

Short abstract
p38α MAP kinase signaling in macrophages plays context‐dependent pro‐ and anti‐inflammatory roles, with the latter dependent on IL‐10.
Keywords: IBD, colitis, autoimmunity, pharmacologic inhibitor, drug
Abstract
The p38 MAPK pathway was originally identified as a master regulator of proinflammatory cytokine production by myeloid cells. Numerous drugs targeting this kinase showed promise in preclinical models of inflammatory disease, but so far, none have shown efficacy in clinical trials. The reasons behind this are unclear, but may, in part, be explained by emerging anti‐inflammatory functions of this kinase or overly refined selectivity of second‐generation pharmacologic inhibitors. Here, we show that p38α signaling in macrophages plays pro‐ and anti‐inflammatory functions in vivo and in vitro, with the outcome depending on the stimulus, output, kinetics, or mode of kinase inhibition (genetic vs. pharmacologic). Different pharmacologic inhibitors of p38 exhibit opposing effects, with second‐generation inhibitors acting more specifically but inhibiting anti‐inflammatory functions. Functionally, we show that the anti‐inflammatory functions of p38α in macrophages are critically dependent on production of IL‐10. Accordingly, in the absence of IL‐10, inhibition of p38α signaling in macrophages is protective in a spontaneous model of colitis. Taken together, our results shed light on the limited clinical efficacy of drugs targeting p38 and suggest that their therapeutic efficacy can be significantly enhanced by simultaneous modulation of p38‐dependent anti‐inflammatory mediators, such as IL‐10.
Abbreviations
- B6
C57BL/6J
- BIRB
BIRB796
- BMDM
bone marrow‐derived macrophage
- CD
Crohn's disease
- CKO
conditional knockout
- fl
floxed
- KO
knockout
- LCN2
lipocalin 2
- MKK
MAPK kinase
- MS
multiple sclerosis
- MTB
Mycobacterium tuberculosis
- RA
rheumatoid arthritis
- SB
SB203580
- VX
VX‐702
- WT
wild‐type
Introduction
It is becoming increasingly evident that the etiopathogenesis of most inflammatory autoimmune diseases, such as RA, CD, or MS, is too complex for a treatment targeting a single molecule to be fully effective. Personalized medicine approaches and/or combinatorial therapy are the most likely solutions to this problem [1, 2], and many important lessons can be learned from unsuccessful therapeutic approaches.
The p38 MAPK pathway is activated by inflammatory insults (e.g., TLR ligands, cytokines) and stress stimuli (e.g., UV radiation, osmotic stress, DNA damage), via the upstream kinases MKK3 and MKK6, which are, in turn, regulated by numerous MKKs [3]. Four isoforms of p38 MAPK (p38α/Mapk14, p38β/Mapk11, p38γ/Mapk12, and p38δ/Mapk13) have been identified, each encoded by a separate gene. The ubiquitously expressed p38α is the best characterized isoform, which is thought to be responsible for the vast majority of the inflammatory functions of this serine/threonine kinase family. p38α was initially discovered as a target of several small molecules that could block proinflammatory cytokine production by monocytes/macrophages [4]. Hence, compounds targeting p38 MAPK were thought to be excellent candidates for the treatment of inflammatory diseases, where these cytokines are overproduced. Indeed, p38 MAPK inhibitors showed great promise in preclinical models of RA and CD, and many of these compounds advanced to clinical trials [5], but with limited success, to date. Whereas adverse side‐effects were an initial concern [6], this issue has been resolved with more specific inhibitors [7]. However, the compounds tested so far, despite favorable safety profiles, have displayed only transient suppression of inflammatory parameters, and little overall clinical efficacy in RA and CD [8, 9–10]. Why this was the case is a critical question that will greatly help to improve our basic understanding of regulation of inflammatory processes.
Genetic tools have proved invaluable in understanding the functions of p38. As homozygous deletion of p38α is embryonic lethal [11], the generation of p38α CKO mice [12, 13–14] has been instrumental in dissecting cell‐specific functions of this kinase. Emerging studies describe a noncanonical role for p38α in dampening, rather than promoting inflammation, potentially via induction of autoinhibitory‐feedback mechanisms involving dual‐specificity phosphatase 1 and/or immune‐suppressive cytokines, such as IL‐10 [15, 16, 17–18], although the functional contribution of these pathways has not been formally demonstrated. Moreover, CKO studies have shown that deletion of p38α in different cell types can have opposing effects on inflammation and disease outcome [17, 19, 20–21]. In fact, deletion of p38α in macrophages/myeloid cells aggravated skin inflammation [17, 20] and experimental arthritis [18]. Moreover, we have shown in a mouse model of MS, an autoimmune inflammatory disease of the CNS—that deletion of p38α in myeloid cells can either exacerbate or ameliorate disease in a sex‐specific fashion [22]. Thus, the targeting of p38 MAPK may require a more finely tuned approach, such as directing p38 inhibitors to specific cell types and/or using combinatorial therapy to counteract the anti‐inflammatory effects that are lost upon p38 inhibition.
In this study, we systematically examined the effects of genetic deletion and pharmacologic inhibition of p38α in macrophages. We found that depending on stimulus, genetic deletion of p38α resulted in elevated production of proinflammatory cytokines, late in inflammation—an effect that was phenocopied by second‐generation inhibitors of p38. Surprisingly, the first‐generation classic p38 inhibitor SB uniformly suppressed proinflammatory cytokine production, even in cells lacking p38α. Moreover, inhibition of p38α was accompanied by a profound loss of IL‐10 production, which in turn, was functionally responsible for elevated proinflammatory cytokine production in p38α‐deficient macrophages. Consistent with this, lack of myeloid p38α was protective in spontaneous colitis in IL‐10‐deficient mice.
MATERIALS AND METHODS
Mice
p38α floxed mice on the B6 background (B6.129‐Mapk14tm1.2Otsu) [12] were obtained from RIKEN BioResource Center (Ibaraki, Japan). LysM‐Cre mice on the B6 background [B6.129P2‐Lyz2 tm1 (cre)Ifo/J] [23] and IL‐10KO mice on the B6 background (B6.129P2‐Il10 tm1Cgn/J) [24] were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). B6 p38αCKO‐LysM mice were generated as previously described [22] and then crossed to B6 IL‐10KO mice to generate double‐deficient mice. Both p38αCKO‐LysM and IL‐10KO mice were originally created using 129Sv embryonic stem cells and subsequently, extensively backcrossed to B6. This was confirmed by genome‐wide single‐nucleotide polymorphism genotyping at the DartMouse Lab (Dartmouth University, Lebanon, NH, USA) using the Illumina GoldenGate Genotyping Assay for p38αCKO‐LysM and IL‐10KO mice, >97 and >95% of the genome was derived from B6, respectively, with 129Sv‐derived intervals remaining around the Mapk14 and Il10 loci, as expected (data not shown). Male and female mice were used at 6–12 wk of age. The experimental procedures used in this study were approved by the Animal Care and Use Committee of the University of Vermont.
