

Short abstract
TLR‐activated FANCC‐deficient macrophages overproduce inflammatory cytokines and sustain DNA damage, where induced DNA damage is not required to sustain aberrant cytokine responses.
Keywords: DNA damage, TLR, TNF‐α
Abstract
The Fanconi anemia proteins participate in a canonical pathway that repairs cross‐linking agent‐induced DNA damage. Cells with inactivated Fanconi anemia genes are universally hypersensitive to such agents. Fanconi anemia‐deficient hematopoietic stem cells are also hypersensitive to inflammatory cytokines, and, as importantly, Fanconi anemia macrophages overproduce such cytokines in response to TLR4 and TLR7/8 agonists. We questioned whether TLR‐induced DNA damage is the primary cause of aberrantly regulated cytokine production in Fanconi anemia macrophages by quantifying TLR agonist‐induced TNF‐α production, DNA strand breaks, crosslinker‐induced chromosomal breakage, and Fanconi anemia core complex function in Fanconi anemia complementation group C‐deficient human and murine macrophages. Although both M1 and M2 polarized Fanconi anemia cells were predictably hypersensitive to mitomycin C, only M1 macrophages overproduced TNF‐α in response to TLR‐activating signals. DNA damaging agents alone did not induce TNF‐α production in the absence of TLR agonists in wild‐type or Fanconi anemia macrophages, and mitomycin C did not enhance TLR responses in either normal or Fanconi anemia cells. TLR4 and TLR7/8 activation induced cytokine overproduction in Fanconi anemia macrophages. Also, although TLR4 activation was associated with induced double strand breaks, TLR7/8 activation was not. That DNA strand breaks and chromosome breaks are neither necessary nor sufficient to account for the overproduction of inflammatory cytokines by Fanconi anemia cells suggests that noncanonical anti‐inflammatory functions of Fanconi anemia complementation group C contribute to the aberrant macrophage phenotype and suggests that suppression of macrophage/TLR hyperreactivity might prevent cytokine‐induced stem cell attrition in Fanconi anemia.
Abbreviations
- CPT
camptothecin
- FA
Fanconi anemia
- FANCC
Fanconi anemia complementation group C
- H2AX
H2A histone family member X
- HSC
hematopoietic stem cell
- MK2
MAPK‐activated protein kinase 2
- MMC
mitomycin C
- NAC
N‐acetylcysteine
- ROS
reactive oxygen species
- T‐shFC
THP‐1 cells expressing short‐hairpin RNA against FANCC
- T‐shNT
THP‐1 cells expressing a nontargeted short‐hairpin RNA
Introduction
FA is an inherited disease characterized by bone marrow failure, developmental defects, and both hematopoietic and epithelial malignancies [1]. The disease is caused by biallelic or X‐chromosome‐linked inactivation of any 1 of 17 different genes, including FANCC [2, 3]. Much research has focused on the canonical role of these gene products, many of which interact in multimeric nuclear complexes in mediating repair of DNA interstrand cross‐links [4]. However, FA proteins interact in subcomplexes outside the nucleus with other molecules not known to be directly involved with nuclear DNA repair [5, 6]. They likely function to mediate cellular responses to ROS [5, 7] and inflammatory stimuli [6, 8, 9–10]. FA hematopoietic stem and progenitor cells are hypersensitive to inflammatory cytokines and repeated cytokine exposure induces stem cell (HSC) exhaustion [11, 12–13]. In addition, FA macrophages overproduce such inflammatory cytokines when provoked by selected TLR ligands [14, 15–16], thereby contributing to the development of the FA marrow failure phenotype.
The role of the canonical DNA damage pathway in the macrophage phenotype is uncertain. Some have suggested that all hematopoietic defects in FA are simply downstream effects of DNA damage accumulation [17]. However, a similar point of view was prevalent in the radiation biology field vis‐á‐vis radiation‐induced marrow failure until more recent work revealed that the loss of HSCs in radiated animals was induced instead by radiation‐induced inflammatory responses ignited by damage to nonhematopoietic sites [18]. Consequently, we sought to directly test the idea that DNA damage is sufficient to account for the characteristically unrestrained cytokine response of FA macrophages. In the present study, by directly quantifying DNA damage and cytokine responses in TLR‐activated FA macrophages, we have shown that TLR‐dependent cytokine overproduction can be uncoupled from differential DNA damage in both human and murine FA cells, findings supporting the view that FA core complex proteins play a fundamental role in modulating the innate immune response.
