

Short abstract
Clinical Review for Basic Researchers: NETs in autoimmune diseases, their potential as disease markers and therapeutic targets.
Keywords: systemic lupus erythematosus, type I IFN, autoimmunity
Abstract
Neutrophil extracellular traps are associated with a unique form of cell death distinct from apoptosis or necrosis, whereby invading microbes are trapped and killed. Neutrophil extracellular traps can contribute to autoimmunity by exposing autoantigens, inducing IFN‐α production, and activating the complement system. The association of neutrophil extracellular traps with autoimmune diseases, particularly systemic lupus erythematosus, will be reviewed. Increased neutrophil extracellular trap formation is seen in psoriasis, antineutrophil cytoplasmic antibody‐associated vasculitis, antiphospholipid antibody syndrome rheumatoid arthritis, and systemic lupus erythematosus. Neutrophil extracellular traps may promote thrombus formation in antineutrophil cytoplasmic antibody‐associated vasculitis and antiphospholipid antibody syndrome. In systemic lupus erythematosus, increased neutrophil extracellular trap formation is associated with increased disease activity and renal disease, suggesting that neutrophil extracellular traps could be a disease activity marker. Neutrophil extracellular traps can damage and kill endothelial cells and promote inflammation in atherosclerotic plaques, which may contribute to accelerated atherosclerosis in systemic lupus erythematosus. As neutrophil extracellular traps induce IFN‐α production, measuring neutrophil extracellular traps may estimate IFN‐α levels and identify which systemic lupus erythematosus patients have elevated levels and may be more likely to respond to emerging anti‐IFN‐α therapies. In addition to anti‐IFN‐α therapies, other novel agents, such as N‐acetyl‐cysteine, DNase I, and peptidylarginine deiminase inhibitor 4, target neutrophil extracellular traps. Neutrophil extracellular traps offer insight into the pathogenesis of autoimmune diseases and provide promise in developing disease markers and novel therapeutic agents in systemic lupus erythematosus. Priority areas for basic research based on clinical research insights will be identified, specifically the potential role of neutrophil extracellular traps as a biomarker and therapeutic target in systemic lupus erythematosus.
Abbreviations
- β2GP1
β‐2‐glycoprotein 1
- ACPA
autoantibodies to citrullinated protein antigen
- ANCA
antineutrophil cytoplasmic antibody
- APS
antiphospholipid antibody syndrome
- C5
complement component 5
- cfDNA
cell‐free DNA
- CRP
C‐reactive protein
- CVD
cardiovascular disease
- EC
endothelial cell
- ENA
extractable nuclear antigen
- ESR
erythrocyte sedimentation rate
- GN
glomerulonephritis
- HCQ
hydroxychloroquine
- LDG
low‐density granulocyte
- MMP
metalloproteinase
- MPO
myeloperoxidase
- mTOR
mammalian target of rapamycin
- NAC
N‐acetyl‐cysteine
- NE
neutrophil elastase
- NET
neutrophil extracellular trap
- NOX
NADPH oxidase
- PAD‐4
peptidylarginine deiminase 4
- pDC
plasmacytoid dendritic cell
- PNH
paroxysmal nocturnal hemoglobinuria
- PR3
proteinase 3
- RA
rheumatoid arthritis
- rh
recombinant human
- rm
recombinant mouse
- ROS
reactive oxygen species
- SLE
systemic lupus erythematosus
- SLEDAI
systemic lupus erythematosus disease activity index
- SRI
systemic lupus erythematosus responder index
Introduction
Neutrophils play a key role in innate immunity with multiple strategies for defending the body against pathogens. Neutrophils initially migrate to the site of infection and phagocytose and kill bacteria with the aid of proteolytic enzymes, antimicrobial proteins, and ROS [1]. Neutrophils can also degranulate to release antimicrobial factors in the extracellular space to kill bacteria [1]. More recently, neutrophils were found to kill bacteria by forming extracellular structures called NETs, which are composed mainly of histones, DNA, and proteases, such as NE. NETs were first discovered in 1996 as a unique form of cell death, distinct from necrosis or apoptosis [2]. This process was further described and termed NETosis in 2004 [1]. In NETosis, the neutrophils extrude large amounts of chromatin and granule proteins, such as NE and MPO, which trap and kill microorganisms. NETs contain the invading microorganism to prevent the spread of infection and use their highly localized concentration of antimicrobial peptides to degrade virulence factors and kill the microorganism [1]. Whereas NETs have a role in pathogen defense, NETs can also lead to toxic effects in the host. Importantly, NETs expose autoantigens, such as nucleic acids and proteins, in an inflammatory milieu that can stimulate an autoimmune response in a susceptible individual. See the related review that highlights the involvement of NETs in the innate immune system and autoimmune diseases [3].
NETosis is a novel form of cell death where neutrophils extrude extracellular fibers composed of chromatin and granule proteins that trap and kill microorganisms.
The association of increased NET formation and autoimmunity was first described in ANCA‐associated vasculitis and subsequently in other autoimmune diseases, including psoriasis, SLE, APS, and RA [4, 5, 6, 7, 8–9]. Proteins found in NETs may be the source of key autoantigens, including MPO and PR3, in ANCA‐associated vasculitis, dsDNA antibodies in SLE, and ACPAs in RA. Not only do the protein contents of NETs serve as the targets for autoantibody and immune complex formation, but also, they induce further NETosis, resulting in a positive‐feedback loop. The exact molecular mechanisms that trigger NETosis are not fully understood. Several pathways have been suggested, such as induction by ROS generated from NOX. MPO and NE then promote chromatin dencondensation with PAD‐4 citrullinating histones [10]. There are conflicting reports as to whether these processes are both required and sufficient for NETosis [10]. NETs were originally described as being activated upon stimulation with IL‐8, PMA, and LPS, along with gram‐positive and ‐negative bacteria, fungi, and parasites [1, 11]. However, in autoimmune diseases, proinflammatory cytokines, such as TNF‐α and IL‐17, and autoantibodies not only stimulate NETosis but also affect the protein contents of NETs [7].
NETs can also promote autoimmunity through production of type I IFNs and activation of the classic and alternative pathways of the complement system. In psoriasis and SLE, the contents of NETs, namely the antimicrobial peptides and the self‐DNA, are able to induce IFN‐α production from the pDC [4]. pDCs are the major producers of IFN‐α and normally do so in response to invading viruses. NETs with their nuclear contents of dsDNA similarly induce IFN‐α from pDCs. IFN‐α can then activate both the innate and adaptive immune systems, specifically inducing a Th type 1 pathway, inhibiting T cell apoptosis, and activating B cells and antibody production [12].
In addition to increased NET formation in patients with autoimmune diseases compared with healthy controls, there is enhanced NET stability with decreased clearance of NETs, particularly in SLE and ANCA‐associated vasculitis [13, 14–15]. There are various mechanisms for the decreased clearance of NETs. One degrader of NETs is an endonuclease called DNase I. In a subset of SLE and ANCA‐associated vasculitis patients, there is low DNase I activity and DNase I inhibitors, resulting in decreased clearance of NETs [13, 14, 15–16]. There are also NET‐protecting antibodies that prevent access of DNase I to the NETs to prevent NET degradation [13, 14, 15–16]. Complement also plays a role in NET degradation. Complement interacts with NETs, with C1q and C3b depositing on the NETs [15]. Deposited C1q inhibits NET degradation through inhibition of DNase I. Furthermore, nondegraded NETs activate complement in vitro [15]. Overall, a positive‐feedback loop exists where NETs activate complement, which then further increases NETs by preventing their degradation. In addition to the role of NETs in autoimmunity, NETs may be a potential disease activity biomarker and a therapeutic target.
ASSOCIATION OF NETs WITH DISEASE ACTIVITY IN AUTOIMMUNE DISEASES
The ability of NETs to induce autoimmunity has been suggested in various autoimmune diseases ( Table 1 and Fig. 1 ). NETs were first implicated in the pathogenesis of ANCA‐associated vasculitis, which is a group of diseases characterized by a systemic necrotizing vasculitis that affects small vessels, predominantly of the lungs and kidneys, and includes granulomatosis with polyangiitis (Wegener's) and microscopic polyangiitis. Patients have ANCAs—antibodies directed against neutrophil granule proteins PR3 or MPO. These autoantibodies are pathogenic in animal models of ANCA‐associated vasculitis [19, 20]. The granule protein contents of NETs may be the antigen source for these pathogenic autoantibodies. ANCAs can then directly stimulate NETosis in vitro [5, 14, 21]. In a study with 38 patients with ANCA‐associated vasculitis, patients’ sera had a high ability to induce NET formation compared with healthy controls [14]. Increased NET formation was associated with increased disease activity [14]. Furthermore, DNase I activity levels were significantly lower, leading to impaired NET degradation in the serum of patients with ANCA‐associated vasculitis vs. healthy controls [14]. Anti‐NET antibodies that can inhibit the DNase I activity were found in some patients and led to a further reduction in the degradation of NETs [14]. Overall, there seems to be a “NET‐ANCA vicious cycle,” where there is excessive formation and decreased clearance of NETs that could lead to ANCA production, which then accelerates further NET formation [14]. Better understanding of the mechanisms of the anti‐NET antibodies and the “NET‐ANCA cycle” could help identify potential therapeutic targets for ANCA‐associated vasculitis.
Table 1.