Cell culture experiments
BMDMs were generated as previously described [25] from age‐ and sex‐matched male and female p38αCKO‐LysM or WT (p38αfl/fl littermate) mice or p38αCKO‐LysM and p38αfl/fl littermates on the B6 Il10−/− background. Differentiated BMDMs were replated in 96‐well plates at 50,000 cells/well (ELISA) or in 12‐well plates at 400,000 cells/well (immunoblotting) and stimulated, as indicated, with Escherichia coli 026:B6 LPS (Sigma‐Aldrich, St. Louis, MO, USA) or sonicated heat‐killed MTB H37Ra (BD Difco; Fisher Scientific, Pittsburgh, PA, USA). Supernatants and/or cell lysates were collected at the indicated time points. In certain experiments, cells were pretreated for 10 min before stimulation with recombinant mouse IL‐10, anti‐IL‐10 neutralizing antibody, or isotype‐matched control IgG (IgG2b; BioLegend, San Diego, CA, USA). For pharmacologic experiments, cells were pretreated for 1 h before stimulation with SB203580 (SB), VX‐702 (VX), or BIRB796 (BIRB) (LC Laboratories, Woburn, MA, USA). The drugs remained in culture throughout the rest of the experiment.
In vivo LPS challenge
Age‐ and sex‐matched male and female p38αCKO‐LysM or WT (p38αfl/fl littermate) mice were injected i.p. with LPS at 1 mg/kg body weight. Blood was collected at the indicated time points, and serum cytokines were analyzed by ELISA.
Cell lysates and immunoblot analysis
Whole‐cell lysates were prepared by lysing adherent macrophages directly in Triton lysis buffer, separated by SDS‐PAGE, and transferred to polyvinylidene difluoride membranes, as described previously [26]. Primary antibodies used for Western blot analysis included anti‐phospho‐p38, anti‐p38α, and anti‐GAPDH (Cell Signaling Technology, Danvers, MA, USA). Anti‐mouse and anti‐rabbit secondary antibodies were conjugated HRP (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Membranes were imaged using a chemiluminescent ECL substrate (Thermo Fisher Scientific, Waltham, MA, USA), exposed to X‐ray film.
Cytokine quantification
For the detection of cytokines in the cell culture supernatants or sera, ELISAs were performed as described previously [26], using the primary capture mAb anti‐TNF‐α, and anti‐IL‐6 and their corresponding biotinylated detection mAb (BioLegend). Recombinant mouse TNF‐α and IL‐6 (BioLegend) were used as standards. Other ELISA reagents included the following: HRP‐conjugated Avidin D (Vector Laboratories, Burlingame, CA, USA) and TMB Microwell Peroxidase Substrate and TMB Stop Solution (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA). IL‐10 was measured using a commercial ELISA kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions.
Colitis in IL‐10KO mice
p38αCKO‐LysM and p38αWT (p38αfl/fl littermate) on the Il10+/− or Il10−/− background were monitored several times per week visually for evidence of rectal prolapse. Mice exhibiting apparent rectal prolapse were euthanized. Body weights were taken weekly. Body weight data were only analyzed up to 6 wk, as at this time point, some of the animals had to be euthanized as a result of rectal prolapse.
For histologic evaluation of colitis severity, mice were euthanized at 4 wk of age; colons were removed, prepared using the “Swiss roll” technique, and fixed overnight in formalin, followed by 70% ethanol. Tissues were paraffin embedded, sectioned, and stained with H&E at the University of Vermont Medical Center histology laboratory. Histologic damage scoring was performed on the basis of a semiquantitative scoring system, as previously described by our laboratory [27]. The following features were considered and scored as follows: extent of destruction of normal mucosal architecture (0 = normal; 3 = maximal damage), presence and degree of cellular infiltration (0 = normal; 3 = maximal infiltration), extent of muscle thickening (0 = normal; 3 = maximal thickness), presence or absence of crypt abscesses (0 = absent; 1 = present), and presence or absence of goblet cell mucus (0 = absent; 1 = present). Scoring was done by a trained, board‐certified pathologist (J.W.C.), blinded to the identity of the samples.
Fecal LCN2 levels were determined using a commercially available ELISA kit (R&D Systems), as follows. Fecal pellets were collected on ice and stored at −20°C. Pellets weighed and PBS with 0.1% Tween 20 were added to achieve 50 mg feces/ml. Silicon carbide beads (BioSpec Products, Bartlesville, OK, USA) were added to enhance homogenization. Samples were homogenized by vortexing for 5 min, insoluble material was pelleted by centrifugation, and supernatants were used for ELISA at serial dilutions ranging from 1:10 to 1:10,000.
RESULTS AND DISCUSSION
p38α in macrophages regulates production of proinflammatory cytokines in a stimulus‐ andtime‐dependent manner
To study the role of p38 MAPK signaling in macrophages, we used mice in which the p38α/Mapk14 gene was floxed [12] and conditionally deleted using Cre recombinase, driven by the LysM/Lyz2 promoter [23] (p38αCKO‐LysM). BMDMs from these mice showed ∼98% efficient deletion of p38α/Mapk14 mRNA compared with WT littermates (p38αfl/fl, Cre‐negative; Fig. 1A ). Similar down‐regulation of p38α was seen at the protein level (Fig. 1B). Accordingly, TLR stimulation of BMDMs with bacterial LPS (a TLR4 ligand) or heat‐killed MTB (containing TLR2 and TLR9 ligands) revealed robust phosphorylation of p38 MAPK (as detected by a nonisoform‐specific antibody), which was almost completely abolished in p38α‐deficient BMDMs (Fig. 1B).
Figure 1.
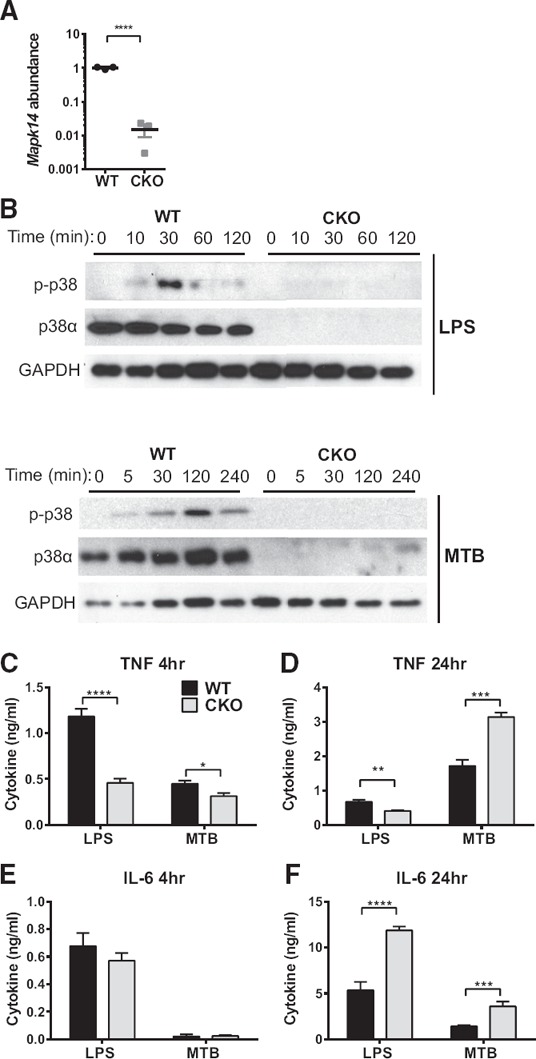
p38α in macrophages regulates production of proinflammatory cytokines in a stimulus‐ and time‐dependent manner.