MATERIALS AND METHODS
Cell culture and reagents
Primary bone marrow‐derived macrophages were prepared from wild‐type and Fancc −/− mice, as described previously [14]. Specifically, low‐density bone marrow mononuclear cells were isolated from 2–3‐month‐old mice and plated into non‐tissue culture‐treated plates in IMDM with 20% FBS and 40 ng/ml GM‐CSF for M1 macrophages or M‐CSF for M2 macrophages. After 7 d of culture, nonadherent cells were removed by PBS washing, and the cells were switched to fresh serum‐free medium with 20 ng/ml GM‐CSF or M‐CSF. After 48 h, nonadherent cells were removed by PBS washing, and macrophages were harvested with Cell Dissociation Buffer (Sigma‐Aldrich, St. Louis, MO, USA). Flow cytometry confirmed that both wild‐type and FA M1 cells were CD14‐positive macrophages [19]. Specifically, wild‐type M1 and M2 cells were 86.9% and 94.2% CD14‐positive, respectively, and Fancc −/− M1 and M2 cells were 88.6% and 90.4% CD14‐positive, respectively (Supplemental Fig. 1). For repolarization experiments, macrophages were treated with either IL‐4 (20 ng/ml) or IFN‐γ (100 ng/ml) during the 48‐h serum‐free treatment. THP‐1 human acute monocytic leukemia cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and THP1‐XBlue cells were purchased from InvivoGen (San Diego, CA, USA). The development and culture conditions of the derivative cell lines we established (T‐shNT and T‐shFC) have been previously described [14, 16].
LPS, NAC, CPT, and MMC were purchased from Sigma‐Aldrich. R848 was purchased from Enzo Life Sciences (Farmingdale, NY, USA). H2O2 was purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Comet Assay
The alkaline Comet Assay (Trevigen, Gaithersburg, MD, USA) was performed according to the manufacturer's protocol, with all cells cultured at 105/ml. Comet images were visualized by fluorescence microscopy (Microphot‐FX; Nikon, Tokyo, Japan) with a fluorescein isothiocyanate filter and photographed using a QImaging Micropublisher camera and QCapture software (QImaging, Surrey, BC, Canada) with ×40 magnification. Images of 100 randomly selected cells were scored using CometScore software (TriTek Corporation, Sumerduck, VA, USA). The percentage of tail DNA (i.e., percentage of tail DNA = 100 − percentage of head DNA) was determined using the imaging software [20].
ELISAs
Mouse bone marrow‐derived macrophages were plated at 105 cells/ml (104 cells/well, 100 µl), and cells of the human monocytic cell line THP‐1 were plated at 106 cells/ml (2 × 105 cells/well, 200 µl) in tissue culture–treated 96‐well dishes and treated with LPS or R848. After 24 h, mouse or human TNF‐α was measured from supernatants using Quantikine ELISA Kits (R&D Systems, Minneapolis, MN, USA). For details regarding cytokine arrays, see Supplemental Materials and Methods.
Chromosomal breakage analysis
Chromosomal breakage analyses were performed as previously described [21]. The cells were treated with the indicated doses of MMC or R848 for 24 h (murine macrophages) or 48 h (THP‐1 cells). The cultures were harvested after a 2‐h exposure to 0.25 μg/ml colcemid (Sigma‐Aldrich) and treated with 3 ml hypotonic solution (75 mM KCl, 5% FBS) for 10 min, and fixed with 3 ml of 3:1 methanol:acetic acid. The slides prepared for metaphase spreads were then stained with 0.2% Wright's stain (Fisher Scientific, Pittsburgh, PA, USA) for 3.5 min. Fifty metaphases for each sample were scored for the number of breaks per cell and radial content on an Eclipse E800 Photoscope (Nikon).
Western blotting
Western blots were performed, as previously described [16]. Anti‐FA complementation group D2 (catalog no. sc‐20022) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐FANCC (catalog no. 2) was obtained from the Fanconi Anemia Research Fund Antibody Project (Portland, OR, USA). Anti‐GAPDH (catalog no. 2118) was purchased from Cell Signaling Technology (Danvers, MA, USA).
Immunofluorescence
Cytopreparations were prepared using the Shandon Elliot cytocentrifuge (Shandon Scientific, London, U.K.), and the slides were stained with anti‐phospho‐histone H2AX (catalog no. 2577; Cell Signaling Technology), anti‐rabbit IgG Fab2 Alexa Fluor 647 (catalog no. 4414; Cell Signaling Technology), and Hoechst‐33342 (catalog no. sc‐200908; Santa Cruz Biotechnology). The cells were visualized using the Axio Imager.M2 microscope (Carl Zeiss, Oberkochen, Germany) using the Plan‐Apochromat 63×/1.40 Oil M27 objective (Carl Zeiss). Images were obtained using the AxioCam MRm camera with ApoTome unit (Carl Zeiss) and Zen software (Carl Zeiss). For details regarding macrophage flow cytometry, see Supplemental Materials and Methods.