Association of NETs with autoimmune diseases
| Study | Disease | NETs measurement | Results | Proposed role of NETs |
|---|---|---|---|---|
| [4] | Psoriasis | In vitro | Endogenous antimicrobial peptide (LL‐37) converts self‐DNA into a potent trigger of pDCs to release IFN‐α in psoriatic skin. | NET protein content LL‐37 drives production of IFN‐α that contributes to autoimmunity. |
| [5] | ANCA‐associated vasculitis | In vivo | NET formation in 15 renal biopsies | The granule protein contents of NET may be the antigen source for pathogenic autoantibodies, ANCAs. |
| [14] | ANCA‐associated vasculitis | In vivo | Association of increased NET formation with increased disease activity, as measured by the Birmingham Vasculitis Activity Score | |
| [17] | ANCA‐associated vasculitis | In vivo | NETs were found in glomerular crescents and in thrombi of patients with ANCA‐associated vasculitis. | Increased NET formation may contribute to thrombus formation in ANCA‐associated vasculitis. |
| [8] | APS | In vitro | Decreased degradation of NETs in sera of patients with APS was associated with increased levels of antibodies against NETs. | |
| [9] | APS | In vitro | Increased circulating levels of NETs were associated with the presence of APS autoantibodies; an APS autoantibody, β2GP1, interacted with neutrophils to stimulate NETosis. | NETs may promote thrombin formation in APS. |
| [7] | RA | In vivo | Increased NET formation was seen in synovial tissue, rheumatoid nodules, and skin; increased percentage of netting neutrophils was associated with increased serum CRP, ESR, ACPA, and IL‐17 | NETs may be the source of citrullinated autoantigens that are pathogenic in RA. |
| [18] | RA | In vitro | Increased NET formation compared with controls |
Figure 1.
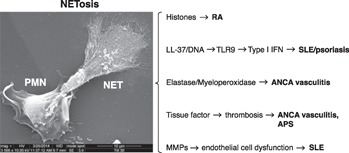
Overview of potential pathogenic roles of NETs in autoimmune diseases. NETs externalize citrullinated histones that act as autoantigens in RA. The contents of NETs, LL‐37 and DNA, induce IFN‐α from pDCs, which contribute to autoimmunity in SLE and psoriasis. The MPO content of NETs is the antigenic source for pathogenic ANCA in ANCA‐associated vasculitis. NETs activate platelets and coagulation factors, such as tissue factor, to promote thrombosis in APS and ANCA‐associated vasculitis. MMP‐9, externalized on NETs, damages ECs to promote vasculopathy and potentially accelerated atherosclerosis in SLE. Electron micrograph of polymorphonuclear neutrophil (PMN) undergoing NETosis courtesy of Drs. Elizabeth Johnson and Leslie Crofford.
ANCA‐associated vasculitis is a group of diseases characterized by a systemic necrotizing vasculitis that affects small vessels, predominantly of the lungs and kidneys, and includes granulomatosis with polyangiitis (Wegener's) and microscopic polyangiitis. Patients frequently have antibodies directed against neutrophil granule proteins PR3 or MPO.
There is also in vivo evidence of NET formation in the affected glomeruli and interstitium of renal biopsies from patients with ANCA‐associated vasculitis [5]. A single case study found abundant NETs in a thrombus from a patient with ANCA‐associated vasculitis [17]. Increased NET formation may cause thrombus formation. Specifically, histones within the NETs can bind platelets and blood coagulants [22, 23]. The role of NETs in thrombosis in ANCA‐associated vasculitis has clinical relevance, as patients with ANCA‐associated vasculitis have an increased risk of venous thrombosis during periods of active disease [24, 25]. More studies are needed to determine whether NETs are a pathogenic mechanism in thrombosis formation in patients with ANCA‐associated vasculitis.
Research Question:
Does increased NET formation lead to thrombus formation in patients with ANCA‐associated vasculitis?
NETs may play a pathogenic role in thrombus formation in another autoimmune disease called APS. In APS, patients are at an increased risk of recurrent arterial and venous thrombosis and pregnancy morbidity. APS can occur in a primary form, where there is no underlying autoimmune disease, or a secondary form, where there is an underlying autoimmune disease, such as SLE. Patients with both primary and secondary APS have autoantibodies to phospholipids and surface proteins, such as lupus anticoagulant, anticardiolipin, and β2GP1, which can promote thrombus formation by activating ECs, monocytes, and platelets [26, 27–28]. β2GP1 can interact with neutrophils to stimulate NETosis in vitro in a process that is dependent on ROS and TLR4 signaling [9]. Furthermore, NETs promoted thrombin generation in vitro [9]. A positive correlation was seen with the presence of β2GP1 and other APS antibodies—lupus anticoagulant and anticardiolipin—with circulating levels of NETs [9]. These increased levels of circulating NETs may be a result of the presence of antiphospholipid autoantibodies but also could result from decreased degradation of NETs, as has been suggested by another in vitro study in APS [8]. Future in vivo studies are needed to confirm the findings of increased NET formation and decreased NET degradation in patients with APS and to characterize the pathways in which autoantibodies in APS may stimulate NETosis and promote thrombus formation.
APS is an autoimmune disease where patients are at increased risk of recurrent arterial and venous thrombosis and pregnancy morbidity, such as miscarriages, fetal loss, and preeclampsia. Patients can have persistently elevated levels of autoantibodies to phospholipids and surface proteins, such as anticardiolipin and β2GP1 and the presence of a circulating lupus anticoagulant.
Research Questions:
What are the mechanisms by which NETs promote thrombus formation in patients with APS? Do levels of NETs increase during clinical events associated with APS, such as acute thrombotic episodes, or pregnancy complications, such as miscarriages or preeclampsia?
NETs have also been implicated in the pathogenesis of RA, which is a chronic autoimmune disease characterized by inflammation and subsequent damage of the synovial joints. Patients with RA have increased levels of inflammatory cytokines, such as TNF‐α and IL‐17, and pathogenic ACPA, years before clinical diagnosis [29]. Citrullination is a post‐translation modification that can generate neoantigens, rendering proteins and immune complexes immunogenic and arthrogenic [30, 31]. Whereas histone citrullation is important in NETosis, it is not sufficient [10]. In an autocrine feed‐forward loop, NETs present these citrullinated histones [32] that serve as proinflammatory and immunostimulatory autoantigens to promote joint inflammation and destruction [6, 33, 34].
Neutrophils from RA cases have increased NET formation in vitro compared with controls [18], with NETs detected in synovial tissue, rheumatoid nodules, and skin [7]. Furthermore, there were significant correlations between the percentage of peripheral blood netting neutrophils and serum levels of CRP, ESR, ACPA, and IL‐17 [7]. Autoantibodies, such as ACPA and rheumatoid factor, and inflammatory cytokines, such as TNF‐α and IL‐17, induced NETosis in RA cases [7]. In addition, NETs externalized citrullinated autoantigens that are key RA autoantigens [7]. This study suggests a cycle where NETs are exposing citrullinated antigens to the immune system with these same citrullinated antigens and inflammatory cytokines, further perpetuating NETosis [7]. In addition, ACPA and inflammatory cytokines, such as TNF‐α, induced distinct protein contents in the NETs. Future studies are needed to confirm these findings but also to investigate how different autoantibodies and inflammatory cytokines stimulate NETs and affect their protein contents.
RA is a chronic autoimmune disease characterized by inflammation and subsequent damage of the synovial joints. Patients with RA can have pathogenic ACPA.
Research Question:
How are the protein contents of NETs influenced by exposure to different autoantibodies and inflammatory cytokines?
ASSOCIATION OF NETs WITH DISEASE ACTIVITY IN SLE
Similar to ANCA‐associated vasculitis and RA, there is in vivo evidence for NETs in SLE ( Table 2 ), which is a chronic autoimmune disease with multisystem organ involvement that can range from a mild disease course, where the skin and joints are affected, to a more severe disease course, involving the kidneys and CNS. The disease is characterized by fluctuating periods of increased disease activity called flares. In SLE, there is a break in self‐tolerance that results in the production of autoantibodies to nucleic acids and associated proteins, such as dsDNA. These autoantibodies can form immune complexes that deposit in the skin and kidneys causing tissue damage.
Table 2.
Association of NETs with SLE
| Study | NETs measurements | Disease measurements | Results | Proposed role of NETs |
|---|---|---|---|---|
| [13] | In vivo, inability to degrade NETs | Presence of SLE nephritis | Inability to degrade NETs correlated with the presence of SLE nephritis. | Decreased ability to degrade NETs may be associated with increased disease activity and renal disease. |
| [15] | In vivo, inability to degrade NETs | Disease activity, as measured by SLEDAI | Inability to degrade NETs correlated with higher SLEDAI scores and the presence of renal involvement and pleurisy. | |
| [35] | In vivo, NET products (cfDNA) | Active nephritis (active lesions on renal biopsy, active urinary sediment, and proteinuria), serum albumin, creatinine clearance rates | Higher concentrations of cfDNA correlated with the presence of active nephritis and with increased levels of 24 h urinary protein and decreased levels of albumin and creatinine clearance rates. | |
| [16] | In vivo, inability to degrade NETs | Disease activity, as measured by SLEDAI, and additional clinical and lab parameters | Decreased ability to degrade NETs was associated with a higher SLEDAI score, nephritis, elevated dsDNA, and low C3. | |
| [14] | In vivo, ability of sera IgG from SLE patients to induce NET formation | Disease activity, as measured by SLEDAI | No significant association between the ability to induce NETs and SLEDAI | |
| [6] | In vivo, percentage of netting neutrophils, LDGs | Activity indices on renal biopsies, dsDNA levels | Class IV SLE nephritis patients had higher percentage of netting neutrophils than class III SLE nephritis patients. Higher percentage of netting neutrophils was associated with higher activity indices on renal biopsies and higher dsDNA levels. LDGs have increased ability to form NETs; LDGs have a propensity to damage and kill ECs and stimulate IFN‐α synthesis by pDCs. | NETs externalize dsDNA, an autoantigen that is immunostimulatory and pathogenic in SLE. NETs promote IFN‐α, production that contributes to autoimmunity in SLE. |
| [36] | In vitro | EC damage by use of HUVECs | MMP‐9 is up‐regulated and externalized on NETs and damages ECs. | NETs damage and kill ECs; MMP‐9, contained in NETs, contributes to EC dysfunction in SLE. |
| [37] | In vitro | HDL oxidation, cholesterol efflux capacity | NETs promote HDL oxidation and decreased cholesterol efflux capacity. | NETs oxidize HDL, rendering it dysfunctional and proatherogenic, possibly contributing to accelerated atherosclerosis in SLE. |
SLE is a chronic autoimmune disease characterized by multisystem organ involvement ranging from skin and joint and renal and CNS involvement with fluctuating episodes of increased disease activity. Patients can have autoantibodies to nucleic acids and associated proteins, including antinuclear antibodies, anti‐Smith, anti‐dsDNA, and antiphospholipid antibodies.
