

Short abstract
Restoration of SPH levels through direct SPH administration, or conversion of increased ceramide to SPH by NC, reduces mortality and mitigates pulmonary infection after burn injury.
Keywords: acid sphingomyelinas, acid ceramidase, ceramide bronchial epithelial cells
Abstract
Burn patients with concomitant pulmonary Pseudomonas aeruginosa (PA) infection have mortality rates as high as 50%, despite antibiotic therapy. Sphingosine is generated from ceramide via ceramidase and has been reported to have antimicrobial properties. We observed a reduction in sphingosine and a concurrent increase in ceramide in bronchial epithelial cells after burn injury. After PA inoculation, these mice had a significant decrease in survival compared to noninjured mice. However, when injured mice were pretreated with sphingosine or neutral ceramidase and subsequently infected, mortality and bacterial levels were robustly reduced. We further observed that sphingosine directly kills PA. Together, these results demonstrate that reduction in sphingosine is associated with an increased susceptibility to pulmonary infection after burn injury. Restoration of sphingosine levels through direct sphingosine administration or conversion of the increased ceramide to sphingosine by neutral ceramidase reduces mortality and mitigates pulmonary infection after burn injury.
Abbreviations
- BALf
= bronchoalveolar lavage fluid
- CBA
= cytometric bead array
- NC
= neutral ceramidase
- PA
= Pseudomonas aeruginosa
- SPH
= sphingosine
Introduction
Infection is the most common cause of burn‐related morbidity and mortality [1, 2]. Significant progress has been made in the control of burn wound infections in severely injured patients, with improvements in wound care, early excision, and antibiotic administration. Because the occurrence of wound infections has significantly decreased, pneumonia has become the most frequent infection in burn‐injured patients and is the primary cause of mortality [2, 3, 4–5]. In particular, burn‐injured patients are highly susceptible to infection with PA, the single most frequently isolated bacterial pathogen [4, 6]. In patients with concomitant burn injury and pulmonary PA infection, mortality rates can be as high as 50%, despite antibiotic therapy [4, 7]. Further, pulmonary PA infections are of major clinical importance in patients with CF and chronic obstructive pulmonary disease [8]. Because of the high mortality rate and antibiotic resistance, there is a need to identify whether innate defense mechanisms are defective after trauma or burn injury and whether these mechanisms can be exploited for novel therapy. Reinforcing endogenous antimicrobial systems would circumvent this clinical problem.
Sphingolipids have been shown to contribute to the innate antimicrobial activity of the skin and mucosal surfaces [9, 10, 11–12]. Specifically, the sphingoid long‐chain base SPH protects skin from bacterial colonization [13] and pulmonary infection in a CF model [14]. SPH can be generated from the membrane lipid ceramide via hydrolysis by the enzyme NC [9]. Increases in ceramide concentration in epithelial cells have been associated with several aspects of pathogenicity, including increased uptake of bacterial pathogens such as Neisseria gonorrhea, Staphylococcus aureus, Escherichia coli, and Listeria monocytogenes, and induction of host cell death [15, 16, 17–18]. Alterations in the SPH/ceramide pathway have demonstrated increased susceptibility to PA infection [14, 19]. However, the role of SPH in the defense against pulmonary infection in burn injury has not been elucidated.
In this study, we investigated SPH in the increased burn‐related susceptibility to pulmonary infection. Specifically, we hypothesized that burn injury alters sphingolipid metabolism such that SPH is reduced, and restoration of SPH via administration of aerosolized SPH or NC will ameliorate pulmonary PA infection and improve survival in burn‐injured mice.
MATERIALS AND METHODS
Mice
Male 6‐wk‐old CF‐1 mice were purchased from Charles River Laboratories (Wilmington, MA, USA) and allowed to acclimate 1 wk before the experiments were conducted. The mice were housed in standard environmental conditions and fed a standard pellet diet and water ad libitum. All murine experiments were performed according to protocols approved by the Institution Animal Care and Use Committee (Protocol number 08‐09‐19‐01) of the University of Cincinnati.