(A) RNA was isolated from naïve BMDM from male WT and p38αCKO (CKO) mice (n = 3), and Mapk14 mRNA abundance was assayed using quantitative RT‐PCR, with the housekeeping gene B2m as a normalizer, and then normalized further to the average expression in WT as equal to 1. BMDMs were stimulated with 100 ng/ml LPS or 50 µg/ml MTB for the indicated lengths of time (minutes). (B) Cells were lysed at the indicated time points (minutes) and subjected to immunoblotting for phosphorylated p38 (p‐p38), total p38α, and GAPDH (as a loading control). (C–F) The production of the TNF‐α (C and D) and IL‐6 (E and F) at the indicated time points (4 and 24 h) in the cell supernatants was quantified by ELISA. Significance of differences was determined by t‐test (A) and 2‐way ANOVA, followed by Sidak's post hoc comparison tests for individual time points, comparing WT with CKO. Symbols indicate a significant difference between WT and CKO, as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The absence of a symbol indicates a lack of significant difference.
We next examined the TLR‐stimulated production of 2 classic macrophage proinflammatory cytokines, TNF‐α and IL‐6, which have been well documented to be dependent on p38 [3]. We found that LPS‐stimulated production of TNF‐α was indeed reduced in p38α‐deficient BMDMs (Fig. 1C and D), consistent with previous reports [28]. However, MTB‐stimulated production of TNF‐α by p38α‐deficient BMDM was only weakly reduced at 4 h poststimulation and was, in fact, enhanced at 24 h (Fig. 1C and D). Moreover, production of IL‐6 by p38α‐deficient BMDM was robustly enhanced at 24 h after either LPS or MTB stimulation (Fig. 1E and F).
To determine whether a similar phenomenon would be observed in vivo, WT and p38αCKO‐LysM mice were challenged by systemic administration of a sublethal dose of LPS (a model of septic shock), followed by examination of serum cytokine production. Similar to what was observed in vitro, LPS‐induced TNF‐α production was reduced in p38αCKO‐LysM mice, whereas IL‐6 production at later time points was significantly elevated ( Fig. 2 ). Taken together, these results suggest that p38α signaling in macrophages can limit the TLR‐induced production of specific proinflammatory cytokines in a time‐ and stimulus‐dependent manner, in vivo and in vitro.
Figure 2.
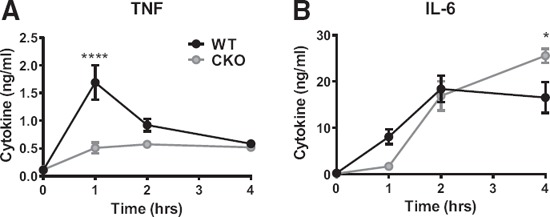
p38α in macrophages regulates systemic production of proinflammatory cytokines in vivo.
WT and p38αCKO‐LysM mice (n = 7/group; WT: 3 females, 4 males; CKO: 4 females, 3 males) received 1 mg/kg LPS i.p. Concentration of TNF‐α (A) and IL‐6 (B) in serum at the indicated time points was determined by ELISA. Data were analyzed by 2‐way ANOVA, followed by Sidak's post hoc comparison tests for individual time points, comparing WT with CKO. Symbols indicate a significant difference between WT and CKO, as follows: *P < 0.05; ****P < 0.0001. The absence of a symbol indicates a lack of significant difference.
Similar context‐dependent opposing functions of p38 MAPK have also been observed in the context of other TLR stimuli. For example, inhibition of p38 enhanced cytokine production by macrophages stimulated with live Borrelia but had the opposite effect if the Borrelia lysate was used for stimulation [29]. With regard to LPS stimulation, our results are partially consistent with previously published data using conditional deletion of p38α in macrophages. Kang et al. [28] showed that TNF‐α production was reduced in p38α‐deficient BMDM or in vivo upon LPS challenge, whereas IL‐6 production was unaffected, the latter observation being partially inconsistent with our results, possibly as a result of the use of a different Cre system (LtrLys‐Cre). However, with the use of the LysM‐Cre system, as used in our study, Kim et al. [17] showed an up‐regulation of IL‐6 in LPS‐stimulated, p38α‐deficient BMDMs, whereas Guma et al. [18] demonstrated elevated IL‐6 production in inflammatory arthritis in myeloid p38α‐deficient mice. More importantly, we showed that stimulation with MTB, unlike LPS, revealed in a robust up‐regulation of both IL‐6 and TNF‐α in p38α‐deficient BMDM (Fig. 1D and F), demonstrating that different TLR stimuli (TLR4 vs. TLR2/9) have differential dependence on the p38 signaling pathway. In this regard, the dependence of “classically” (LPS/TLR4)‐induced TNF‐α production on p38α may have been misleading in identifying this kinase as a broadly anti‐inflammatory drug target.
Two recent studies have also documented the involvement of p38γ and ‐δ signaling in macrophages in LPS‐induced TNF‐α and IL‐6 production, in vivo and in vitro [30, 31]. Simultaneous deletion of these 2 isoforms was shown to result in the reduction of the production of these 2 cytokines, suggesting that p38γ and ‐δ, unlike p38α, play mostly a proinflammatory role. Our results (Fig. 1B) suggest that p38α is the predominant p38 MAPK isoform that is activated by phosphorylation in response to TLR signals in macrophages. We examined the expression of the other 3 isoforms of p38 (β, γ, and δ) in BMDM and found that all 3 were expressed below the limit of detection, and their expression was not affected by deletion of p38α (Supplemental Fig. 1A), similar to previously published results [17]. Similar results were obtained using peritoneal macrophages that exhibited low but detectable levels of the p38β, ‐γ, and ‐δ, which were not affected by p38α deficiency (Supplemental Fig. 1B). However, our results do not completely rule out compensatory or parallel roles of p38γ and ‐δ in BMDM.
Divergent effects of pharmacological inhibitors of p38 MAPK in macrophages
The vast majority of studies implicating p38 MAPK in inflammatory responses have relied on small molecule inhibitors. Such inhibitors have not succeeded in the clinic, and the understanding of why requires the elucidation of the underlying molecular mechanisms. To this end, we compared the effects of genetic deletion of p38α with treatment with 3 different inhibitors of p38: SB, VX, and BIRB. SB is a widely used, first‐generation, competitive inhibitor of p38α and p38β [32]. VX, a second‐generation, competitive inhibitor, has high selectivity for p38α over other isoforms [7]. BIRB, a second‐generation, high‐affinity compound that inhibits p38 MAPKs by binding allosterically [33], inhibits all 4 isoforms of p38, albeit with higher affinity for p38α and p38β [34]. Interestingly, both VX and BIRB have been tested in clinical trials for inflammatory diseases, and both have shown little to no efficacy [8, 10].