Statistical analysis
For the ELISA experiments, each numerical value was the average of 3 biologic replicates in which the concentration of TNF‐α was measured. Error bars represent the sd of these values. For the comet assays, each value represents the average percentage of DNA in the comet tail from 100 randomly selected cells from each condition. The error bars in the comet assays represent the sem of these values [22, 23, 24–25]. For quantification of phospho‐H2AX foci, each value represents the total number of foci counted from 12 images per condition (approximately 20 cells per image) divided by the total number of cells counted on the 12 images. Nuclear foci quantification and measurement were performed by an observer (G.C.B.) who was unaware of the culture conditions. Error bars represent the sem foci per cell. All P values were calculated using the paired Student t test.
RESULTS
TLR4‐induced overproduction of TNF‐α and DNA strand breaks in M1 FA macrophages but not in M2 FA macrophages
The phenotype of macrophages is profoundly affected by their microenvironment [26, 27] and by “polarizing” growth factors and cytokines [28]. Two well‐studied phenotypes, M1 and M2, are commonly generated using culture conditions that induce reversible functional changes [29, 30–31]. Activation of type M1 cells leads to the production of more proinflammatory cytokines, and activated M2 cells produce more anti‐inflammatory cytokines [30]. Both M1 and M2 cells produce TNF‐α in response to LPS, although the production of TNF‐α is higher in M1 cells [30]. The responsiveness of both cell types provided us with an opportunity to test the sufficiency of crosslinker‐induced DNA damage in each of those cell types. To determine whether the cytokine overproduction phenotype in FA macrophages is common to both M1 and M2 macrophages, we produced each type from low‐density bone marrow cells of Fancc −/− and wild‐type mice using 9‐day exposures to GM‐CSF (type M1) or M‐CSF (type M2) [29]. M1 macrophages from Fancc −/− mice produced approximately twofold more TNF‐α in response to the TLR4 agonist LPS than did M1 cells from wild‐type mice ( Fig. 1A ). However, M2 cells from Fancc −/− and wild‐type mice produced identical levels of TNF‐α in response to LPS (Fig. 1A). When Fancc −/− and wild‐type M2 macrophages were “repolarized” to the M1 type by treatment with IFN‐γ, the cells from the Fancc −/− mice overproduced TNF‐α by approximately 1.5‐fold in response to LPS (Fig. 1B). In contrast, the LPS‐induced overproduction of TNF‐α by M1 macrophages from Fancc −/− mice was partially suppressed (from 2‐fold [Fig. 1A] to 1.5‐fold) by their “repolarization” to the M2 type by treatment with IL‐4 (Fig. 1C). Neither IL‐4 nor IFN‐γ affected TNF‐α production by M2 cells from either Fancc −/− or wild‐type mice (Fig. 1B and C). Therefore, the TNF‐α production phenotype was not “fixed” and sorted with the M1 phenotype, ruling out that the increased TNF‐α production resulted from acquired genetic or fixed epigenetic changes during ex vivo culture.
Figure 1.