Scald burn injury
Mice underwent full‐thickness scald injury [20]. In brief, the sham and scald procedures were always conducted between 8 and 11 AM. The mice were weighed and then anesthetized with 4.5% isoflurane in oxygen. Hair was clipped from their dorsal surface, and they were then placed in a template that exposed 28% of their total body surface area and immersed in 90°C water for 9 s resulting in a full‐thickness scald injury. After injury, mice received 1.5 ml of 0.9% saline, intraperitoneally and were then placed on a 42°C heating pad and allowed to recover for 3 h. Sham‐treated mice received the same treatment, except that they were immersed in room temperature water.
Bacterial growth and inoculation
The PA01 strain was grown for 14 h on tryptic soy agar plates (BD Biosciences, Franklin Lakes, NJ, USA). Bacteria were transferred to an Erlenmeyer flask containing 40 ml warmed tryptic soy broth (BD Biosciences), grown for 60 min at 37°C, resuspended in PBS (Thermo Fischer Scientific; Waltham, MA, USA), and diluted with PBS to the desired concentration of 1 × 106/20 μl with a standard bacterial growth curve. Mice were anesthetized with 3% isoflurane in oxygen and then were intranasally inoculated with 20 μl bacteria with a 31‐gauge 1 ml syringe.
Sphingosine and NC administration
Mice inhaled 800 μl of 0.9% NaCl containing 125 μM SPH (Avanti Polar Lipids, Inc., Alabaster, AL, USA) via a Vios Compressor and Nebulizer (Model 310B0003; PARI Respiratory Equipment, Midlothian, VA, USA). A second group of mice were intranasally inoculated with 0.15 μg of recombinant mouse NC (3558‐AH; R&D Systems, Minneapolis, MN, USA) in 20 μl PBS with a 31‐gauge 1 ml syringe. SPH or NC were administered 60 min before inoculation with PA.
Immunohistochemical analysis of SPH and NC
Mice were euthanized and the lungs were harvested, fixed in 10% neutral‐buffered formalin (Thermo Fisher Scientific), and stained with either Cy3‐coupled anti‐SPH antibodies (clone NHSPH; Alfresa Pharma Corporation, Osaka, Japan) or anti‐ceramide antibodies (clone S58‐9; Glycobiotech, Borstel, Germany)[14]. Immunofluorescence was analyzed using Leica software 2.61 (Leica Microsystems, Wetzlar, Germany) of 10 areas per sample, by measuring the fluorescence in the apical one‐third of the bronchial epithelial cells.
Bacteria incubation with SPH
PA was grown for 14 h on tryptic soy agar plates. Bacteria were diluted in PBS to a concentration of 1 × 103 CFU/ml. Solutions of 25 and 100 μM SPH were prepared and added to the bacteria. Solutions were incubated for 2 h at 37°C at 125 rpm. Subsequently, 100 μl of solution was plated on agar plates, incubated overnight at 37°C, the CFUs enumerated.
Bacterial load determination
Mice were euthanized, and BALf was harvested 4 h after bacterial inoculation. Normal saline was instilled through the trachea into the lungs with a 20‐gauge catheter, and 2 ml of fluid was removed. Analysis of BAL enabled quantification of cell type and number by flow cytometry and direct association of the data with the bacterial CFU. Samples were serially diluted in sterile saline and cultured on tryptic soy agar plates. Plates were incubated overnight at 37°C, and colony enumeration was performed.
Cytokine measurements by cytometric bead array
BALf samples were harvested as described and centrifuged at 450 g for 10 min. Cell‐free BALf was used to determine IL‐6 and ‐10 (BD Biosciences) levels by CBA, performed according to the manufacturer's protocol. The following antibodies were used to enumerate neutrophil numbers and activation: Ly‐6G (clone 1A8; BD Biosciences) and CD11b (clone M1/70, BD Biosciences). Cell and CBA assays were analyzed on Attune Acoustic Focusing Cytometer using Attune Cytometric Software v2 (Thermo Fisher Scientific).