We focused on the stimuli and time points where the loss of p38α resulted in enhanced cytokine production (Fig. 1D and F). WT and p38α‐deficient BMDMs were pretreated with either a low (0.5 µM) or high (5 µM) dose of VX, SB, or BIRB, followed by stimulation with LPS or MTB. Data were displayed either as concentrations of cytokines ( Fig. 3A–C ) or to facilitate visualization of inhibitor effects, were normalized to the untreated control levels for WT and p38α‐deficient BMDM separately (Fig. 3D–F). We found that treatment of WT BMDM with VX and BIRB enhanced the production of TNF‐α and IL‐6 to levels approaching that of control (no inhibitor), p38α‐deficient BMDM (Fig. 3). As expected, treatment of p38α‐deficient BMDM with VX or BIRB generally resulted in no effect (Fig. 3). The one exception was MTB‐stimulated release of TNF‐α, where high‐dose BIRB inhibited cytokine production (Fig. 3A and D), suggesting possible off‐target effects at higher doses or potential involvement of p38β, ‐γ, and/or ‐δ (see above). Overall, these results suggest that VX and low‐dose BIRB target p38α specifically and efficiently inhibit its anti‐inflammatory functions.
Figure 3.
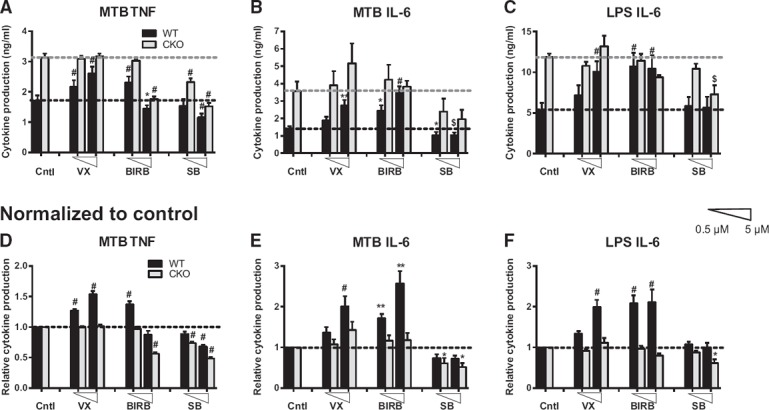
Divergent effects of pharmacological inhibitors of p38 MAPK in macrophages.
WT (black bars) or p38α‐deficient (CKO; gray bars) BMDMs from male mice were pretreated with 0.5 or 5 µM (low or high; as indicated by triangles) of the indicated inhibitors for 30 min, followed by stimulation with 50 µg/ml MTB (A, B, D, and E) or 100 ng/ml LPS (C and F) for 24 h. Production of TNF‐α (A and D) and IL‐6 (B, C, E, and F) in the supernatants was determined by ELISA. Data are displayed as concentration values (A–C) or are normalized relative to the control (Cntl; no inhibitor) production values for WT or CKO (D–F) to facilitate the visualization of the effects of the inhibitors. Data were analyzed by 2‐way ANOVA, followed by Sidak's post hoc test comparing each of the inhibitor‐treated conditions with untreated control for a given genotype (WT and CKO). Symbols indicate a significant difference from control, as follows: *P < 0.05; **P < 0.01; $P < 0.001; #P < 0.0001. The absence of a symbol indicates a lack of significant difference.
Unexpectedly, we found that treatment of WT BMDM with SB resulted in the opposite phenotype compared with VX and BIRB, i.e., reduction of TNF‐α and IL‐6 production. Moreover, treatment of p38α‐deficient BMDM with SB also reduced TNF‐α and IL‐6 production (Fig. 3). Taken together, these results suggest that the widely used, first‐generation SB inhibitor inhibits proinflammatory functions in macrophages, likely by targeting other kinases, in addition to p38α, whereas VX and BIRB—2 drugs that have shown no efficacy in clinical trials—target p38α specifically and abolish its anti‐inflammatory functions.
Many studies implicating p38 MAPK signaling in various physiologic processes have relied solely on the use of classic small molecule inhibitors, such as SB. The specificity of these inhibitors remains unclear. Numerous studies have shown that these inhibitors exhibit off‐target effects on additional kinases [35, 36–37]. These studies are contrasted by elegant chemical genetics studies, in which expression of an SB‐resistant mutant allele of p38α abrogated most of this inhibitor's anti‐inflammatory actions [38, 39]. Importantly, this latter study demonstrates the requirement but not the sufficiency for p38α inhibition in these phenotypes, as concomitant inhibition of kinases other than p38α cannot be ruled out. Consistent with this notion, our results showing that SB affects WT and p38αCKO BMDM similarly support the notion that this drug may inhibit other kinases, in addition to p38α, whereas second‐generation inhibitors, VX and BIRB, are more specific for p38α. Importantly, our results suggest that this improved specificity may have enhanced inhibition of anti‐inflammatory functions of p38α and thus, resulted in lack of efficacy of these drugs in clinical trials.
IL‐10 is required for anti‐inflammatory functions of p38α
We next sought to identify potential downstream mediators for anti‐inflammatory functions of p38α in macrophages. IL‐10 is a critical anti‐inflammatory cytokine that is produced by macrophages in response to TLR stimulation, and its production has been documented by us [22] and others [17] to be dependent on p38α. However, whether IL‐10 is functionally required for anti‐inflammatory functions of p38α has not been directly addressed. We found that, as expected, p38α‐deficient BMDM produced significantly reduced amounts of IL‐10 after 24 h of MTB or LPS stimulation ( Fig. 4A ). No detectable IL‐10 was produced at earlier time points (data not shown). Moreover, all 3 p38 inhibitors uniformly blocked IL‐10 production in WT but not p38α‐deficient BMDM, although not to the same extent as genetic deletion (Fig. 4B and C). Furthermore, in response to LPS challenge in vivo, p38αCKO‐LysM mice produced profoundly reduced amounts of IL‐10 (Fig. 4D).
Figure 4.
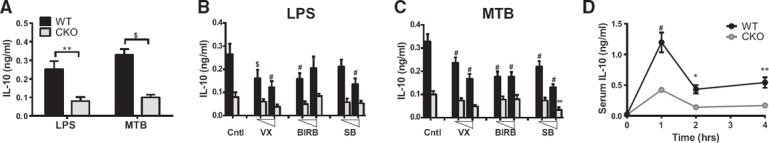
Production of IL‐10 by macrophages is highly dependent on p38α in vitro and in vivo.
