

Abstract
The innate immune response of phagocytes to microbes has long been known to depend on the core signaling cascades downstream of pattern recognition receptors (PRRs), which lead to expression and production of inflammatory cytokines that counteract infection and induce adaptive immunity. Cell‐autonomous responses have recently emerged as important mechanisms of innate immunity. Either IFN‐inducible or constitutive, these processes aim to guarantee cell homeostasis but have also been shown to modulate innate immune response to microbes and production of inflammatory cytokines. Among these constitutive cell‐autonomous responses, autophagy is prominent and its role in innate immunity has been well characterized. Other stress responses, such as metabolic stress, the ER stress/unfolded protein response, mitochondrial stress, or the DNA damage response, seem to also be involved in innate immunity, although the precise mechanisms by which they regulate the innate immune response are not yet defined. Of importance, these distinct constitutive cell‐autonomous responses appear to be interconnected and can also be modulated by microbes and PRRs, which add further complexity to the interplay between innate immune signaling and cell‐autonomous responses in the mediation of an efficient innate immune response.
Keywords: autophagy, mTOR, ER stress/UPR, mitochondrial stress, DNA damage response, stress granules
Short abstract
Review of constitutive cell‐autonomous responses such as autophagy, metabolic stress, ER stress/UPR, and mitochondrial stress emerge as decisive regulators of innate immune response to microbes.
Abbreviations
- ATF
activating transcription factor
- ATG
autophagy‐related
- CHOP
C/EBP homologous protein
- DC
dendritic cell
- DDR
DNA damage response
- dsDNA
double‐tranded DNA
- ER
endoplasmic reticulum
- IRE1α
inositol‐requiring enzyme 1α
- IRF
IFN regulatory factor
- mTOR
mammalian target of rapamycin
- mTORC1/2
mammalian target of rapamycin complex 1/2
- mtROS
mitochondrial reactive oxygen species
- PAMP
pathogen‐associated molecular pattern
- PERK
protein kinase RNA‐like endoplasmic reticulum kinase
- PRR
pattern recognition receptor
- RLR
RIG‐I–like receptor
- ROS
reactive oxygen species
- UPR
unfolded protein response
- XBP‐1
X‐box binding protein 1
Introduction
Decades of work have shown that recognition of microbes by the innate immune system relies on the detection of PAMPs by PRRs expressed by cells, notably, professional phagocytes [1, 2]. Among these PRRs, surface molecules, such as TLRs or C‐type lectin receptors, and cytoplasmic molecules, such as NOD‐like receptors or RLRs, are specifically involved in the recognition of particular microbial components [3]. Each of these receptors mobilizes specific signaling cascades that modify gene expression to create an innate immune response that involves phagocytosis and degradation of the microbe, production of inflammatory cytokines, and establishment of an adaptive immune response [3, 4].
Beyond this well‐characterized PAMP engagement of PRRs and downstream signaling pathways, innate immunity also depends on cell‐autonomous responses, which include both IFN‐inducible and constitutive processes that help degrade microbes and counteract infection [5, 6]. Constitutive cell‐autonomous immunity mobilizes preexisting processes and molecules to quickly target microbes and defend the cell and the host against infection and is therefore considered to be the first line of immune defense. On the contrary, IFN‐inducible cell‐autonomous defense mechanisms involve detection of PAMPs by compartment‐specific PRRs, production of IFN, and activation of IFN‐inducible genes and, therefore, act at a longer period after infection. These IFN‐inducible mechanisms include oxidative and nitrosative defense as well as nutrient‐restrictive and membranolytic activities to defend the host cells [5].
Here, we examine the role of constitutive cell‐autonomous responses, whose involvement in the innate immune response to microbes has only been appreciated within the last few years, and we focus particularly on cellular stress responses. What is the nature of these responses? How do microbes modulate them? How can they, in turn, interact with PRR signaling cascades and regulate the innate immune response to infection, production of cytokines, and degradation of microbes?
AUTOPHAGY
Mechanisms and functions of autophagy
The first cell‐autonomous process that has been associated with innate immunity is macroautophagy, which is commonly referred to as autophagy. Autophagy is a highly conserved process in eukaryotic cells that maintains cell homeostasis by degrading and recycling cytoplasmic components, such as defective organelles or protein aggregates. Autophagy relies on the formation of a characteristic double‐membrane structure called an autophagosome, which sequesters cytosolic autophagy targets and finally fuses with a lysosome, leading to degradation of autophagosomal contents [7, 8]. Autophagy relies on the activity of at least 30 conserved ATG proteins that act sequentially in 3 macromolecular complexes involved in the 3 successive stages of autophagy: initiation, which involves the ULK1‐ATG13‐FIP200 complex; membrane nucleation, which requires the Beclin1‐PI3K complex; and membrane elongation, with the involvement of ATG8/LC3 lipidation [9]. ATG8/LC3 lipidation is a hallmark of autophagy and is established by a covalent linkage of cytosolic LC3 to the lipid phosphatidylethanolamine on the surface of the autophagosome, which enables autophagosome elongation and recruitment of autophagy targets [8, 9]. Autophagy is a selective process as a result of the involvement of autophagy receptors (e.g., NDP52, p62, and optineurin), which physically link ubiquitinated autophagy targets to ubiquitin‐like proteins of the ATG8/LC3 family displayed on the autophagosomal membrane [10, 11]. Of note, autophagy can be divided into subtypes depending on the organelle that is targeted for autophagic degradation. For instance, mitophagy corresponds to autophagy of mitochondria, whereas ER‐phagy corresponds to autophagy of the ER [12, 13].
In addition to macroautophagy, 2 other types of autophagy have been described: microautophagy and chaperone‐mediated autophagy. Unlike autophagy, microautophagy does not involve the autophagosome‐dependent degradation of cytoplasmic components but, rather, relies on the direct engulfment of cytoplasmic material into lysosomes, which leads to their degradation [14, 15]. Chaperone‐mediated autophagy corresponds to the translocation of cytosolic proteins into lysosomes via a process that requires protein interaction with the chaperone Hsc70 and association of Hsc70 with the lysosomal protein LAMP‐2A [14, 16].
Modulation of autophagy by microbes
Autophagy has also long been known to be co‐opted by cells to defend the cytoplasm in case of microbial infection, to attack bacteria‐containing phagosomes, and to degrade pathogen‐derived inclusion bodies [5, 6, 17]. Indeed, autophagy participates in the degradation of viruses or bacteria such as Mycobacterium or Salmonella, which invade the cytoplasm during the infection process and is therefore sometimes called xenophagy [17, 18–19]. Once in the cytoplasm, these bacteria undergo active ubiquitination—for example, by the E3 ubiquitin ligase LRSAM1 [17, 20]—interact with autophagy receptors NDP52, p62, and optineurin [11, 21, 22–23], and are therefore recruited to the LC3‐containing autophagosomes where they are degraded ( Fig. 1 ). A fast ubiquitin‐independent autophagy has recently been proposed in light of observations that show that the cytosolic protein galectin‐8 is recruited to Salmonella‐containing phagosomes upon exposure of intraphagosomal glycans and subsequently interacts with autophagy receptor NDP52 to recruit the autophagy machinery [24]. In the case of viral infection, proteins from capsids were shown to bind directly to autophagy receptor p62, which led to virus degradation by autophagy [25].
Figure 1.