Statistical analysis
Statistical comparisons were performed with the Kaplan Meier Log‐Rank Test (survival), Student t test (2 groups), or 1‐way ANOVA with the Tukey post hoc comparison (>2 groups), using Prism 6.0 (GraphPad Software, La Jolla, CA, USA). Data are expressed as means ± sem for experiments containing multiple data points. P ≤ 0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Characterization of SPH in the upper respiratory tract of burn‐injured mice
Although it is a long‐standing clinical observation that burn injury results in a marked susceptibility to pulmonary PA infection, the molecular mechanisms of this infection are still unclear. We have recently shown that SPH is a critical innate response mechanism of the upper respiratory tract that is defective in CF [14]. However, the effect of burn injury on the SPH/ceramide system in the lungs has not been elucidated. We hypothesized that the systemic stress response after burn injury reduces SPH expression in large airways. Immunohistochemical analysis using an anti‐SPH antibody and fluorescence microscopy demonstrated that SPH was abundantly expressed in bronchial epithelial cells of control mice ( Fig. 1A ). However, 24 h after burn injury, SPH expression was reduced by 4‐fold (Fig. 1B, C). At present, we assume that acid ceramidase is inhibited in epithelial cells of bronchi after burn injury, resulting in a decrease in SPH. However, the reduction is difficult to prove, because bronchi are not accessible to directly measure the activity of the acid ceramidase. Novel technologies are needed to address the local activity of ceramidase in bronchi.
Figure 1.
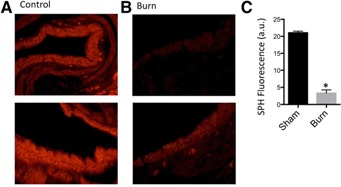
Sphingosine expression in bronchial epithelial cells of burn‐ and sham‐injured mice. Representative fluorescent staining of bronchial epithelial cells for Cy3‐coupled anti‐SPH antibodies from (A) sham and (B) burn‐injured mice are depicted. The images show the range of response of SPH levels before (control) after burn injury (burn). (C) Quantification of SPH expression (n = 4 mice/group). *P < 0.05 vs. sham group.
Studies have shown that tracheal and bronchial SPH levels are necessary for the defense against PA bronchopneumonia [14, 19]. Therefore, we next examined the role of reduced SPH in susceptibility to pulmonary infection after burn injury.
Role of SPH in bacterial pulmonary infection after burn injury
After intranasal infection with PA, burn‐injured mice had 100% mortality by 48 h, whereas infected sham mice had a significantly reduced mortality rate (28.6%; Fig. 2A ). Further, burn‐injured mice had increased sensitivity to PA, as demonstrated by a 3‐log increase in PA CFUs in BALf (Fig. 2B). Burn‐infected mice have a significant increase in the release of cytokines such as IL‐6 (Fig. 2D) and ‐10 (Fig. 2E), the release of which have been described as an important host response to PA [21]. The increased mortality and susceptibility of burn‐injured mice to pulmonary PA infection correlated with the observed reduction in SPH in bronchial epithelial cells.
Figure 2.
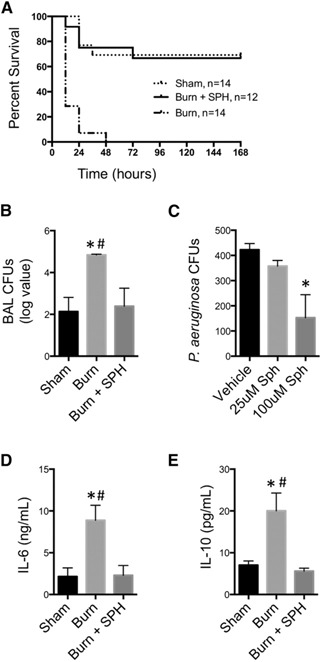
Survival, bacterial growth, and cytokine expression following SPH treatment. Sham‐, burn‐, or burn+SPH‐treated mice were inoculated with 1 × 106 CFU PA 24 h after injury. (A) Kaplan‐Meier survival curve is shown after inoculation (n = 12–14 mice/group). (B) Four hours after infection, BAL was harvested and CFUs quantified. (n = 8‐14 mice/group). (C) SPH was applied to PA in an in vitro assay and the CFUs enumerated. BALf was harvested 4 h after inoculation and IL‐6 (D) and ‐10 (E) were measured (n = 4–7 mice/group). *P < 0.05 vs. sham, # P < 0.05 vs. burn + SPH.