(A–C) WT (black bars) or p38α‐deficient (CKO; gray bars) BMDMs from male mice were stimulated with 100 ng/ml LPS (A and B) or 50 µg/ml MTB (A and C) for 24 h. (B and C) BMDMs were pretreated with 0.5 or 5 µM of the indicated inhibitors for 30 min before stimulation. Production of IL‐10 in the supernatants was determined by ELISA. (D) WT and p38αCKO‐LysM mice (n = 7/group; WT: 3 females, 4 males; CKO: 4 females, 3 males) received 1 mg/kg LPS i.p. Concentration of IL‐10 in serum at the indicated time points was determined by ELISA. Data were analyzed by 2‐way ANOVA, followed by Sidak's post hoc comparison tests comparing WT with CKO. Symbols indicate a significant difference either between WT and CKO (A and D) or between control and inhibitor‐treated conditions (B and C), as follows: *P < 0.05; **P < 0.01; $P < 0.001; #P < 0.0001. The absence of a symbol indicates a lack of significant difference.
To address whether the reduction of autocrine IL‐10 in p38α‐deficient BMDM was responsible for enhanced production of TNF‐α and IL‐6, we undertook 3 different approaches: 1) restoration of IL‐10 levels by addition of exogenous IL‐10, 2) neutralization of IL‐10, or 3) genetic deletion of IL‐10. Treatment of p38α‐deficient BMDM with a modest level of exogenous IL‐10 (0.2 ng/ml; corresponding to the reduction in IL‐10 levels as a result of p38α deletion; see Fig. 4A) suppressed the production of TNF‐α and IL‐6 to the levels of approximately that of untreated WT BMDM ( Fig. 5A–C ). Conversely, neutralization of IL‐10 in WT BMDM restored TNF‐α and IL‐6 production to similar levels as those of p38α‐deficient BMDM, either partially or completely (Fig. 5D–F). To exclude the possibility that the anti‐IL‐10 neutralizing antibody caused ligation of FcR in macrophages, which can itself activate p38 and modulate IL‐10 production [40], we treated macrophages (stimulated as in Fig. 5) with isotype control antibody and found that this treatment did not affect IL‐10 production or the difference in TNF‐α and IL‐6 production between WT and p38α‐deficient BMDM (Supplemental Fig. 2). Lastly, genetic ablation of IL‐10 by crossing p38αCKO‐LysM mice to IL‐10KO mice completely eliminated any difference in TNF‐α or IL‐6 production between WT and p38α‐deficient BMDM (Fig. 5H–I). Taken together, these results demonstrate that anti‐inflammatory functions of p38α require production of IL‐10.
Figure 5.
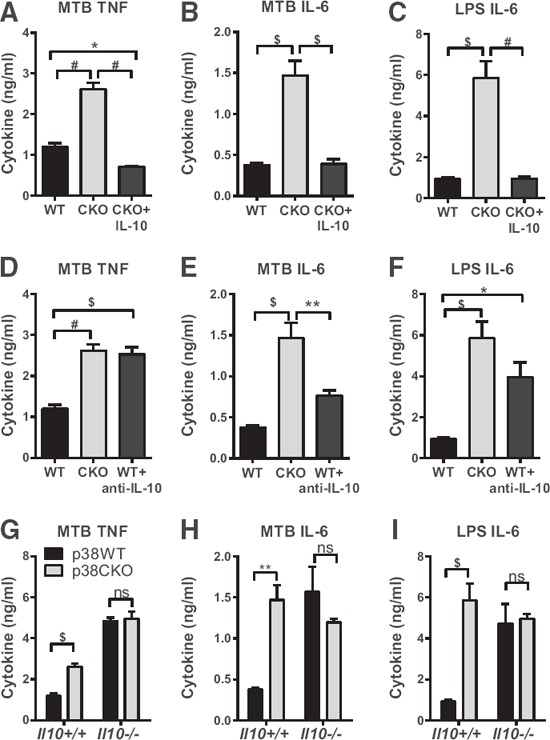
IL‐10 is required for anti‐inflammatory functions of p38α.
WT (black bars) or p38α‐deficient (CKO; gray bars) BMDMs from male and female mice (WT: 4 females, 4 males; CKO: 2 females, 3 males) were stimulated with 50 µg/ml MTB (A, B, D, E, G, and H) or 100 ng/ml LPS (C, F, and I) for 24 h. Production of TNF‐α (A, D, and G) and IL‐6 (B, C, E, F, H, and I) in the supernatants was determined by ELISA. (A–C) BMDMs were also pretreated with 0.2 ng/ml recombinant IL‐10 (blue bars) for 10 min before stimulation. (D–F) BMDMs were also pretreated with 5 µg/ml anti‐IL‐10 neutralizing antibody (blue bars) for 10 min before stimulation. (G–I) Il10+/+ and Il10−/− BMDMs from male mice were used, either p38αWT or p38α deficient (CKO), as indicated. (A–F) Data were analyzed by 2‐way ANOVA, followed by Sidak's post hoc individual comparison tests for all 3 groups. (G–I) Data were analyzed by 2‐way ANOVA, followed by Sidak's post hoc individual comparisons for differences between p38αWT and p38αCKO. Symbols indicate a significant difference among the indicated groups, as follows: *P < 0.05; **P < 0.01; $P < 0.001; #P < 0.0001. The absence of a symbol indicates a lack of significant difference.
These findings are fully consistent with the documented role of the IL‐10‐negative feedback loop downstream of kinases MAPK‐activated protein kinase 2 and mitogen‐ and stress‐activated protein kinase 1/2, both of which are activated by p38α [41, 42–43]. Interestingly, Mosser and colleagues [44] have shown that production of IL‐12, another proinflammatory cytokine, by LPS‐stimulated macrophages (in the presence or absence of FCRγ ligation) is enhanced upon inhibition of p38α, correlating with a reduction in IL‐10. However, the effect of p38 on IL‐12, unlike what we demonstrated here for IL‐6 and TNF, was independent of IL‐10 [44], suggesting that these two phenotypes are distinct and context and stimulus dependent.
Deletion of myeloid p38α in the absence of IL‐10 is protective in spontaneous colitis
Inhibitors of p38 have shown a lack of efficacy in treating inflammatory bowel disease in humans. Although the role of p38 MAPK has been studied in models of chemically induced colitis, its roles in spontaneous colitis have not been addressed. IL‐10KO mice exhibit varying degrees of spontaneous colitis, depending on the facility where the mice are housed [24, 45]. We found a high incidence of rectal prolapse—a hallmark of severe colitis in B6 IL‐10KO mice [45, 46]—in our colony of IL‐10KO mice, and we took advantage of this model to address the role of myeloid p38α in this disease. Heterozygous Il10+/− mice did not exhibit any overt signs of colitis and gained weight normally ( Fig. 6A and B ). Homozygous IL‐10KO mice with an intact p38α gene (Il10−/− p38f/f) exhibited a high incidence of rectal prolapse, which was accompanied by a failure to gain weight, whereas myeloid‐specific deletion of p38α on the IL‐10‐deficient background (Il10−/− p38αCKO) reduced rectal prolapse incidence and ameliorated the associated weight loss (Fig. 6A and B). Histologic damage scoring of colitis‐associated pathology supported these observations, with p38α deficiency resulting in significantly lower colitis scores in Il10−/− mice (Fig. 6C and Supplemental Fig. 3). As a complementary quantitative molecular approach, we measured the levels of fecal LCN2, a widely used dynamic marker of intestinal inflammation [47], and found that p38α deficiency reduced the levels of fecal LCN2 in Il10−/− mice (Fig. 6D). These results demonstrate that in the absence of IL‐10, inhibition of p38α can ameliorate inflammation in vivo.