Netting neutrophils were present in 6 of 9 renal biopsies from SLE patients with class III or IV SLE nephritis [6]. Patients with class IV SLE nephritis, a diffuse‐proliferative form, typically with active inflammatory lesions, had a higher percentage of netting neutrophils infiltrating the glomeruli compared with patients with class III SLE nephritis, a focal‐proliferative form, typically with active inflammatory lesions. Patients with higher activity biopsy indices and anti‐dsDNA levels had more netting neutrophils infiltrating the glomeruli compared with patients with lower activity biopsy indices and anti‐dsDNA levels. NETs were also found in skin lesions in SLE patients with various forms of cutaneous lupus [6]. In addition, there are multiple studies investigating the association of NET degradation with the presence of SLE nephritis and disease activity. One study with 94 SLE patients confirmed that sera from a subset of patients failed to degrade NETs [15]. In a retrospective analysis of 61 patients, the inability to degrade NETs correlated with the presence of SLE nephritis and increased disease activity [13], as measured by the SLEDAI [38]. Renal involvement and pleurisy, as measured by SLEDAI, were also associated with the inability to degrade NETs [13]. SLEDAI is a validated and commonly used disease activity measure, where the physician assigns points to clinical and lab parameters that represent active SLE based on their presence or absence in the prior 10 d.
SLEDAI is a validated disease activity measure, where a physician assigns points to a list of 24 items, 16 of which are based on clinical symptoms, and 8 are laboratory measures based on their presence or absence within the prior 10 d. More severe organ involvement, such as renal involvement, is weighted with more points. The points are then summed with a possible score ranging from 0 to 105, with higher scores indicating higher disease activity. A score of 6 or more is consistent with active disease that requires therapy.
With the use of a different measure to assess NET formation, one cross‐sectional study assessed levels of cfDNA, products of NETs [35]. Compared with controls, SLE patients had significantly higher mean concentrations of cfDNA [35]. SLE patients with active nephritis, defined as active urinary sediment, proteinuria, or active lesions on a renal biopsy, had higher concentrations of cfDNA than SLE patients with inactive nephritis and no history of nephritis. cfDNA positively correlated with 24 h urinary protein and negatively correlated with albumin levels and the creatinine clearance rate. In summary, with the use of 3 different measures to assess NETs, there are multiple studies supporting the association between increased NET formation and increased disease activity and renal involvement in patients with SLE.
To show more convincingly the association of NETs and SLE disease activity, one prospective longitudinal study conducted an analysis of 69 SLE patients. The authors measured the degradation of NETs, along with other serologic and clinical parameters, every 2 mo for a mean 2.5 y [16]. To define normal vs. decreased degradation of NETs, the ability of healthy controls to degrade NETs was measured. If a SLE patient had NET degradation at a cut‐off set as 3 sd or lower below the mean NET degradation of controls, then the SLE patient was defined as having a “decreased” ability to degrade NETs. Among the 69 SLE patients, 28 (41%) displayed, at least once, a decreased ability to degrade NETs, with 2 SLE patients having a reduced ability to degrade NETs at all time points measured. SLE patients who had decreased NET degradation at least once were more likely to be dsDNA positive compared with SLE patients with normal NET degradation. In an attempt to describe temporal associations of clinical and laboratory parameters with NET degradation, this study analyzed items predominantly from the SLEDAI to assess disease activity that occurred 2 mo before, concomitantly, and 2 mo after NET degradation measurement. An increase in SLEDAI was significantly associated with decreased NET degradation and seen before, at, and after sampling of NET degradation. Furthermore, manifestations of active GN, such as cellular casts, hematuria, and proteinuria, were all significantly associated with decreased NET degradation and seen at the time of sampling. Proteinuria was associated with decreased NET degradation and seen before, during, and after NET sampling. In addition, lab parameters, such as elevated dsDNA, CRP, low levels of C3, and leukopenia, were all associated with decreased NET degradation and seen at the time of sampling. This prospective longitudinal study showed that a subset of SLE patients had a decreased ability to degrade NETs, which was associated with a higher SLEDAI score, GN, and known disease activity markers, such as elevated dsDNA and low C3 levels. Decreased NET degradation even preceded an increase in SLEDAI and proteinuria. This study suggests that NET degradation may have value as a biomarker of both predicting and detecting disease activity and renal involvement in SLE. However, larger prospective longitudinal studies that assess NET degradation and disease activity are needed.
Research Questions:
Do all patients with SLE have increased NETosis, or does only a subset of patients with specific autoantibodies have increased NETosis? In which subsets of patients with SLE does increased NETosis correlate with increased disease activity?
NETs AS AUTOIMMUNE DISEASE BIOMARKERS
Multiple cross‐sectional studies and 1 longitudinal prospective study suggest that the measurement of NET degradation may be useful as a biomarker to predict and assess disease activity and renal disease. However, there is a recent, small study with 23 patients with SLE that did not find an association between increased NETs and disease activity, as measured by SLEDAI [14]. Of note, this study assessed for the presence of NETs differently by measuring the ability of sera IgG from SLE patients to induce NET formation instead of measuring NET degradation, as done in the previously discussed studies. These contrasting results highlight the importance of standardizing and validating the way NETs are measured in SLE patients. Whereas more studies in the literature, including a prospective one, tend to use NET degradation as the measure of increased NET formation or NETosis, it is not clear if this method is more accurate to capture NETosis compared with measuring the ability for sera to induce NETs or measuring NET products, such as cfDNA. Another consideration is how to define “normal” versus increased NET formation. Some studies do not define specific cutoffs, whereas others define decreased NET degradation, for example, as 3 sd or lower than the mean for healthy controls [16]. The standardization of what is defined as decreased NET degradation or increased NET formation is needed. In addition, autoantibodies to NETs could interfere with serum NETs measurements using an ELISA. Any measurement technique proposed as a biomarker should also be relatively easy to perform in a clinical lab setting. Prospective longitudinal studies with larger sample sizes are needed to measure the test characteristics of any measure of NETosis to predict and correlate with disease activity and renal involvement. As illustrated by current disease activity markers, such as complement and dsDNA levels, the clinical heterogeneity of SLE may limit which biomarkers are useful in assessing for disease activity and renal involvement. For example, not all SLE patients have anti‐dsDNA antibodies. Furthermore, SLE patients with anti‐dsDNA antibodies may not have increased levels associated with increased disease activity. This limitation can also be seen with complement levels with the lack of association between low complements and increased disease activity. Potentially, not all SLE patients will have increased NETosis or NETosis that correlates with increased disease activity. Future studies would need to clearly define in which SLE patients it may be beneficial to use NET measurement to help clinicians with predicting and assessing disease activity and renal involvement.
Research Questions:
What is the best method (ability of sera to induce NETs, NET degradation, NET products, such as cfDNA) to measure increased NET formation in SLE patients? How will we define normal NET formation versus increased NET formation? Which measurement of NETosis could be most easily performed in clinical labs?
In addition to SLE, the understanding of NETs in ANCA‐associated vasculitis has led to the identification of potential biomarkers for disease activity and treatment response. In a recent study, increased expression of a granulocyte gene‐expression signature was associated with increased disease activity and decreased treatment response in patients with ANCA‐associated vasculitis [39]. The source of this signature was a subset of neutrophils called LDGs, which are proinflammatory [6, 36, 40] and able to undergo spontaneous NETosis without stimulation [40]. This study was the first to show the presence of LDGs in patients with ANCA‐associated vasculitis, as LDGs had previously been shown in patients with SLE [40]. Furthermore, the LDGs in the patients with ANCA‐associated vasculitis, produced NETs containing MPO and PR3, major autoantigens in ANCA‐associated vasculitis. Future studies are needed to assess the potential pathogenic role of LDGs and NETs in ANCA‐associated vasculitis.
Research Questions:
What is the role of LDGs in the pathogenesis of ANCA‐associated vasculitis? Could LDGs be potential biomarkers for ANCA‐associated vasculitis and SLE?
In SLE, measures of molecular signatures or mRNA expression of type I IFN are used as pharmacodynamic markers in developing new SLE treatments. Type I IFNs are a cytokine family of IFN‐α, IFN‐β, IFN‐Τ, IFN‐κ, and IFN‐ω [41]. Whereas individual type I IFNs, such as IFN‐α, are difficult to measure, as a result of the small quantities in peripheral blood, the type I IFNs as a family are easier to measure in terms of mRNA expression. In SLE, there is an overexpression of type I IFN‐inducible mRNAs in blood and in involved tissues in ∼60% of patients [41]. Increased expression of mRNAs induced by type I IFN is associated with increased disease activity [41]. The measurement of the type I IFN signature is a potential pharmacodynamic marker to evaluate the inhibition of anti‐IFN therapies, as well as select SLE patients with an increased type I IFN signature that may benefit from these therapies.
Research Question:
Does NETosis correlate with an increased type I IFN signature and increased IFN‐α levels in patients with SLE?