To further elucidate whether there was an association between the reduction in SPH in large airways and increased mortality and reduced bacterial clearance, we gave mice aerosolized SPH. Inhalation of SPH 1 h before PA inoculation, protected burn‐injured mice from pulmonary infection. Burn‐injured mice that received SPH treatment before inoculation had improved survival to that equivalent to sham (Fig. 2A). Further, bacterial loads and cytokine release in BALf were significantly reduced from infected burn‐injured mice and were equivalent to levels in infected sham mice (Fig. 2B, D, E). We further observed that there was no significant difference in survival or BAL bacterial load in infected sham mice treated with SPH as compared to untreated infected sham (data not shown). These data are consistent with research using a CF model that unhealthy or injured lungs are deficient in SPH and highly susceptible to PA [14]. Further, SPH has been demonstrated to have antimicrobial activity against bacteria found on the skin and oral mucosa [9, 11, 22]. Our data suggest that SPH is a critical first‐line defense of healthy airways against PA. After injury, SPH is reduced and allows for the invasion of PA and development of severe pulmonary infection. However, restoration of SPH in injured airways can mitigate pulmonary infection and improve survival in susceptible burn‐injured mice.
SPH kills bacteria directly
To verify the antimicrobial activity of SPH in the bacteria used in the present study under the described conditions, we performed in vitro assay with low micromolar concentrations of SPH. A stepwise reduction in bacterial growth was observed with increased SPH concentrations (Fig. 2C), confirming published data [14]. We have demonstrated that SPH kills PA directly, but the mechanism by which it kills bacterial pathogens is unknown. Recent studies suggest that SPH may cause ultrastructural damage in E. coli and S. aureus [23] or cause upregulation of porin proteins in the bacterial membrane [24]; however, further research is necessary to elucidate its mechanism of action.
Together, these data indicate that in healthy airways, high concentrations of SPH kill invading pathogens and protect from infection whereas low SPH, as occurs after burn injury, abrogate killing of invading bacterial pathogens and allow the development of PA infection. In addition, SPH is able to directly kill bacteria. Moreover, restoration of SPH through administration of aerosolized SPH protects against pulmonary PA infection and reduces mortality.
Characterization of ceramide in the upper respiratory tract of burn injured mice
SPH is generated from ceramide by the activity of acid or NC [25]. We hypothesized that with the reduction in SPH, we would observe a concurrent increase in ceramide expression in bronchial epithelial cells after burn injury. Immunohistochemical staining and fluorescence microscopy demonstrated that in healthy mice, ceramide concentrations are low in bronchial epithelial cells ( Fig. 3A ). After burn injury, there is a 4‐fold increase in ceramide expression (Fig. 3B, C). Thus, in burn injury the ceramide increase is concurrent to decreased SPH. This increase in ceramide after burn injury is consistent with previous research that has demonstrated accumulation of ceramide rich membranes following application of various stressful stimuli [26, 27–28]. As ceramide is a precursor to SPH, we sought to exploit the observed pathologic increase in ceramide by treatment with NC to increase SPH and ameliorate pulmonary infection. We used NC, because the pH in the airways is slightly acidic to neutral, and lower amounts of the enzyme are sufficient to achieve the same effect than that previously reported for the acid ceramidase [14].
Figure 3.
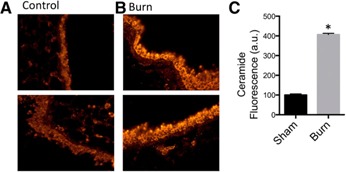
Ceramide expression in bronchial epithelial cells of burn‐ and sham‐injured mice. Representative fluorescent staining of bronchial epithelial cells with anti‐ceramide antibody from (A) sham‐ and (B) burn‐injured mice. (C) Quantification of ceramide expression (n = 4 mice/group). *P < 0.05 vs. sham group.
