

Abstract
The liver contains 2 transcriptionally distinct group 1 ILC subsets: CD49a+ ILC1s and CD49b+ NK cells. However, little is known about how group 1 ILCs contribute to hepatic immune responses. Therefore, we characterized murine liver‐resident group 1 ILCs and found that CD49a+ ILC1s express high levels of the inhibitory receptor NKG2A and localize near DCs in perivascular spaces surrounding the portal triads. Upon hepatic viral infection, NKG2A signaling in group 1 ILCs, especially in CD49a+ ILC1s, inhibits CXCL9 expression required for robust accumulation of IFN‐γ+CD49b+ NK cells. As a consequence, NKG2A−/− mice showed increased numbers of IFN‐γ‐producing NK cells that preferentially activate liver CD103+ DCs, leading to the sustained proliferation of adoptively transferred, virus‐specific CD8+ T cells. Collectively, these data suggest that group 1 ILCs play a role in maintaining the liver as a tolerogenic site by limiting the recruitment of peripheral NK cells during the early phase of viral infection. Furthermore, our findings implicate that the inhibition of NKG2A signaling on group 1 ILCs may be a novel vaccine strategy to induce robust CD8+ T cell responses against persistent liver pathogens.
Keywords: immunity, viruses, natural killer cells, T lymphocytes
Short abstract
Intersection of innate and adaptive immunity in viral infection and implications for vaccine design.
Abbreviations
- Ad
adenovirus
- B6
C57BL/6
- DC
dendritic cell
- dpi
days postinfection
- HCV
hepatitis C virus
- hpi
hours postinfection
- ILC
innate lymphoid cell
- i.v.
intravenous
- MFI
mean fluorescence intensity
- MHC‐I/II
MHC class I/II
- PLP
periodate‐lysine‐paraformaldehyde
- qRT‐PCR
quantitative RT‐PCR
- SH2
Src homology 2
- Tg
transgenic
Introduction
The liver lies as the intersection of portal and systemic circulation. As a result, it maintains a tolerogenic environment that dampens responses to highly immunogenic material arriving in intestinal blood. Numerous pathogens, including the HCV and malaria, exploit the immunosuppressive liver microenvironment to establish persistent infections that can lead to the development of hepatocellular carcinoma [1]. Persistence of hepatotropic pathogens is strongly correlated to impaired T cell responses in the liver microenvironment [2]. However, despite our knowledge of cellular and molecular mechanisms contributing to T cell dysfunction in chronic hepatic infections, attempts at developing prophylactic or therapeutic vaccines have failed.
Induction and maintenance of efficient T cell responses are highly dependent on priming by DCs [3]. Immature DCs are activated by broadly conserved pattern recognition receptors (i.e., TLRs), along with proinflammatory cytokines (i.e., IFN‐γ) [4, 5]. Upon activation/maturation, DCs up‐regulate antigen‐presenting molecules (i.e., MHC‐I and ‐II) and costimulatory molecules (i.e., CD80 and CD86) necessary for optimal T cell activation [4]. The liver includes diverse DC populations, including conventional tissue‐resident CD11b+ and CD103+ subsets [6]. During acute viral infection, hepatic CD103+ DCs up‐regulate expression of costimulatory molecules and preferentially present viral antigen on MHC‐I to naïve CD8+ T cells [6]. However, during chronic viral infection, liver‐resident DCs may exhibit inefficient activation/maturation, limiting their capacity to generate robust CD8+ T cell responses capable of clearing virus.
Recently, ILCs have emerged as important regulators of acute and chronic inflammatory responses and are classified into 3 groups [7]. Of these, group 1 ILCs, which are further divided into T‐bet+Eomes−CD49a+ ILC1s and T‐bet+Eomes+CD49b+ conventional NK cells, are similar to the CD4+ TH1 lineage, in that they produce IFN‐γ upon stimulation [8, 9]. Group 1 ILCs regulate DC function by direct killing of immature DCs via cytolytic molecules (i.e., granzymes, perforin) or conversely, enhancing DC activation via production of cytokines (i.e., IFN‐γ, TNF‐α) [10]. Compared with the spleen, hepatic group 1 ILCs exhibit limited Ly49 receptor expression and reduced capacity to produce IFN‐γ upon IL‐12/IL‐18 stimulation [11]. Parabiosis studies have further distinguished the 2 subsets, where CD49a+ ILC1s were found to be sessile, perhaps indicating their differentiation from a liver‐resident precursor, whereas CD49b+ NK cells migrate through blood, suggesting that they are extrahepatically derived, likely from the bone marrow [12, 13].
Initial recruitment of NK cells into the liver during viral infection is dependent on the chemokine CCL3 [14]. This initial influx of NK cells into the infected organ increases local expression of IFN‐γ, which triggers production of CXCL9 required for more robust, subsequent infiltration of NK cells [15, 16]. This sequence of chemokine production is likely tightly regulated during viral infection. Importantly, recruited IFN‐γ+CD49b+ NK cells have been demonstrated to cross talk with DCs, leading to strong anti‐viral T cell responses required for efficient viral clearance [10]. However, the molecular and cellular signals suppressing effective NK–DC crosstalk within the liver’s tolerogenic immune environment have yet to be elucidated.
In this study, we sought to determine the regulatory role of CD49a+ ILC1s in suppressing hepatic DC activation during infection. Ad is an important human pathogen and has been used as a candidate vaccine against HIV and HCV [17, 18]. With the use of the hepatotropic Ad engineered to express OVA (Ad‐OVA), we demonstrate that group 1 ILCs, particularly CD49a+ ILC1s, signal through the inhibitory receptor NKG2A to limit the production of chemokines required for recruitment of peripheral IFN‐γ+CD49b+ NK cells. Our study identifies hepatic NKG2A+CD49a+ ILC1 group 1 ILCs as the primary cells regulating the activation of liver‐resident APCs, namely CD103+ DCs, which in turn, dictate anti‐viral CD8+ T cell responses to pathogens invading the liver and potential pathogens draining the gut. Moreover, these findings implicate that ILC1s are a potential cellular target in designing vaccine strategies and therapeutic venues for chronic bloodborne hepatic viral infections (e.g., HCV).
MATERIALS AND METHODS
Mice
All experiments used 8‐ to 12‐wk‐old gender/age‐matched male and female mice. Thy1.2+ B6 (H‐2b) and Thy1.1+ OT‐I TCR Tg (OT‐I, H‐2b) mice were purchased from Taconic Biosciences (Hudson, NY, USA). NKG2A−/− mice were kindly provided by Dr. Richard Enelow (Dartmouth College, Hanover, New Hampshire, USA) [19]. These mice were generated on the 129/SJ genetic background and back crossed onto the B6 background by speed congenics using high‐density single nucleotide polymorphism chips. Mice were handled according to protocols approved by the University of Virginia Institutional Animal Care and Use Committee.