Similar to the IFN signature, NETs are increased in the blood and involved tissues of SLE patients. Increased NET formation and decreased NET degradation are associated with increased disease activity. NETs are able to induce IFN‐α production from pDCs. Therefore, the measurement of NETs may allow for an indirect measure of IFN‐α. The measurement of the IFN signature captures multiple type I IFNs, not just IFN‐α. The measurement of NETs may be a more specific way to estimate IFN‐α levels and identify which SLE patients at baseline in clinical trials have elevated IFN‐α levels and may be more likely to respond to anti‐IFN‐α therapies.
ASSOCIATION OF NETosis WITH EC DYSFUNCTION AND ATHEROSCLEROSIS IN AUTOIMMUNE DISEASES
NETs may play a role in the pathogenesis of premature CVD in SLE. SLE patients have substantially increased morbidity and mortality from CVD, with the incidence of myocardial infarction 5 times higher in patients with SLE compared with the general population [42]. CVD occurs at earlier ages in SLE patients than in the general population, with traditional and disease‐related risk factors not fully accounting for this increased risk [43, 44]. One proposed mechanism is an altered innate immune response that contributes to EC dysfunction and damage leading to plaque formation [37].
SLE patients have EC dysfunction with evidence for accelerated EC apoptosis [45], which results in loss of NO release, leading to impaired vasodilation, which may potentiate atherosclerosis [45]. Increased EC apoptosis in SLE patients correlated with reduced endothelial function, as measured by brachial artery flow‐mediated dilation [45]. In addition, increased EC apoptosis in SLE patients correlated with elevated tissue factor, which is associated with a prothrombogenic phenotype [45]. Neutrophils and NETs may play a central role in increased EC apoptosis in SLE. There is evidence that neutrophils and NETs enhance EC killing and death in vitro [6]. In summary, NETs accelerate EC death that may contribute to both atherosclerotic and thrombotic events in SLE patients.
Other mechanisms have also been implicated in the role of NETs in EC damage and the pathogenesis of accelerated atherosclerosis in SLE. MMP‐9 is up‐regulated and externalized on the surface of the NETs and able to damage ECs [36]. Furthermore, the MMPs from the NETs cause secretion of MMP‐2 from the ECs, resulting in further EC dysfunction. NETs may also mediate HDL oxidation in SLE patients [37]. Compared with controls, SLE patients have increased levels of oxidized HDL, which is a proinflammatory lipoprotein with impaired cholesterol efflux capacity. NET‐derived MPO, NOX, and NOS promote HDL oxidation and decrease cholesterol efflux capacity in vitro [37]. In summary, NETs lead to the generation of a dysfunctional HDL that impairs cholesterol efflux capacity, further promoting proinflammatory responses in the vascular system that may contribute to accelerated atherosclerosis in SLE.
Recently, an inflammatory role of neutrophils and macrophages in the pathogenesis of atherosclerosis has been described. Specifically, NETs were found in atherosclerotic plaques in apolipoprotein E‐deficient mice [46]. Cholesterol crystals induced neutrophils to undergo NETosis, which primed macrophages to release pro‐IL‐1β [46]. In addition, the cholesterol crystals, bound to the cell surface of the macrophages, activated the inflammasome, which further propagated inflammation [46]. In vitro human studies showed that cholesterol crystals triggered NETosis [46]. Whether neutrophils, NETs, and macrophages interact in human plaques to generate an inflammatory state needs further human in vivo studies.
Research Questions:
Do NETs propagate inflammation in atherosclerotic lesions in humans? Is the NET‐mediated inflammatory process in atherosclerosis more pronounced in patients with autoimmune diseases, such as SLE, compared with the general population?
NETs AS POTENTIAL THERAPEUTIC TARGETS
NETs represent a novel target for therapy in SLE and other autoimmune diseases. The literature currently focuses on targeting NETs in developing SLE therapies, but potential therapies may be applicable to other autoimmune diseases. There are both conventional medications already used in SLE, as well as novel agents that target NETs ( Table 3 and Fig. 2 ). As a defense against invading pathogens, NETs release ROS, with ROS important in NET formation. Treatment of neutrophils with NAC blocks ROS and NET formation in vitro [47]. NAC also blocks the mTOR in SLE T cells in vitro and in vivo [49, 65]. In 2 small studies, NAC improved SLE disease activity [48, 49]. Future randomized, controlled clinical trials are needed to assess fully the efficacy of NAC in reducing disease activity in SLE.
Table 3.
Therapies that target NETs in SLE
| Drug | Mechanism of action | Study | Results | Outstanding questions |
|---|---|---|---|---|
| NAC | Antioxidant reduces ROS and NET formation. | [47] | NAC blocks NET formation in vitro. | What dose and frequency of NAC should be used? |
| [48] | NAC reduces SLE disease activity and attention deficit hyperactivity disorder scores. | Will NAC be effective in reducing disease activity in SLE in controlled trials? | ||
| [49] | NAC reduces disease activity and blocks mTOR in T lymphocytes. | |||
| DNase I | Endonuclease to digest extracellular DNA and degrade NETs | [50] | In mice, rmDNase I prolonged survival and improved kidney histology. | What are the in vivo effects of DNase I in SLE patients? |
| [51] | In mice, DNase I had no effect on survival and kidney disease. | |||
| [52] | In mice, DNase I reduced reactive B cells, production of type I IFN, and immune complex deposition in the kidneys. | |||
| [53] | In a phase Ib study, there were no significant differences in adverse reactions in the treatment versus placebo groups. Serum markers of disease activity were unchanged in the treatment group. | |||
| PAD‐4 inhibitor | Deiminates histones (NET contents) | [54] | In mice, PAD‐4 decreased autoimmune responses and protected against vascular damage. | What are the in vitro and in vivo effects of PAD‐4 inhibitors in SLE patients? |
| [55] | In mice, PAD‐4 decreased proteinuria, improved skin involvement, and improved EC dysfunction. | |||
| Eculizumab | rhmAb to C5 may block NET formation. | [56] | In PNH patients, eculizumab inhibited markers of NET formation in vivo. | Does eculizumab block NET formation in SLE patients? |
| [57] | In a phase I study, there were no significant differences in adverse reactions in the treatment versus placebo groups, with complement inhibition persisting for 10 d; there were no significant changes in SLEDAI scores. | In larger SLE studies, will eculizumab be shown to be safe and effective in reducing disease activity? | ||
| Vitamin D (calcitriol) | Unknown, reduces NET formation | [58] | In vitro, vitamin D reduced NET formation and decreased rates of EC apoptosis in SLE patients with low vitamin D. | Will vitamin D affect NET formation and improve EC dysfunction in larger studies? |
| [59] | In vivo, vitamin D improved endothelial function in SLE patients with low vitamin D. | Will vitamin D reduce CVD in SLE patients? | ||
| Antimalarials | Blocks processing of NETs through TLR9 in pDCs | [37] | In vitro, chloroquine inhibited NET formation in SLE neutrophils. | Will antimalarials inhibit NET formation in SLE in vivo? |
| Sifalimumab | Human mAb to IFN‐α | [60] | In a phase Ia placebo‐controlled study, there was no safety signal seen with dose‐dependent inhibition of the type I IFN signature with sifalimumab; there were trends toward improvement in SLEDAI with sifalimumab. | Is sifalimumab generally safe in SLE patients? |
| [61] | In a phase Ib, placebo‐controlled study, adverse events were similar in both groups; however, more discontinuations and deaths were seen with sifalimumab; there was dose‐dependent neutralization of the type I IFN signature with no difference in SLEDAI scores. | Is sifalimumab only effective for SLE patients with increased type I IFN signatures, or can it be used in all SLE patients? | ||
| [62] | In a phase IIb, placebo‐controlled study, adverse events were similar in both groups, except with higher rates of herpes zoster seen with sifalimumab. There was a significantly higher percentage of patients with an SRI response at d 365 with sifalimumab. | Will sifalimumab reduce disease activity in larger phase II and III studies? | ||
| Rontalizumab | Human mAb to IFN‐α | [63] | In a phase I, placebo‐controlled study, adverse events were similar, except a higher infection rate with rontalizumab; rontalizumab reduced levels of the type I IFN signature but did not reduce levels of IFN‐inducible proteins or anti‐dsDNA levels. | Will infection risk, such as from viral reactivation, limit rontalizumab and sifalimumab's use? |
| [64] | In a phase IIb, placebo‐controlled study, response rates to BILAG and SRI were similar; prespecified subgroup analyses showed a trend toward improvement in flare rates and a decrease in corticosteroid use. | Are other subsets of type I IFN, which are not blocked by sifalimumab or rontalizumab, important drivers of clinical disease in SLE? |
BILAG, British Isles Lupus Assessment Group.
Figure 2.

Overview of potential therapies that target NETs. Neutrophils release NETs, which contain histones, complexes of DNA and LL‐37, an antimicrobial peptide, elastase, and MPO. NAC is an antioxidant that can reduce ROS, which promotes NETosis. DNase I digests extracellular DNA and degrades NETs. PAD‐4 inhibitor deiminates histones, an important component of NETs. Eculizumab, a mAb to C5, can block complement activation that stimulates NETosis. The target by which vitamin D reduces NET formation is unknown. Antimalarials block processing of NETs through TLR9 in pDCs. Sifalimumab and rontalizumab are mAb to IFN‐α, which is a product of NETosis that can stimulate further NETosis. Electron micrograph of polymorphonuclear neutrophil (PMN) undergoing NETosis courtesy of Drs. Elizabeth Johnson and Leslie Crofford.