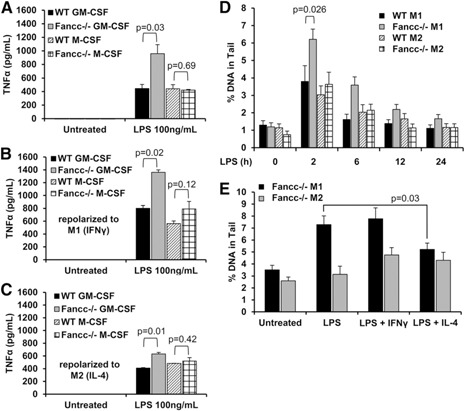
LPS‐induced overproduction of TNF‐α and DNA damage in FA macrophages requires M1 polarization. (A–C) Type M1 (GM‐CSF) and type M2 (M‐CSF) bone marrow‐derived macrophages from wild‐type (WT) and Fancc −/− mice were plated at a concentration of 105 cells/ml and treated with LPS (100 ng/ml) for 24 h. Secreted TNF‐α was measured in the conditioned media by ELISA (mean ± sd) from 1 representative experiment of 3 performed, each of which included 3 biologic replicates. Before LPS stimulation, some cells were repolarized toward the M1 type with IFN‐γ (100 ng/ml) (B) or toward the M2 type with IL‐4 (20 ng/mL) (C) for 48 h. (D) M1 and M2 bone marrow‐derived macrophages from wild‐type and Fancc −/− mice were plated at a concentration of 105 cells/ml and treated with LPS (100 ng/ml) for 2, 6, 12, or 24 h. The zero time point represents cells not treated with LPS. Five thousand cells from each condition were used in alkaline comet assays as described in Materials and Methods. Images of 100 randomly selected cells were scored using CometScore software (TriTek Corporation). Shown is the mean ± sem percentage of tail DNA. (E) M1 and M2 bone marrow‐derived macrophages from Fancc −/− mice were repolarized toward the M1 type with IFN‐γ (100 ng/ml), toward the M2 type with IL‐4 (20 ng/ml), or not repolarized for 48 h, as indicated. Cells were plated at a concentration of 105 cells/ml and treated with LPS (100 ng/ml) for 2 h. Five thousand cells from each condition were used in comet assays. Shown is the mean ± sem percentage of tail DNA. Each of 3 experiments contained pooled cells from 2 wild‐type mice and 2 Fancc −/− mice. P values were calculated using the 2‐tailed paired Student t test.
LPS has been shown to induce DNA damage in several cell types [32]. To determine whether LPS‐induced production of TNF‐α correlated directly with DNA damage, we performed single cell gel electrophoresis (comet assays) on DNA from M1 and M2 macrophages of Fancc −/− and wild‐type mice to quantify the DNA strand breaks. Untreated M1 and M2 macrophages from Fancc −/− and wild‐type mice contained equivalent amounts (1%) of DNA in the comet tail; however, treatment with LPS for 2 h induced DNA strand breaks in all 4 cell types. M1 macrophages from Fancc −/− mice contained approximately 2‐fold more DNA strand breaks than did M1 macrophages from wild‐type mice after 2 h of LPS treatment (Fig. 1D). M2 macrophages from Fancc −/− and wild‐type mice treated with LPS for 2 h contained similar levels of comet formation. In all 4 cell types, DNA strand breaks declined after 6 h and returned to background (untreated) levels after 24 h (Fig. 1D). To examine the possibility that greater amounts of DNA damage occurred earlier after LPS exposure, we measured LPS‐induced DNA damage in M1 macrophages after time points shorter than 2 h. LPS induced comet formation after 1 h; however, only after 2 h was significantly more DNA damage present in cells from Fancc −/− mice (Supplemental Fig. 2). Repolarization of Fancc‐deficient M1 macrophages to the M2 type by treatment with IL‐4 abrogated differential LPS‐induced strand breaks (Fig. 1E). In summary, although LPS induced DNA strand breaks in both M1 and M2 cell types, only FA M1 cells overproduced TNF‐α and they were the only cells that exhibited higher levels of DNA strand breaks than were seen in wild‐type M1 cells in response to LPS.
M1 and M2 macrophages from Fancc −/− mice are equally hypersensitive to mitomycin C
If the classic FA phenotype of crosslinker hypersensitivity was sufficient to cause TNF‐α overproduction, we reasoned that FA M2 macrophages (which produce normal amounts of TNF‐α in response to LPS) should have fewer chromosomal breaks in response to MMC, a DNA crosslinking agent routinely used in establishing the diagnosis of FA [1]. However, MMC induced chromosomal breaks in both M1 and M2 macrophages from Fancc −/− mice ( Table 1 ), indicating that DNA damage induced by a canonical challenge to FA cells was not sufficient to account for the differences observed in M1 and M2 FA cells. These findings suggest that the differential responses of M1 and M2 macrophages either resulted from a noncanonical function of Fancc in M1 macrophages or that the FA phenotype was observable only in the M1 cells because a greater load of double strand breaks (but not crosslinks) was induced by LPS. To distinguish between these 2 possibilities, we quantified LPS‐induced double strand breaks in response to LPS.
Table 1.