Virus
Replication‐defective recombinant Ad type 5 expressing OVA under the human CMV promoter and lacking E1 and E3 genes was purchased from the Viral Vector Core Facility, University of Iowa (Iowa City, IA, USA). Mice were injected intravenously with 2.5 × 107 IU Ad‐OVA.
Liver and spleen leukocyte isolation
Livers were perfused with PBS via the portal vein until blanched and then put in IMDM, supplemented with 10% newborn calf serum. Whole livers were passed through a metal spleen screen and digested with 0.05% collagenase IV (Sigma‐Aldrich, St. Louis, MO, USA) for 30 min at 37°C. Intrahepatic mononuclear cells were purified on a 21% Histodenz (Sigma‐Aldrich) gradient after centrifugation at 1250 g for 20 min without braking. Spleens were passed through a mesh spleen screen, followed by RBC lysis. All samples were resuspended in IMDM plus serum. Leukocytes were counted on a hemocytometer.
Flow cytometry and intracellular staining
Cells were labeled with antibodies against CD45, CD3ɛ, NKp46, NK1.1, CD49a, CD49b, NKG2A‐B6, NKG2A/C/E, CD94, T‐bet, Eomes, CD69, B220, I‐A/I‐E, CD11c, CD11b, CD8α, CD103, CD80, CD86, Thy1.1, Thy1.2, and IFN‐γ (all obtained from eBioscience, San Diego, Ca, USA) and CXCL9 (from BioLegend, San Diego, CA, USA). For cell‐surface labeling, 1 × 106 cells were blocked with anti‐CD16/CD32 (2.4G2; University of Virginia, Charlottesville, VA, USA) and incubated with the corresponding antibodies for 30 min at 4°C in staining buffer (PBS with 2% FBS and 0.1% NaN3). For cytokine and chemokine staining, 1 × 106 cells were incubated for 5 h in IMDM, supplemented with 10% HyClone FBS, 10 U/ml penicillin G/streptomycin, 2 mM l‐glutamine, 5 mM 2‐ME, and 1 μl/ml GolgiPlug/GolgiStop (BD Biosciences, San Jose, CA, USA). OT‐I cells and CD8+ T cells were restimulated with 2 μg/ml SIINFEKL peptide (AnaSpec, Fremont, CA, USA). Group 1 ILCs were restimulated with 5 μg/ml plate‐bound anti‐NKp46 for IFN‐γ expression and left unstimulated for CXCL9 expression. After incubation, the cells were surface labeled as described above and fixed using Cytofix/Cytoperm (BD Biosciences), according to the manufacturer’s instructions before intracellular IFN‐γ or CXCL9 staining. All samples were run on a BD FACSCanto II (BD Biosciences) and analyzed using FlowJo software 8.8.6 (Tree Star, Ashland, OR, USA).
Microscopic studies
Mouse liver tissues were perfused with 1× PBS and PLP fixative, excised, incubated in PLP for 3 h at 4°C, and equilibrated in graded sucrose solutions (5–30%). Tissues were frozen in OCT medium, sectioned at 5 μm thickness, blocked with 2.4G2 solution (2.4G2 supernatant containing anti‐CD16/32; 10% each of chicken, donkey, and horse serum; and 0.1% NaN3), and stained with Alexa Fluor‐conjugated goat anti‐NKp46 (R&D Systems, Minneapolis, MN, USA), goat anti‐mouse CD31 (R&D Systems), and hamster anti‐CD49a (clone Ha31/8; BD Biosciences) antibodies. mAb labeling kits were used for fluorescence tag conjugation (Thermo Fisher Scientific Life Sciences, Waltham, MA, USA). Confocal microscopy was performed on a Zeiss LSM‐700, and data were analyzed using Zen 2009 Light Edition software (Carl Zeiss Microscopy, Thornwood, NY, USA).
In vivo chemokine blockade
At the time of infection, 8‐ to 12‐wk‐old B6 mice were treated with 200 μg, i.v. anti‐CXCL9 antibody (BioLegend) or goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Liver mononuclear cells were harvested at 12 hpi for analysis by flow cytometry.
In vitro chemotaxis assay
NK cells were magnetically isolated from the spleens of naïve B6 and NKG2A−/− mice by positive selection for CD49b (Stemcell Technologies, Vancouver, BC, Canada) and assessed for migration. In brief, 2 × 105 cells in 100 μl were plated in the upper chamber of a 5 μm Transwell filter in a 24‐well plate. The lower chambers contained 600 μl medium containing 0, 10, 20, or 40 ng/ml recombinant CXCL9 (R&D Systems). After 3 h at 37°C, cells were harvested from the lower chamber and stained for FACS analysis.
qRT‐PCR analysis
RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA, USA) and reverse transcribed using the high‐capacity RNA‐to‐cDNA kit (Thermo Fisher Scientific). QuantiTect primers for qRT‐PCR for H2‐T23 (Qa‐1b) were purchased from Qiagen. Amplification was performed on a StepOnePlus Real‐Time PCR system and detected by SYBR Green incorporation (Thermo Fisher Scientific).
Adoptive transfer of TCR Tg T cells
CD8+ T cells were isolated from the spleens and mesenteric lymph nodes of Thy1.1+OT‐I+ mice using anti‐CD8α antibody‐conjugated magnetic bead separation kits (Miltenyi Biotec, San Diego, CA, USA). Cells were labeled with 1.8 μM CFSE for 8 min at room temperature and transferred by intravenous injection into naïve Thy1.2+ recipients.
DC and group 1 ILC isolation
Liver DCs and group 1 ILCs were isolated from animals (7–10 B6 or NKG2A−/− mice) infected with 2.5 × 107 IU Ad‐OVA for 12 h before harvest. For DC isolation, CD11c+ cells were first enriched by positive selection (Miltenyi Biotec). DCs were stained using antibodies for CD45, MHC‐II, CD11c, B220, CD11b, and CD103. Group 1 ILCs were stained using antibodies for CD45, CD3ɛ, NKp46, CD49a, and CD49b. Cells were sorted by flow cytometry using a BD Influx cell sorter at the Flow Cytometry Core Facility (University of Virginia).
In vitro priming of naïve OT‐I T cells
FACS‐sorted liver DCs, isolated 12 hpi from livers of infected B6 or NKG2A−/− mice, were cultured with CFSE‐labeled OT‐I T cells at a ratio of 1:10 for 4 d at 37°C. All DC:T cell coculture media included IMDM supplemented with 10% heat‐inactivated HyClone FBS, 10 U/ml penicillin G/streptomycin, 2 mM l‐glutamine, 5 mM 2‐ME, 20 mM HEPES, and 100 μg/ml gentamycin (all from Thermo Fisher Scientific).