Neutral ceramidase mitigates PA pulmonary infection in burn‐injured mice
Ceramide accumulation is associated with increased susceptibility to bacterial infection [14, 29] and destruction of ceramide lipid membrane rafts prevents internalization of PA [30]. In the current study, we demonstrated that infected burn mice treated with NC, 1 h before PA inoculation significantly improved survival as compared to untreated infected burn mice ( Fig. 4A ). Further, treatment with inhaled NC results in a 2‐log reduction in PA CFUs in BAL fluid of infected burn mice (Fig. 4B). In addition, enumeration of PA CFUs demonstrates that there was no difference between infected burn mice treated with NC and infected sham mice. Furthermore, infected burn mice that received NC treatment had a significant reduction in cytokine release as compared to infected burn‐injured mice that was equivalent to levels in infected sham mice (Fig. 4C, D). These data indicate that an appropriate balance between ceramide and SPH is a key mechanism mediating important pathophysiological aspects of pulmonary bacterial infection in burns. The pathologic elevation in ceramide concentration in bronchial epithelial cells after burn injury can be exploited by the enzyme NC to reduce ceramide and therefore susceptibility to bacterial infection.
Figure 4.
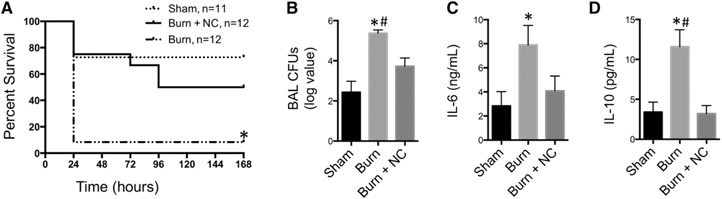
Survival, bacterial growth, and cytokine expression after NC treatment. Sham‐, burn‐, or burn+NC‐treated mice were inoculated with 1 × 106 CFU PA 24 h after injury. (A) Kaplan‐Meier survival curve after inoculation (n = 11–12 mice/group). (B) Four hours after infection, BAL was harvested and CFUs quantified (n = 12–13 mice/group). (C) IL‐6 and (D) IL‐10 were measured (n = 4–7 mice/group). *P < 0.05 vs. sham; # P < 0.05 vs. burn+NC.
In summary, these data establish an unexplored paradigm—namely, that SPH in tracheal and bronchial epithelial cells is critical in preventing pulmonary bacterial infection. The constitutive presence of high SPH concentrations in healthy bronchial epithelial cells directly kills bacterial pathogens. Meanwhile, a decrease in SPH and an increase in ceramide, as occurs under pathologic conditions, abrogates killing of invading pathogens, and facilitates the development of pulmonary PA infection. The discovery that burn injury affects sphingolipids by decreasing SPH and increasing ceramide may explain the high infection susceptibility of burn patients.
We also quantified neutrophils, inflammatory monocytes, and lymphocytes at the 4 h time point, but did not find a significant difference.
Ceramide and SPH are not the only molecules that contribute to the defense against bacterial infection. However, we demonstrated that normalizing SPH concentrations, through direct administration of SPH or using NC to exploit the pathologic increase in ceramide, prevents pulmonary infection in otherwise susceptible burn‐injured mice. This finding may provide a guide for novel treatment options in pulmonary infection. It is of great clinical significance, as many strains of PA are multidrug resistant, resulting in pulmonary infections that are difficult to treat. SPH has been shown to kill Moraxella catarrhalis, Borrelia burgdorferi, and Haemophilus influenzae [14], expanding its potential therapeutic benefit. Our data may motivate further investigations into sphingolipid biology to identify injuries or pathologies in which SPH is reduced or defective, and restoration has the potential to reduce morbidity and mortality by offering a novel antimicrobial therapy. Such patients would then have the opportunity to receive therapy to prevent pulmonary infection rather than postinfection treatment strategies.
AUTHORSHIP
T.C.R, contributed to the study's design and performed the experiments; A.P.S. performed the experiments; M.J.E. and C.C.C. contributed to the study's conception and design; and E.G. contributed to the study's conception and design and performed the experiments.
DISCLOSURES
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by the Shriners Hospitals for Children Grant 85600‐CIN‐16 (to C.C.C.). The project described was supported by U.S. National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS) Grants R01 GM100913 (to C.C.C.) and T32 GM08478 (to T.C.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the NIH.
