

Short abstract
CLU facilitates proliferation and IFN‐γ production of murine NK cells stimulated with suboptimal dose of IL‐2, without affecting natural cytotoxicity.
Keywords: cell proliferation, NK activation markers, natural cytotoxicity, immune function
Abstract
CLU is a secreted, multifunctional protein implicated in several immunologic and pathologic conditions. As the level of serum CLU was shown to be elevated during inflammatory responses, we questioned if CLU might interact with circulating lymphocytes leading to functional consequences. To assess this possibility directly, mouse splenocytes and purified NK cells were cultured with varying dose of CLU, and its effect on cell proliferation was examined. Our data showed that CLU up‐regulated DNA synthesis and expansion of NK cells significantly in response to a suboptimal, but not maximal, dose of IL‐2, and CLU alone did not exhibit such effects. This CLU‐mediated synergy required the copresence of CLU at the onset of IL‐2 stimulation and needed a continuous presence during the rest of the culture. Importantly, NK cells stimulated with CLU showed increased formation of cell clusters and a CD69 activation receptor, representing a higher cellular activation status compared with those from the control group. Furthermore, these NK cells displayed elevated IFN‐γ production upon RMA/S tumor target exposures, implying that CLU regulates not only NK cell expansion but also effector function of NK cells. Collectively, our data present a previously unrecognized function of CLU as a novel regulator of NK cells via providing costimulation required for cell proliferation and IFN‐γ secretion. Therefore, the role of CLU on NK cells should be taken into consideration for the previously observed, diverse functions of CLU in chronic inflammatory and autoimmune conditions.
Abbreviations
- CLU
clusterin
- Cr
chromium
- dpm
distintegrations per minute
- RA
rheumatoid arthritis
- RMA/S
a T cell lymphoma derived from the Rauscher murine leukemia virus‐induced RBL‐5 cell line
Introduction
CLU, known as apolipoprotein J, is a secreted glycoprotein expressed ubiquitously in a wide variety of tissues [1]. Since its discovery in 1983 as a protein enhancing cell aggregation in vitro [2], CLU has been implicated in several diverse, physiological processes including lipid transportation, complement inhibition, tissue remodeling, cell–cell and cell–substratum interactions, promotion, and inhibition of apoptosis. Furthermore CLU has been shown to facilitate carcinogenesis and tumor formation [3], demonstrating its function as a growth regulator.
Recent data provided an additional facet for the role of CLU in regulating inflammation and immunity [4, –, 6, 7, 8, 9]. CLU was shown to protect tissues or cells from damages by complement attack via complexing with the membrane attack complex [7]. CLU deposition was detected in the renal glomeruli of patients with glomerulonephritis, and a significant increase of CLU mRNA was demonstrated in lupus‐like nephritis, presumably as a defense mechanism from tissue damages [8]. Moreover, the fact that intracellular CLU inhibited NF‐κB expression in synoviocytes suggests that the action of CLU might be closely linked to the pathogenesis of inflammatory RA [4]. For example, Devauchelle et al. [9] have shown that CLU expression was lacking in RA patients and hence, resulted in enhanced IκB degradation and prolonged activation of NF‐κB. Consistent with these results, CLU‐deficient mice showed accelerated and lasting arthritis [9] and more severe inflammation in a myosin‐induced autoimmune myocarditis model [5, 6]. Furthermore, secretion of proinflammatory cytokines, IL‐6 and IL‐8, was enhanced in cultured fibroblast‐like synoviocytes when CLU expression was silenced with small interfering RNAs [9]. Collectively, these data underscore a potential implication of CLU in regulating chronic inflammation and autoimmunity.
Belonging to innate immunity, NK cells constitute circulating lymphocytes that provide a bodyˈs first line of defense against infections and cancers [10]. Via secreting proinflammatory cytokines and stimulating cytotoxicity directly [11], NK cells eliminate infected and transformed cells in the body. Additionally, human NK cells have been shown to control autoimmune conditions, e.g., psoriasis or atopic dermatitis [12], although a precise molecular mechanism underlying this regulation remains elusive. Like CLU, the role of NK cells is diverse and may be controlled by environmental conditions and the progression status of the disease.
NK cells become activated immediately after pathogen recognition and further activated by IL‐12 and IL‐18 produced by macrophages and DCs [13]. Interestingly, DCs expressed CLU at their immature states, whose expression increased over 30‐fold upon maturation [4]. Similarly, CLU in the human serum (35–105 μg/ml) was shown to undergo significant up‐regulation during inflammation [4]. As elevated CLU may likely come in contact with circulating lymphocytes, it raises a question of whether interaction of CLU with NK cells in the peripheral blood may result in any functional consequences. Therefore, we set up this study to investigate the relationship between CLU and NK cells in vitro using freshly isolated mouse NK cells. Our data demonstrate that CLU could indeed affect the NK cell activation process via facilitating 2 aspects of immune responses, proliferation and IFN‐γ secretion, in response to IL‐2. This effect required the copresence of CLU at the onset of IL‐2 stimulation and was only visible at the suboptimal dose of IL‐2. Therefore, our data suggest that CLU serves as a novel, costimulatory ligand in NK cells.
MATERIALS AND METHODS
Purification of soluble CLU
CLU was purified from fresh, normal human plasma, which was precipitated using 12–23% polyethylene glycol (MW 3350; Sigma Chemical Co., St. Louis, MO, USA) overnight at 4°C. This precipitate was dissolved and subjected to DEAE‐Sepharose and heparin‐Sepharose column chromatography (GE Healthcare Life Sciences, Piscataway, NJ, USA), as described previously [14, –, 16, 17, 18]. CLU‐positive fractions were then applied to a CLU mAb (1G8)–affinity chromatography column [15]. The anti‐CLU mAb (1G8) was generated using recombinant human full‐length CLU expressed in Escherichia coli as an antigen and covalently conjugated to cyanogen bromide‐activated Sepharose 4B (Sigma Chemical Co.). Eluted proteins were dialyzed against PBS and stored at –80°C prior to use. As human CLU shares 75% aa identity with mouse CLU [19] and shows cross‐reactivity with its rodent counterpart [20], experiments were performed using purified human CLU. The purity and homogeneity of purified CLU (>95%) were assessed by SDS‐PAGE under reducing and nonreducing conditions and visualized by Coomassie brilliant blue staining. The identity of CLU was analyzed further by Western blot analysis using clone B5 or M18 anti‐CLU Ab (Supplemental Fig. 3). The endotoxin level of purified CLU was below detection level, as measured by the Limulus amoebocyte lysate kinetic turbidimetric assay (Endosafe®, Charles River Laboratory, Korea).