Another possible target is treatment with DNase I, which is an endonuclease that is secreted into the extracellular space and found in the blood and gastrointestinal tract with a presumed function of digesting extracellular DNA [66]. With their DNA contents, DNase I is an important degrader of NETs. In SLE patients, sera levels of DNase I are decreased [13, 15, 16]. With these decreased levels, it was hypothesized that administration of DNase I would reduce the extracellular DNA found in SLE that triggers multiple autoimmune pathways leading to inflammation and tissue damage. DNase I was administered to multiple models of lupus‐prone mice with mixed results [66]. One group that uses rmDNase I showed improvement in histologic changes in kidneys and survival in the NZB/NZW F1 mouse model [50]. However, with the use of the same model, another group showed that DNase I had no effect in improving disease in the mice [51]. With the use of an estrogen‐induced mouse model of lupus, DNase I treatment did improve disease in the mice [52]. rhDNase I was given to 17 SLE nephritis patients in a phase Ib, randomized, double‐blinded, placebo‐controlled trial to determine the drug's safety and pharmacokinetics [53]. rhDNase I was well tolerated without significant adverse events. However, serum markers of disease activity were unchanged. Future studies with more SLE patients are needed to assess the safety and efficacy of rhDNase I.
Research Question:
What are the in vivo effects of DNase I in SLE patients?
Another potential drug target also involves inhibiting a major component of NETs. PAD‐4 is a hydrolase that deiminates histones, which are prominent in NETs. Without PAD‐4, NETs cannot form, as illustrated by PAD‐4‐deficient mice [32, 67]. In one lupus‐prone mouse model—the New Zealand Mixed 2328—which is prone to proliferative GN and a prominent type I IFN signature, PAD inhibitors decreased autoimmune responses and protected against NET‐mediated vascular damage [54]. In lupus‐prone MRL/Mp‐lpr/lpr mice with proliferative GN and a much less prominent type I IFN signature, PAD inhibitors blocked NET formation, decreased immune complex deposition in the kidneys and proteinuria, and improved skin involvement [55]. Treatment with the PAD inhibitors also improved EC dysfunction [55]. PAD inhibitors represent an anti‐NET target that may be an effective, novel therapy in SLE to treat not only disease manifestations but also to improve endothelial dysfunction. To date, trials have not been conducted in humans.
Research Question:
What are the in vitro and in vivo effects of PAD‐4 inhibitors in SLE patients?
Another potential target of NETs is the complement system. Complement deposits on NETs and is important in regulating NETs by preventing their degradation. Eculizumab is a rhmAb that is a terminal complement inhibitor and approved for the treatment of PNH, a rare, acquired hemolytic anemia, where the complement system attacks RBCs, and atypical hemolytic uremic syndrome—a hemolytic anemia that can result in acute kidney failure and a low platelet count. Specifically, eculizumab binds to the terminal C5 and inhibits the cleavage of C5 to C5a and C5b by the C5 convertasae, which prevents the formation of the terminal complement complex. Eculizumab inhibited NET formation in PNH patients [56]. Eculizumab has been studied in 24 SLE patients in a phase I, randomized, placebo‐controlled, double‐blind trial to evaluate its safety, pharmacodynamic, and pharmacokinetic properties [57]. There were no significant differences in adverse events in the treatment vs. placebo groups. Complement inhibition of >80% was observed in the treatment group and persisted for 10 d but then returned to baseline at 14 d. There were no significant changes in disease activity scores, as measured by SLEDAI, in exploratory analyses. However, this initial, small controlled study shows promising safety data to justify future, larger phase I and II studies.
Research Question:
Does eculizumab block NET formation in vitro and in vivo in SLE patients?
In addition to the above novel agents, known agents used in SLE also represent NET‐targeted therapy. In a small study of 5 SLE patients, vitamin D (calcitriol) reduced NET formation in neutrophils from SLE patients with low vitamin D [58]. In addition, vitamin D decreased rates of apoptosis in ECs [58]. This study suggests that vitamin D reduces the cytotoxic effects of NETs on ECs. A small in vivo study of 22 stable SLE patients with vitamin D deficiency (<20 ng/ml) showed that vitamin D improved endothelial function [59]. These studies would need to be expanded in a larger SLE population with vitamin D tested in controlled trials to determine if improved endothelial function translates to improved CVD outcomes.
Research Question:
Does vitamin D block NET formation and improve EC dysfunction in vivo in SLE patients?
Antimalarials, such as HCQ, are the cornerstone of drug therapy in SLE. Antimalarials have the potential to block the processing of NETs through TLR9 in pDCs. Chloroquine, an antimalarial, significantly inhibited NET formation in control and SLE neutrophils in vitro [37]. However, this potential mechanism has not been investigated in vivo. Commonly used medications in SLE, such as azathioprine, mycophenolate mofetil, and cyclophosphamide, have effects on neutrophils. However, currently, there are no studies investigating the effects of these medications on NET formation. Furthermore, it is not clear if corticosteroids affect NETs. Both in vitro and in vivo studies show that corticosteroids do not affect the activation of pDCs and thus, do not reduce IFN‐α levels [68]. As NETs promote autoimmunity through IFN‐α production, it is not surprising that 1 study suggests that corticosteroids do not block NET formation in vitro [34]. These findings have not been replicated or studied in vivo in SLE patients.
Research Question:
Do antimalarials and other commonly used medications in SLE, such as azathioprine, mycophenolate mofetil, and cyclophosphamide, block NET formation in vivo in SLE patients?
With NETs being potent inducers of IFN‐α, drugs that target IFN, specifically IFN‐α, have been studied. In SLE, there are multiple studies that establish the presence of an IFN signature, an overexpression of type I IFN‐induced genes [69]. These increased gene‐expression levels correlate with increased SLE disease activity [69]. As it is difficult to measure the low quantities of IFN‐α, the gene‐expression levels are typically measured. pDCs are the major producers of IFN‐α and normally do so in response to invading viruses. NETs with their nuclear contents of dsDNA similarly induce IFN‐α from pDCs. IFN‐α can then activate the innate and adaptive immune systems, specifically inducing a Th type 1 pathway, inhibiting T cell apoptosis, and activating B cells and antibody production [12].
Several drugs have been developed that block IFN‐α, including sifalimumab (MEDI‐545) and rontalizumab (RG7415), as well as other similar neutralizing antibodies to IFN‐α and IFN‐γ [70]. Sifalimumab and rontalizumab are both fully human mAb that bind to IFN‐α and prevent IFN‐α from signaling to its receptor. However, sifalimumab and rontalizumab target slightly different IFN‐α subtypes. In a phase Ia, randomized, double‐blind, placebo‐controlled study, the safety profile, tolerability, and pharmacokinetics of sifalimumab were examined in 69 patients with SLE [60]. The pharmacodynamics of the type I IFN signature were assessed by measuring the expression level of type I IFN‐inducible mRNAs in whole blood, along with the expression of 2 IFN‐α/β‐inducible protein products in affected skin biopsies. Adverse event rates were similar between the sifalimumab and placebo groups. The SLE patients, with an elevated type I IFN signature at baseline, had a dose‐dependent inhibition of the type I IFN signature with sifalimumab. In exploratory analyses, there were consistent trends of improvement in disease activity in the sifalimumab versus placebo groups. However, statistical significance was not attained. In summary, this study established the safety of sifalimumab in SLE. Sifalimumab neutralized the overexpression of the type I IFN signature and may reduce disease activity.
Sifalimumab was examined further in a phase Ib, randomized, controlled study [61]. In this study, 161 adult SLE patients with moderate‐to‐severe disease were randomized to receive IV sifalimumab and 40 SLE patients to placebo. Blood samples at screening and at multiple time points during treatment and follow‐up were evaluated for the expression of 21 type I‐inducible genes. The median fold change in expression of the type I‐inducible genes was used as a pharmacodynamic biomarker [71]. There were similar frequencies of adverse events in the sifalimumab and placebo groups. However, 11 SLE patients in the sifalimumab group with adverse events had to permanently discontinue the drug. There were 4 deaths in the sifalimumab group versus 1 death in the placebo group, with infection contributing to 2 of the deaths in the sifalimumab group. Similar to the phase Ia study, sifalimumab showed dose‐dependent neutralization of the type IFN gene signature in the SLE patients with overexpression of the type I IFN signature at baseline. In post hoc analyses, there was a trend toward improvement in the disease activity measures in patients with increased baseline type I IFN signature in the sifalimumab group. In summary, this phase Ib study showed an adequate safety profile, although the increased deaths in the sifalimumab vs. placebo group warrant investigation and the need for longer follow‐up. In a phase IIb, randomized, double‐blind, placebo‐controlled study, 431 moderate‐to‐severe SLE patients were randomized to receive sifalimumab or placebo for 1 y [62, 70]. The primary endpoint was a response at 1 y on the SRI, a composite disease activity measure [72]. There were similar adverse events in the sifalimumab vs. placebo groups; however, in the sifalimumab group, there were more frequent Herpes zoster infections. A significantly higher percentage of patients met the primary endpoint in all dose groups of sifalimumab vs. placebo. Whereas sifalimumab inhibits most but not all type I IFN‐α subtypes, it does not inhibit IFN‐β or IFN‐δ. Whether these other IFN subtypes are clinically important and need to be targeted remains unanswered.