M1 and M2 macrophages from Fancc −/− mice are equally hypersensitive to MMC in chromosomal breakage analysis
| Cell type | Treatment | Concentration (ng/ml) | Radials (%) | Breaks per cell |
|---|---|---|---|---|
| WT GM‐CSF | Control | 0 | 0.02 | |
| MMC | 10 | 0 | 0.09 | |
| MMC | 20 | 2 | 0.16 | |
| WT M‐CSF | Control | 0 | 0.11 | |
| MMC | 10 | 0 | 0.09 | |
| MMC | 20 | 2 | 0.12 | |
| Fancc −/− GM‐CSF | Control | 3 | 0.71 | |
| MMC | 10 | 6 | 0.58 | |
| MMC | 20 | 8 | 1.38 | |
| Fancc −/− M‐CSF | Control | 2 | 0.55 | |
| MMC | 10 | 6 | 1.22 | |
| MMC | 20 | 10 | 2.00 |
M1 and M2 bone marrow‐derived macrophages from wild‐type and Fancc −/− mice were treated with MMC (10 or 20 ng/ml) for 24 h. MMC induced more breaks per cell and more radial forms in both M1 (GM‐CSF) and M2 (M‐CSF) cells from Fancc −/− mice than in cells from wild‐type mice. WT, wild type.
LPS‐induced TNF‐α overproduction and DNA damage in Fancc −/− M1 macrophages are dependent on ROS
Early work seeking to define unique cellular features of FA described an important role for oxidative stress in the development of the chromosomal aberrations characteristic of this disease [33]. In light of previous observations by Sejas et al [12] that LPS induces both high‐level ROS and TNF‐α in Fancc‐deficient mice, we considered that the DNA strand breaks evolved in M1 cells as a consequence of LPS‐induced ROS. NAC suppressed LPS‐induced production of TNF‐α by both M1 and M2 macrophages in a dose‐dependent manner ( Fig. 2A ). NAC also suppressed LPS‐induced comet formation in both M1 and M2 macrophages from Fancc −/− and wild‐type mice (Fig. 2B). That the lowest dose of NAC completely abrogated the differences between the wild‐type and Fancc‐deficient macrophages in both comet and TNF‐α assays is compatible with the view that hypersensitivity to ROS or excessive production of ROS by mutant cells leads to strand breaks and that TNF‐α overproduction is a consequence of DNA damage in mutant cells. However, because NAC can directly suppress NF‐κB signaling, irrespective of its antioxidant effects [34], additional experiments are required to confirm a direct relationship. Considering that the LPS‐induced double strand breaks might be limited to the TLR4 pathway, we sought to identify an alternative TLR agonist that might not induce ROS and DNA strand breaks.
Figure 2.
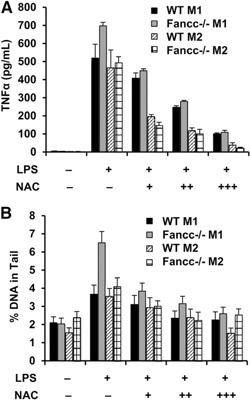
NAC suppresses LPS‐induced TNF‐α production and DNA strand breaks. (A) M1 and M2 bone marrow‐derived macrophages from wild‐type (WT) and Fancc −/− mice were plated (105 cells/ml) and treated with LPS (100 ng/ml) for 24 h with and without NAC (1, 5, and 10 mM). Secreted TNF‐α was measured in the conditioned media by ELISA (mean ± sd). Shown is 1 representative experiment of 3, each of which were performed using 3 biologic replicates and pooled cells from 2 wild‐type mice and 2 Fancc −/− mice. (B) M1 and M2 bone marrow‐derived macrophages from wild‐type and Fancc −/− mice (105 cells/ml) were treated with LPS (100 ng/ml) for 2 h with and without NAC (1, 5, and 10 mM). Five thousand cells from each condition were used in the comet assay. Mean ± sem percentage of tail DNA is shown.
R848‐induced TNF‐α overproduction by Fancc‐ and FANCC‐deficient macrophages is not ROS dependent
When treated with R848 (a TLR7/8 agonist), M1 and M2 macrophages from Fancc −/− mice produced more TNF‐α than did cells from wild‐type mice ( Fig. 3A ). However, R848 did not induce differential DNA strand breaks (Supplemental Fig. 3), and NAC failed to suppress R848‐induced TNF‐α production by these cells (Fig. 3B). To examine R848 responses in human cells and confirm that our results were not species specific, we used previously described control (T‐shNT) and FANCC‐deficient (T‐shFC) THP‐1 human monocytic leukemia cell lines [14, 15–16]. We first reconfirmed [16] that the T‐shFC cells exhibited the classic FA phenotype in chromosomal breakage assays. Specifically, we treated these cells with either MMC or R848 and measured the chromosomal breakage and radial formation. MMC induced more chromosomal breaks and radials in T‐shFC cells than in T‐shNT cells ( Table 2 ). However, R848 induced neither chromosomal breaks nor radial forms (Table 2) in either cell type. Moreover, that MMC, but not R848, induced FA complementation group D2 ubiquitination confirmed that R848 does not activate the canonical FA pathway in normal cells (Fig. 3C).