Statistical analysis
Student’s t tests (2‐tailed) or 1‐way ANOVA with Tukey’s post‐test were used as appropriate to evaluate the significance of the differences. A value of P < 0.05 was regarded as statistically significant.
RESULTS
CD49a+ ILC1s are the major liver‐resident ILC subset in the steady state and preferentially localize to the perivascular spaces near the portal triads
Recent studies describing transcriptional differences between the liver‐resident group 1 ILC subsets have helped establish their key defining features; however, there is contrasting data as to the accurate representation of these cells in the steady state [12, 20, 21]. To define a physiologically relevant representation of liver‐resident group 1 ILCs, we isolated mononuclear cells from murine livers using Histodenz gradient centrifugation following tissue digestion with collagenase IV. When naïve livers are subjected to enzymatic digestion for increasing time intervals, the frequency and cellularity of isolated CD49a+ ILC1s increase proportionally to the digestion time ( Fig. 1A and B ). CD49a+ ILC1s thus represent the dominant hepatic‐resident ILC subset liberated after 30 min digestion. In addition, we used confocal microscopy to determine the location of group 1 ILCs in relation to the liver vasculature and stroma. With the use of CD31 (blue) to demarcate liver sinusoidal endothelial cells, we found that NKp46+CD49a+ ILC1s (yellow) primarily reside in the perivascular spaces surrounding the portal areas, whereas NKp46+CD49a+ ILC1s (yellow) and NKp46+CD49a− NK cells (green) are equally dispersed throughout the liver parenchyma (Fig. 1C). Taken together, these results suggest that only CD49a+ ILC1s localize along the portal tracts of the liver in the steady state. Further analysis of additional NK cell‐surface markers, including the inhibitory receptor NKG2A, on hepatic group 1 ILCs indicated that compared with conventional NK cells, CD49a+ ILC1s preferentially express CD11c, CD69, and NKG2A (Fig. 1D).
Figure 1.

CD49a+ ILC1s are the major liver‐resident ILC subset in the steady state. Eight‐ to 12‐wk‐old B6 livers were perfused and digested with collagenase IV at 37°C and the mononuclear cells isolated by density gradient centrifugation. Spleens were treated with RBC lysis solution. (A) With the use of flow cytometry, representative resident CD3ɛ−NK1.1+ group 1 ILC populations, isolated from 30 min digested livers or RBC lysed spleens, are gated from debris and doublet‐excluded CD45+ cells and discriminated using CD49a and CD49b surface expression. (B) Representative flow plots of group 1 ILCs isolated from livers digested with collagenase IV for 0, 10, 20, or 30 min. (C) Fluorescence microscopy of a naïve liver section stained for CD31 (blue), NKp46 (green), and CD49a (red); P indicates the portal vein. (D) Representative flow plots of liver group 1 ILCs stained for CD49a and CD11c, CD69, or NKG2A expression. Data are representative of 2 independent experiments with 6–8 mice/experiment.
Liver‐resident CD49a+ ILC1 numbers are constant before and during Ad infection
Given the notable presence of CD49a+ ILC1s in the liver compared with the spleen in the steady state, it was of interest to assess changes in group 1 ILC populations (i.e., CD49a+ ILC1s and CD49b+ NK cells) during viral infection. Therefore, we administered intravenously the hepatotropic Ad‐OVA and analyzed group 1 ILCs at early time points after infection. Interestingly, there was a transient appearance of a CD49a+CD49b+ ILC population starting at 3 hpi, which declined to levels similar to the steady state by 48 hpi ( Fig. 2A ). Although the frequency of CD49a+ ILC1s decreases during Ad‐OVA infection, their cellularity remains constant through 48 hpi. In contrast, both frequency and cellularity of CD49b+ NK cells and CD49a+CD49b+ ILCs follow a similar pattern of influx and contraction (Fig. 2B). We next evaluated surface expression of CD11c, CD69, and NKG2A at 12 hpi on group 1 ILCs (Fig. 2C). These surface proteins were detectable at higher levels on CD49a+ ILC1s than CD49b+ NK cells. Notably, CD49a+CD49b+ ILCs express intermediate levels of CD11c, CD69, and NKG2A at 12 hpi, thus transitioning from CD49b+ NK cells to a phenotype more similar to CD49a+ ILC1s. This intermediate phenotype of CD49a+CD49b+ ILCs was also evident in the expression of lineage‐specific transcription factors. T‐bet, which is necessary for the development of all group 1 ILCs, was expressed at increasing levels in CD49a+ ILC1s, CD49a+CD49b+ ILCs, and CD49b+ NK cells (Fig. 2C). In contrast, expression of Eomes, which is more specific to the NK cell lineage, was more robust in CD49a+CD49b+ ILCs and CD49b+ NK cells (Fig. 2C). The transient increase in their number, as well as their intermediate pattern of expression of cell‐surface markers and transcription factors, suggests that CD49a+CD49b+ ILCs may originate from infiltrating CD49b+ NK cells by up‐regulating liver‐resident markers in response to signals from the local liver microenvironment.
Figure 2.

CD49b+ NK cells up‐regulate CD49a surface expression in the liver during Ad infection. Eight‐ to 12‐wk‐old B6 mice were administered with 2.5 × 107 IU Ad‐OVA (intravenously) for 3, 6, 12, 24, or 48 h. The livers were perfused and digested with collagenase IV at 37°C for 30 min and the mononuclear cells isolated by density gradient centrifugation. (A) With the use of flow cytometry, representative resident CD3ɛ−NK1.1+ group 1 ILC populations are discriminated using CD49a and CD49b surface expression. (B) Quantified cellularity of CD49a+ ILC1 (red), CD49a+CD49b+ ILCs (green), and CD49b+ NK cells (blue) from uninfected and 3, 6, 12, 24, or 48 h‐infected livers. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Representative histograms of CD11c, CD69, NKG2A, T‐bet, and Eomes expression by liver CD49a+ ILC1s (red), CD49a+CD49b+ ILCs (green), and CD49b+ NK cells (blue) isolated at 12 hpi. Data are representative of means ± sem of 2 independent experiments with 3 mice/experiment.