In addition to sifalimumab, rontalizumab has been developed as an IFN‐α‐targeted therapy in SLE. Rontalizumab is a humanized IgG1 mAb that binds to and neutralizes all known subtypes of human IFN‐α. A phase I, placebo‐controlled, double‐blind study was conducted in 60 SLE patients to assess the pharmacokinetics and pharmacodynamic activity [63]. The IFN signature was measured via microarray analyses and quantitative RT‐PCR. IFN‐inducible proteins and autoantibodies were also measured. There were similar rates of adverse events in the treatment and placebo groups. However, infections did occur at a higher rate in the rontalizumab versus placebo groups. Rontalizumab reduced the levels of the IFN signature in a dose‐dependent fashion, with higher doses reducing the gene expression by >50%. However, there was no decline in the levels of the IFN‐inducible proteins or in levels of anti‐dsDNA or anti‐ENA autoantibodies. Rontalizumab was fairly well tolerated and generally safe, although with a concern for increased infection risk. Whereas rontalizumab reduced the levels of the IFN signature in SLE patients with an elevated signature at baseline, these levels were not reduced to levels seen in healthy controls or even to baseline levels detected in SLE patients with low‐level signatures. Furthermore, rontalizumab did not reduce IFN‐inducible protein, anti‐dsDNA, or anti‐ENA autoantibody levels. In a phase II, placebo‐controlled trial, moderate‐to‐severe SLE patients were randomized to receive rontalizumab or placebo [64, 70]. Response rates were similar in the rontalizumab and placebo groups. However, in prespecified groups, rontalizumab was associated with improvement in flare rates and lower corticosteroid use at wk 24. In summary, rontalizumab seems to reduce the IFN signature in SLE, but further studies are needed to establish its safety in terms of infection risks and efficacy.
With the development of multiple therapies targeting IFN‐α, questions remain about the safety and efficacy of this drug class. Whether IFN signature levels need to be suppressed to a certain level to have clinical effect has not been established. Specifically, it is not known if other types of type I IFN‐s are important drivers in clinical disease. As the majority of anti‐IFN therapies target various subtypes of IFN‐α, it will be important to delineate the contribution of other type I IFNs to disease activity in SLE. In addition, the published safety and efficacy data of the sifalimumab and rontalizumab phase II trials will need to be examined to decide on moving forward with phase III trials. NETs may be an important part of this developing story of anti‐IFN‐α therapies in SLE as a potential tool to estimate IFN‐α levels and select patients that may be more likely to respond to these therapies. In addition, NETs could be measured in current trials assessing the impact of NAC on disease activity [73] and a phase I trial of an anti‐C5aR antibody in SLE [74].
Research Question:
Are there other subsets of type I IFN, in addition to IFN‐α, that are important drivers of SLE disease activity?
CONCLUSIONS
NETs play an important role in the pathogenesis of multiple autoimmune diseases. NETs may be the source of key autoantigens, as well as potent inducers of IFN‐α. Increased NET formation, as assessed by multiple methods, has been associated with increased disease activity in multiple autoimmune diseases. Increased NET formation may be useful as a disease activity biomarker in ANCA‐associated vasculitis and SLE. However, standard and validated methods need to be established in measuring NETs along with prospective, longitudinal studies describing the test characteristics of NET measurements compared with established disease activity measures. By better understanding the role of NETs in EC dysfunction and atherosclerosis, therapies could potentially be designed to target NETs and potentially reduce the burden of atherosclerosis in SLE and other autoimmune diseases. The literature currently focuses on targeting NETs in developing SLE therapies, but potential therapies may be applicable to other autoimmune diseases. Both known agents, such as antimalarials, and novel agents, such as NAC, DNase I, PAD inhibitors, and anti‐IFN‐α therapies, may target NETs in SLE. Anti‐IFN‐α therapies have shown some promise in phase I and II trials. However, questions remain regarding their clinical efficacy in SLE and whether these therapies may only be helpful in a subset of SLE patients with increased type I IFN signatures ( Table 4 ). As these new therapies emerge in future clinical trials, measurement of NETs may be helpful in assessing levels of IFN‐α. In summary, NETs offer insight into the pathogenesis of multiple autoimmune diseases, as well as provide promise in developing disease biomarkers and novel therapeutic agents.
Table 4.
Summary of questions for future clinical‐translational research
| • Does increased NET formation lead to thrombus formation in patients with ANCA‐associated vasculitis? |
| • What are the mechanisms by which NETs promote thrombus formation in patients with APS? |
| • Do levels of NETs increase during clinical events associated with APS, such as acute thrombotic episodes, or pregnancy complications, such as miscarriages or preeclampsia? |
| • How are the protein contents of NETs influenced by exposure to different autoantibodies and inflammatory cytokines? |
| • Do all patients with SLE have increased NETosis, or does only a subset of patients with specific autoantibodies have increased NETosis? |
| • In which subsets of patients with SLE does increased NETosis correlate with increased disease activity? |
| • What is the best method (ability of sera to induce NETs, NET degradation, NET products, such as cfDNA) to measure increased NET formation in SLE patients? |
| • How will we define normal NET formation versus increased NET formation? |
| • Which measurement of NETosis could be most easily performed in clinical labs? |
| • What is the role of LDGs in the pathogenesis of ANCA‐associated vasculitis? |
| • Could LDGs be potential biomarkers for ANCA‐associated vasculitis and SLE? |
| • Does NETosis correlate with an increased type I IFN signature and increased IFN‐α levels in patients with SLE? |
| • Do NETs propagate inflammation in atherosclerotic lesions in humans? |
| • Is the NET‐mediated inflammatory process in atherosclerosis more pronounced in patients with autoimmune diseases, such as SLE, compared with the general population? |
| • What are the in vivo effects of DNase I in SLE patients? |
| • What are the in vitro and in vivo effects of PAD‐4 inhibitors in SLE patients? |
| • Does eculizumab block NET formation in vitro and in vivo in SLE patients? |
| • Does vitamin D block NET formation and improve EC dysfunction in vivo in SLE patients? |
| • Do antimalarials and other commonly used medications in SLE, such as azathioprine, mycophenolate mofetil, and cyclophosphamide, block NET formation in vivo in SLE patients? |
| • Are there other subsets of type I IFN, in addition to IFN‐α, that are important drivers of SLE disease activity? |
DISCLOSURES
The authors declare no conflicts of interest.
Footnotes
SEE CORRESPONDING ARTICLE ON PAGE 253
REFERENCES
- 1. Brinkmann, V. , Reichard, U. , Goosmann, C. , Fauler, B. , Uhlemann, Y. , Weiss, D.S. , Weinrauch, Y. , Zychlinsky, A. (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. [DOI] [PubMed] [Google Scholar]
- 2. Takei, H. , Araki, A. , Watanabe, H. , Ichinose, A. , Sendo, F. (1996) Rapid killing of human neutrophils by the potent activator phorbol 12‐myristate 13‐acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 59, 229–240. [DOI] [PubMed] [Google Scholar]
- 3. Grayson, P.C. , Kaplan, M.J. (2016) At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J. Leukoc. Biol. 99, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lande, R. , Gregorio, J. , Facchinetti, V. , Chatterjee, B. , Wang, Y.H. , Homey, B. , Cao, W. , Wang, Y.H. , Su, B. , Nestle, F.O. , Zal, T. , Mellman, I. , Schröder, J.M. , Liu, Y.J. , Gilliet, M. (2007) Plasmacytoid dendritic cells sense self‐DNA coupled with antimicrobial peptide. Nature 449, 564–569. [DOI] [PubMed] [Google Scholar]
- 5. Kessenbrock, K. , Krumbholz, M. , Schönermarck, U. , Back, W. , Gross, W.L. , Werb, Z. , Gröne, H.J. , Brinkmann, V. , Jenne, D.E. (2009) Netting neutrophils in autoimmune small‐vessel vasculitis. Nat. Med. 15, 623–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Villanueva, E. , Yalavarthi, S. , Berthier, C.C. , Hodgin, J.B. , Khandpur, R. , Lin, A.M. , Rubin, C.J. , Zhao, W. , Olsen, S.H. , Klinker, M. , Shealy, D. , Denny, M.F. , Plumas, J. , Chaperot, L. , Kretzler, M. , Bruce, A.T. , Kaplan, M.J. (2011) Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 187, 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khandpur, R. , Carmona‐Rivera, C. , Vivekanandan‐Giri, A. , Gizinski, A. , Yalavarthi, S. , Knight, J.S. , Friday, S. , Li, S. , Patel, R.M. , Subramanian, V. , Thompson, P. , Chen, P. , Fox, D.A. , Pennathur, S. , Kaplan, M.J. (2013) NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 5, 178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leffler, J. , Stojanovich, L. , Shoenfeld, Y. , Bogdanovic, G. , Hesselstrand, R. , Blom, A.M. (2014) Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin. Exp. Rheumatol. 32, 66–70. [PubMed] [Google Scholar]