Isolation and culture of NK cells
Female C57BL/6 mice between 6 and 8 weeks of age were purchased from Nara Biotech (Seoul, Korea) and used under the protocol approved by the Korea University Institutional Animal Care and Use Committee (KUIACUC‐2009‐126). NK cells were enriched from B cell‐depleted splenocytes via negative selection using a mixture of Ab including biotin‐conjugated anti‐CD3 (145‐2C11), anti‐CD4 (GK1.5), anti‐CD8 (53‐6.7), and anti‐CD19 (MB19‐1) and streptavidin‐conjugated microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), as described previously [21]. After depletion of non‐NK cells, the purity of CD3–DX5+ NK cells was 70–80%. In some experiments, CD3–DX5+ NK cells were enriched up to 95% using a mouse NK isolation kit (Cat. No. 130‐090‐864, Miltenyi Biotec, Auburn, CA, USA), which contained biotin‐conjugated Ab cocktail (anti‐CD4, ‐CD5, ‐CD8a, and ‐CD19, Gr‐1, Ter‐119) and antibiotin microbeads. Enriched NK cells were cultured in RPMI‐1640 (Welgene, Daegu, Korea) supplemented with 5% FBS (Lonza Walkersville Inc., Walkersville, MD, USA) and human rIL‐2 (Novartis Pharmaceuticals, East Hanover, NJ, USA) in the presence or absence of CLU at the doses indicated in each figure.
Western blot assay
Cell lysates were resolved by SDS‐PAGE and transferred to nitrocellulose membranes (Schleicher and Schuell, Dassel, Germany). Membranes were then blocked with 5% skim milk in TBST (20 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 0.1% Tween‐20) overnight at 4°C and probed with rabbit polyclonal anti‐CLU Ab (C‐18 or M‐18, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). After incubation with anti‐rabbit secondary Ab coupled to HRP (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA), immunoreactive CLU proteins were visualized using SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL, USA), according to the manufacturerˈs protocol.
Ab and flow cytometry
FITC‐conjugated anti‐CD69 (H1.2F3), anti‐CD3 (145‐2C11), and anti‐LFA (M17/4); allophycocyanin‐conjugated anti‐CD49b (DX5) and anti‐ICAM‐1 (YN1/1.7.4); and PE‐conjugated anti‐CD44 (IM7), anti‐CD11c (N418), anti‐NKG2D (CX5), anti‐I‐Ab (M5/114.15.2), anti‐CD27 (LG.7F9), and anti‐CD3 (145‐2C11) mAb were purchased from eBioscience (San Diego, CA, USA). Isolated NK cells were resuspended in 100 μl FACS buffer (PBS containing 2% FBS and 0.02% sodium azide) and incubated with anti‐CD16/CD32 mAb to block FcγRIII/II. Without washing, cells were incubated with mAb, indicated in Fig. 5, for 20 min at 4°C. After washing with FACS buffer, cells were fixed in 200 μl 1% paraformaldehyde in PBS. Flow cytometry was performed with a FACSCalibur (BD Biosciences, San Diego, CA, USA) and the data analyzed with CellQuest software (BD PharMingen, San Jose, CA, USA). Fifty thousand lymphocyte populations gated by forward‐/side‐scatter were analyzed [22].
Figure 5.
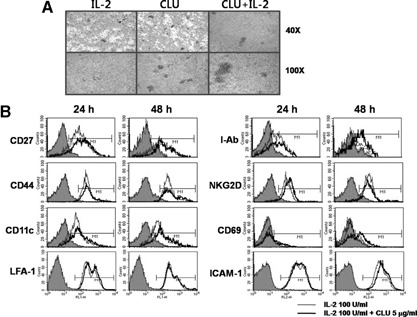
CLU causes aggregation of NK cells without significantly altering surface NK receptors. (A) Purified NK cells (7.5×105) were incubated with 5 μg/ml CLU in the presence or absence of IL‐2 (100 U/ml) for 5 days prior to taking photos using an inverted microscope (40× and 100× original magnification). (B) Surface expression of CD27, CD44, CD11c, LFA‐1, I‐Ab, NKG2D, CD69, or ICAM‐1 was monitored by FACS using NK cells cultured with 100 U/ml IL‐2 in the presence or absence of 5 μg/ml CLU. The filled line represents isotype control, thin line IL‐2 alone, and thick line IL‐2 + CLU‐cultured NK cells. The results of this particular experiment represent those from 3 other independent experiments.
CFSE dilution assay
Proliferation of NK cells was evaluated by the CFSE (Molecular Probes, Inc., Eugene, OR, USA) dividing method. Briefly, freshly isolated NK cells suspended in PBS were labeled with 5 μM CFSE at 37°C for 10 min. Reaction was quenched by adding an equal volume of FBS. Cells were washed twice in PBS, and CFSE‐loaded NK cells at 1 × 105/well were incubated with 100 U/ml IL‐2 in the presence or absence of 5 μg/ml CLU. After incubation, CFSE‐labeled cells were harvested, and their division was assayed. Flow cytometry was performed with a FACSCalibur and the data analyzed with CellQuest software.
3H‐Thymidine uptake assay
Live cell numbers were counted using a hemocytometer (Improved Neubauer, Hawksley Co., Lansing, UK) by trypan blue exclusion.
To assess the effect of CLU, 2 × 105 total splenocytes or 1 × 105 purified NK cells/well were incubated for 3 days in a 96‐well plate in the presence or absence of an indicated concentration of CLU or IL‐2. 3H‐Thymidine (1 μCi; Perkin Elmer, Boston, MA, USA) was added to the well and incubated for an additional 12 h. Cells were harvested onto filter paper with a Micro96 harvester (Skatron, Lier, Norway), and dpm was counted using a β‐counter (Packard Instrument Co., Meriden, CT, USA).