Group 1 ILCs in NKG2A−/− livers produce CXCL9 that promotes peripheral NK cell infiltration into the liver
NKG2A is a member of the inhibitor NK receptor family. Engagement of the CD94/NKG2A heterodimer transduces a negative signal through the 2 intracytoplasmic ITIMs on NKG2A. CD49a+ ILC1s express higher levels of NKG2A when compared with CD49b+ NK cells (Fig. 2C). Therefore, we hypothesized that NKG2A+CD49a+ ILC1s exert an immunoregulatory role in hepatic immune responses during viral infection. To this end, we used NKG2A−/− mice [19] and first verified that liver‐resident group 1 ILCs lack NKG2A transcript and protein expression (Supplemental Fig. 1A and B). Although comparable at steady state, the livers of NKG2A−/− mice exhibit a greater frequency and cellularity in group 1 ILCs than B6 livers at 12 and 24 hpi ( Fig. 3A and B ). Interestingly, only CD49b+ NK cells and CD49a+CD49b+ ILCs in the NKG2A−/− livers exhibit increased cellularity at 12 hpi (Fig. 3C and D). In contrast, CD49a+ ILC1s in both the B6 and NKG2A−/− livers display no differences in cellularity at 12 hpi or later time points assessed (Fig. 3D; data not shown).
Figure 3.
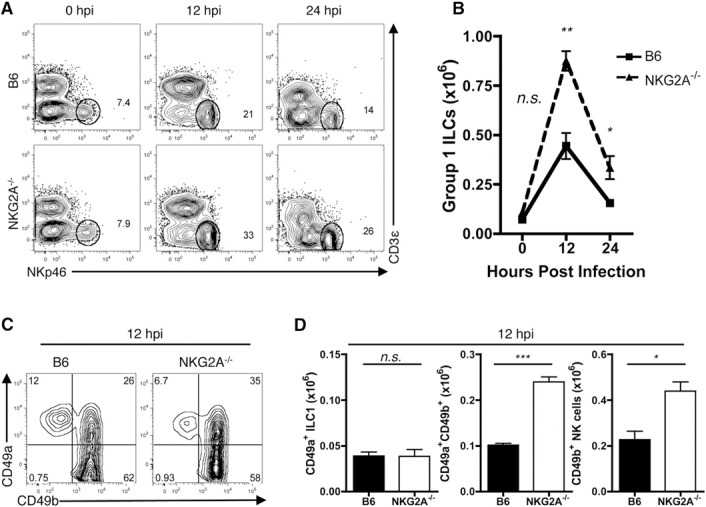
Similar CD49a+ ILC1 but enhanced CD49b+ NK cellularity in the NKG2A−/− livers during Ad infection. Eight‐ to 12‐wk‐old B6 and NKG2A−/− mice were administered with PBS or 2.5 × 107 IU Ad‐OVA (intravenously) for 12 or 24 h. The livers were perfused and digested with collagenase IV at 37°C for 30 min and the mononuclear cells isolated by density gradient centrifugation. (A and B) Dot plots and quantified cellularity of total group 1 ILCs (representative of 2–5 independent experiments; n = 3–4 mice/experiment). (C and D) Dot plots and quantified cellularity of CD49a+ ILC1s, CD49a+CD49b+ ILCs, and CD49b+ NK cells at 12 hpi. Data are representative of means ± sem of 5 independent experiments with 3–4 mice/experiment. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant.
As previously reported, CD49b+ NK cells are recruited to the liver during viral infection [16]. To test the possibility that the enhanced number of CD49b+ NK cells observed in the NKG2A−/− mice following infection is a result of recruitment of these cells, we determined the expression level of CXCL9, a chemokine involved in NK cell recruitment, in liver‐resident group 1 ILCs. When compared with B6 counterparts, NKG2A−/− group 1 ILCs expressed more CXCL9 on a per‐cell basis ( Fig. 4A ). In particular, ILC1s were more sensitive to the loss of NKG2A than NK cells, as the difference in MFI of CXCL9 between B6 and NKG2A−/− ILC1s was greater than the difference in MFI between B6 and NKG2A−/− NK cells (Fig. 4A). Furthermore, the difference in MFI between B6 and NKG2A−/− CD49a+CD49b+ ILCs recapitulated the intermediate phenotype of this population. Importantly, the subsequent recruitment of NK cells was blocked by administration of anti‐CXCL9 antibody, verifying the importance of this chemokine in NK cell recruitment to the liver (Fig. 4B). The enhanced recruitment of NK cells to the livers of NKG2A−/− mice was not a result of intrinsic differences in chemotaxis, as the migratory capacity of NK cells was comparable between B6 and NKG2A−/− mice (Fig. 4C).
Figure 4.
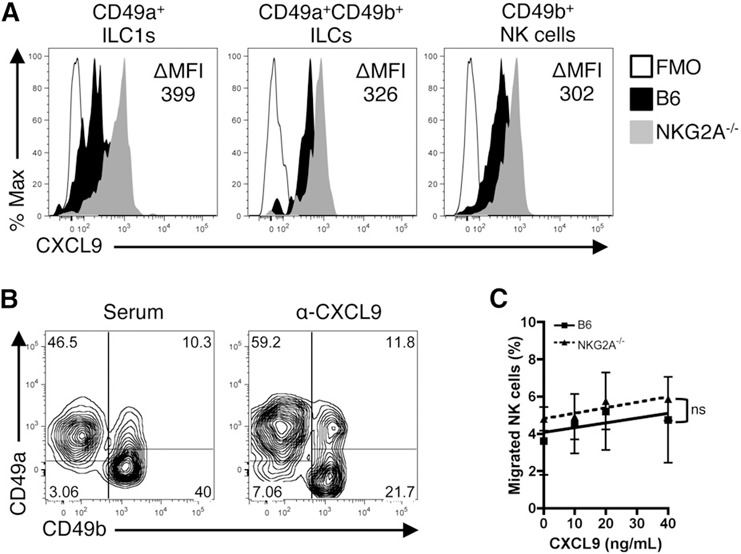
Group 1 ILCs produce increased levels of NK cell chemoattractant CXCL9 in NKG2A−/− livers during Ad infection. Eight‐ to 12‐wk‐old B6 and NKG2A−/− mice were administered with PBS or 2.5 × 107 IU Ad‐OVA (intravenously) for 12 h. The livers were perfused and digested with collagenase IV at 37°C for 30 min and the mononuclear cells isolated by density gradient centrifugation. (A) Representative histogram of CXCL9 expression in hepatic group 1 ILCs at 12 hpi. The difference in MFI (ΔMFI) of CXCL9 between NKG2A−/− and B6 mice is indicated for each population of group 1 ILCs. Data are representative 2 independent experiments with 3 mice per experiment. FMO, Fluorescence minus one. (B) Representative flow plot of group 1 ILCs in mice that received 200 μg anti‐CXCL9 antibody or goat serum. Data are representative of 2 mice per group. (C) In vitro NK cell migration in response to CXCL9. Magnetically sorted, splenic NK cells from naïve B6 and NKG2A−/− mice were assessed for migration in response to CXCL9 across a Transwell filter. Data are representative of 2 independent experiments with 2–3 mice/experiment.