Figure 3.
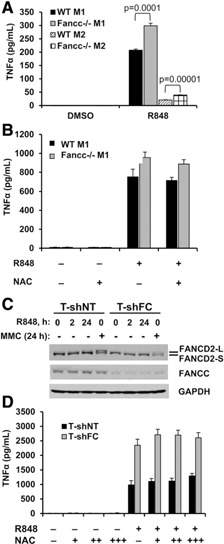
NAC does not suppress R848‐induced TNF‐α production and R848 does not induce FA complementation group D2 ubiquitination. (A) M1 and M2 bone marrow‐derived macrophages from wild‐type (WT) and Fancc −/− mice were plated at a concentration of 105 cells/ml and stimulated with R848 (3 µM) for 24 h. (B) M1 bone marrow‐derived macrophages from wild‐type and Fancc −/− mice were plated at a concentration of 105 cells/ml and pretreated with NAC (1 mM) for 2 h before stimulation with R848 (3 µM) for 24 h. (C) T‐shNT and T‐shFC cells were plated at a concentration of 106 cells/ml treated with R848 (30 μM) for 0, 2, or 24 h or with MMC (120 nM) for 24 h. Whole cell extracts were prepared and subjected to Western blot analysis with antibodies against FA complementation group D2, FANCC, and GAPDH (loading control). MMC (positive control) induced wholesale conversion of FA complementation group D2‐S to FA complementation group D2‐L in T‐shNT cells, but this effect was not observed in the same cells exposed to R848 (lane 3). The suppression of FANCC in T‐shFC cells was also confirmed and did not change with either R848 or MMC treatment. (D) THP‐1 cells expressing short‐hairpin RNA directed against FANCC (T‐shFC) or a nontargeted short‐hairpin RNA (T‐shNT) were plated at a concentration of 106 cells/ml and pretreated with NAC (10 and 100 µM and 1 mM) for 2 h before stimulation with R848 (30 μM) for 24 h. Secreted TNF‐α was measured in the conditioned media by ELISA (mean ± sd from 1 representative experiment of 3 performed with 3 biologic replicates). P values were calculated using a paired 2‐tailed Student t test. FANCD2‐L, mono‐ubiquitinated FA complementation group D2; FANCD2‐S, unmodified FA complementation group D2.
Table 2.
MMC induces excessive chromosomal breaks and radial formation in T‐shFC cells
| Cell type | Treatment | Concentration | Radials (%) | Breaks per cell |
|---|---|---|---|---|
| T‐shNT | Control | 2 | 0.1 | |
| MMC | 60 ng/ml | 4 | 0.18 | |
| MMC | 80 ng/ml | 4 | 0.2 | |
| T‐shFC | Control | 0 | 0 | |
| MMC | 60 ng/ml | 8 | 0.44 | |
| MMC | 80 ng/ml | 10 | 0.46 | |
| T‐shNT | Control | 0 | 0.02 | |
| R848 | 3 µM | 0 | 0.02 | |
| R848 | 30 µM | 0 | 0.12 | |
| T‐shFC | Control | 0 | 0.02 | |
| R848 | 3 µM | 0 | 0.02 | |
| R848 | 30 µM | 0 | 0 |
In response to MMC (48 h), T‐shFC cells acquired more breaks per cell and radial forms than did T‐shNT cells, but R848 treatment (48 h) failed to induce breaks or radials in either cell type.
As expected [14, 15–16], R848 induced TNF‐α overproduction by T‐shFC cells but the response to this TLR7/8 activator was not suppressed by NAC in either T‐shNT or T‐shFC cells (Fig. 3D). Therefore, although TLR4‐dependent TNF‐α overproduction by Fancc‐deficient macrophages might depend on LPS‐induced ROS and DNA double strand breaks, excessive TLR7/8‐dependent TNF‐α production induced by R848 in FANCC‐deficient human and Fancc‐deficient murine cells is ROS and DNA strand break independent.
DNA damage is not sufficient to induce cytokine production in normal or FA macrophages
To formally test the capacity of DNA damage per se to induce TNF‐α gene expression, we treated T‐shNT and T‐shFC cells with R848 or several doses of MMC, CPT, and H2O2 and measured TNF‐α production ( Table 3 ). Although R848 induced TNF‐α overproduction by T‐shFC cells, no dose of MMC, CPT, or H2O2 induced detectable TNF‐α production in either T‐shNT or T‐shFC cells (Table 3). Similar results were observed in murine Fancc −/− and wild‐type M1 and M2 macrophages (data not shown).