51Cr release assay
A standard Cr release assay was performed with minor modifications. In brief, RMA/S target cells were labeled with 51Cr (Perkin Elmer, Boston, MA, USA) at 50 μCi/5 × 105 cells. Five thousand RMA/S cells were mixed with serially diluted NK cells at the indicated E:T ratio for 4 h at 37°C [21]. The γ‐scintillation of the supernatant was measured by a γ‐counter (Perkin Elmer). Percent of specific lysis was calculated as follows: 100 × (experimental release–spontaneous release)/(maximum release–spontaneous release).
Statistical analysis
Nonparametric one‐way or two‐way ANOVA was performed with SPSS Version 12.0 software (SPSS, Chicago, IL, USA), depending on the data. Where the P value was <0.05, the result was considered significant.
RESULTS
CLU synergizes with IL‐2 for the proliferation of whole splenocytes and purified NK cells
Previous data have shown that CLU expression was absent in T lymphocytes [23] but found to be expressed at a low level in a subset of T cells called Th‐17 cells [24]. However, no data were available regarding the expression of CLU in murine splenocytes and NK cells. Therefore, we first examined the expression of CLU on whole splenocytes containing mixtures of lymphocytes, e.g., T, B, and NK cells. Data from the Western blot analysis using 2 polyclonal anti‐CLU Ab, C‐18 or M‐18, showed CLU expression on whole splenocytes (Fig. 1 A). However, little or no expression of CLU was detected in purified NK cells (Fig. 1A). A major portion of endogenous CLU detected in whole splenocytes was found to come from B cells, as depletion of B cells abrogated CLU expression from the splenocyte fraction (data not shown). These data demonstrate that murine NK cells do not express a significant level of endogenous CLU.
Figure 1.
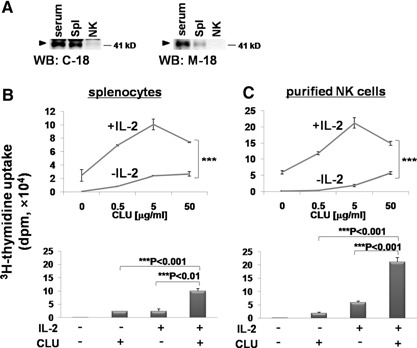
Effect of CLU on the proliferation of whole splenocytes and purified NK cells. (A) Endogenous expression of CLU in total splenocytes and purified NK cells was monitored using Western blots (WB), as described in Materials and Methods. Cell lysates (20 μg), prepared from whole splenocytes (Spl) or purified NK cells, were subjected to SDS‐PAGE and immunoblotting with anti‐CLU Ab (C‐18 or M‐18). Diluted (1:10) C57BL/6 mouse serum (10 μl) was loaded as a positive control (CLU). (B and C) Total splenocytes (2×105; B) or 1 × 105 purified NK cells (C) were cultured for 3 days with 0, 0.5, 5, or 50 μg/ml CLU in the presence or absence of 100 U/ml IL‐2. 3H‐Thymidine (1 μCi) was added for an additional 12 h to count radioactivity incorporated into the DNA. The lower bar graphs are re‐plots of the upper graphs to compare 3H‐thymidine incorporation at 5 μg/ml CLU in the presence or absence of 100 U/ml of IL‐2. The data are representatives from 5 independent experiments. Error bars represent sd (**P<0.01, ***P<0.001).
We next determined if the interaction of exogenous CLU with NK cells affected proliferation of NK cells using a 3H‐thymidine incorporation assay. When whole splenocytes were cultured only with CLU, little or no significant uptake of 3H‐thymidine was observed for up to 50 μg/ml CLU (Fig. 1B, upper panel). However, addition of CLU in the presence of a suboptimal dose of IL‐2 markedly potentiated cell proliferation by IL‐2. This CLU‐mediated augmentation of DNA synthesis started to appear at 0.5 μg/ml and reached maximal at 5 μg/ml, where CLU increased IL‐2‐induced proliferation for up to 4.35 ± 1.19‐fold (Fig. 1B, lower panel; 2.43±0.85×104 dpm in the IL‐2 group vs. 10.06±0.81×104 dpm in the IL‐2+CLU group). As IL‐2‐responsive cells present in whole splenocytes were likely to be mostly NK cells, we next purified NK cells from the spleen and examined the effect of CLU on NK cell proliferation. As seen in Fig. 1C, CLU alone did not result in substantial proliferation of purified NK cells. However, in the presence of IL‐2, CLU augmented NK cell proliferation significantly (Fig. 1C), similar to that observed from whole splenocytes (Fig. 1B). Compared with splenocytes, the level of 3H‐thymidine incorporated into purified NK cells was substantially higher, confirming that a major population of IL‐2‐responsive cells in the spleen was NK cells. At 5 μg/ml, CLU‐enhanced, ∼3.61 ± 0.03‐fold of DNA synthesis resulted from IL‐2 stimulation alone (Fig. 1C, lower panel; 5.88±0.50×104 dpm vs. 21.22±1.63×104 dpm). Therefore, these data demonstrate that CLU alone is not sufficient to drive NK cells into the cell cycle; however, it can promote NK cell proliferation substantially in the presence of IL‐2.
CLU stimulates NK cell proliferation at the low dose of IL‐2
We next investigated if the effect of CLU on IL‐2‐induced proliferation could be seen at the higher doses of IL‐2. To test this, we cultured purified NK cells with 5 μg/ml CLU in the presence of IL‐2 ranging from 10 to 1000 U/ml. As seen in Fig. 2 A, NK cells displayed augmentation of their 3H‐thymidine uptake when cells were cultured with up to 500 U/ml IL‐2. This synergy on DNA synthesis by CLU became noticeable as low as 10 U/ml IL‐2, maximal at 100 U/ml, and saturated at 500 U/ml. Increasing IL‐2 over 500 U/ml did not enhance IL‐2‐induced NK cell proliferation further by CLU, indicating that the effect of CLU on NK cell proliferation reached their maximal capacity at 500 U/ml. To confirm further the role of CLU on NK cell proliferation, we performed a CFSE dilution assay, which allows monitoring of the percentage of cells undergoing cell division by FACS. As shown in Fig. 2B, CLU‐mediated facilitation of cell division became noticeable from 3 days following IL‐2 addition (the percentage undergoing cell division was 2.67% in the IL‐2 group and 5.51% in the IL‐2+CLU group). This synergy lasted for up to 5 days, but thereafter, it was no longer detectible (Fig. 2B). When the percentage of cells on division was compared, CLU promoted ∼2.06‐fold, 1.88‐fold, and 2.44‐fold of the control at Days 3, 4, and 5, respectively. When daily CFSE data were averaged, CLU‐mediated synergy in cell proliferation was calculated to be 2.13 ± 0.29‐fold. Consistently, the number of cells expanded by 6 days showed a 2.29 ± 0.16‐fold increase by CLU [5.05±0.92×105 cells in the IL‐2 group and 11.48±0.13×105 cells in the IL‐2+CLU group (Fig. 2C)]. This synergy in NK cell proliferation by CLU was likely a result of its direct effect on NK cells not working on the non‐NK cells (<20%) contaminated in cultures, as similar effects were observed in NK cells isolated with >95% purity (Supplemental Fig. 2). Collectively, these data demonstrate that CLU synergizes with IL‐2 for facilitating NK cell proliferation and expansion at the suboptimal dose of IL‐2.