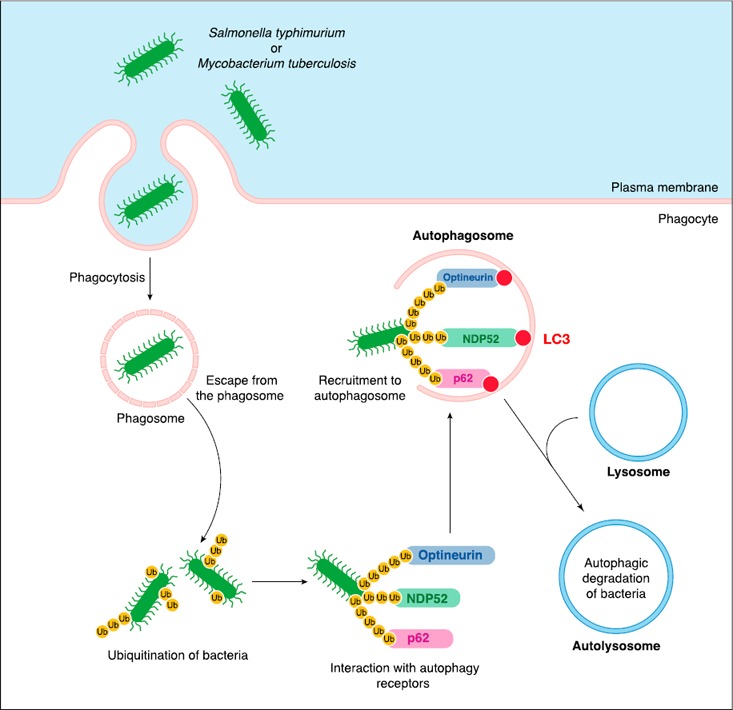
Autophagy is a defense mechanism against infection. After phagocytosis, invasive bacteria, such as S. typhimurium or Mycobacterium tuberculosis, escape the phagosome through the action of virulence factors. Once released into the cytoplasm, bacteria are targeted by the host ubiquitination machinery, which enables the interaction with autophagy receptors p62, NDP52, and optineurin and the recruitment of bacteria to preformed LC3‐labeled autophagosomes. Fusion of mature autophagosomes with lysosomes leads to autophagic degradation of bacteria.
Physiologic basal levels of autophagy in cells may lead to microbial degradation, but whether and how microbes can directly activate or modulate autophagy remains less well understood and may well be context dependent. Several PRRs have been shown to link microbial detection to autophagy induction depending on the microbe/host cell considered. For example, TLR signaling pathways have been shown to induce autophagy, particularly via TLR signaling adaptor TRIF (TIR (Toll/IL‐1R) domain‐containing adapter inducing IFN‐β) [26, 27, 28–29]. Other cytosolic PRRs, such as the NOD‐like receptor NOD2 and its effector kinase RIPK2 (receptor‐interacting serine/threonine protein kinase 2), can induce autophagy in phagocytes via interaction with autophagy protein ATG16 and direct induction of membrane nucleation [30]. RLRs, such as RIG‐I, were also proposed to modulate autophagy, although the relationship between RLRs and the autophagy machinery seems reciprocal and complex [31, 32]. The precise molecular mechanisms by which PRRs modulate autophagy remain largely unknown ( Fig. 2 ).
Figure 2.
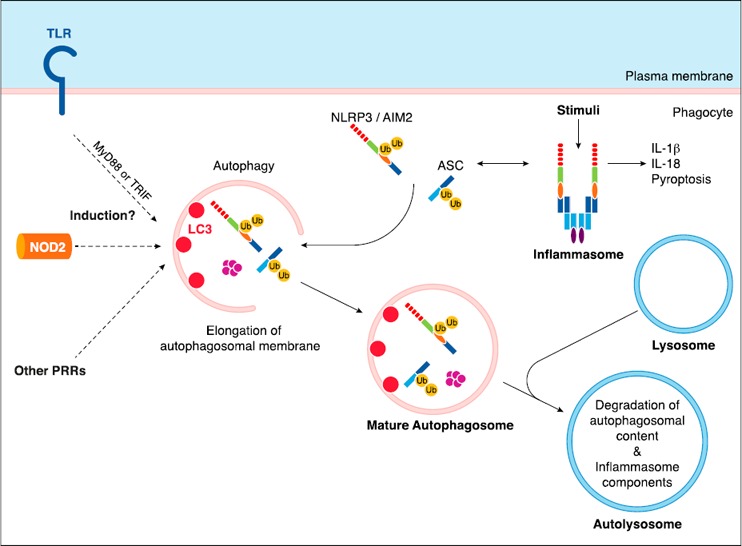
Autophagy: induction by PRRs and inhibition of inflammasomes. PRRs, such as TLRs or NOD2, can induce autophagy via unknown molecular mechanisms. In response to diverse stimuli, NLRP3 and AIM2 inflammasomes are assembled and lead to production of proinflammatory cytokines IL‐1β and IL‐18. Autophagy is known to inhibit inflammasome activity. Inflammasome components NLRP3, AIM2, and ASC have been shown to be ubiquitinated (Ub) and degraded via autophagosomes, which explains inflammasome inhibition by autophagy.
Some microbes have evolved mechanisms to directly inhibit or escape the autophagy response within the cytoplasm. Although Mycobacteria and Salmonella can be degraded by autophagy, these bacteria have evolved mechanisms that directly inhibit autophagy initiation and, therefore, counteract their degradation by host cells [33, 34]. Alternatively, Shigella flexneri has been shown to mask its bacterial surface, thus avoiding ubiquitination and recognition by autophagy machinery [35]. Another example concerns Listeria monocytogenes, although targeted and degraded by autophagy, which takes advantage of 2 independent virulence factors to escape the autophagy machinery: i) ActA, which enables actin polymerization and mediates movement of bacteria inside the cell, thus escaping recruitment to autophagosomes; and ii) IlnK, which enables decoration of bacteria with the major vault protein, thus avoiding ubiquitination and counteracting autophagic degradation [36, 37].
Involvement of autophagy in the innate immune response
Similar to mechanisms of autophagy induction by microbes, the impact of autophagy on the innate immune response to microbes, in particular on the production of inflammatory cytokines by phagocytes, seems to be context dependent. Autophagy has been shown to inhibit both NLRP3 and AIM2 inflammasome activation and subsequent production of proinflammatory cytokines IL‐1β and IL‐18 in response to diverse infectious stimuli both in vitro and in vivo and primarily in response to gram‐negative bacteria [38, 39]. A possible proposed mechanism is that inflammasome components and pro–IL‐1β are subjected to ubiquitination and subsequent degradation by autophagy, thereby leading to functional inactivation of inflammasomes [40, 41] (Fig. 2). Furthermore, autophagy has previously been linked to induction of type I IFN responses to viruses, such as vesicular stomatitis virus or Sendai virus, by plasmacytoid DCs, whereby autophagy facilitates delivery of cytosolic PAMPs to endosomal TLR7 and therefore enables TLR7‐dependent type I IFN production [42]. In addition, LC3‐associated phagocytosis, which corresponds to recruitment of autophagy machinery components around phagosomes [28], has been shown to promote TLR9‐MyD88‐IRF‐7–dependent IFN‐α production in response to DNA‐immune complexes [43], whereby LC3‐associated phagocytosis enables rapid phagosome acidification required for type I IFN signaling downstream of TLRs [28, 44]. Alternatively, studies have also revealed that autophagy inhibits type I IFN production by RLRs RIG‐I and MDA‐5 in mouse embryonic fibroblasts that are infected with vesicular stomatitis virus. Indeed, it was shown that ATG5–ATG12 conjugates—essential components of autophagosome elongation—block RLR signaling by direct interaction with RIG‐I and IPS‐1, which results in inhibition of type I IFN production [31, 45]. Thus, autophagy has been shown to have opposing effects on cytokine production depending on the context, either inducing or inhibiting their production, which may reflect the subcellular compartments in which PRRs are engaged.
Despite the fact that autophagy is the best‐known cell‐autonomous response in innate immunity and has clearly been shown to counteract microbial infection, much is left unclear regarding the specific modulation of autophagy by microbes, the precise interaction of autophagy with innate immune signaling cascades, and the cooperation between autophagy and other physiologic cell autonomous processes during microbial infection.