- 9. Yalavarthi, S. , Gould, T.J. , Rao, A.N. , Mazza, L.F. , Morris, A.E. , Núñez‐Álvarez, C. , Hernández‐Ramírez, D. , Bockenstedt, P.L. , Liaw, P.C. , Cabral, A.R. , Knight, J.S. (2015) Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 67, 2990–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Branzk, N. , Papayannopoulos, V. (2013) Molecular mechanisms regulating NETosis in infection and disease. Semin. Immunopathol. 35, 513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ermert, D. , Urban, C.F. , Laube, B. , Goosmann, C. , Zychlinsky, A. , Brinkmann, V. (2009) Mouse neutrophil extracellular traps in microbial infections. J. Innate Immun. 1, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rönnblom L. (2010) Potential role of IFNα in adult lupus. Arthritis Res. Ther. 12 (Suppl 1), S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hakkim, A. , Fürnrohr, B.G. , Amann, K. , Laube, B. , Abed, U.A. , Brinkmann, V. , Herrmann, M. , Voll, R.E. , Zychlinsky, A. (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 107, 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakazawa, D. , Shida, H. , Tomaru, U. , Yoshida, M. , Nishio, S. , Atsumi, T. , Ishizu, A. (2014) Enhanced formation and disordered regulation of NETs in myeloperoxidase‐ANCA‐associated microscopic polyangiitis. J. Am. Soc. Nephrol. 25, 990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leffler, J. , Martin, M. , Gullstrand, B. , Tydén, H. , Lood, C. , Truedsson, L. , Bengtsson, A.A. , Blom, A.M. (2012) Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 188, 3522–3531. [DOI] [PubMed] [Google Scholar]
- 16. Leffler, J. , Gullstrand, B. , Jönsen, A. , Nilsson, J.A. , Martin, M. , Blom, A.M. , Bengtsson, A.A. (2013) Degradation of neutrophil extracellular traps co‐varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res. Ther. 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakazawa, D. , Tomaru, U. , Yamamoto, C. , Jodo, S. , Ishizu, A. (2012) Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front. Immunol. 3, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sur Chowdhury, C. , Giaglis, S. , Walker, U.A. , Buser, A. , Hahn, S. , Hasler, P. (2014) Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 16, R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao, H. , Heeringa, P. , Hu, P. , Liu, Z. , Zhao, M. , Aratani, Y. , Maeda, N. , Falk, R.J. , Jennette, J.C. (2002) Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J. Clin. Invest. 110, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiao, H. , Heeringa, P. , Liu, Z. , Huugen, D. , Hu, P. , Maeda, N. , Falk, R.J. , Jennette, J.C. (2005) The role of neutrophils in the induction of glomerulonephritis by anti‐myeloperoxidase antibodies. Am. J. Pathol. 167, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelley, J.M. , Monach, P.A. , Ji, C. , Zhou, Y. , Wu, J. , Tanaka, S. , Mahr, A.D. , Johnson, S. , McAlear, C. , Cuthbertson, D. , Carette, S. , Davis, J.C., Jr. , Dellaripa, P.F. , Hoffman, G.S. , Khalidi, N. , Langford, C.A. , Seo, P. , St Clair, E.W. , Specks, U. , Stone, J.H. , Spiera, R.F. , Ytterberg, S.R. , Merkel, P.A. , Edberg, J.C. , Kimberly, R.P. (2011) IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc. Natl. Acad. Sci. USA 108, 20736–20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu, J. , Zhang, X. , Pelayo, R. , Monestier, M. , Ammollo, C.T. , Semeraro, F. , Taylor, F.B. , Esmon, N.L. , Lupu, F. , Esmon, C.T. (2009) Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuchs, T.A. , Brill, A. , Duerschmied, D. , Schatzberg, D. , Monestier, M. , Myers, D.D., Jr. , Wrobleski, S.K. , Wakefield, T.W. , Hartwig, J.H. , Wagner, D.D. (2010) Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 107, 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Von Scheven, E. , Lu, T.T. , Emery, H.M. , Elder, M.E. , Wara, D.W. (2003) Thrombosis and pediatric Wegener's granulomatosis: acquired and genetic risk factors for hypercoagulability. Arthritis Rheum. 49, 862–865. [DOI] [PubMed] [Google Scholar]
- 25. Wegener's Granulomatosis Etanercept Trial (WGET) Research Group. (2005) Etanercept plus standard therapy for Wegener's granulomatosis. N. Engl. J. Med. 352, 351–361. [DOI] [PubMed] [Google Scholar]
- 26. Ma, K. , Simantov, R. , Zhang, J.C. , Silverstein, R. , Hajjar, K.A. , McCrae, K.R. (2000) High affinity binding of beta 2‐glycoprotein I to human endothelial cells is mediated by annexin II. J. Biol. Chem. 275, 15541–15548. [DOI] [PubMed] [Google Scholar]
- 27. Allen, K.L. , Fonseca, F.V. , Betapudi, V. , Willard, B. , Zhang, J. , McCrae, K.R. (2012) A novel pathway for human endothelial cell activation by antiphospholipid/anti‐β2 glycoprotein I antibodies. Blood 119, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sorice, M. , Circella, A. , Griggi, T. , Garofalo, T. , Nicodemo, G. , Pittoni, V. , Pontieri, G.M. , Lenti, L. , Valesini, G. (1996) Anticardiolipin and anti‐beta 2‐GPI are two distinct populations of autoantibodies. Thromb. Haemost. 75, 303–308. [PubMed] [Google Scholar]
- 29. Deane, K.D. , Norris, J.M. , Holers, V.M. (2010) Preclinical rheumatoid arthritis: identification, evaluation, and future directions for investigation. Rheum. Dis. Clin. North Am. 36, 213–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kidd, B.A. , Ho, P.P. , Sharpe, O. , Zhao, X. , Tomooka, B.H. , Kanter, J.L. , Steinman, L. , Robinson, W.H. (2008) Epitope spreading to citrullinated antigens in mouse models of autoimmune arthritis and demyelination. Arthritis Res. Ther. 10, R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lundberg, K. , Nijenhuis, S. , Vossenaar, E.R. , Palmblad, K. , van Venrooij, W.J. , Klareskog, L. , Zendman, A.J. , Harris, H.E. (2005) Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res. Ther. 7, R458–R467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li, P. , Li, M. , Lindberg, M.R. , Kennett, M.J. , Xiong, N. , Wang, Y. (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin, A.M. , Rubin, C.J. , Khandpur, R. , Wang, J.Y. , Riblett, M. , Yalavarthi, S. , Villanueva, E.C. , Shah, P. , Kaplan, M.J. , Bruce, A.T. (2011) Mast cells and neutrophils release IL‐17 through extracellular trap formation in psoriasis. J. Immunol. 187, 490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lande, R. , Ganguly, D. , Facchinetti, V. , Frasca, L. , Conrad, C. , Gregorio, J. , Meller, S. , Chamilos, G. , Sebasigari, R. , Riccieri, V. , Bassett, R. , Amuro, H. , Fukuhara, S. , Ito, T. , Liu, Y.J. , Gilliet, M. (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self‐DNA‐peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang, S. , Lu, X. , Shu, X. , Tian, X. , Yang, H. , Yang, W. , Zhang, Y. , Wang, G. (2014) Elevated plasma cfDNA may be associated with active lupus nephritis and partially attributed to abnormal regulation of neutrophil extracellular traps (NETs) in patients with systemic lupus erythematosus. Intern. Med. 53, 2763–2771. [DOI] [PubMed] [Google Scholar]
- 36. Carmona‐Rivera, C. , Zhao, W. , Yalavarthi, S. , Kaplan, M.J. (2015) Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase‐2. Ann. Rheum. Dis. 74, 1417–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith, C.K. , Vivekanandan‐Giri, A. , Tang, C. , Knight, J.S. , Mathew, A. , Padilla, R.L. , Gillespie, B.W. , Carmona‐Rivera, C. , Liu, X. , Subramanian, V. , Hasni, S. , Thompson, P.R. , Heinecke, J.W. , Saran, R. , Pennathur, S. , Kaplan, M.J. (2014) Neutrophil extracellular trap‐derived enzymes oxidize high‐density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 66, 2532–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bombardier, C. , Gladman, D.D. , Urowitz, M.B. , Caron, D. , Chang, C.H. ; The Committee on Prognosis Studies in SLE. (1992) Derivation of the SLEDAI. A disease activity index for lupus patients. Arthritis Rheum. 35, 630–640. [DOI] [PubMed] [Google Scholar]
- 39. Grayson, P.C. , Carmona‐Rivera, C. , Xu, L. , Lim, N. , Gao, Z. , Asare, A.L. , Specks, U. , Stone, J.H. , Seo, P. , Spiera, R.F. , Langford, C.A. , Hoffman, G.S. , Kallenberg, C.G. , St Clair, E.W. , Tchao, N.K. , Ytterberg, S.R. , Phippard, D.J. , Merkel, P.A. , Kaplan, M.J. , Monach, P.A. ; Rituximab in ANCA‐Associated Vasculitis‐Immune Tolerance Network Research Group. (2015) Neutrophil‐related gene expression and low‐density granulocytes associated with disease activity and response to treatment in antineutrophil cytoplasmic antibody‐associated vasculitis. Arthritis Rheumatol. 67, 1922–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Denny, M.F. , Yalavarthi, S. , Zhao, W. , Thacker, S.G. , Anderson, M. , Sandy, A.R. , McCune, W.J. , Kaplan, M.J. (2010) A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 184, 3284–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yao, Y. , Higgs, B.W. , Richman, L. , White, B. , Jallal, B. (2010) Use of type I interferon‐inducible mRNAs as pharmacodynamic markers and potential diagnostic markers in trials with sifalimumab, an anti‐IFNα antibody, in systemic lupus erythematosus. Arthritis Res. Ther. 12 (Suppl 1), S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manzi, S. , Meilahn, E.N. , Rairie, J.E. , Conte, C.G. , Medsger, T.A., Jr. , Jansen‐McWilliams, L. , D'Agostino, R.B. , Kuller, L.H. (1997) Age‐specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am. J. Epidemiol. 145, 408–415. [DOI] [PubMed] [Google Scholar]