Figure 2.
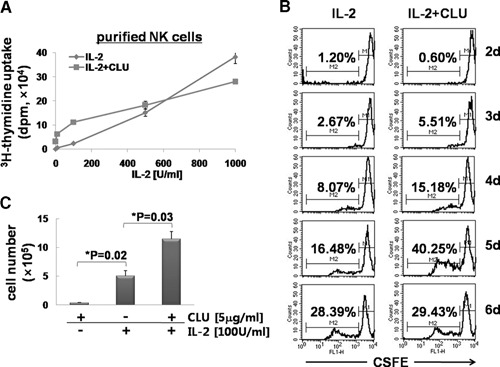
CLU synergizes with IL‐2 for the proliferation of NK cells. (A) Purified NK cells were plated at 1 × 105 cells/well and incubated for 3 days with various concentrations of IL‐2 (10, 100, 500, and 1000 U/ml) in the presence or absence of 5 μg/ml CLU. 3H‐Thymidine (1 μCi) was added for an additional 12 h to count radioactivity incorporated into the DNA. (B) Purified NK cells (5×105) were labeled with 5 μM CFSE for 10 min, and cell division was analyzed by FACS, as described in Materials and Methods. Numbers in each graph indicate the percentage of cells undergoing cell division. FL1‐H, Fluorescence 1‐height. (C) Purified NK cells (1×106) were cultured in the presence or absence of 5 μg/ml CLU and/or 100 U/ml IL‐2 for 6 days. Live cells were counted using a hemocytometer. The data shown are representatives of a minimum of 3 independent experiments. Error bars represent sd (*P<0.05).
CLU‐mediated synergy with IL‐2 occurs early at the onset of NK cell activation and requires a continuous presence of CLU throughout the culture
As our CFSE data show that the synergy between CLU and IL‐2 for NK cell proliferation started to appear at Day 3 following IL‐2 stimulation (Fig. 2B), we examined if the effect of CLU could still be observed when CLU was added later during the IL‐2‐induced activation process. For this, we added CLU at 1 or 2 days after addition of IL‐2 and compared the level of DNA synthesis with the group added at the beginning of the culture (Fig. 3 A). When CLU was added at 1 day following activation (Fig. 3A, 24 h), a dramatic increase of DNA synthesis, as seen in Fig. 1C, was no longer observed. Rather, a slight but statistically significant increase in DNA synthesis (2.54±0.03×104 dpm) was detected as compared with that of IL‐2 alone (1.45±0.24×104 dpm). These data suggest that CLU could display its ability to facilitate NK cell proliferation even after cells had entered the cell cycle. However, this effect was small compared with the one added simultaneously with IL‐2 and became abrogated if CLU were added at 2 days after IL‐2 stimulation (Fig. 3A; 48 h: 1.77±0.53×104 dpm). Therefore, CLU had to be in contact with NK cells at the onset of IL‐2 stimulation to exhibit a maximal growth‐promoting effect.
Figure 3.
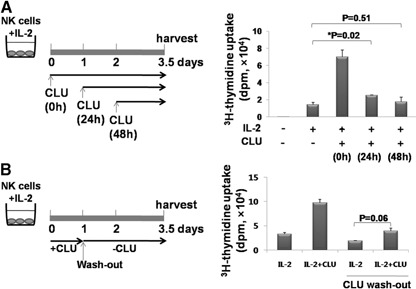
CLU stimulates NK cell proliferation at the early onset. (A) Purified NK cells (1×105; >95% purity) were cultured for a total 3.5 days with 100 U/ml IL‐2 in the absence or presence of 5 μg/ml CLU, which was added at the same time (0 h), 24 h, or 48 h following IL‐2 stimulation, as depicted in the left panel. Total incubation time with IL‐2 was 84 h in all conditions. 3H‐Thymidine incorporation into the DNA of NK cells was plotted as a bar graph (right panel). (B) Purified NK cells (1×105; >95% purity) were cultured with 100 U/ml IL‐2 in the absence or presence of 5 μg/ml CLU. Twenty‐four hours later, media were removed and replaced with new ones containing 100 U/ml IL‐2 but without CLU (left panel). As controls, cells were cultured in the continuous presence of IL‐2, with or without 5 μg/ml CLU (left two bars). 3H‐Thymidine (1 μCi) was added at 2 days following washout and cultured for an additional 12 h, and radioactivity was compared. The level of 3H‐thymidine incorporated into NK cells was plotted as a bar graph (right panel). The data shown are representatives of 3 independent experiments. Error bars represent sd (*P<0.05).
We next determined if CLU‐mediated synergy in NK cell proliferation could still be observed when cells were exposed to CLU only briefly at the onset of IL‐2 stimulation (Fig. 3B). For this, NK cells were cultured with IL‐2 in the presence of CLU as in Fig. 3A. After 24 h, supernatants were removed and replaced with fresh ones containing only IL‐2 (Fig. 3B). When the level of cell proliferation was measured at 3.5 days, NK cells exposed to CLU only for the initial 24 h proliferated significantly less (3.90±0.62×104 dpm) as compared with the control group (9.72±0.73×104 dpm). These data demonstrate unequivocally that CLU was required to be copresent with IL‐2 for the entire culture to exert its maximal stimulatory effects on NK cell proliferation.