METABOLIC STRESS AND THE METABOLIC SENSOR mTOR
Mechanisms and functions of the mTOR pathway
mTOR is considered to be the central sensor and regulator of cell metabolism and is associated with 2 different multiprotein complexes: mTORC1 and mTORC2 [46]. Of note, mTORC1 is activated by signaling downstream of growth factor receptors via the PI3K‐AKT‐TSC pathway, or by sensing of amino acids within the cytoplasm [47]. Signaling downstream of mTORC1 leads to activation of protein translation via phosphorylation of mTOR targets S6K and 4EBP1, but also to lipid or nucleic acid synthesis, as mTORC1 is considered to be a strong anabolic pathway [46, 48] (see Fig. 3 ). Of importance, mTORC1 is known as the main cytoplasmic inhibitor of autophagy by acting on the ULK1‐ATG13‐FIP200‐ATG101 initiation complex [46, 49, 50] (Fig. 3). Therefore, the mTORC1 pathway is of particular interest in the context of cell‐autonomous responses during microbial infection, and recent studies have revealed its involvement in the regulation of innate immunity.
Figure 3.
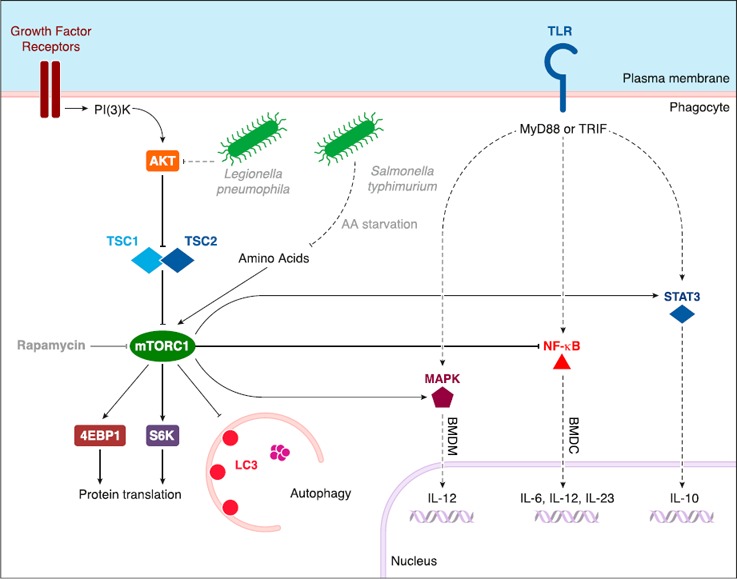
The mTORC1 pathway: effect on TLR signaling cascades and modulation by bacteria. mTORC1 is activated primarily downstream of growth factor receptors (receptor tyrosine kinase) via the PI3K‐AKT‐TSC pathway or by amino acids in the cytoplasm. mTORC1 activates 4EBP1 and S6K, which promote protein translation. mTORC1 is also the main cytosolic inhibitor of autophagy. mTORC1 regulates TLR signaling via activation of MAPK and STAT3 or inhibition of NF‐κB. The mTORC1 pathway is modulated by pathogenic bacteria through the inhibition of AKT (in response to L. pneumophila) or amino acid starvation (in response to S. typhimurium). TRIF = TIR (Toll/IL‐1R) domain‐containing adapter inducing IFN‐β.
Involvement of the mTOR pathway in the innate immune response
Implication of the mTOR pathway in innate immunity has mainly been shown via treatment of cells with rapamycin to inhibit mTOR. Such a treatment was shown to have a proinflammatory effect on phagocytic cells upon stimulation with LPS, which increased their capacity to produce cytokines, such as IL‐12, IL‐23, and IL‐6, and decreased production of anti‐inflammatory cytokine IL‐10. Indeed, mTOR was shown to inhibit NF‐κB activity—involved in expression of proinflammatory cytokines—and to activate STAT3, which is necessary for expression of IL‐10 [51] (Fig. 3). In addition, inhibition of the mTOR pathway with rapamycin also has immunostimulatory effects in murine cell models after bacterial infection [51, 52]. To circumvent potential rapamycin‐induced nonspecific effects or nonphysiologic consequences, genetic models have been established. Inactivation of mTORC1 by genetic deletion of Rptor, which encodes the mTORC1 component RAPTOR, in mouse intestinal DCs induces a severe inflammatory response to enteric bacteria after treatment with the intestinal epithelium–damaging agent dextran sodium sulfate [53]. Conversely, activation of mTORC1 via deletion of Tsc1—member of the mTOR inhibitory complex TSC—leads to a reduction of IL‐12 production in bone marrow–derived DCs [54]. However, deletion of Tsc1 leads to increased IL‐12 production upon LPS stimulation of bone marrow–derived macrophages as a result of the activation of JNK1 and/or JNK2 [54] (Fig. 3). Although IL‐12 transcription is broadly regulated by JNK1/2, the molecular basis for this specific activation of JNK1/2 in macrophages, and not in DCs, is unknown and suggests some context‐dependent regulation of immune signaling pathways by mTORC1 [54]. Altogether, these observations show that extrinsic or pharmacologic modulation of metabolic sensor mTOR is important for the regulation of innate immune response, although the precise mechanisms and involvement of autophagy in these responses are still unclear.
Modulation of the mTOR pathway by microbes
Similar to the undefined nature of the mechanisms by which mTOR regulates innate immune response, mechanisms that underlie the modulation of mTOR by microbes in the case of infection are also uncertain. A few studies have proposed that mTOR activity can be modulated by microbes, such as Legionella pneumophila, that inhibit the PI3K‐AKT‐mTOR pathway via ubiquitin‐dependent degradation of proteins that are upstream mTOR, which leads to an enhanced proinflammatory cytokine response in macrophages independent of autophagy modulation [52]. In addition, Shigella flexneri or Salmonella typhimurium were shown to trigger amino acid starvation in epithelial cells, presumably via membrane damage that activates the GCN2 (general control nondepressible 2)/ATF3‐dependent amino acid starvation, which results in mTORC1 inhibition and activation of autophagic clearance of bacteria [34] (Fig. 3). However, examples of such a regulation of mTORC1 activity are rare and the mechanisms that are involved in mTOR modulation, either in the upstream PI3K‐AKT cascade or in the modulation of amino acid content, as well as the involvement of autophagy, have not yet been identified.
ER STRESS/UPR
Mechanisms and functions of ER stress/UPR
ER stress corresponds to the accumulation of unfolded proteins in the ER and leads to mobilization of a UPR via activation of 3 different ER transmembrane proteins: PERK, IRE1α, and ATF6 [55]. Subsequently, PERK and IRE1α are autophosphorylated, while ATF6 is exported to the Golgi apparatus and undergoes 2 successive proteolytic cleavages. Signaling downstream of these 3 proteins establishes a selective program of protein translation and transcriptional activation of UPR target genes that are involved in the resolution of ER stress, of note, via ER‐associated degradation [55, 56–57] ( Fig. 4 ). It also leads to cell death through apoptosis, primarily via expression of the PERK‐dependent UPR target CHOP [57, 58].
Figure 4.
