microbes Infect. 1, 745–751. [DOI] [PubMed] [Google Scholar]
- 53. Vallejo, A. N. , Yang, H. , Klimiuk, P. A. , Weyand, C. M. , Goronzy, J. J. (2003) Synoviocyte‐mediated expansion of inflammatory T cells in rheumatoid synovitis is dependent on CD47‐thrombospondin 1 interaction. J. Immunol. 171, 1732–1740. [DOI] [PubMed] [Google Scholar]
- 54. Yang, K. , Vega, J. L. , Hadzipasic, M. , Schatzmann Peron, J. P. , Zhu, B. , Carrier, Y. , Masli, S. , Rizzo, L. V. , Weiner, H. L. (2009) Deficiency of thrombospondin‐1 reduces Th17 differentiation and attenuates experimental autoimmune encephalomyelitis. J. Autoimmun. 32, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Racke, M. K. , Scott, D. E. , Quigley, L. , Gray, G. S. , Abe, R. , June, C. H. , Perrin, P. J. (1995) Distinct roles for B7‐1 (CD‐80) and B7‐2 (CD‐86) in the initiation of experimental allergic encephalomyelitis. J. Clin. Invest. 96, 2195–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Theien, B. E. , Vanderlugt, C. L. , Eagar, T. N. , Nickerson‐Nutter, C. , Nazareno, R. , Kuchroo, V. K. , Miller, S. D. (2001) Discordant effects of anti‐VLA‐4 treatment before and after onset of relapsing experimental autoimmune encephalomyelitis. J. Clin. Invest. 107, 995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hünig, T. (2012) The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nat. Rev. Immunol. 12, 317–318. [DOI] [PubMed] [Google Scholar]
- 58. Suntharalingam, G. , Perry, M. R. , Ward, S. , Brett, S. J. , Castello‐Cortes, A. , Brunner, M. D. , Panoskaltsis, N. (2006) Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355, 1018–1028. [DOI] [PubMed] [Google Scholar]