Figure 6.
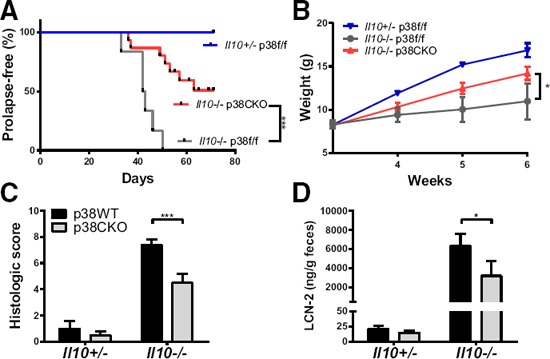
Myeloid‐specific deletion of p38α is protective in spontaneous colitis.
Sex‐matched littermate mice of the indicated genotypes [Il10+/− p38αfl/fl (2 females and 2 males), Il10−/− p38αfl/fl, (4 females and 2 males), and Il10−/− p38αCKO (5 females and 8 males)] were monitored for rectal prolapse (A) and weighed (B) on a weekly basis. (C) Colons were collected from a second cohort of mice at 4 wk of age [Il10+/− p38αfl/fl (5 females and 3 males), Il10+/− p38αCKO (7 females and 4 males), Il10−/− p38αfl/fl, (4 females and 8 males), and Il10−/− p38αCKO (4 females and 2 males)], followed by semiquantitative histologic assessment of colitis severity (see Materials and Methods). (D) Fecal samples were collected from the second cohort of mice at 3 wk of age, and levels of fecal LCN2 were determined (see Materials and Methods). Data were analyzed to test for the effect of p38α deficiency as follows: (A) 2‐way ANOVA (for all time points); (B) Mantel‐Cox test for survival analysis; (C) Mann‐Whitney U test; (D) 2‐way ANOVA with Sidak's multiple comparisons. Symbols indicate a significant difference, as follows: *P < 0.05; ***P < 0.001. The absence of a symbol indicates a lack of significant difference.
The role of p38α in chemically induced colitis has been described in several studies. Unexpectedly, deletion of p38α in the intestinal epithelium was shown to exacerbate disease [19, 48, 49]. However, in line with our results, myeloid‐specific deletion of p38α was shown to be protective, although the production and contribution of IL‐10 were not assessed [19]. As new genetic data strongly implicate dysregulation of the IL‐10 pathway as causative in human colitis [50, 51–52], our use of the IL‐10‐deficient, spontaneous colitis model is particularly relevant. Taken together, these results suggest that cell‐specific targeting of p38α in myeloid cells may have therapeutic potential in treating colitis and possibly other inflammatory diseases, particularly if combined with restoration of IL‐10 levels. In contrast, deletion of p38α in myeloid cells can aggravate skin inflammation [17, 20], experimental arthritis [18], and autoimmune CNS inflammation [22], suggesting that perhaps protective functions of myeloid p38α are colitis specific. This notion is supported by the exquisite sensitivity of colitis to the absence of IL‐10 and the critical requirement for IL‐10 to maintain normal intestinal homeostasis [50, 53, 54].
Taken together, our results indicate that p38α signaling in myeloid cells can play pro‐ or anti‐inflammatory roles, depending on the context. Moreover, drugs targeting p38 exhibit divergent effects on myeloid cells, with second‐generation inhibitors acting more specifically but inhibiting anti‐inflammatory functions. Additionally, we show, to our knowledge, for the first time, that p38α‐dependent production of IL‐10 is required for anti‐inflammatory functions of this kinase in vitro and in vivo. Therefore, we conclude that successful therapeutic targeting of p38α in inflammatory disease would require 2 parameters: 1) cell type specificity and 2) concomitant targeting of anti‐inflammatory pathways dependent on p38α.
AUTHORSHIP
G.M.M., C.T., and D.N.K. conceived of the project and designed the experiments. A.R., J.W.C., M.M.M., and D.N.K. carried out experiments and analyzed data. D.N.K. wrote the manuscript with assistance from A.R. and C.T. Funding was secured by C.T. and D.N.K.
DISCLOSURES
D.N.K. and C.T. are inventors of a patent, p38 MAPK Pathway Inhibitors as Female‐Specific Therapeutics for Autoimmune Disorders of the CNS (US20140127231 A1). The other authors declare no conflicts of interest.
Supporting information
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files
ACKNOWLEDGMENTS
This work was supported, in part, by a FISAR grant from UVM OVPR to D.N.K., the National Multiple Sclerosis Society (Research Grants RG‐1602‐07780 to D.N.K. and RG‐5170A6/1 to C.T.), and U.S. National Institutes of Health (Grants NS069628, NS095007, AI128595, and NS076200 to C.T. and DK62267 to G.M.M.).