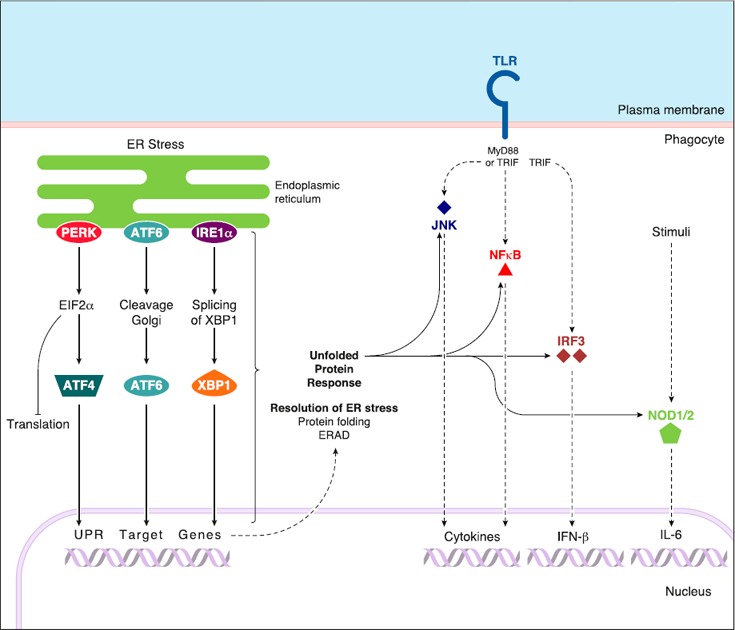
ER stress/UPR: effects on innate immunity. ER stress starts with the accumulation of unfolded proteins in the lumen of the ER. It activates a UPR via engagement of 3 UPR sensors and their respective signaling cascades: PERK‐EIF2α‐ATF4, ATF6, and IRE1α‐XBP‐1 pathways. These 3 UPR pathways lead to a global inhibition of protein translation and to expression of UPR target genes that are involved in the resolution of ER stress via protein folding processes or ER‐associated degradation of unfolded proteins (ERAD). ER stress/UPR was proposed to activate different immune signaling cascades, of note downstream of TLRs, such as JNK, NF‐κB, and IRF3 pathways, and, therefore, to promote the production of inflammatory cytokines. ER stress/UPR also activates NOD1/2‐dependent inflammatory cytokine production.
Of importance, ER stress/UPR seems to be linked to autophagy in as much as chemical induction of ER stress was shown to induce autophagy and genes that encode for autophagy‐related proteins, such as Atg12, are found among UPR target genes [59]. Moreover, a tight bidirectional relationship has been proposed between ER stress and the mTORC1 pathway [60] ( Fig. 5 ). Of note, JNK kinase, which has been described to inhibit the mTOR pathway [61], is activated downstream of IRE1α during ER stress/UPR [62], which makes IRE1α signaling a possible inhibitor of mTORC1 in the case of ER stress. Therefore, ER stress/UPR seems to be a possible cell‐autonomous defense mechanism against microbial infection.
Figure 5.
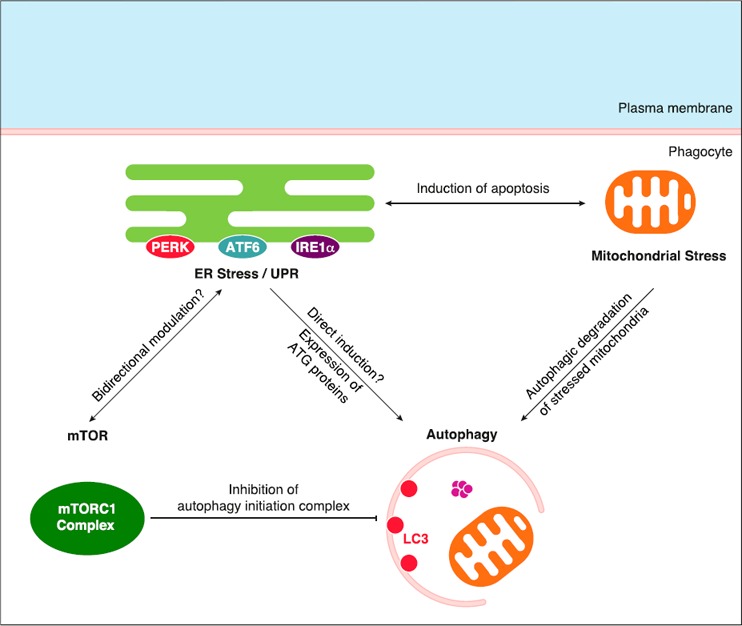
Connections between ER stress/UPR, mTORC1 pathway, autophagy, and mitochondrial stress. mTORC1 complex is known to inhibit autophagy via the inhibitory phosphorylation of components of the autophagy initiation complex. ER stress/UPR might promote autophagy directly or via expression of ATG proteins found among UPR target genes. Moreover, a bidirectional modulation seems to connect ER stress/UPR and the mTOR pathway, although no precise mechanism has been described. ER stress/UPR cooperates with mitochondrial stress (via Ca2+ flux and ROS production) to induce apoptosis. Stressed/damaged mitochondria are subjected to ubiquitination and recruitment to the autophagy machinery, which leads to their autophagic degradation.
Involvement of ER stress/UPR in the innate immune response
It has been proposed that ER stress/UPR intersects at various levels with innate inflammatory pathways and leads to the activation of NF‐κB, JNK, and IRF‐3 [63] (Fig. 4). For example, concomitant chemical activation of UPR with several TLR ligands clearly amplifies the cytokine responses of cells, such as i) type I IFN and IL‐6 production in mouse embryonic fibroblasts in a CHOP‐dependent manner; ii) IL‐23 production in DCs in a CHOP‐dependent manner; or iii) IL‐6, IL‐8, and TNF‐α production in macrophages in an IRE1α‐dependent manner that also involves XBP‐1, which is a target transcription factor downstream of the IRE1α‐dependent pathway [64, 65–66]. Similarly, ER stress inducers thapsigargin and DTT stimulate production of the proinflammatory cytokine IL‐6 in a NOD1/2‐dependent manner [67] (Fig. 4).
Of note, ER stress/UPR has been associated with inflammatory bowel disease [68]. Loss of XBP‐1 in intestinal epithelial or Paneth cells leads to severe enteritis associated with a considerable increase in activation of NF‐κB [68]. Of interest, polymorphisms in XBP1 have been found in patients with Crohn’s disease and ulcerative colitis, which suggests that improper function of major UPR branches could contribute to the inflammatory pathology that is typically observed in inflammatory bowel disease [69].
Modulation of ER stress/UPR by microbes
Although activation of TLRs affects UPR signaling pathways [66, 70], whether microbes can directly modulate ER stress/UPR through activation of PRRs remains rarely documented. Viruses, such as hepatitis C virus, have been proposed to modulate ER stress/UPR during the process of infection [71]. Bacteria, such as Listeria monocytogenes, which induces ER expansion before entry into host cells, or Brucella abortus, which replicates in ER‐derived structures, were also shown to induce an ER stress/UPR via respective virulence factors [67, 72, 73]. However, whether this direct physiologic modulation of ER stress/UPR by microbes impacts the innate immune response and production of cytokines is unknown, which keeps open many interesting questions regarding the contribution of cell‐autonomous responses to innate immunity.
MITOCHONDRIAL STRESS
Mechanisms and functions of mitochondrial stress
Mitochondria are required to maintain cellular energy levels; therefore, their health is crucial for cell viability. Mitochondrial dysfunction and stress result in excessive production of ROS and release of oxidized mitochondrial DNA in the cytosol, which triggers cell‐autonomous pathways that lead to apoptosis or to an inflammatory response.
Links have been made between ER and mitochondrial stress, in particular in the case of apoptosis induction by both ER stress and ROS generated by mitochondrial stress. Indeed, stressed ER and mitochondria cooperate to signal apoptosis, notably via the ER stress–induced Ca2+ flux from ER to mitochondria that results in calcium overload, generates mitochondrial depolarization, leads to ROS production, and subsequently induces apoptosis [74] (Fig. 5). In addition to this functional cooperation, some physical interactions between ER and mitochondria have also been observed during infection with Listeria monocytogenes, where Listeria causes mitochondrial fragmentation in a listeriolysin O–dependent manner, a process that is facilitated by the ER, and which is proposed to mechanically favor constriction and scission of mitochondria [75, 76].
Of importance, mitochondrial stress is also related to autophagy: mitochondrial dysfunction provokes recruitment of E3 ubiquitin ligase Parkin to the mitochondrial protein PINK1, which leads to polyubiquitination of mitochondrial proteins and recruitment of damaged mitochondria to the preformed LC3‐containing autophagosomes [77, 78] (Fig. 5). Altogether, these observations make mitochondrial stress a possible cell‐autonomous process that is involved in the regulation of innate immunity.