To assess if CLU exerted its action via stimulating non‐NK cell populations contaminated in the NK culture, we stimulated fresh NK cells (>95% purity) using IL‐2 or IL‐2 + CLU in the presence of preactivated culture supernatants collected 24 h following IL‐2 or IL‐2 + CLU stimulation (Fig. 4 A). When cell division was measured 1 and 2 days following stimulation, no significant increase was observed in the group containing preactivated culture supernatants as compared with the control groups (Fig. 4B). These data preclude the possibility of the contribution of non‐NK cells or cytokines secreted by contaminating cell populations in promoting NK cell proliferation. Therefore, the synergistic effect of NK cell proliferation by CLU was likely a result of its direct action on NK cells. Collectively, CLU induced its synergistic proliferation at the early onset of IL‐2 stimulation via working directly on NK cells in a continuous manner.
Figure 4.
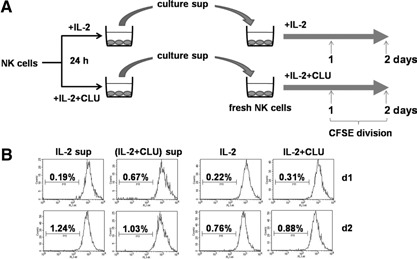
Addition of prestimulated culture supernatants to the freshly isolated NK cells did not facilitate proliferation. (A) Purified NK cells (1×105; with 80% purity) were stimulated with 100 U/ml IL‐2 in the absence or presence of 5 μg/ml CLU. Twenty‐four hours later, culture supernatants (sup) were collected and added to the freshly isolated NK cells (>95% purity) labeled with 5 μM CFSE. (B) Cells were cultured for 2 days in the presence of IL‐2, with or without 5 μg/ml CLU, and their divisions were analyzed by FACS, as described in Materials and Methods.
CLU causes cell‐to‐cell clustering and up‐regulation of CD69 in NK cells
As CLU was identified originally for its ability to induce cell aggregation [2], we next examined if NK cells formed clusters in the presence of CLU. Upon activation with IL‐2, murine NK cells tended to form small aggregates as shown in Fig. 5 A (left panel). Addition of CLU caused NK cells to form clusters (Fig. 5A, middle panel), slightly larger than those seen from IL‐2 cultures. When CLU was added together with IL‐2, the frequency to form clusters did not change; however, the size of clusters became much bigger (Fig. 5A, right panel) than those observed from CLU or IL‐2 alone. As cell‐to‐cell clustering occurs through binding of surface adhesion receptors to their counterparts on the adjacent cells, we explored the possibility that CLU might have affected the expression of surface adhesion receptors on NK cells. As can be seen in Fig. 5B, expression of CD27, CD44, CD11c, LFA‐1, and ICAM‐1 adhesion receptors did not show any significant changes upon CLU addition at 24 and 48 h following IL‐2 stimulation. These data demonstrate that CLU promotes NK cell clustering without affecting their surface adhesion receptors.
We next examined if CLU facilitated up‐regulation of other NK‐activating receptors. When measured by FACS, we found that surface expression of I‐Ab, NKG2D, and CD69 was not altered significantly by addition of CLU (Fig. 5B, right panel). However, a subset of NK cells showed elevation of CD69 at 48 h following IL‐2 stimulation. As CD69 was considered to be an earliest inducible cell surface glycoprotein for T and NK cells [25], it is tempting to speculate that CLU‐mediated up‐regulation of CD69 reflected a higher cellular activation status leading to form cell clusters, DNA synthesis, and hence, proliferation. Together, these data demonstrate that cell‐to‐cell clustering and up‐regulation of CD69 preceded CLU‐mediated stimulation of NK cells.
CLU promotes IFN‐γ secretion but leaves NK cytotoxic function unaffected
As CLU functioned as a costimulator for NK cell proliferation, it was likely to affect NK effector functions, cytokine secretion and cytotoxicity. To determine the effect of CLU on cytokine secretion, we mixed NK cells, cultured with IL‐2 alone or IL‐2 + CLU, with RMA/S tumor target cells, and the expression of IFN‐γ was measured by intracellular FACS (Fig. 6 A). Upon interaction with tumor targets, production of IFN‐γ was noticeable in a subset of IL‐2‐cultured NK cells (1.80%). Strikingly, NK cells cultured in the presence of CLU showed an approximate 4‐fold increase of IFN‐γ secretion (7.49%) as compared with that of IL‐2 cultures. The production of IFN‐γ was primarily from CD3–NK1.1+ NK cells, not from CD3+NK1.1+ NKT cells present <1% in the culture (Supplemental Fig. 3). These data indicate that CLU rendered NK cells to mount elevated secretory function when encountered with tumor targets. On the contrary, NK cytotoxicity against RMA/S targets did not appear to be affected substantially by CLU, as specific lysis obtained at various E:T ratios was comparable between the IL‐2 group and the IL‐2 + CLU group (Fig. 6B). These data demonstrate that CLU affects the effector function of NK cells selectively, facilitating IFN‐γ secretion without altering cytotoxic capacity.
Figure 6.
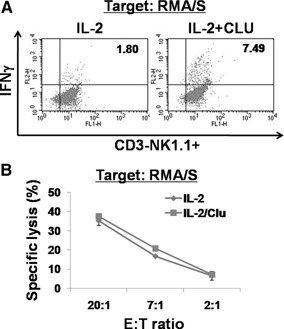
CLU promotes IFN‐γ secretion without affecting NK cytotoxicity. (A) Purified NK cells were incubated with 100 U/ml IL‐2 and/or 5 μg/ml CLU for 5 days. At the end of culture, cells were coincubated with RMA/S target cells in a 1:1 ratio for 6 h in the presence of Golgi stop, and intracellular staining for IFN‐γ was performed (n=3). Cells were gated on CD3–NK1.1+ NK cells. (B) 51Cr‐labeled RMA/S cells were mixed with NK cells cultured for 5 days with IL‐2 alone or IL‐2 + CLU at indicated E:T ratios (n=6). Radioactivity released into the supernatant after 4 h was counted by using a γ‐counter. Percent of specific lysis was calculated as follows: 100 × (experimental release–spontaneous release)/(maximum release–spontaneous release).