As group 1 ILCs are major producers of the anti‐viral cytokine IFN‐γ, we next assessed the influence of NKG2A on group 1 ILC IFN‐γ production. Surprisingly, following restimulation of total mononuclear cells isolated from livers at 12 hpi with plate‐bound NKp46 antibody, CD49a+CD49b+ ILCs and CD49b+ NK cells, but not CD49a+ ILC1s, produced IFN‐γ in both B6 and NKG2A−/− mice ( Fig. 5A and B ). Although we find no differences in IFN‐γ protein expression between the B6 and NKG2A−/− group 1 ILCs, the cellularity of IFN‐γ+CD49b+ NK cells is increased in NKG2A−/− livers at 12 hpi (Fig. 5C). Of note, we were able to detect IFN‐γ+CD49a+ ILC1s upon IL‐12/IL‐18 restimulation, although at significantly lower levels than CD49b+ NK cells; however, similar to restimulation with NKp46, there were no differences in IFN‐γ production in any of the subsets between the B6 and NKG2A−/− livers at 12 hpi (data not shown). These results suggest that CD49a+ ILC1s express chemokines that promote the recruitment of peripheral CD49b+ NK cells yet exhibit significantly less potential to produce IFN‐γ than their conscripted counterparts.
Figure 5.
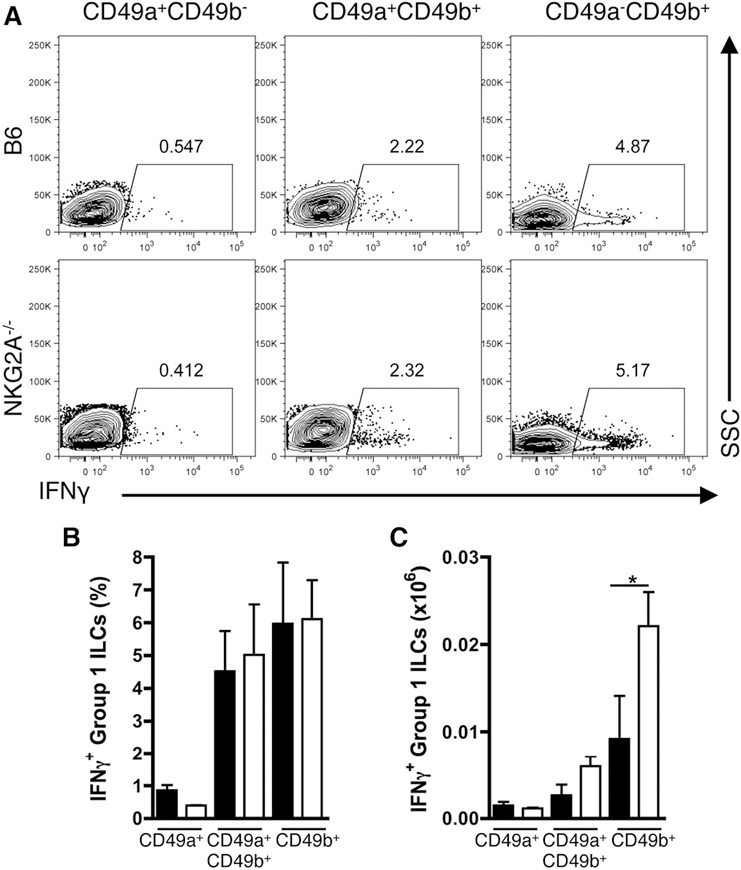
NKG2A does not affect the intrinsic production of IFN‐γ in group 1 ILCs. (A–C) Representative flow plots of IFN‐γ expression, aggregate frequency of IFN‐γ+ cells, and quantified IFN‐γ+ cellularity from liver CD49a+ ILC1s, CD49a+CD49b+ ILCs, and CD49b+ NK cells isolated at 12 hpi and stimulated with plate‐bound anti‐NKp46 for 5 h. Data are representative of 2 independent experiments with 3 mice/experiment. SSC, side‐scatter. *P < 0.05.
Increased priming of OT‐I T cells with improved effector function is detectable in NKG2A−/− mice compared with B6 mice
It is well established that NK cells are important in controlling DC fates during viral insult. DCs, in turn, are necessary for effective priming of anti‐viral CD8+ T cells. As a consequence, it was of interest to determine if the enhanced infiltration of NK cells in NKG2A−/− livers influenced CD8+ T cell responses during viral infection. To explore the impact of NKG2A deficiency in group 1 ILCs on naïve CD8+ T cells responding to Ad‐OVA infection in vivo, we adoptively transferred naïve, CFSE‐labeled OT‐I T cells into B6 and NKG2A−/− mice, 1 d before intravenous Ad‐OVA infection and analyzed the livers at 3 dpi for both the accumulation of OT‐I T cells and their CFSE dilution profiles. Infected NKG2A−/− T cell recipients displayed increased frequency and total numbers of responding OT‐I T cells in the livers at 3 dpi ( Fig. 6A and C ). Moreover, the CFSE dilution profile of OT‐I T cells in B6 and NKG2A−/− recipients revealed a more rapid division in the NKG2A−/− livers (Fig. 6A). In addition to an accelerated rate of proliferation, activated (responding) OT‐I T cells isolated from the livers of NKG2A−/− mice at 3 dpi are able to secrete more IFN‐γ, as measured by both frequency and MFI upon in vitro stimulation with cognate antigen (Fig. 6B and D).
Figure 6.
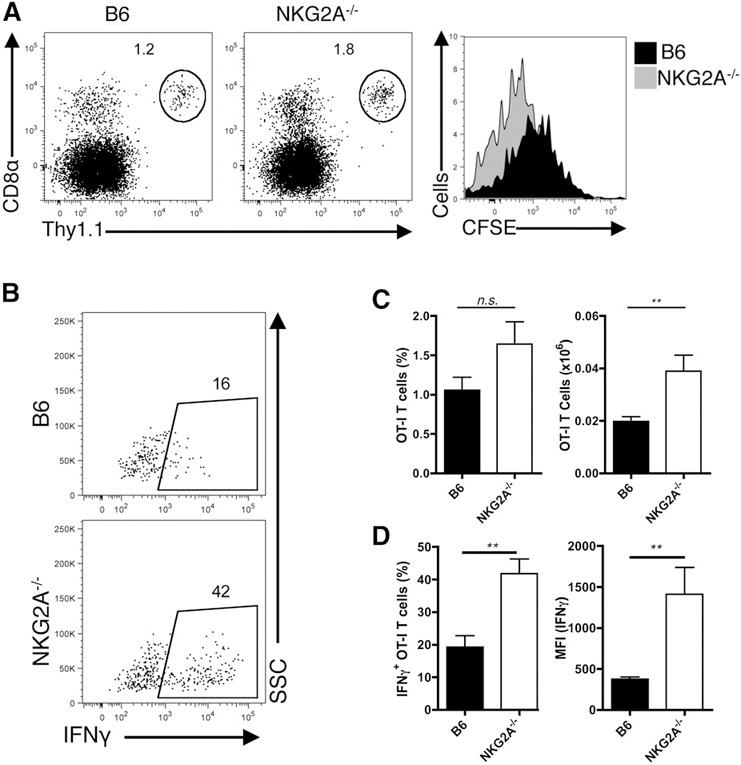
Enhanced antigen‐specific CD8+ T cells in NKG2A−/− livers during Ad infection. (A–D) Naïve, CFSE+ OT‐I cells (2 × 105) were transferred via the tail vein into B6 and NKG2A−/− mice, 1 d before 2.5 × 107 IU Ad‐OVA infection (intravenously). Data are representative of means ± sem of 3 independent experiments with n = 3 mice/experiment. (A) Representative CD45+CD8α+Thy1.1+ OT‐I flow plots and CFSE dilution histograms from B6 (black) and NKG2A−/− (gray) livers at 3 dpi. (B) Representative IFN‐γ+ OT‐I flow plots from B6 and NKG2A−/− livers at 3 dpi. (C) Quantified OT‐I CD8+ T cell frequencies and cellularity from B6 (black) and NKG2A−/− (white) livers at 3 dpi. (D) Frequencies and MFI of IFN‐γ from B6 (black) and NKG2A−/− (white) livers at 3 dpi. **P < 0.01.