Involvement of mitochondrial stress in the innate immune response
In the case of mitochondrial dysfunction, mtROS released in the cytoplasm were shown to drive proinflammatory cytokine production. Indeed, mtROS influence the transcription of proinflammatory cytokines, such as IL‐6, by increasing the levels of active JNK and p38 [79]. mtROS are also involved in activation of the NLRP3 inflammasome and, thus, in production of IL‐1β upon stimulation of macrophages with an experimental stimulus, LPS and ATP [80, 81]. Of importance, mtROS‐producing damaged mitochondria also activate mitophagy, and the autophagy machinery then inhibits NLRP3 inflammasome activity, which restricts the inflammatory host response and maintains cell homeostasis [82]. Recently, it was also shown that mitochondrial DNA stress (i.e., the release of oxidized mitochondrial DNA in the cytosol upon mitochondrial stress) leads to enhanced expression of IFN‐stimulated genes via activation of the PRR cGAS [83]. Although the direct modulation of mitochondrial function by microbes is still unknown, mitochondrial stress seems to be a crucial process in the regulation of innate immune responses.
THE DDR
Mechanisms and functions of the DDR
Maintaining the integrity of nuclear DNA is required to ensure proper cellular function and viability as well as the transmission of genetic information to the progeny. Therefore, to avoid damage to nuclear DNA that imperils cell viability, cells have developed an arsenal of DNA damage sensors, signal transducers, and DNA repair systems, the aim of which is to sense DNA lesions, propagate the DDR, and eventually to repair the damaged DNA template [84]. Depending on the type of DNA lesions, different DNA damage sensors are involved. For example, DNA double strand breaks are recognized by the Mre11‐Rad50‐Nbs1 complex [85], UV‐induced DNA lesions are sensed by the XPC‐RAD23‐CETN2 complex [86], and mismatched and damaged bases are detected by MutS proteins and DNA glycosylases, respectively [87, 88]. After DNA damage recognition, the DDR is primarily mediated by proteins of the PI3K‐like protein kinase family, such as ATM, ATR, and DNA‐PK, and by members of the poly(ADP‐ribose) polymerase family [84]. Then, at least 6 DNA repair mechanisms are involved: base excision repair, nucleotide excision repair, mismatch repair, homologous recombination, nonhomologous end joining, and DNA interstrand cross‐link repair [89].
Involvement of the DDR in the innate immune response
Implication of the DDR in innate immunity has been revealed these last few years, as sensors of DNA damage were also characterized as proper PRRs involved in recognition of microbial DNA in the cytoplasm. For instance, Ku70, a DNA‐binding repair protein that is required for nonhomologous end joining, was identified as a cytosolic DNA sensor that induces IFN‐λ1 activation [90]. In addition, DNA‐PK, which forms a heterotrimer with Ku proteins (Ku70/Ku80), also acts as a DNA‐sensing PRR in the cytoplasm to induce transcription of IFN‐β via the STING–TBK1–IRF‐3 pathway [91]. Similarly, the DNA damage sensor Mre11 has also been shown to be involved in dsDNA‐mediated type I IFN production, acting upstream of the STING–TBK1–IRF‐3 axis. Of note, the DNA damage sensor Rad50, a protein that binds Mre11, is required for Mre11‐dependent recognition of dsDNA and activation of the STING pathway [92]. More recently, Rad50 was also involved in another innate immune pathway that was independent of STING. Indeed, Rad50 was shown to interact with the adaptor CARD9, and the subsequent dsDNA‐Rad50‐CARD9 signaling complex was required to activate NF‐κB and control the expression of pro–IL‐1β, either in the presence of dsDNA or in the case of infection of DCs with a DNA virus [93].
Of interest, DDR regulators ATM and ATR were also recently shown to inhibit the autophagy‐dependent degradation of the transcription factor GATA4 via unknown mechanisms, which resulted in accumulation of GATA4 and led to both inflammation through activation of NF‐κB and to cellular senescence [94]. This last example highlights an exciting connection between DDR and autophagy, the functions of which remain to be clarified.
Altogether, these recent findings show that DDR pathways intersect with DNA‐induced innate immune signaling and can modulate the production of cytokines; however, the precise mechanisms by which DNA damage sensors switch to innate immune recognition of microbial DNA are not yet understood. This raises interesting questions about the possible modulation of these sensors by microbes and innate immune pathways, which will assuredly be assessed in the next few years.
STRESS GRANULES
Under such stress conditions as oxidative stress or nutrient starvation, cells form dense aggregates in the cytosol that are composed of both mRNAs and RNA‐binding proteins and are called stress granules [95]. Stress granule formation is considered to be a mechanism to prevent the generation of abnormal proteins by transient stalling of translation, in particular translation initiation.
Among other stress conditions, viral infection has been shown to induce formation of stress granules, in particular in the case of many RNA viruses, such as Poliovirus [96]. The mechanisms of stress granule induction by viruses have been identified and involve activation of the eukaryotic translation initiation factor eIF2α kinases PKR and GCN2 (general control nondepressible 2), which leads to eIF2α phosphorylation and, therefore, to translation inhibition. Of importance, both kinases can be triggered by detection of viral RNA in the cytoplasm [96, 97]. Of note, modulation of eIF2α activity is a common mechanism that is shared by stress granule formation and ER stress/UPR, where PERK leads to eIF2α phosphorylation and, therefore, to translation inhibition (Fig. 4), which suggests the existence of possible cross‐talk between these two stress responses. Depending on the context, viruses can induce stable, transient, or oscillating/alternating stress granules. Transient stress granule formation is explained by the dissociation of crucial components of stress granules by viral proteins. For example, the viral 3C protease of several picornaviruses is responsible for the cleavage of stress granule protein G3BP1 (Ras‐GAP SH3 domain binding protein‐1), which leads to stress granule dissociation, whereas ectopic expression of a cleavage‐resistant form of G3BP1 stabilizes the picornaviruses‐induced stress granules [98, 99–100].
Of note, some viruses do not induce stress granules, and several lines of evidence suggest that these viruses actually inhibit stress granule formation. For instance, Mengovirus and Theiler’s murine encephalomyelitis virus do not generate stress granules in infected cells [101]. This occurs via a total inhibition of stress granule formation by the viral nonstructural L protein (leader protein), although the precise mechanism of this inhibition remains unknown [101]. Similarly, stress granules do not form in response to measles virus [102]. This inhibition is a result of the viral C protein, as C‐deficient measles virus highly induces stress granules, via an unidentified process [102]. Of importance, stress granules seem to have an antiviral role, and many viruses, such as Mengovirus or measles virus, clearly benefit from inhibition of stress granule formation by viral proteins. Indeed, an inverse correlation has been described between stress granule formation and viral propagation [96]. For instance, Japanese encephalitis virus inhibits stress granules via the viral core protein, which directly interacts with the stress granule component caprin 1 [103]. In response to a mutant virus in which viral core protein is not able to interact with caprin 1, stress granules are formed and viral propagation is impaired both in vitro and in vivo, which suggests that stress granules somehow inhibit viral replication. Of importance, studies have shown that some RLRs localize to stress granules that are formed after viral infection and propose that stress granules might act as a platform for viral RNA sensing and activation of downstream signaling pathways [104]. So far, however, there is no evidence that formation or inhibition of stress granules in the case of viral infection impacts the production of inflammatory cytokines and the innate immune response. Further work is required to determine whether stress granules directly impact innate immunity and whether they can be considered to be a crucial stress response that counteracts microbial infection.