REFERENCES
- 1. Bluestone, J. A. , Bour‐Jordan, H. (2012) Current and future immunomodulation strategies to restore tolerance in autoimmune diseases. Cold Spring Harb. Perspect. Biol. 4, a007542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goris, A. , Liston, A. (2012) The immunogenetic architecture of autoimmune disease. Cold Spring Harb. Perspect. Biol. 4, a007260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rincón, M. , Davis, R. J. (2009) Regulation of the immune response by stress‐activated protein kinases. Immunol. Rev. 228, 212–224. [DOI] [PubMed] [Google Scholar]
- 4. Lee, J. C. , Laydon, J. T. , McDonnell, P. C. , Gallagher, T. F. , Kumar, S. , Green, D. , McNulty, D. , Blumenthal, M. J. , Heys, J. R. , Landvatter, S. W. , Strickler, J. E. , McLaughlin, M. M. , Siemens, I. R. , Fisher, S. M. , Livi, G. P. , White, J. R. , Adams, J. L. , Young, P. R. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746. [DOI] [PubMed] [Google Scholar]
- 5. Schindler, J. F. , Monahan, J. B. , Smith, W. G. (2007) p38 Pathway kinases as anti‐inflammatory drug targets. J. Dent. Res. 86, 800–811. [DOI] [PubMed] [Google Scholar]
- 6. Dambach, D. M. (2005) Potential adverse effects associated with inhibition of p38alpha/beta MAP kinases. Curr. Top. Med. Chem. 5, 929–939. [DOI] [PubMed] [Google Scholar]
- 7. Damjanov, N. , Kauffman, R. S. , Spencer‐Green, G. T. (2009) Efficacy, pharmacodynamics, and safety of VX‐702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double‐blind, placebo‐controlled clinical studies. Arthritis Rheum. 60, 1232–1241. [DOI] [PubMed] [Google Scholar]
- 8. Genovese, M. C. (2009) Inhibition of p38: has the fat lady sung? Arthritis Rheum. 60, 317–320. [DOI] [PubMed] [Google Scholar]
- 9. Hammaker, D. , Firestein, G. S. (2010) “Go upstream, young man”: lessons learned from the p38 saga. Ann. Rheum. Dis. 69 (Suppl 1), i77–i82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arthur, J. S. , Ley, S. C. (2013) Mitogen‐activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692. [DOI] [PubMed] [Google Scholar]
- 11. Adams, R. H. , Porras, A. , Alonso, G. , Jones, M. , Vintersten, K. , Panelli, S. , Valladares, A. , Perez, L. , Klein, R. , Nebreda, A. R. (2000) Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 6, 109–116. [PubMed] [Google Scholar]
- 12. Nishida, K. , Yamaguchi, O. , Hirotani, S. , Hikoso, S. , Higuchi, Y. , Watanabe, T. , Takeda, T. , Osuka, S. , Morita, T. , Kondoh, G. , Uno, Y. , Kashiwase, K. , Taniike, M. , Nakai, A. , Matsumura, Y. , Miyazaki, J. , Sudo, T. , Hongo, K. , Kusakari, Y. , Kurihara, S. , Chien, K. R. , Takeda, J. , Hori, M. , Otsu, K. (2004) p38alpha Mitogen‐activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 24, 10611–10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ventura, J. J. , Tenbaum, S. , Perdiguero, E. , Huth, M. , Guerra, C. , Barbacid, M. , Pasparakis, M. , Nebreda, A. R. (2007) p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 39, 750–758. [DOI] [PubMed] [Google Scholar]
- 14. Hui, L. , Bakiri, L. , Mairhorfer, A. , Schweifer, N. , Haslinger, C. , Kenner, L. , Komnenovic, V. , Scheuch, H. , Beug, H. , Wagner, E. F. (2007) p38alpha Suppresses normal and cancer cell proliferation by antagonizing the JNK‐c‐Jun pathway. Nat. Genet. 39, 741–749. [DOI] [PubMed] [Google Scholar]
- 15. Clark, A. R. , Dean, J. L. (2012) The p38 MAPK pathway in rheumatoid arthritis: a sideways look. Open Rheumatol. J. 6, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark, A. R. , Dean, J. L. , Saklatvala, J. (2009) The p38 MAPK pathway mediates both antiinflammatory and proinflammatory processes: comment on the article by Damjanov and the editorial by Genovese. Arthritis Rheum. 60, 3513–3514. [DOI] [PubMed] [Google Scholar]
- 17. Kim, C. , Sano, Y. , Todorova, K. , Carlson, B. A. , Arpa, L. , Celada, A. , Lawrence, T. , Otsu, K. , Brissette, J. L. , Arthur, J. S. , Park, J. M. (2008) The kinase p38 alpha serves cell type‐specific inflammatory functions in skin injury and coordinates pro‐ and anti‐inflammatory gene expression. Nat. Immunol. 9, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guma, M. , Hammaker, D. , Topolewski, K. , Corr, M. , Boyle, D. L. , Karin, M. , Firestein, G. S. (2012) Antiinflammatory functions of p38 in mouse models of rheumatoid arthritis: advantages of targeting upstream kinases MKK‐3 or MKK‐6. Arthritis Rheum. 64, 2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otsuka, M. , Kang, Y. J. , Ren, J. , Jiang, H. , Wang, Y. , Omata, M. , Han, J. (2010) Distinct effects of p38alpha deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology 138, 1255–1265.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ritprajak, P. , Hayakawa, M. , Sano, Y. , Otsu, K. , Park, J. M. (2012) Cell type‐specific targeting dissociates the therapeutic from the adverse effects of protein kinase inhibition in allergic skin disease. Proc. Natl. Acad. Sci. USA 109, 9089–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gupta, J. , Nebreda, A. R. (2015) Roles of p38a mitogen‐activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 282, 1841–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krementsov, D. N. , Noubade, R. , Dragon, J. A. , Otsu, K. , Rincon, M. , Teuscher, C. (2014) Sex‐specific control of central nervous system autoimmunity by p38 mitogen‐activated protein kinase signaling in myeloid cells. Ann. Neurol. 75, 50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clausen, B. E. , Burkhardt, C. , Reith, W. , Renkawitz, R. , Förster, I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277. [DOI] [PubMed] [Google Scholar]
- 24. Kühn, R. , Löhler, J. , Rennick, D. , Rajewsky, K. , Müller, W. (1993) Interleukin‐10‐deficient mice develop chronic enterocolitis. Cell 75, 263–274. [DOI] [PubMed] [Google Scholar]
- 25. Zhang, X. , Goncalves, R. , Mosser, D. M. (2008) The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter 14, Unit 14.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noubade, R. , Milligan, G. , Zachary, J. F. , Blankenhorn, E. P. , del Rio, R. , Rincon, M. , Teuscher, C. (2007) Histamine receptor H1 is required for TCR‐mediated p38 MAPK activation and optimal IFN‐gamma production in mice. J. Clin. Invest. 117, 3507–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Linden, D. R. , Chen, J. X. , Gershon, M. D. , Sharkey, K. A. , Mawe, G. M. (2003) Serotonin availability is increased in mucosa of guinea pigs with TNBS‐induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G207–G216. [DOI] [PubMed] [Google Scholar]
- 28. Kang, Y. J. , Chen, J. , Otsuka, M. , Mols, J. , Ren, S. , Wang, Y. , Han, J. (2008) Macrophage deletion of p38alpha partially impairs lipopolysaccharide‐induced cellular activation. J. Immunol. 180, 5075–5082. [DOI] [PubMed] [Google Scholar]