The enhanced cellularity and effector function seen in OT‐I T cells in the NKG2A−/− livers are likely a result of greater activation/maturation of hepatic DCs in these mice. Indeed, despite modest differences in frequencies between B6 and NKG2A−/− livers, the cellularity of total, CD11b+, and CD103+ DCs are increased in NKG2A−/− livers at 0, 12, and 24 hpi compared with those from B6 mice (Supplemental Fig. 2A–C). This increase was not a result of intrinsic influence of NKG2A signaling on liver‐resident DCs, as cells that did not express NK1.1, including hepatic DCs, lacked surface expression of NKG2A (data not shown). Interestingly, the expression of the Qa‐1b transcript, the IFN‐γ‐inducible ligand for NKG2A, is enhanced in sorted CD11b+ and CD103+ DCs from the NKG2A−/− livers at 12 hpi; nonetheless, CD11b+ and CD103+ DCs transcribe Qa‐1b to similar levels in both B6 and NKG2A−/− livers (Supplemental Fig. 2D). Thus, the global increase in IFN‐γ production by infiltrating CD49b+ NK cells is likely mediating proinflammatory signaling events on multiple local immune cells.
We also evaluated the maturation status of CD11b+ and CD103+ DCs via expression of the costimulatory receptors CD80 and CD86. Although equal in the naïve livers, both the CD11b+ and CD103+ DCs in the NKG2A−/− express higher levels of CD86 when compared with the corresponding DC subsets from B6 mice at 12 and 24 hpi (Supplemental Fig. 2E and F). In contrast, although the CD103+ DCs express enhanced CD80 expression in the NKG2A−/− livers at 12 hpi, as measured by MFI, the CD11b+ DC subset exhibits no differences between the B6 and NKG2A−/− mice (Supplemental Fig. 2F). This transient up‐regulation of costimulatory receptors was reduced upon administration of anti‐CXCL9 antibody (Supplemental Fig. 3), underscoring the importance of NK cells in recruiting and activating DC subsets in hepatic viral infection.
Given that CD11b+ and CD103+ DCs from the NKG2A−/− livers express high levels of costimulatory receptors at 12 hpi, we examined their capacity to activate antigen‐specific CD8+ T cells. To this end, we sorted liver DCs from B6 and NKG2A−/− mice infected with Ad‐OVA for 12 h and analyzed their ability to support the accumulation of OT‐I CD8+ T cells in vitro. As assessed by CFSE dilution, NKG2A−/− CD103+ DCs led to a more potent, sustained OT‐1 accumulation than B6 CD103+ DCs ( Fig. 7A ). This enhanced stimulatory capacity of NKG2A−/− CD103+ DCs was also evident from the elevated numbers of total OT‐I CD8+ T cells recovered at the end of the in vitro coculture (Fig. 7B). In contrast to the differences in OT‐I CD8+ T cell number seen with the CD103+ DC subsets, the CD11b+ DCs from both the B6 and NKG2A−/− livers supported similar low‐level accumulation in OT‐I CD8+ T cells (Fig. 7A and B). Taken together, these findings demonstrate that CD103+ but not CD11b+ DCs from the NKG2A−/− livers are intrinsically better at stimulating naïve, antigen‐specific CD8+ T cells, perhaps by extending their survival, when compared with CD103+ and CD11b+ DCs sorted from B6 livers.
Figure 7.
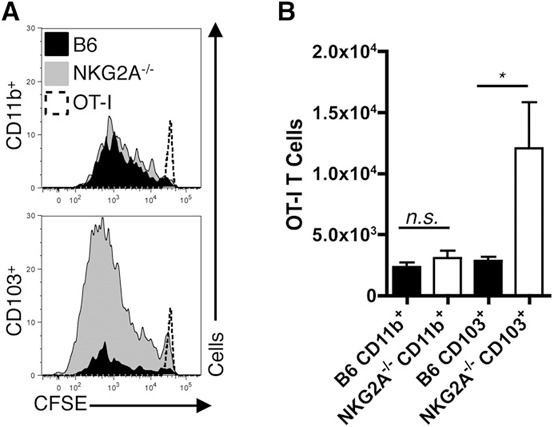
CD103+ DC priming of antigen‐specific CD8+ T cells is heightened in NKG2A−/− livers. (A and B) Representative OT‐I CFSE dilution histograms and quantified cell numbers (via counting beads; Spherotech, Lake Forest, IL, USA) from 4 d of coculture at 37°C using a ratio of 2 × 104 cell‐sorted CD11b+ or CD103+ DCs isolated from 2.5 × 107 IU Ad‐OVA (intravenously) administered B6 and NKG2A−/− livers at 12 hpi to 2 × 105 sorted OT‐I T cells. Data are representative of 2 independent experiments done in triplicate. *P < 0.05.
DISCUSSION
In the present study, we examined the immunoregulatory role of liver‐resident group 1 ILCs in modulating host immune responses to bloodborne hepatic viral infection. Our analyses demonstrate that liver‐resident group 1 ILCs play a role in activating CD103+ DCs, leading to the enhanced induction of antigen‐specific, anti‐viral CD8+ T cells responses. We identified the transient presence of a hepatic NKG2A+CD49a+CD49b+ ILC population during the early phase of viral infection, which likely arises from CD49b+ NK cells through a process that may be mediated by TGF‐β signaling [22]. Most importantly, we determined that liver‐resident NKG2A+ group 1 ILCs play a critical role in suppressing the recruitment of peripheral NK cells, resulting in a loss of IFN‐γ production crucial for the enhanced priming of CD8+ T cells in hepatic Ad infection.