CONCLUSION
Recent studies provide evidence that constitutive cell‐autonomous stress responses—the initial goal of which is to maintain cellular homeostasis and ensure cell integrity—are also crucial processes in the innate immune response to microbes. Autophagy is the best‐known example of such a cell‐autonomous defense mechanism, with involvement in response to various viral or bacterial stimuli and an effect on production of several inflammatory cytokines and in the proper degradation of microbes. Other physiologic stress responses, such as mTOR, ER stress/UPR, and mitochondrial stress, have been proposed to modulate innate immune responses. Other mechanisms, such as DDR or stress granules, are also host stress responses that intersect with innate immune pathways that are involved during microbial infection, although less is known about their modulation in innate immune cells. More work is needed to understand the clear involvement of all these processes in response to microbial infection and to determine the mechanisms by which microbes could directly modulate these processes (perhaps upon their detection by PRRs) and by which these stress responses, in turn, modify the production of cytokines.
Strikingly, these cell‐autonomous responses, which are all highly conserved among eukaryotic species, seem to be interconnected and interdependent. Again, autophagy appears in the center of all cell‐autonomous stress responses, which may reflect its central role in the maintenance of cell homeostasis, depending on metabolic stress (via mTOR pathway), oxidative stress (via mitochondrial stress), or ER stress. Furthermore, this interconnection suggests the existence of a complex and cooperative network of stress processes to draw an efficient innate immune response and contain the infection. There is no doubt that understanding the connections between constitutive cell‐autonomous and innate immune responses will provide considerable insights into the fight against microbial infection.
AUTHORSHIP
J.M. and J.M.B. wrote the review.
DISCLOSURES
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by institutional seed funds to J.M.B. J.M. was supported in part by an award from the Philippe Foundation. J.M.B. and her laboratory are supported by U.S. National Institutes of Health Grants DK072201, AI095245, and AI123284; the Burroughs Wellcome Fund; and a Leukemia and Lymphoma Society Scholar Award.
REFERENCES
- 1. Janeway, C. A., Jr. (1989) Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54, 1–13. [DOI] [PubMed] [Google Scholar]
- 2. Janeway, C. A., Jr. (1992) The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today 13, 11–16. [DOI] [PubMed] [Google Scholar]
- 3. Thompson, M. R. , Kaminski, J. J. , Kurt‐Jones, E. A. , Fitzgerald, K. A. (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moretti, J. , Blander, J. M. (2014) Insights into phagocytosis‐coupled activation of pattern recognition receptors and inflammasomes. Curr. Opin. Immunol. 26, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. MacMicking, J. D. (2012) Interferon‐inducible effector mechanisms in cell‐autonomous immunity. Nat. Rev. Immunol. 12, 367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Randow, F. , MacMicking, J. D. , James, L. C. (2013) Cellular self‐defense: how cell‐autonomous immunity protects against pathogens. Science 340, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. He, C. , Klionsky, D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizushima, N. (2007) Autophagy: process and function. Genes Dev. 21, 2861–2873. [DOI] [PubMed] [Google Scholar]
- 9. Glick, D. , Barth, S. , Macleod, K. F. (2010) Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kirkin, V. , McEwan, D. G. , Novak, I. , Dikic, I. (2009) A role for ubiquitin in selective autophagy. Mol. Cell 34, 259–269. [DOI] [PubMed] [Google Scholar]
- 11. Kraft, C. , Peter, M. , Hofmann, K. (2010) Selective autophagy: ubiquitin‐mediated recognition and beyond. Nat. Cell Biol. 12, 836–841. [DOI] [PubMed] [Google Scholar]
- 12. Bernales, S. , Schuck, S. , Walter, P. (2007) ER‐phagy: selective autophagy of the endoplasmic reticulum. Autophagy 3, 285–287. [DOI] [PubMed] [Google Scholar]
- 13. Youle, R. J. , Narendra, D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boya, P. , Reggiori, F. , Codogno, P. (2013) Emerging regulation and functions of autophagy. Nat. Cell Biol. 15, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li, W. W. , Li, J. , Bao, J. K. (2012) Microautophagy: lesser‐known self‐eating. Cell. Mol. Life Sci. 69, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaushik, S. , Bandyopadhyay, U. , Sridhar, S. , Kiffin, R. , Martinez‐Vicente, M. , Kon, M. , Orenstein, S. J. , Wong, E. , Cuervo, A. M. (2011) Chaperone‐mediated autophagy at a glance. J. Cell Sci. 124, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levine, B. , Mizushima, N. , Virgin, H. W. (2011) Autophagy in immunity and inflammation. Nature 469, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang, J. , Brumell, J. H. (2014) Bacteria‐autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 12, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jo, E. K. , Yuk, J. M. , Shin, D. M. , Sasakawa, C. (2013) Roles of autophagy in elimination of intracellular bacterial pathogens. Front. Immunol. 4, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huett, A. , Heath, R. J. , Begun, J. , Sassi, S. O. , Baxt, L. A. , Vyas, J. M. , Goldberg, M. B. , Xavier, R. J. (2012) The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin‐dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe 12, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thurston, T. L. , Ryzhakov, G. , Bloor, S. , von Muhlinen, N. , Randow, F. (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin‐coated bacteria. Nat. Immunol. 10, 1215–1221. [DOI] [PubMed] [Google Scholar]
- 22. Wild, P. , Farhan, H. , McEwan, D. G. , Wagner, S. , Rogov, V. V. , Brady, N. R. , Richter, B. , Korac, J. , Waidmann, O. , Choudhary, C. , Dötsch, V. , Bumann, D. , Dikic, I. (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng, Y. T. , Shahnazari, S. , Brech, A. , Lamark, T. , Johansen, T. , Brumell, J. H. (2009) The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916. [DOI] [PubMed] [Google Scholar]
- 24. Thurston, T. L. , Wandel, M. P. , von Muhlinen, N. , Foeglein, A. , Randow, F. (2012) Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orvedahl, A. , MacPherson, S. , Sumpter, R., Jr. , Tallóczy, Z. , Zou, Z. , Levine, B. (2010) Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Delgado, M. A. , Deretic, V. (2009) Toll‐like receptors in control of immunological autophagy. Cell Death Differ. 16, 976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jabir, M. S. , Ritchie, N. D. , Li, D. , Bayes, H. K. , Tourlomousis, P. , Puleston, D. , Lupton, A. , Hopkins, L. , Simon, A. K. , Bryant, C. , Evans, T. J. (2014) Caspase‐1 cleavage of the TLR adaptor TRIF inhibits autophagy and β‐interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe 15, 214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanjuan, M. A. , Dillon, C. P. , Tait, S. W. , Moshiach, S. , Dorsey, F. , Connell, S. , Komatsu, M. , Tanaka, K. , Cleveland, J. L. , Withoff, S. , Green, D. R. (2007) Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257. [DOI] [PubMed] [Google Scholar]
- 29. Shi, C. S. , Kehrl, J. H. (2008) MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 283, 33175–33182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cooney, R. , Baker, J. , Brain, O. , Danis, B. , Pichulik, T. , Allan, P. , Ferguson, D. J. , Campbell, B. J. , Jewell, D. , Simmons, A. (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97. [DOI] [PubMed] [Google Scholar]