DISCUSSION
CLU is a multifaceted protein implicated in several physiologic processes and multiple pathological conditions [3]. Although the precise molecular mechanisms underlying the extremely diverged functions of CLU are not understood completely, it was found to depend largely on the cellular genetic background and the nature of the stimuli [26]. Additionally, this diversity can be attributed to the existence of 2 alternatively spliced forms of the CLU gene that encode secreted CLU or nuclear CLU [9, 27, 28], which have been shown to exert distinct functions. Furthermore, intracellular CLU was found to inhibit NF‐κB activity in human synoviocytes, human neuroblastoma cells, and murine embryonic fibroblasts [9, 28], and secretory CLU appeared to stimulate NF‐κB expression in murine NK cells as it facilitated production of IFN‐γ in response to IL‐2. These data highlight the nonoverlapping role of intracellular versus secretory CLU in the NF‐κB pathway. As murine NK cells were not found to express endogenous CLU, CLU‐dependent regulation of NF‐κB was absent, and thus, the action of CLU was largely dependent on an extracellular secreted form of CLU.
In this study, we present previously unrecognized functions of CLU in promoting cell proliferation and IFN‐γ production of murine NK cells. A similar synergistic effect was also observed in human NK cells (Supplemental Fig. 4), highlighting its significance as a costimulatory molecule across the species. As CLU was found to be up‐regulated in the human plasma during inflammation [4, –, 6, 7, 8, 9], it could interact with circulating NK cells and affect the course of immune responses by facilitating expansion of NK cells and promoting IFN‐γ expression. This synergism was only evident at the suboptimal dose of IL‐2 and required copresence of CLU and IL‐2 at the onset of NK cell activation, suggesting that CLU lowered the threshold for NK cell activation when the strength of the mitogenic signal was weak. This effect represents the concept of costimulation, seen widely in lymphocyte signaling [29]. For example, CD28 on T cells binds to CD80 or CD86 expressed on APCs and provides costimulation for T cell activation (Signal 2) initiated from TCR signaling (Signal 1) [29].
The molecular mechanism by which CLU drives NK cell activation still remains unclear. However, the fact that CLU facilitated DNA synthesis without inhibiting apoptosis (Supplemental Fig. 5) suggests that the growth‐promoting effect of CLU was largely a result of facilitating cell proliferation rather than inhibiting apoptotic events. In fact, CLU appeared to increase the rate of apoptosis slightly, presumably as a result of activation‐induced cell death following IL‐2‐induced stimulation. Thus, the antiapoptotic and prosurvival effect of CLU seen previously in prostate, bladder, and breast cancer cells [30, –, 32] did not seem to operate in NK cells. Moreover, CLU‐mediated NK cell costimulation was not found to be through binding to plasma IgG or signaling through FcγRIII/II, shown previously [33], as addition of IgG or blocking anti‐CD16/CD32 mAb did not significantly affect the level of DNA synthesis by IL‐2 or IL‐2 + CLU (Supplemental Fig. 6).Nonetheless, cell‐to‐cell crosstalk among NK cells might have been increased drastically and functioned to costimulate IL‐2‐stimualted NK cells, as NK cells formed large clusters in the presence of CLU. This homotypic cell‐to‐cell interaction, previously demonstrated by our group, was shown to be mediated primarily by binding of 2B4 on NK cells with its ligand CD48 on the neighboring NK cells [34]. 2B4/CD48 binding among NK cells was found to be necessary for optimal expansion, lytic potential, and cytokine secretion in murine NK cells. Therefore, it is tempting to speculate that CLU functioned to facilitate homotypic interactions via 2B4/CD48 and/or other receptor/ligand pairs. Interestingly, although CLU promoted cell‐to‐cell clusters and adhesion, it did not appear to alter surface expression of NK adhesion receptors, CD27, CD44, LFA‐1, ICAM‐1, or CD11c. Furthermore, it did not change the expression of I‐Ab MHC class I molecules or NKG2D activation receptors. These data imply that CLU‐mediated activation events occurred independently of up‐regulation of surface adhesion or activation receptors in NK cells.
On a closer look, we did observe a subset of NK cells expressing a higher level of CD69 in the presence of CLU. As CD69 was found to be involved in lymphocyte proliferation and function as a signal‐transmitting receptor [25], up‐regulation of CD69 by CLU might represent an early signaling event for NK cell activation. In line with this, we have seen that CLU‐mediated synergy required p60Src and MEK activation, as treatment of NK cells with PP2 or PD98059, pharmacological inhibitors of p60Src or ERK1/2, respectively, brought down the level of 3H‐thymidine incorporation to that observed in the IL‐2‐alone group (data not shown). These data demonstrate and confirm our previous findings in rat astrocytes that protein tyrosine phosphorylation and ERK activity were critical for CLU‐induced proliferation [15, 16]. Similarly, we have also observed the growth factor‐like property of CLU in insulinoma and pancreatic duct cells [35]. Unlike these cells, NK cells did not undergo proliferation by CLU alone. This might have been a result of the fact that activation of lymphocytes needed to be controlled more tightly than other cells in the body, possibly to prevent their hyperactivation against foreign and even self‐antigens. Therefore, CLU‐mediated costimulation events provide an additional checkpoint to assure proper activation of NK cells in response to weak mitogenic stimuli (Signal 1), such as a low dose of IL‐2. In the absence of Signal 1, cells remain unactivated even in the presence of a high dose of CLU (Signal 2). Together, these data underscore multifunctional features of CLU by providing stimulation (Signal 1) or costimulation (Signal 2) to the cells of multiple origins through a mechanism involving cell‐to‐cell crosstalk.
At present, the receptors for CLU on NK cells are not known. Although CLU was shown to stimulate proliferation of primary astrocytes via activating EGFR [16], this appeared to be indirect stimulation of EGFR by CLU [16]. Moreover, megalin, which causes endocytosis of surface receptors into the specialized membrane microdomains, such as lipid rafts, and potentiates intracellular signaling [36], was the only known binding partner for CLU so far. However, it is expressed mostly in the kidney and brain [37]. Therefore, other receptors, yet to be discovered, may be operational in NK cells. Experiments are currently underway to identify the receptors for CLU on NK cells using the yeast 2‐hybrid system and coimmunoprecipitation methods.
Collectively, our findings of synergism between CLU and IL‐2 provide important meanings in the NK cell biology and innate immunity. Up‐regulation of CLU by cytokines in vivo, e.g., IL‐1β and IL‐2 [38], results in protection from cellular and tissue damages and concurrently stimulates NK cell proliferation and IFN‐γ secretion to facilitate immune responses. Increased NK cell numbers with elevated IFN‐γ could promote further activation of macrophages and T cells and hence, link to the activation of adaptive immunity. As CLU up‐regulated IFN‐γ without affecting cytotoxic function of NK cells, it would become therapeutically useful in the treatment of autoimmune and chronic inflammatory diseases. However, as a result of the multifaceted features of CLU, its application into the treatment of inflammation and autoimmune diseases awaits complete understanding of cellular and molecular mechanisms in the particular disease setting.