Table 3.
DNA damage does not induce TNF‐α production by macrophages
| Cell line | Treatment | TNF‐α (pg/ml) | sd |
|---|---|---|---|
| T‐shNT | DMSO | Undetectable | NA |
| T‐shFC | DMSO | Undetectable | NA |
| T‐shNT | R848 (30 μM) | 984 | 147 |
| T‐shFC | R848 (30 μM) | 2349 | 208 |
| T‐shNT | MMC (100 nM) | Undetectable | NA |
| T‐shFC | MMC (100 nM) | Undetectable | NA |
| T‐shNT | MMC (300 nM) | Undetectable | NA |
| T‐shFC | MMC (300 nM) | Undetectable | NA |
| T‐shNT | MMC (1 μM) | Undetectable | NA |
| T‐shFC | MMC (1 μM) | Undetectable | NA |
| T‐shNT | H2O2 (0.00003%) | Undetectable | NA |
| T‐shFC | H2O2 (0.00003%) | Undetectable | NA |
| T‐shNT | H2O2 (0.0003%) | Undetectable | NA |
| T‐shFC | H2O2 (0.0003%) | Undetectable | NA |
| T‐shNT | H2O2 (0.003%) | Undetectable | NA |
| T‐shFC | H2O2 (0.003%) | Undetectable | NA |
| T‐shNT | CPT (100 nM) | Undetectable | NA |
| T‐shFC | CPT (100 nM) | Undetectable | NA |
| T‐shNT | CPT (1 μM) | Undetectable | NA |
| T‐shFC | CPT (1 μM) | Undetectable | NA |
T‐shNT and T‐shFC cells were plated at a concentration of 106 cells/ml and treated with R848 (30 µM), MMC (100 nM, 300 nM, or 1 µM), H2O2 (0.00003%, 0.0003%, or 0.003%), or CPT (100 nM or 1 µM) for 24 h. Mean (TNF‐α) ± sd was derived from 3 biologic replicates for each condition. NA, not available.
R848 and CPT synergistically enhance DNA damage and TNF‐α overproduction in FANCC‐deficient macrophages
Although R848 alone was incapable of inducing DNA damage ( Fig. 4A ), we tested the idea that R848 might enhance damage induced by a well‐known inducer of double strand breaks, CPT. CPT induced slightly more DNA damage in T‐shFC cells than in T‐shNT cells, and R848 enhanced that response in both cell types by approximately 20% (Fig. 4B). At the highest dose, CPT enhanced R848‐induced TNF‐α production by T‐shFC cells by approximately 20% but did not affect production by T‐shNT cells (Fig. 4C). MMC had no influence on R848‐induced TNF‐α production by either cell type (Fig. 4C). As long as the TLR7/8 agonist was included in the macrophage cultures, the quantitative differences in TNF‐α production that distinguished the FANCC‐deficient from the control cells were maintained in all experiments, irrespective of exposure to CPT or MMC.
Figure 4.
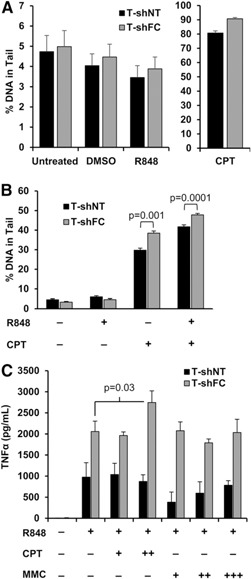
R848 alone does not induce DNA strand breaks, but R848 and CTP synergistically enhance strand breaks and TNF‐α overproduction in FANCC‐deficient mononuclear phagocytes. (A) T‐shNT and T‐shFC cells were plated at a concentration of 105 cells/ml and treated with R848 (30 μM) or CPT (10 µM) for 2 h. (B) T‐shNT and T‐shFC cells were plated at a concentration of 105 cells/ml and pretreated with CPT (10 µM) for 2 h before stimulation with R848 (30 μM) for 2 h. Five thousand cells from each condition were analyzed using the comet assay. The mean ± sem percentage of tail DNA is shown. (C) T‐shNT and T‐shFC cells were plated at a concentration of 106 cells/ml and pretreated with CPT (100 nM or 1 µM) or MMC (100 mM, 300 mM, or 1 µM) before stimulation with R848 (30 μM) for 24 h. Secreted TNF‐α was measured in the conditioned media by ELISA. Data shown (mean ± sd) were derived from 3 biologic replicates for each condition, and P values were calculated using the paired 2‐tailed Student t test. CPT enhanced TNF‐α production only in R848‐stimulated T‐shFC cells but MMC had no such effect.