- 43. Asanuma, Y. , Oeser, A. , Shintani, A.K. , Turner, E. , Olsen, N. , Fazio, S. , Linton, M.F. , Raggi, P. , Stein, C.M. (2003) Premature coronary‐artery atherosclerosis in systemic lupus erythematosus. N. Engl. J. Med. 349, 2407–2415. [DOI] [PubMed] [Google Scholar]
- 44. Esdaile, J.M. , Abrahamowicz, M. , Grodzicky, T. , Li, Y. , Panaritis, C. , du Berger, R. , Cote, R. , Grover, S.A. , Fortin, P.R. , Clarke, A.E. , Senécal, J.L. (2001) Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 44, 2331–2337. [DOI] [PubMed] [Google Scholar]
- 45. Rajagopalan, S. , Somers, E.C. , Brook, R.D. , Kehrer, C. , Pfenninger, D. , Lewis, E. , Chakrabarti, A. , Richardson, B.C. , Shelden, E. , McCune, W.J. , Kaplan, M.J. (2004) Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood 103, 3677–3683. [DOI] [PubMed] [Google Scholar]
- 46. Warnatsch, A. , Ioannou, M. , Wang, Q. , Papayannopoulos, V. (2015) Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349, 316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patel, S. , Kumar, S. , Jyoti, A. , Srinag, B.S. , Keshari, R.S. , Saluja, R. , Verma, A. , Mitra, K. , Barthwal, M.K. , Krishnamurthy, H. , Bajpai, V.K. , Dikshit, M. (2010) Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide 22, 226–234. [DOI] [PubMed] [Google Scholar]
- 48. Garcia, R.J. , Francis, L. , Dawood, M. , Lai, Z.W. , Faraone, S.V. , Perl, A. (2013) Attention deficit and hyperactivity disorder scores are elevated and respond to N‐acetylcysteine treatment in patients with systemic lupus erythematosus. Arthritis Rheum. 65, 1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lai, Z.W. , Hanczko, R. , Bonilla, E. , Caza, T.N. , Clair, B. , Bartos, A. , Miklossy, G. , Jimah, J. , Doherty, E. , Tily, H. , Francis, L. , Garcia, R. , Dawood, M. , Yu, J. , Ramos, I. , Coman, I. , Faraone, S.V. , Phillips, P.E. , Perl, A. (2012) N‐Acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum. 64, 2937–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Macanovic, M. , Sinicropi, D. , Shak, S. , Baughman, S. , Thiru, S. , Lachmann, P.J. (1996) The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin. Exp. Immunol. 106, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Verthelyi, D. , Dybdal, N. , Elias, K.A. , Klinman, D.M. (1998) DNAse treatment does not improve the survival of lupus prone (NZB x NZW)F1 mice. Lupus 7, 223–230. [DOI] [PubMed] [Google Scholar]
- 52. Venkatesh, J. , Yoshifuji, H. , Kawabata, D. , Chinnasamy, P. , Stanevsky, A. , Grimaldi, C.M. , Cohen‐Solal, J. , Diamond, B. (2011) Antigen is required for maturation and activation of pathogenic anti‐DNA antibodies and systemic inflammation. J. Immunol. 186, 5304–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davis, J.C., Jr. , Manzi, S. , Yarboro, C. , Rairie, J. , Mcinnes, I. , Averthelyi, D. , Sinicropi, D. , Hale, V.G. , Balow, J. , Austin, H. , Boumpas, D.T. , Klippel, J.H. (1999) Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus 8, 68–76. [DOI] [PubMed] [Google Scholar]
- 54. Knight, J.S. , Zhao, W. , Luo, W. , Subramanian, V. , O'Dell, A.A. , Yalavarthi, S. , Hodgin, J.B. , Eitzman, D.T. , Thompson, P.R. , Kaplan, M.J. (2013) Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Invest. 123, 2981–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Knight, J.S. , Subramanian, V. , O'Dell, A.A. , Yalavarthi, S. , Zhao, W. , Smith, C.K. , Hodgin, J.B. , Thompson, P.R. , Kaplan, M.J. (2015) Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus‐prone MRL/lpr mice. Ann. Rheum. Dis. 74, 2199–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zeerleder, S. , van Bijnen, S. , Wouters, D. , van Mierlo, G.J. , Muus, P. (2013) Neutrophil extracelluar trap formation in PNH patients with and without a history of thrombosis—effects of eculizumab. Blood 122, 1235. [Google Scholar]
- 57. Furie, R. , Matis, L. , Rollins, S.A. , Mojcik, C.F. (2001) A single dose, placebo‐controlled, double blind, phase I study of the humanized anti‐C5 antibody hbG1. 1 in patients with systemic lupus erythematosus. 2001 Innovative Therapies in Autoimmune Diseases, American College of Rheumatology, San Francisco. [Google Scholar]
- 58. Wahono, C.S. , Rusmini, H. , Soelistyoningsih, D. , Hakim, R. , Handono, K. , Endharti, A.T. , Kalim, H. , Widjajanto, E. (2014) Effects of 1,25(OH)2D3 in immune response regulation of systemic lupus erithematosus (SLE) patient with hypovitamin D. Int. J. Clin. Exp. Med. 7, 22–31. [PMC free article] [PubMed] [Google Scholar]
- 59. Reynolds, J. , Ray, D. , Alexander, M.Y. , Bruce, I. (2015) Role of vitamin D in endothelial function and endothelial repair in clinically stable systemic lupus erythematosus. Lancet 385 (Suppl 1), S83. [DOI] [PubMed] [Google Scholar]
- 60. Merrill, J.T. , Wallace, D.J. , Petri, M. , Kirou, K.A. , Yao, Y. , White, W.I. , Robbie, G. , Levin, R. , Berney, S.M. , Chindalore, V. , Olsen, N. , Richman, L. , Le, C. , Jallal, B. , White, B. ; Lupus Interferon Skin Activity (LISA) Study Investigators. (2011) Safety profile and clinical activity of sifalimumab, a fully human anti‐interferon α monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double‐blind randomised study. Ann. Rheum. Dis. 70, 1905–1913. [DOI] [PubMed] [Google Scholar]
- 61. Petri, M. , Wallace, D.J. , Spindler, A. , Chindalore, V. , Kalunian, K. , Mysler, E. , Neuwelt, C.M. , Robbie, G. , White, W.I. , Higgs, B.W. , Yao, Y. , Wang, L. , Ethgen, D. , Greth, W. (2013) Sifalimumab, a human anti‐interferon‐α monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose‐escalation study. Arthritis Rheum. 65, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Khamashta, M. Merrill, J.T. , Werth, V.P. , Furie, R. , Kalunian, K. , Illei, G.G. , Drappa, J. , Wang, L. , Greth, W. (2014) Safety and efficacy of sifalimumab, an anti IFN‐alpha monoclonal antibody, in a phase 2b study of moderate to severe systemic lupus erythematosus. 2014 American College of Rheumatology and Association of Reproductive Health Professionals Annual Meeting, L4 (abs.).
- 63. McBride, J.M. , Jiang, J. , Abbas, A.R. , Morimoto, A. , Li, J. , Maciuca, R. , Townsend, M. , Wallace, D.J. , Kennedy, W.P. , Drappa, J. (2012) Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo‐controlled, double‐blind, dose‐escalation study. Arthritis Rheum. 64, 3666–3676. [DOI] [PubMed] [Google Scholar]
- 64. Kalunian, K. , Merrill, J.T. , Maciuca, R. , Ouyang, W. , McBride, J. , Townsend, M.J. , Park, E. (2012) Efficacy and safety of rontalizumab (anti‐interferon alpha) in SLE subjects with restricted immunosuppressant use: results of a randomized, double‐blind, placebo‐controlled phase 2 study. Arthritis Rheum. 64 (Suppl 10), 2622 (abs.). [Google Scholar]
- 65. Fernandez, D.R. , Telarico, T. , Bonilla, E. , Li, Q. , Banerjee, S. , Middleton, F.A. , Phillips, P.E. , Crow, M.K. , Oess, S. , Muller‐Esterl, W. , Perl, A. (2009) Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES‐1/Rab4‐regulated lysosomal degradation. J. Immunol. 182, 2063–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Frese, S. , Diamond, B. (2011) Structural modification of DNA—a therapeutic option in SLE? Nat. Rev. Rheumatol. 7, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hemmers, S. , Teijaro, J.R. , Arandjelovic, S. , Mowen, K.A. (2011) PAD4‐mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One 6, e22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guiducci, C. , Gong, M. , Xu, Z. , Gill, M. , Chaussabel, D. , Meeker, T. , Chan, J.H. , Wright, T. , Punaro, M. , Bolland, S. , Soumelis, V. , Banchereau, J. , Coffman, R.L. , Pascual, V. , Barrat, F.J. (2010) TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465, 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lauwerys, B.R. , Ducreux, J. , Houssiau, F.A. (2014) Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford) 53, 1369–1376. [DOI] [PubMed] [Google Scholar]
- 70. Mathian, A. , Hie, M. , Cohen‐Aubart, F. , Amoura, Z. (2015) Targeting interferons in systemic lupus erythematosus: current and future prospects. Drugs 75, 835–846. [DOI] [PubMed] [Google Scholar]
- 71. Yao, Y. , Richman, L. , Higgs, B.W. , Morehouse, C.A. , de los Reyes, M. , Brohawn, P. , Zhang, J. , White, B. , Coyle, A.J. , Kiener, P.A. , Jallal, B. (2009) Neutralization of interferon‐alpha/beta‐inducible genes and downstream effect in a phase I trial of an anti‐interferon‐alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 60, 1785–1796. [DOI] [PubMed] [Google Scholar]
- 72. Furie, R.A. , Petri, M.A. , Wallace, D.J. , Ginzler, E.M. , Merrill, J.T. , Stohl, W. , Chatham, W.W. , Strand, V. , Weinstein, A. , Chevrier, M.R. , Zhong, Z.J. , Freimuth, W.W. (2009) Novel evidence‐based systemic lupus erythematosus responder index. Arthritis Rheum. 61, 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. State University of New York—Upstate Medical University. (2008) Treatment of systemic lupus erythematosus with N‐acetylcysteine (NAC). ClinicalTrials.gov, NCT00775476. Retrieved October 2, 2015, from http://clinicaltrials.gov/ct2/show/NCT00775476?term=lupus+and+N+Acetyl+cysteine&rank=1.
- 74. Novo Nordisk A/S. (2009) An investigation of NNC 0151‐0000‐0000 in subjects with systemic lupus erythematosus. ClinicalTrials.gov, NCT01018238. Retrieved October 2, 2015, from http://clinicaltrials.gov/ct2/show/NCT01018238?term#lupus+and+c5&rank#1.