AUTHORSHIP
C.H.S. performed experiments, analyzed the data, and wrote the paper; Y‐B.Y., Y‐J.H., and Y‐J.S. performed the research; J.A.B. analyzed the data and wrote the paper; B‐H.M. and K‐M.L. designed the research, analyzed the data, and wrote the paper.
Supporting information
Supplementary Material Files
ACKNOWLEDGMENTS
This work was supported by grants from KICOS (K20704000007‐09A0500‐00710, K20601000002‐09E0100‐00210), the National Nuclear R&D Program (grant BAERI), the Seoul R&BD Program (10920), KRF 205‐2004‐1‐E00029, and the Innovative Research Institute for Cell Therapy (A062260). B‐H.M. was supported by a Korea University grant.
Contributor Information
Bon‐Hong Min, Email: bhmin@korea.ac.kr.
Kyung‐Mi Lee, Email: bhmin@korea.ac.kr.
REFERENCES
- 1.Trougakos, I. P., Gonos, E. S. (2006) Regulation of clusterin/apolipoprotein J, a functional homologue to the small heat shock proteins, by oxidative stress in aging and age‐related diseases. Free Radic. Res. 40, 1324–1334. [DOI] [PubMed] [Google Scholar]
- 2.Blaschuk, O., Burdzy, K., Fritz, I. B. (1983) Purification and characterization of a cell‐aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 258, 7714–7720. [PubMed] [Google Scholar]
- 3.Shannan, B., Seifert, M., Leskov, K., Willis, J., Boothman, D., Tilgen, W., Reichrath, J. (2006) Challenge and promise: roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 13, 12–19. [DOI] [PubMed] [Google Scholar]
- 4.Falgarone, G., Chiocchia, G. (2009) Chapter 8: clusterin: a multifacet protein at the crossroad of inflammation and autoimmunity. Adv. Cancer Reis. 104, 139–170. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin, L., Zhu, G., Mistry, M., Ley‐Ebert, C., Stuart, W. D., Florio, C. J., Groen, P. A., Witt, S. A., Kimball, T. R., Witte, D. P., Harmony, J. A., Aronow, B. J. (2000) Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J. Clin. Invest. 106, 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swertfeger, D. K., Witte, D. P., Stuart, W. D., Rockman, H. A., Harmony, J. A. (1996) Apolipoprotein J/clusterin induction in myocarditis: a localized response gene to myocardial injury. Am. J. Pathol. 148, 1971–1983. [PMC free article] [PubMed] [Google Scholar]
- 7.Choi, N. H., Mazda, T., Tomita, M. (1989) A serum protein SP40,40 modulates the formation of membrane attack complex of complement on erythrocytes. Mol. Immunol. 26, 835–840. [DOI] [PubMed] [Google Scholar]
- 8.Moll, S., Menoud, P. A., French, L., Sappino, A. P., Pastore, Y., Schifferli, J. A., Izui, S. (1998) Tubular up‐regulation of clusterin mRNA in murine lupus‐like nephritis. Am. J. Pathol. 152, 953–962. [PMC free article] [PubMed] [Google Scholar]
- 9.Devauchelle, V., Essabbani, A., De Pinieux, G., Germain, S., Tourneur, L., Mistou, S., Margottin‐Goguet, F., Anract, P., Migaud, H., Le Nen, D., Lequerre, T., Saraux, A., Dougados, M., Breban, M., Fournier, C., Chiocchia, G. (2006) Characterization and functional consequences of underexpression of clusterin in rheumatoid arthritis. J. Immunol. 177, 6471–6479. [DOI] [PubMed] [Google Scholar]
- 10.Lodoen, M. B., Lanier, L. L. (2006) Natural killer cells as an initial defense against pathogens. Curr. Opin. Immunol. 18, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fehniger, T. A., Carson, W. E., Mrozek, E., Caligiuri, M. A. (1997) Stem cell factor enhances interleukin‐2‐mediated expansion of murine natural killer cells in vivo. Blood 90, 3647–3653. [PubMed] [Google Scholar]
- 12.Schmid‐Ott, G., Jaeger, B., Adamek, C., Koch, H., Lamprecht, F., Kapp, A., Werfel, T. (2001) Levels of circulating CD8(+) T lymphocytes, natural killer cells, and eosinophils increase upon acute psychosocial stress in patients with atopic dermatitis. J. Allergy Clin. Immunol. 107, 171–177. [DOI] [PubMed] [Google Scholar]
- 13.Walzer, T., Dalod, M., Robbins, S. H., Zitvogel, L., Vivier, E. (2005) Natural‐killer cells and dendritic cells: “l'union fait la force”. Blood 106, 2252–2258. [DOI] [PubMed] [Google Scholar]
- 14.Kim, B. M., Kim, S. Y., Lee, S., Shin, Y. J., Min, B. H., Bendayan, M., Park, I. S. (2006) Clusterin induces differentiation of pancreatic duct cells into insulin‐secreting cells. Diabetologia 49, 311–320. [DOI] [PubMed] [Google Scholar]
- 15.Shin, Y. J., Kang, S. W., Jeong, S. Y., Shim, Y. J., Kim, Y. H., Kim, B. M., Kee, S. H., Park, J. J., Park, I. S., Min, B. H. (2006) Clusterin enhances proliferation of primary astrocytes through extracellular signal‐regulated kinase activation. Neuroreport 17, 1871–1875. [DOI] [PubMed] [Google Scholar]
- 16.Shim, Y. J., Shin, Y. J., Jeong, S. Y., Kang, S. W., Kim, B. M., Park, I. S., Min, B. H. (2009) Epidermal growth factor receptor is involved in clusterin‐induced astrocyte proliferation. Neuroreport 20, 435–439. [DOI] [PubMed] [Google Scholar]
- 17.Kim, J. H., Yu, Y. S., Kim, K. W., Min, B. H. (2007) The role of clusterin in in vitro ischemia of human retinal endothelial cells. Curr. Eye Res. 32, 693–698. [DOI] [PubMed] [Google Scholar]
- 18.Kim, J. H., Jun, H. O., Yu, Y. S., Min, B. H., Park, K. H., Kim, K. W. (2010) Protective effect of clusterin from oxidative stress‐induced apoptosis in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 51, 561–566. [DOI] [PubMed] [Google Scholar]