- 31. Jounai, N. , Takeshita, F. , Kobiyama, K. , Sawano, A. , Miyawaki, A. , Xin, K. Q. , Ishii, K. J. , Kawai, T. , Akira, S. , Suzuki, K. , Okuda, K. (2007) The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 104, 14050–14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu, B. , Hur, S. (2015) How RIG‐I like receptors activate MAVS. Curr. Opin. Virol. 12, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shin, D. M. , Jeon, B. Y. , Lee, H. M. , Jin, H. S. , Yuk, J. M. , Song, C. H. , Lee, S. H. , Lee, Z. W. , Cho, S. N. , Kim, J. M. , Friedman, R. L. , Jo, E. K. (2010) Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox‐dependent signaling. PLoS Pathog. 6, e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tattoli, I. , Sorbara, M. T. , Vuckovic, D. , Ling, A. , Soares, F. , Carneiro, L. A. , Yang, C. , Emili, A. , Philpott, D. J. , Girardin, S. E. (2012) Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575. [DOI] [PubMed] [Google Scholar]
- 35. Ogawa, M. , Yoshimori, T. , Suzuki, T. , Sagara, H. , Mizushima, N. , Sasakawa, C. (2005) Escape of intracellular Shigella from autophagy. Science 307, 727–731. [DOI] [PubMed] [Google Scholar]
- 36. Dortet, L. , Mostowy, S. , Cossart, P. (2012) Listeria and autophagy escape: involvement of InlK, an internalin‐like protein. Autophagy 8, 132–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Py, B. F. , Lipinski, M. M. , Yuan, J. (2007) Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3, 117–125. [DOI] [PubMed] [Google Scholar]
- 38. Meunier, E. , Dick, M. S. , Dreier, R. F. , Schürmann, N. , Kenzelmann Broz, D. , Warming, S. , Roose‐Girma, M. , Bumann, D. , Kayagaki, N. , Takeda, K. , Yamamoto, M. , Broz, P. (2014) Caspase‐11 activation requires lysis of pathogen‐containing vacuoles by IFN‐induced GTPases. Nature 509, 366–370. [DOI] [PubMed] [Google Scholar]
- 39. Saitoh, T. , Fujita, N. , Jang, M. H. , Uematsu, S. , Yang, B. G. , Satoh, T. , Omori, H. , Noda, T. , Yamamoto, N. , Komatsu, M. , Tanaka, K. , Kawai, T. , Tsujimura, T. , Takeuchi, O. , Yoshimori, T. , Akira, S. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 456, 264–268. [DOI] [PubMed] [Google Scholar]
- 40. Harris, J. , Hartman, M. , Roche, C. , Zeng, S. G. , O'Shea, A. , Sharp, F. A. , Lambe, E. M. , Creagh, E. M. , Golenbock, D. T. , Tschopp, J. , Kornfeld, H. , Fitzgerald, K. A. , Lavelle, E. C. (2011) Autophagy controls IL‐1beta secretion by targeting pro‐IL‐1beta for degradation. J. Biol. Chem. 286, 9587–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi, C. S. , Shenderov, K. , Huang, N. N. , Kabat, J. , Abu‐Asab, M. , Fitzgerald, K. A. , Sher, A. , Kehrl, J. H. (2012) Activation of autophagy by inflammatory signals limits IL‐1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee, H. K. , Lund, J. M. , Ramanathan, B. , Mizushima, N. , Iwasaki, A. (2007) Autophagy‐dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401. [DOI] [PubMed] [Google Scholar]
- 43. Henault, J. , Martinez, J. , Riggs, J. M. , Tian, J. , Mehta, P. , Clarke, L. , Sasai, M. , Latz, E. , Brinkmann, M. M. , Iwasaki, A. , Coyle, A. J. , Kolbeck, R. , Green, D. R. , Sanjuan, M. A. (2012) Noncanonical autophagy is required for type I interferon secretion in response to DNA‐immune complexes. Immunity 37, 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sasai, M. , Linehan, M. M. , Iwasaki, A. (2010) Bifurcation of Toll‐like receptor 9 signaling by adaptor protein 3. Science 329, 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deretic, V. , Saitoh, T. , Akira, S. (2013) Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laplante, M. , Sabatini, D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laplante, M. , Sabatini, D. M. (2009) mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Copp, J. , Manning, G. , Hunter, T. (2009) TORC‐specific phosphorylation of mammalian target of rapamycin (mTOR): phospho‐Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 69, 1821–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jung, C. H. , Jun, C. B. , Ro, S. H. , Kim, Y. M. , Otto, N. M. , Cao, J. , Kundu, M. , Kim, D. H. (2009) ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jung, C. H. , Ro, S. H. , Cao, J. , Otto, N. M. , Kim, D. H. (2010) mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weichhart, T. , Costantino, G. , Poglitsch, M. , Rosner, M. , Zeyda, M. , Stuhlmeier, K. M. , Kolbe, T. , Stulnig, T. M. , Hörl, W. H. , Hengstschläger, M. , Müller, M. , Säemann, M. D. (2008) The TSC‐mTOR signaling pathway regulates the innate inflammatory response. Immunity 29, 565–577. [DOI] [PubMed] [Google Scholar]
- 52. Ivanov, S. S. , Roy, C. R. (2013) Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat. Immunol. 14, 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ohtani, M. , Hoshii, T. , Fujii, H. , Koyasu, S. , Hirao, A. , Matsuda, S. (2012) Cutting edge: mTORC1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL‐10 production. J. Immunol. 188, 4736–4740. [DOI] [PubMed] [Google Scholar]
- 54. Pan, H. , O'Brien, T. F. , Zhang, P. , Zhong, X. P. (2012) The role of tuberous sclerosis complex 1 in regulating innate immunity. J. Immunol. 188, 3658–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hetz, C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. [DOI] [PubMed] [Google Scholar]
- 56. Schröder, M. (2008) Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 65, 862–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tabas, I. , Ron, D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Szegezdi, E. , Logue, S. E. , Gorman, A. M. , Samali, A. (2006) Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep. 7, 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kouroku, Y. , Fujita, E. , Tanida, I. , Ueno, T. , Isoai, A. , Kumagai, H. , Ogawa, S. , Kaufman, R. J. , Kominami, E. , Momoi, T. (2007) ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine‐induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239. [DOI] [PubMed] [Google Scholar]
- 60. Appenzeller‐Herzog, C. , Hall, M. N. (2012) Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 22, 274–282. [DOI] [PubMed] [Google Scholar]
- 61. Qi, M. , Zhou, H. , Fan, S. , Li, Z. , Yao, G. , Tashiro, S. , Onodera, S. , Xia, M. , Ikejima, T. (2013) mTOR inactivation by ROS‐JNK‐p53 pathway plays an essential role in psedolaric acid B induced autophagy‐dependent senescence in murine fibrosarcoma L929 cells. Eur. J. Pharmacol. 715, 76–88. [DOI] [PubMed] [Google Scholar]
- 62. Urano, F. , Wang, X. , Bertolotti, A. , Zhang, Y. , Chung, P. , Harding, H. P. , Ron, D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. [DOI] [PubMed] [Google Scholar]
- 63. Hasnain, S. Z. , Lourie, R. , Das, I. , Chen, A. C. , McGuckin, M. A. (2012) The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 90, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Clavarino, G. , Cláudio, N. , Couderc, T. , Dalet, A. , Judith, D. , Camosseto, V. , Schmidt, E. K. , Wenger, T. , Lecuit, M. , Gatti, E. , Pierre, P. (2012) Induction of GADD34 is necessary for dsRNA‐dependent interferon‐β production and participates in the control of Chikungunya virus infection. PLoS Pathog. 8, e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goodall, J. C. , Wu, C. , Zhang, Y. , McNeill, L. , Ellis, L. , Saudek, V. , Gaston, J. S. (2010) Endoplasmic reticulum stress‐induced transcription factor, CHOP, is crucial for dendritic cell IL‐23 expression. Proc. Natl. Acad. Sci. USA 107, 17698–17703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Martinon, F. , Chen, X. , Lee, A. H. , Glimcher, L. H. (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Keestra‐Gounder, A. M. , Byndloss, M. X. , Seyffert, N. , Young, B. M. , Chávez‐Arroyo, A. , Tsai, A. Y. , Cevallos, S. A. , Winter, M. G. , Pham, O. H. , Tiffany, C. R. , de Jong, M. F. , Kerrinnes, T. , Ravindran, R. , Luciw, P. A. , McSorley, S. J. , Bäumler, A. J. , Tsolis, R. M. (2016) NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Adolph, T. E. , Tomczak, M. F. , Niederreiter, L. , Ko, H. J. , Böck, J. , Martinez‐Naves, E. , Glickman, J. N. , Tschurtschenthaler, M. , Hartwig, J. , Hosomi, S. , Flak, M. B. , Cusick, J. L. , Kohno, K. , Iwawaki, T. , Billmann‐Born, S. , Raine, T. , Bharti, R. , Lucius, R. , Kweon, M. N. , Marciniak, S. J. , Choi, A. , Hagen, S. J. , Schreiber, S. , Rosenstiel, P. , Kaser, A. , Blumberg, R. S. (2013) Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Adolph, T. E. , Niederreiter, L. , Blumberg, R. S. , Kaser, A. (2012) Endoplasmic reticulum stress and inflammation. Dig. Dis. 30, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Woo, C. W. , Kutzler, L. , Kimball, S. R. , Tabas, I. (2012) Toll‐like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat. Cell Biol. 14, 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. He, B. (2006) Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 13, 393–403. [DOI] [PubMed] [Google Scholar]