Footnotes
SEE CORRESPONDING EDITORIAL ON PAGE 1227
References
- 1. Coban, Y. K. (2012) Infection control in severely burned patients. World J. Crit. Care Med. 1, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pruitt, B. A., Jr. , McManus, A. T. (1992) The changing epidemiology of infection in burn patients. World J. Surg. 16, 57–67. [DOI] [PubMed] [Google Scholar]
- 3. Canadian Critical Care Trials Group ; Canadian Critical Care Society . (2004) Evidence‐based clinical practice guideline for the prevention of ventilator‐associated pneumonia. Ann. Intern. Med. 141, 305–313. [DOI] [PubMed] [Google Scholar]
- 4. Morrison, A. J., Jr. , Wenzel, R. P. (1984) Epidemiology of infections due to Pseudomonas aeruginosa. Rev. Infect. Dis. 6 (Suppl 3), S627–S642. [DOI] [PubMed] [Google Scholar]
- 5. National Nosocomial Infections Surveillance (NNIS) System Report . (2001) Data summary from January 1992‐June 2001, issued August 2001. Am. J. Infect. Control 29, 404–421. [DOI] [PubMed] [Google Scholar]
- 6. Cook, D. J. , Kollef, M. H. (1998) Risk factors for ICU‐acquired pneumonia. JAMA 279, 1605–1606. [DOI] [PubMed] [Google Scholar]
- 7. Vidal, F. , Mensa, J. , Almela, M. , Martínez, J. A. , Marco, F. , Casals, C. , Gatell, J. M. , Soriano, E. , Jimenez de Anta, M. T. (1996) Epidemiology and outcome of Pseudomonas aeruginosa bacteremia, with special emphasis on the influence of antibiotic treatment: analysis of 189 episodes. Arch. Intern. Med. 156, 2121–2126. [PubMed] [Google Scholar]
- 8. Ratjen, F. , Döring, G. (2003) Cystic fibrosis. Lancet 361, 681–689. [DOI] [PubMed] [Google Scholar]
- 9. Fischer, C. L. , Drake, D. R. , Dawson, D. V. , Blanchette, D. R. , Brogden, K. A. , Wertz, P. W. (2012) Antibacterial activity of sphingoid bases and fatty acids against Gram‐positive and Gram‐negative bacteria. Antimicrob. Agents Chemother. 56, 1157–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cameron, D. J. , Tong, Z. , Yang, Z. , Kaminoh, J. , Kamiyah, S. , Chen, H. , Zeng, J. , Chen, Y. , Luo, L. , Zhang, K. (2007) Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int. J. Biol. Sci. 3, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drake, D. R. , Brogden, K. A. , Dawson, D. V. , Wertz, P. W. (2008) Thematic review series. Skin lipids: antimicrobial lipids at the skin surface. J. Lipid Res. 49, 4–11. [DOI] [PubMed] [Google Scholar]
- 12. Smith, K. R. , Thiboutot, D. M. (2008) Thematic review series. Skin lipids: sebaceous gland lipids—friend or foe? J. Lipid Res. 49, 271–281. [DOI] [PubMed] [Google Scholar]
- 13. Bibel, D. J. , Aly, R. , Shinefield, H. R. (1992) Antimicrobial activity of sphingosines. J. Invest. Dermatol. 98, 269–273. [DOI] [PubMed] [Google Scholar]
- 14. Pewzner‐Jung, Y. , Tavakoli Tabazavareh, S. , Grassmé, H. , Becker, K. A. , Japtok, L. , Steinmann, J. , Joseph, T. , Lang, S. , Tuemmler, B. , Schuchman, E. H. , Lentsch, A. B. , Kleuser, B. , Edwards, M. J. , Futerman, A. H. , Gulbins, E. (2014) Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol. Med. 6, 1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Esen, M. , B. Schreiner, V. Jendrossek, F. Lang, K. Fassbender, H. Grassme, E. Gulbins . 2001. Mechanisms of Staphylococcus aureus induced apoptosis of human endothelial cells. Apoptosis 6, 431–439. [DOI] [PubMed] [Google Scholar]
- 16. Faulstich, M. , Hagen, F. , Avota, E. , Kozjak‐Pavlovic, V. , Winkler, A. C. , Xian, Y. , Schneider‐Schaulies, S. , Rudel, T. (2015) Neutral sphingomyelinase 2 is a key factor for PorB‐dependent invasion of Neisseria gonorrhoeae. Cell. Microbiol. 17, 241–253. [DOI] [PubMed] [Google Scholar]