- 29. Sahay, B. , Patsey, R. L. , Eggers, C. H. , Salazar, J. C. , Radolf, J. D. , Sellati, T. J. (2009) CD14 signaling restrains chronic inflammation through induction of p38‐MAPK/SOCS‐dependent tolerance. PLoS Pathog. 5, e1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. González‐Terán, B. , Cortés, J. R. , Manieri, E. , Matesanz, N. , Verdugo, Á. , Rodríguez, M. E. , González‐Rodríguez, Á. , Valverde, Á. M. , Martín, P. , Davis, R. J. , Sabio, G. (2013) Eukaryotic elongation factor 2 controls TNF‐α translation in LPS‐induced hepatitis. J. Clin. Invest. 123, 164–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Risco, A. , del Fresno, C. , Mambol, A. , Alsina‐Beauchamp, D. , MacKenzie, K. F. , Yang, H. T. , Barber, D. F. , Morcelle, C. , Arthur, J. S. , Ley, S. C. , Ardavin, C. , Cuenda, A. (2012) p38γ and p38δ Kinases regulate the Toll‐like receptor 4 (TLR4)‐induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc. Natl. Acad. Sci. USA 109, 11200–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cuenda, A. , Rouse, J. , Doza, Y. N. , Meier, R. , Cohen, P. , Gallagher, T. F. , Young, P. R. , Lee, J. C. (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin‐1. FEBS Lett. 364, 229–233. [DOI] [PubMed] [Google Scholar]
- 33. Pargellis, C. , Tong, L. , Churchill, L. , Cirillo, P. F. , Gilmore, T. , Graham, A. G. , Grob, P. M. , Hickey, E. R. , Moss, N. , Pav, S. , Regan, J. (2002) Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 9, 268–272. [DOI] [PubMed] [Google Scholar]
- 34. Kuma, Y. , Sabio, G. , Bain, J. , Shpiro, N. , Máirquez, R. , Cuenda, A. (2005) BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 280, 19472–19479. [DOI] [PubMed] [Google Scholar]
- 35. Godl, K. , Wissing, J. , Kurtenbach, A. , Habenberger, P. , Blencke, S. , Gutbrod, H. , Salassidis, K. , Stein‐Gerlach, M. , Missio, A. , Cotten, M. , Daub, H. (2003) An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc. Natl. Acad. Sci. USA 100, 15434–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Menon, M. B. , Kotlyarov, A. , Gaestel, M. (2011) SB202190‐induced cell type‐specific vacuole formation and defective autophagy do not depend on p38 MAP kinase inhibition. PLoS One 6, e23054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao, Y. , Davies, S. P. , Augustin, M. , Woodward, A. , Patel, U. A. , Kovelman, R. , Harvey, K. J. (2013) A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem. J. 451, 313–328. [DOI] [PubMed] [Google Scholar]
- 38. O'Keefe, S. J. , Mudgett, J. S. , Cupo, S. , Parsons, J. N. , Chartrain, N. A. , Fitzgerald, C. , Chen, S. L. , Lowitz, K. , Rasa, C. , Visco, D. , Luell, S. , Carballo‐Jane, E. , Owens, K. , Zaller, D. M. (2007) Chemical genetics define the roles of p38alpha and p38beta in acute and chronic inflammation. J. Biol. Chem. 282, 34663–34671. [DOI] [PubMed] [Google Scholar]
- 39. Guo, X. , Gerl, R. E. , Schrader, J. W. (2003) Defining the involvement of p38alpha MAPK in the production of anti‐ and proinflammatory cytokines using an SB 203580‐resistant form of the kinase. J. Biol. Chem. 278, 22237–22242. [DOI] [PubMed] [Google Scholar]
- 40. Lucas, M. , Zhang, X. , Prasanna, V. , Mosser, D. M. (2005) ERK activation following macrophage FcgammaR ligation leads to chromatin modifications at the IL‐10 locus. J. Immunol. 175, 469–477. [DOI] [PubMed] [Google Scholar]
- 41. Elcombe, S. E. , Naqvi, S. , Van Den Bosch, M. W. , MacKenzie, K. F. , Cianfanelli, F. , Brown, G. D. , Arthur, J. S. (2013) Dectin‐1 regulates IL‐10 production via a MSK1/2 and CREB dependent pathway and promotes the induction of regulatory macrophage markers. PLoS One 8, e60086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. MacKenzie, K. F. , Van Den Bosch, M. W. , Naqvi, S. , Elcombe, S. E. , McGuire, V. A. , Reith, A. D. , Blackshear, P. J. , Dean, J. L. , Arthur, J. S. (2013) MSK1 and MSK2 inhibit lipopolysaccharide‐induced prostaglandin production via an interleukin‐10 feedback loop. Mol. Cell. Biol. 33, 1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ehlting, C. , Trilling, M. , Tiedje, C. , Le‐Trilling, V. T. K. , Albrecht, U. , Kluge, S. , Zimmermann, A. , Graf, D. , Gaestel, M. , Hengel, H. , Häussinger, D. , Bode, J. G. (2016) MAPKAP kinase 2 regulates IL‐10 expression and prevents formation of intrahepatic myeloid cell aggregates during cytomegalovirus infections. J. Hepatol. 64, 380–389. [DOI] [PubMed] [Google Scholar]
- 44. Yang, Z. , Zhang, X. , Darrah, P. A. , Mosser, D. M. (2010) The regulation of Th1 responses by the p38 MAPK. J. Immunol. 185, 6205–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kullberg, M. C. , Ward, J. M. , Gorelick, P. L. , Caspar, P. , Hieny, S. , Cheever, A. , Jankovic, D. , Sher, A. (1998) Helicobacter hepaticus triggers colitis in specific‐pathogen‐free interleukin‐10 (IL‐10)‐deficient mice through an IL‐12‐ and gamma interferon‐dependent mechanism. Infect. Immun. 66, 5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matta, R. , Barnard, J. A. , Wancket, L. M. , Yan, J. , Xue, J. , Grieves, J. , Frazier, W. J. , Nelin, L. , Cato, A. C. , Liu, Y. (2012) Knockout of Mkp‐1 exacerbates colitis in Il‐10‐deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G1322–G1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chassaing, B. , Srinivasan, G. , Delgado, M. A. , Young, A. N. , Gewirtz, A. T. , Vijay‐Kumar, M. (2012) Fecal lipocalin 2, a sensitive and broadly dynamic non‐invasive biomarker for intestinal inflammation. PLoS One 7, e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Caballero‐Franco, C. , Choo, M. K. , Sano, Y. , Ritprajak, P. , Sakurai, H. , Otsu, K. , Mizoguchi, A. , Park, J. M. (2013) Tuning of protein kinase circuitry by p38a is vital for epithelial tissue homeostasis. J. Biol. Chem. 288, 23788–23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gupta, J. , del Barco Barrantes, I. , Igea, A. , Sakellariou, S. , Pateras, I. S. , Gorgoulis, V. G. , Nebreda, A. R. (2014) Dual function of p38α MAPK in colon cancer: suppression of colitis‐associated tumor initiation but requirement for cancer cell survival. Cancer Cell 25, 484–500. [DOI] [PubMed] [Google Scholar]
- 50. Shouval, D. S. , Ouahed, J. , Biswas, A. , Goettel, J. A. , Horwitz, B. H. , Klein, C. , Muise, A. M. , Snapper, S. B. (2014) Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv. Immunol. 122, 177–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Engelhardt, K. R. , Grimbacher, B. (2014) IL‐10 in humans: lessons from the gut, IL‐10/IL‐10 receptor deficiencies, and IL‐10 polymorphisms. Curr. Top. Microbiol. Immunol. 380, 1–18. [DOI] [PubMed] [Google Scholar]
- 52. Louis, E. , Libioulle, C. , Reenaers, C. , Belaiche, J. , Georges, M. (2009) Genetics of ulcerative colitis: the come‐back of interleukin 10. Gut 58, 1173–1176. [DOI] [PubMed] [Google Scholar]
- 53. Keubler, L. M. , Buettner, M. , Häger, C. , Bleich, A. (2015) A multihit model: colitis lessons from the interleukin‐10‐deficient mouse. Inflamm. Bowel Dis. 21, 1967–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mantovani, A. , Marchesi, F. (2014) IL‐10 and macrophages orchestrate gut homeostasis. Immunity 40, 637–639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Files
Supplementary Material Files
Supplementary Material Files