- 72. Celli, J. , Tsolis, R. M. (2015) Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat. Rev. Microbiol. 13, 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pillich, H. , Loose, M. , Zimmer, K. P. , Chakraborty, T. (2012) Activation of the unfolded protein response by Listeria monocytogenes . Cell. Microbiol. 14, 949–964. [DOI] [PubMed] [Google Scholar]
- 74. Malhotra, J. D. , Kaufman, R. J. (2011) ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb. Perspect. Biol. 3, a004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lebreton, A. , Stavru, F. , Cossart, P. (2015) Organelle targeting during bacterial infection: insights from Listeria . Trends Cell Biol. 25, 330–338. [DOI] [PubMed] [Google Scholar]
- 76. Stavru, F. , Bouillaud, F. , Sartori, A. , Ricquier, D. , Cossart, P. (2011) Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 108, 3612–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Narendra, D. , Tanaka, A. , Suen, D. F. , Youle, R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wild, P. , Dikic, I. (2010) Mitochondria get a Parkin’ ticket. Nat. Cell Biol. 12, 104–106. [DOI] [PubMed] [Google Scholar]
- 79. Bulua, A. C. , Simon, A. , Maddipati, R. , Pelletier, M. , Park, H. , Kim, K. Y. , Sack, M. N. , Kastner, D. L. , Siegel, R. M. (2011) Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nakahira, K. , Haspel, J. A. , Rathinam, V. A. , Lee, S. J. , Dolinay, T. , Lam, H. C. , Englert, J. A. , Rabinovitch, M. , Cernadas, M. , Kim, H. P. , Fitzgerald, K. A. , Ryter, S. W. , Choi, A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhou, R. , Yazdi, A. S. , Menu, P. , Tschopp, J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. [DOI] [PubMed] [Google Scholar]
- 82. Zhong, Z. , Umemura, A. , Sanchez‐Lopez, E. , Liang, S. , Shalapour, S. , Wong, J. , He, F. , Boassa, D. , Perkins, G. , Ali, S. R. , McGeough, M. D. , Ellisman, M. H. , Seki, E. , Gustafsson, A. B. , Hoffman, H. M. , Diaz‐Meco, M. T. , Moscat, J. , Karin, M. (2016) NF‐κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. West, A. P. , Khoury‐Hanold, W. , Staron, M. , Tal, M. C. , Pineda, C. M. , Lang, S. M. , Bestwick, M. , Duguay, B. A. , Raimundo, N. , MacDuff, D. A. , Kaech, S. M. , Smiley, J. R. , Means, R. E. , Iwasaki, A. , Shadel, G. S. (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ciccia, A. , Elledge, S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Grenon, M. , Gilbert, C. , Lowndes, N. F. (2001) Checkpoint activation in response to double‐strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat. Cell Biol. 3, 844–847. [DOI] [PubMed] [Google Scholar]
- 86. Nishi, R. , Okuda, Y. , Watanabe, E. , Mori, T. , Iwai, S. , Masutani, C. , Sugasawa, K. , Hanaoka, F. (2005) Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Mol. Cell. Biol. 25, 5664–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Krokan, H. E. , Bj⊘rås, M. (2013) Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Peña‐Diaz, J. , Jiricny, J. (2012) Mammalian mismatch repair: error‐free or error‐prone? Trends Biochem. Sci. 37, 206–214. [DOI] [PubMed] [Google Scholar]
- 89. Chatzinikolaou, G. , Karakasilioti, I. , Garinis, G. A. (2014) DNA damage and innate immunity: links and trade‐offs. Trends Immunol. 35, 429–435. [DOI] [PubMed] [Google Scholar]
- 90. Zhang, X. , Brann, T. W. , Zhou, M. , Yang, J. , Oguariri, R. M. , Lidie, K. B. , Imamichi, H. , Huang, D. W. , Lempicki, R. A. , Baseler, M. W. , Veenstra, T. D. , Young, H. A. , Lane, H. C. , Imamichi, T. (2011) Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 186, 4541–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ferguson, B. J. , Mansur, D. S. , Peters, N. E. , Ren, H. , Smith, G. L. (2012) DNA‐PK is a DNA sensor for IRF‐3‐dependent innate immunity. eLife 1, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kondo, T. , Kobayashi, J. , Saitoh, T. , Maruyama, K. , Ishii, K. J. , Barber, G. N. , Komatsu, K. , Akira, S. , Kawai, T. (2013) DNA damage sensor MRE11 recognizes cytosolic double‐stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 110, 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Roth, S. , Rottach, A. , Lotz‐Havla, A. S. , Laux, V. , Muschaweckh, A. , Gersting, S. W. , Muntau, A. C. , Hopfner, K. P. , Jin, L. , Vanness, K. , Petrini, J. H. , Drexler, I. , Leonhardt, H. , Ruland, J. (2014) Rad50‐CARD9 interactions link cytosolic DNA sensing to IL‐1β production. Nat. Immunol. 15, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kang, C. , Xu, Q. , Martin, T. D. , Li, M. Z. , Demaria, M. , Aron, L. , Lu, T. , Yankner, B. A. , Campisi, J. , Elledge, S. J. (2015) The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Buchan, J. R. , Parker, R. (2009) Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Onomoto, K. , Yoneyama, M. , Fung, G. , Kato, H. , Fujita, T. (2014) Antiviral innate immunity and stress granule responses. Trends Immunol. 35, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Berlanga, J. J. , Ventoso, I. , Harding, H. P. , Deng, J. , Ron, D. , Sonenberg, N. , Carrasco, L. , de Haro, C. (2006) Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J. 25, 1730–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fung, G. , Ng, C. S. , Zhang, J. , Shi, J. , Wong, J. , Piesik, P. , Han, L. , Chu, F. , Jagdeo, J. , Jan, E. , Fujita, T. , Luo, H. (2013) Production of a dominant‐negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria‐associated protective stress granules during CVB3 infection. PLoS One 8, e79546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ng, C. S. , Jogi, M. , Yoo, J. S. , Onomoto, K. , Koike, S. , Iwasaki, T. , Yoneyama, M. , Kato, H. , Fujita, T. (2013) Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 87, 9511–9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. White, J. P. , Cardenas, A. M. , Marissen, W. E. , Lloyd, R. E. (2007) Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2, 295–305. [DOI] [PubMed] [Google Scholar]
- 101. Borghese, F. , Michiels, T. (2011) The leader protein of cardioviruses inhibits stress granule assembly. J. Virol. 85, 9614–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Okonski, K. M. , Samuel, C. E. (2013) Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J. Virol. 87, 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Katoh, H. , Okamoto, T. , Fukuhara, T. , Kambara, H. , Morita, E. , Mori, Y. , Kamitani, W. , Matsuura, Y. (2013) Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with Caprin‐1 and facilitates viral propagation. J. Virol. 87, 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Onomoto, K. , Jogi, M. , Yoo, J. S. , Narita, R. , Morimoto, S. , Takemura, A. , Sambhara, S. , Kawaguchi, A. , Osari, S. , Nagata, K. , Matsumiya, T. , Namiki, H. , Yoneyama, M. , Fujita, T. (2012) Critical role of an antiviral stress granule containing RIG‐I and PKR in viral detection and innate immunity. PLoS One 7, e43031. [DOI] [PMC free article] [PubMed] [Google Scholar]