- 17. Grassmé, H. , Gulbins, E. , Brenner, B. , Ferlinz, K. , Sandhoff, K. , Harzer, K. , Lang, F. , Meyer, T. F. (1997) Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell 91, 605–615. [DOI] [PubMed] [Google Scholar]
- 18. Utermöhlen, O. , Karow, U. , Löhler, J. , Krönke, M. (2003) Severe impairment in early host defense against Listeria monocytogenes in mice deficient in acid sphingomyelinase. J. Immunol. 170, 2621–2628. [DOI] [PubMed] [Google Scholar]
- 19. Grassmé, H. , Jendrossek, V. , Riehle, A. , von Kürthy, G. , Berger, J. , Schwarz, H. , Weller, M. , Kolesnick, R. , Gulbins, E. (2003) Host defense against Pseudomonas aeruginosa requires ceramide‐rich membrane rafts. Nat. Med. 9, 322–330. [DOI] [PubMed] [Google Scholar]
- 20. Adediran, S. G. , Dauplaise, D. J. , Kasten, K. R. , Tschöp, J. , Dattilo, J. , Goetzman, H. S. , England, L. G. , Cave, C. M. , Robinson, C. T. , Caldwell, C. C. (2010) Early infection during burn‐induced inflammatory response results in increased mortality and p38‐mediated neutrophil dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R918–R925. [DOI] [PubMed] [Google Scholar]
- 21. Schultz, M. J. , Rijneveld, A. W. , Florquin, S. , Edwards, C. K. , Dinarello, C. A. , van der Poll, T. (2002) Role of interleukin‐1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L285–L290. [DOI] [PubMed] [Google Scholar]
- 22. Fayet, B. , Koster, H. , Benabderrazik, S. , Bernard, J. A. , Pouliquen, Y. (1991) Six canalicular stenoses after 34 punctal plugs. Eur. J. Ophthalmol. 1, 154–155. [DOI] [PubMed] [Google Scholar]
- 23. Fischer, C. L. , Walters, K. S. , Drake, D. R. , Blanchette, D. R. , Dawson, D. V. , Brogden, K. A. , Wertz, P. W. (2013) Sphingoid bases are taken up by Escherichia coli and Staphylococcus aureus and induce ultrastructural damage. Skin Pharmacol. Physiol. 26, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. LaBauve, A. E. , Wargo, M. J. (2014) Detection of host‐derived sphingosine by Pseudomonas aeruginosa is important for survival in the murine lung. PLoS Pathog. 10, e1003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henry, B. , Ziobro, R. , Becker, K. A. , Kolesnick, R. , Gulbins, E. (2013) Acid sphingomyelinase. Handbook Exp. Pharmacol. 215, 77–88. [DOI] [PubMed] [Google Scholar]
- 26. Charruyer, A. , Grazide, S. , Bezombes, C. , Müller, S. , Laurent, G. , Jaffrézou, J. P. (2005) UV‐C light induces raft‐associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J. Biol. Chem. 280, 19196–19204. [DOI] [PubMed] [Google Scholar]
- 27. Lacour, S. , Hammann, A. , Grazide, S. , Lagadic‐Gossmann, D. , Athias, A. , Sergent, O. , Laurent, G. , Gambert, P. , Solary, E. , Dimanche‐Boitrel, M. T. (2004) Cisplatin‐induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 64, 3593–3598. [DOI] [PubMed] [Google Scholar]
- 28. Rotolo, J. A. , Zhang, J. , Donepudi, M. , Lee, H. , Fuks, Z. , Kolesnick, R. (2005) Caspase‐dependent and ‐independent activation of acid sphingomyelinase signaling. J. Biol. Chem. 280, 26425–26434. [DOI] [PubMed] [Google Scholar]
- 29. Seitz, A. P. , Grassmé, H. , Edwards, M. J. , Pewzner‐Jung, Y. , Gulbins, E. (2015) Ceramide and sphingosine in pulmonary infections. Biol. Chem. 396, 611–620. [DOI] [PubMed] [Google Scholar]
- 30. Kowalski, M. P. , Pier, G. B. (2004) Localization of cystic fibrosis transmembrane conductance regulator to lipid rafts of epithelial cells is required for Pseudomonas aeruginosa‐induced cellular activation. J. Immunol. 172, 418–425. [DOI] [PubMed] [Google Scholar]