CD49b+ NK cells and CD49a+CD49b+ ILCs follow similar expansion and contraction kinetics in the liver during the first 48 h of Ad infection. Moreover, NKG2A is up‐regulated on CD49a+CD49b+ ILCs in the first 12 h of infection, suggesting that its inhibitory signals may contribute to the CD49a+CD49b+ phenotype during early viral insult (Fig. 2C; data not shown). It is interesting that liver‐resident CD49a+ ILC1s do not exhibit divergent cellularity at any time points analyzed in B6 mice during Ad infection, positing that CD49b+ NK cells are uniquely recruited to the liver (Fig. 2B).
Despite constant cellularity of CD49a+ ILC1s, they may play a critical role in orchestrating the infiltration of CD49b+ NK cells. When compared with CD49b+ NK cells, liver‐resident group 1 ILCs from NKG2A−/− mice expressed higher levels of the IFN‐γ‐inducible NK cell chemoattractant CXCL9 at 12 hpi (Fig. 4A). Indeed, cell division in response to IL‐15 and staining for the proliferation marker Ki67 in NK cells exhibited no differences between the B6 and NKG2A−/− mice, suggesting that enhanced CD49b+ NK cellularity is solely a result of increased recruitment in NKG2A−/− livers (Supplemental Fig. 4). NKG2A signaling, via recruitment of the phosphatases SH2 domain‐containing phosphatase 1 and 2/SH2 domain‐containing inositol 5′‐phosphatase, may limit phosphorylated Jak1‐ or STAT1‐mediated production of CXCL9 in group 1 ILCs, especially in CD49a+ ILC1s, which express high levels of NKG2A [23]. In addition, NKG2A−/− mice retain some (<2%) founding 129 alleles as a result of high‐linkage disequilibrium in the NK complex. Consequently, the enhanced chemokine expression during infection in NKG2A−/− group 1 ILCs may stem from NKG2A and additional signaling events.
IFN‐γ is used by DCs and macrophages for their activation and survival [24, 25, 26–27]. Moreover, expression of Qa‐1b, a nonclassic MHC that serves as the ligand for NKG2A, is induced upon IFN‐γ signaling [28]. The Qa‐1b transcript is elevated in hepatic DCs from NKG2A−/− mice at 12 hpi, suggesting that the increased influx of CD49b+ NK cells and CD49a+CD49b+ ILCs into NKG2A−/− livers enhances the IFN‐γ input received by DCs. It has been reported that the concentration of inflammatory cytokines required to initiate a robust immune response in stressed tissues is not linear but sigmoidal, where a specific concentration of cytokine overcomes inhibition, leading to robust immune responses [29]. Thus, NKG2A signaling on hepatic ILC1s reins in local IFN‐γ production, preventing liver‐resident DCs from reaching the threshold of activation necessary for initiating a robust immune response.
At present, there is an urgent need for vaccines capable of inducing robust and sustainable CD8+ T cell responses to chronic or persistent hepatic infections. It has previously been shown that NKG2A signaling in activated CD8+ T cells suppresses their effector functions, suggesting that global blockade of NKG2A may be beneficial in generating robust anti‐viral CD8+ T cell responses [19]. In this report, we examine the role of NKG2A on innate hematopoietic cells, while excluding the role of NKG2A on activated CD8+ T cells. We demonstrate NKG2A‐competent OT‐I T cell accumulation, with significantly higher levels of IFN‐γ in the NKG2A−/− livers at 3 dpi (Fig. 6A–D). CD11b+ DCs have been reported to play crucial roles in the production of memory anti‐viral CD8+ T cells in the influenza infection model [30]. Although CD103+ DCs contribute to the activation and accumulation of CD8+ T cells early in Ad infection, it is possible that CD11b+ DCs are regulating an enhanced memory CD8+ T cell response in the NKG2A−/− livers. Therefore, the exploration of how NKG2A+ group 1 ILCs may uniquely control each liver‐resident DC subset and their respective contribution to generation of anti‐viral CD8+ T cells during hepatotropic viral infections may provide novel insights into hepatic immunobiology. Indeed, expression of NKG2A on group 1 ILCs is increased in patients chronically infected with HCV [31]. Furthermore, the absence of NKG2A expression on group 1 ILCs is associated with increased resistance to HCV infection [32]. Therefore, the blocking of NKG2A on group 1 ILCs—in particular, CD49a+ ILC1s, in parallel with targeted antigen delivery to hepatic‐resident DCs—could significantly improve vaccine efficacy to generate sustainable CD8+ T cell responses required for the elimination of elusive hepatic pathogens.
AUTHORSHIP
P.D.K., S.N., M.G.B., and Y.S.H. designed the study. P.D.K., S.N., F.A.S., and S‐S.J.S. conducted experiments. P.D.K., S.N., and Y.S.H. wrote the manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
Supporting information
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases Grants F30‐DK104562 (to S.N.) and DK063222 (to Y.S.H.); and NIH National Institute of Allergy and Infectious Diseases Grant U19‐AI083024 (to Y.S.H.). The authors thank the members of the Hahn laboratory for providing advice and constructive criticism on this work. The authors are also grateful to Jeff Teoh for providing technical expertise and assistance.