- 19.Jordan‐Starck, T. C., Lund, S. D., Witte, D. P., Aronow, B. J., Ley, C. A., Stuart, W. D., Swertfeger, D. K., Clayton, L. R., Sells, S. F., Paigen, B., et al. (1994) Mouse apolipoprotein J: characterization of a gene implicated in atherosclerosis. J. Lipid Res. 35, 194–210. [PubMed] [Google Scholar]
- 20.Huber, C., Thielen, C., Seeger, H., Schwarz, P., Montrasio, F., Wilson, M. R., Heinen, E., Fu, Y. X., Miele, G., Aguzzi, A. (2005) Lymphotoxin‐β receptor‐dependent genes in lymph node and follicular dendritic cell transcriptomes. J. Immunol. 174, 5526–5536. [DOI] [PubMed] [Google Scholar]
- 21.Lee, K. M., McNerney, M. E., Stepp, S. E., Mathew, P. A., Schatzle, J. D., Bennett, M., Kumar, V. (2004) 2B4 acts as a non‐major histocompatibility complex binding inhibitory receptor on mouse natural killer cells. J. Exp. Med. 199, 1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, H., Hong, C., Shin, J., Oh, S., Jung, S., Park, Y. K., Hong, S., Lee, G. R., Park, S. H. (2009) The presence of CD8+ invariant NKT cells in mice. Exp. Mol. Med. 41, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Silva, H. V., Harmony, J. A., Stuart, W. D., Gil, C. M., Robbins, J. (1990) Apolipoprotein J: structure and tissue distribution. Biochemistry 29, 5380–5389. [DOI] [PubMed] [Google Scholar]
- 24.Santarlasci, V., Maggi, L., Capone, M., Frosali, F., Querci, V., De Palma, R., Liotta, F., Cosmi, L., Maggi, E., Romagnani, S., Annunziato, F. (2009) TGF‐β indirectly favors the development of human Th17 cells by inhibiting Th1 cells. Eur. J. Immunol. 39, 207–215. [DOI] [PubMed] [Google Scholar]
- 25.Lanier, L. L., Buck, D. W., Rhodes, L., Ding, A., Evans, E., Barney, C., Phillips, J. H. (1988) Interleukin 2 activation of natural killer cells rapidly induces the expression and phosphorylation of the Leu‐23 activation antigen. J. Exp. Med. 167, 1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cochrane, D. R., Wang, Z., Muramaki, M., Gleave, M. E., Nelson, C. C. (2007) Differential regulation of clusterin and its isoforms by androgens in prostate cells. J. Biol. Chem. 282, 2278–2287. [DOI] [PubMed] [Google Scholar]
- 27.Essabbani, A., Margottin‐Goguet, F., Chiocchia, G. (2010) Identification of clusterin domain involved in NF‐κB pathway regulation. J. Biol. Chem. 285, 4273–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santilli, G., Aronow, B. J., Sala, A. (2003) Essential requirement of apoli‐poprotein J (clusterin) signaling for IκB expression and regulation of NF‐κB activity. J. Biol. Chem. 278, 38214–38219. [DOI] [PubMed] [Google Scholar]
- 29.Lenschow, D. J., Walunas, T. L., Bluestone, J. A. (1996) CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14, 233–258. [DOI] [PubMed] [Google Scholar]
- 30.July, L. V., Akbari, M., Zellweger, T., Jones, E. C., Goldenberg, S. L., Gleave, M. E. (2002) Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate 50, 179–188. [DOI] [PubMed] [Google Scholar]
- 31.Ranney, M. K., Ahmed, I. S., Potts, K. R., Craven, R. J. (2007) Multiple pathways regulating the anti‐apoptotic protein clusterin in breast cancer. Biochim. Biophys. Acta 1772, 1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamanaka, K., Gleave, M., Muramaki, M., Hara, I., Miyake, H. (2005) Enhanced radiosensitivity by inhibition of the anti‐apoptotic gene clusterin using antisense oligodeoxynucleotide in a human bladder cancer model. Oncol. Rep. 13, 885–890. [PubMed] [Google Scholar]
- 33.Wilson, M. R., Easterbrook‐Smith, S. B. (1992) Clusterin binds by a multivalent mechanism to the Fc and Fab regions of IgG. Biochim. Biophys. Acta 1159, 319–326. [DOI] [PubMed] [Google Scholar]
- 34.Lee, K. M., Forman, J. P., McNerney, M. E., Stepp, S., Kuppireddi, S., Guzior, D., Latchman, Y. E., Sayegh, M. H., Yagita, H., Park, C. K., Oh, S. B., Wulfing, C., Schatzle, J., Mathew, P. A., Sharpe, A. H., Kumar, V. (2006) Requirement of homotypic NK‐cell interactions through 2B4(CD244)/CD48 in the generation of NK effector functions. Blood 107, 3181–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min, B. H., Kim, B. M., Lee, S. H., Kang, S. W., Bendayan, M., Park, I. S. (2003) Clusterin expression in the early process of pancreas regeneration in the pancreatectomized rat. J. Histochem. Cytochem. 51, 1355–1365. [DOI] [PubMed] [Google Scholar]
- 36.Bento‐Abreu, A., Velasco, A., Polo‐Hernandez, E., Lillo, C., Kozyraki, R., Tabernero, A., Medina, J. M. (2009) Albumin endocytosis via megalin in astrocytes is caveola‐ and Dab‐1 dependent and is required for the synthesis of the neurotrophic factor oleic acid. J. Neurochem. 111, 49–60. [DOI] [PubMed] [Google Scholar]
- 37.Zlokovic, B. V., Martel, C. L., Matsubara, E., McComb, J. G., Zheng, G., McCluskey, R. T., Frangione, B., Ghiso, J. (1996) Glycoprotein 330/ megalin: probable role in receptor‐mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid β at the blood‐brain and blood‐cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. USA 93, 4229–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zwain, I. H., Grima, J., Cheng, C. Y. (1994) Regulation of clusterin secretion and mRNA expression in astrocytes by cytokines. Mol. Cell. Neurosci. 5, 229–237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Files