REFERENCES
- 1. Klenerman, P. , Thimme, R. (2012) T Cell responses in hepatitis C: the good, the bad and the unconventional. Gut 61, 1226–1234. [DOI] [PubMed] [Google Scholar]
- 2. Schmidt, J. , Blum, H. E. , Thimme, R. (2013) T‐Cell responses in hepatitis B and C virus infection: similarities and differences. Emerg. Microbes Infect. 2, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heath, W. R. , Carbone, F. R. (2009) Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 10, 1237–1244. [DOI] [PubMed] [Google Scholar]
- 4. Merad, M. , Sathe, P. , Helft, J. , Miller, J. , Mortha, A. (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 31, 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Münz, C. , Steinman, R. M. , Fujii, S. (2005) Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J. Exp. Med. 202, 203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krueger, P. D. , Kim, T. S. , Sung, S. S. , Braciale, T. J. , Hahn, Y. S. (2015) Liver‐resident CD103+ dendritic cells prime antiviral CD8+ T cells in situ. J. Immunol. 194, 3213–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sonnenberg, G. F. , Artis, D. (2015) Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 21, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cortez, V. S. , Robinette, M. L. , Colonna, M. (2015) Innate lymphoid cells: new insights into function and development. Curr. Opin. Immunol. 32, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Serafini, N. , Vosshenrich, C. A. , Di Santo, J. P. (2015) Transcriptional regulation of innate lymphoid cell fate. Nat. Rev. Immunol. 15, 415–428. [DOI] [PubMed] [Google Scholar]
- 10. Moretta, A. (2002) Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat. Rev. Immunol. 2, 957–964. [DOI] [PubMed] [Google Scholar]
- 11. Lassen, M. G. , Lukens, J. R. , Dolina, J. S. , Brown, M. G. , Hahn, Y. S. (2010) Intrahepatic IL‐10 maintains NKG2A+Ly49‐ liver NK cells in a functionally hyporesponsive state. J. Immunol. 184, 2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peng, H. , Jiang, X. , Chen, Y. , Sojka, D. K. , Wei, H. , Gao, X. , Sun, R. , Yokoyama, W. M. , Tian, Z. (2013) Liver‐resident NK cells confer adaptive immunity in skin‐contact inflammation. J. Clin. Invest. 123, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sojka, D. K. , Plougastel‐Douglas, B. , Yang, L. , Pak‐Wittel, M. A. , Artyomov, M. N. , Ivanova, Y. , Zhong, C. , Chase, J. M. , Rothman, P. B. , Yu, J. , Riley, J. K. , Zhu, J. , Tian, Z. , Yokoyama, W. M. (2014) Tissue‐resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife 3, e01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salazar‐Mather, T. P. , Orange, J. S. , Biron, C. A. (1998) Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP‐1alpha)‐dependent pathways. J. Exp. Med. 187, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pak‐Wittel, M. A. , Yang, L. , Sojka, D. K. , Rivenbark, J. G. , Yokoyama, W. M. (2013) Interferon‐γ mediates chemokine‐dependent recruitment of natural killer cells during viral infection. Proc. Natl. Acad. Sci. USA 110, E50–E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salazar‐Mather, T. P. , Hamilton, T. A. , Biron, C. A. (2000) A chemokine‐to‐cytokine‐to‐chemokine cascade critical in antiviral defense. J. Clin. Invest. 105, 985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Brien, K. L. , Liu, J. , King, S. L. , Sun, Y. H. , Schmitz, J. E. , Lifton, M. A. , Hutnick, N. A. , Betts, M. R. , Dubey, S. A. , Goudsmit, J. , Shiver, J. W. , Robertson, M. N. , Casimiro, D. R. , Barouch, D. H. (2009) Adenovirus‐specific immunity after immunization with an Ad5 HIV‐1 vaccine candidate in humans. Nat. Med. 15, 873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barnes, E. , Folgori, A. , Capone, S. , Swadling, L. , Aston, S. , Kurioka, A. , Meyer, J. , Huddart, R. , Smith, K. , Townsend, R. , Brown, A. , Antrobus, R. , Ammendola, V. , Naddeo, M. , O'Hara, G. , Willberg, C. , Harrison, A. , Grazioli, F. , Esposito, M. L. , Siani, L. , Traboni, C. , Oo, Y. , Adams, D. , Hill, A. , Colloca, S. , Nicosia, A. , Cortese, R. , Klenerman, P. (2012) Novel adenovirus‐based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Transl. Med. 4, 115ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ely, K. H. , Matsuoka, M. , DeBerge, M. P. , Ruby, J. A. , Liu, J. , Schneider, M. J. , Wang, Y. , Hahn, Y. S. , Enelow, R. I. (2014) Tissue‐protective effects of NKG2A in immune‐mediated clearance of virus infection. PLoS One 9, e108385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daussy, C. , Faure, F. , Mayol, K. , Viel, S. , Gasteiger, G. , Charrier, E. , Bienvenu, J. , Henry, T. , Debien, E. , Hasan, U. A. , Marvel, J. , Yoh, K. , Takahashi, S. , Prinz, I. , de Bernard, S. , Buffat, L. , Walzer, T. (2014) T‐bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med. 211, 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Immunological Genome Consortium . (2015) Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cortez, V. S. , Cervantes‐Barragan, L. , Robinette, M. L. , Bando, J. K. , Wang, Y. , Geiger, T. L. , Gilfillan, S. , Fuchs, A. , Vivier, E. , Sun, J. C. , Cella, M. , Colonna, M. (2016) Transforming growth factor‐β signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity 44, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu, T. R. , Hong, Y. K. , Wang, X. D. , Ling, M. Y. , Dragoi, A. M. , Chung, A. S. , Campbell, A. G. , Han, Z. Y. , Feng, G. S. , Chin, Y. E. (2002) SHP‐2 is a dual‐specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J. Biol. Chem. 277, 47572–47580. [DOI] [PubMed] [Google Scholar]
- 24. Gerosa, F. , Baldani‐Guerra, B. , Nisii, C. , Marchesini, V. , Carra, G. , Trinchieri, G. (2002) Reciprocal activating interaction between natural killer cells and dendritic cells. J. Exp. Med. 195, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldszmid, R. S. , Caspar, P. , Rivollier, A. , White, S. , Dzutsev, A. , Hieny, S. , Kelsall, B. , Trinchieri, G. , Sher, A. (2012) NK cell‐derived interferon‐γ orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 36, 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinez, F. O. , Helming, L. , Gordon, S. (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483. [DOI] [PubMed] [Google Scholar]
- 27. Walzer, T. , Dalod, M. , Robbins, S. H. , Zitvogel, L. , Vivier, E. (2005) Natural‐killer cells and dendritic cells: “l'union fait la force”. Blood 106, 2252–2258. [DOI] [PubMed] [Google Scholar]
- 28. Gustafson, K. S. , Ginder, G. D. (1996) Interferon‐gamma induction of the human leukocyte antigen‐E gene is mediated through binding of a complex containing STAT1alpha to a distinct interferon‐gamma‐responsive element. J. Biol. Chem. 271, 20035–20046. [DOI] [PubMed] [Google Scholar]
- 29. Smith, K. A. (2004) The quantal theory of how the immune system discriminates between “self and non‐self”. Med. Immunol. 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim, T. S. , Gorski, S. A. , Hahn, S. , Murphy, K. M. , Braciale, T. J. (2014) Distinct dendritic cell subsets dictate the fate decision between effector and memory CD8(+) T cell differentiation by a CD24‐dependent mechanism. Immunity 40, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nattermann, J. , Feldmann, G. , Ahlenstiel, G. , Langhans, B. , Sauerbruch, T. , Spengler, U. (2006) Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut 55, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thoens, C. , Berger, C. , Trippler, M. , Siemann, H. , Lutterbeck, M. , Broering, R. , Schlaak, J. , Heinemann, F. M. , Heinold, A. , Nattermann, J. , Scherbaum, N. , Alter, G. , Timm, J. (2014) KIR2DL3+NKG2A– natural killer cells are associated with protection from productive hepatitis C virus infection in people who inject drugs. J. Hepatol. 61, 475–481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files