R848 does not induce phospho‐H2AX nuclear foci in FANCC‐deficient mononuclear phagocytes
Phosphorylated histone H2AX (also known as γ‐H2AX) nuclear foci formation is a sensitive, albeit an indirect and a nonspecific, marker of DNA damage [35]. Although we did not observe differences in chromosomal breakage (Table 2) or comet formation (Fig. 4A) between R848‐treated T‐shNT and T‐shFC cells, we questioned whether R848 or MMC could induce phospho‐H2AX nuclear foci formation. We counted nuclear phospho‐H2AX foci (diameter ≥1 µM) in unstimulated (DMSO), R848‐treated, and MMC‐treated T‐shNT and T‐shFC cells. MMC equally induced (approximately 3 per nucleus) phospho‐H2AX foci per cell in both T‐shNT and T‐shFC cells ( Fig. 5A and B ). We observed approximately 1.7‐fold more nuclear foci in unstimulated T‐shFC than in unstimulated T‐shNT cells but R848 did not significantly enhance focus formation in either cell type (Fig. 5B). As expected [14, 15–16], T‐shFC cells produced approximately 9‐fold more TNF‐α in response to R848 than did the T‐shNT cells, and MMC did not induce TNF‐α production (Fig. 5C).
Figure 5.
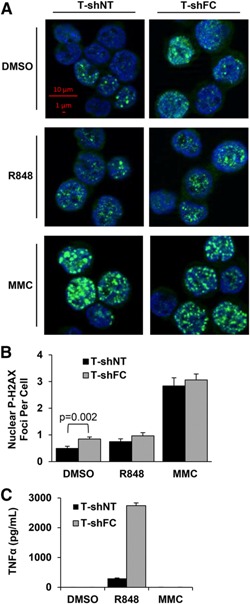
R848 does not induce phospho‐H2AX foci. (A) T‐shNT and T‐shFC cells were plated at a concentration of 106/ml and treated with R848 (30 μM) or MMC (120 nM) for 24 h. Cytopreparations were made from each of 3 biologic replicates for each condition, and the cells were stained with anti‐phospho‐H2AX (green) and Hoechst‐33342 (blue). Scale bars (red) indicate 1 and 10 μm (total magnification, ×630). Images were obtained using the Carl Zeiss Axio Imager.M2 microscope with the Plan‐Apochromat 63x/1.40 Oil M27 objective and AxioCam MRm camera with ApoTome unit. Foci were counted from four 142.1‐µm × 102.48‐µm fields of each replicate for a total of 12 fields per condition, and each image contained approximately 20 cells with visible phopho‐H2AX staining. Shown is 1 representative cropped image of each condition that contained 3 biologic replicates from 1 experiment. (B) Nuclear phospho‐H2AX foci (≥1 µM in diameter) were counted using images of unstimulated T‐shNT (258 cells) and T‐shFC (287 cells), R848‐treated T‐shNT (170 cells) and T‐shFC (256 cells), and MMC‐treated T‐shNT (195 cells) and T‐shFC (295 cells). Values represent pooled total foci counted from all 12 images from each condition divided by the total number of cells. Data shown (mean ± sem) indicate number of foci per cell. P values were calculated using the paired 2‐tailed Student t test. (C) Secreted TNF‐α was measured in the conditioned media by ELISA. Data shown (mean ± sd) are derived from 3 biologic replicates for each condition.
DISCUSSION
Chemical cross‐linking agents induce chromosomal breakage, G2/M arrest, and cell death in normal cells [36]. Somatic cells from patients with FA are highly sensitive to the DNA‐damaging effects of such agents, even at doses that will not injure normal cells [37]. The response to cross‐linking agents, in fact, forms the basis of the most commonly used diagnostic test for this disease. Based on recently published genetic experiments in mice, it has been proposed that the genomic hypersensitivity to endogenous cross‐linking agents (aldehydes) underlies the stem cell injury and leads to one of the most common and life‐threatening clinical phenotypes in this disease, that of bone marrow failure [38].
However, noncanonical FA protein subcomplexes exist [39], and some of the 17 FA proteins have rather extensive intermolecular interactions with the proteins involved in signal transduction processes in extranuclear sites [5, 6, 40, 41–42]. In vitro and in vivo studies have also revealed that hematopoietic cells deficient in FA proteins exhibit other quantifiable defects that contribute to marrow failure and even the evolution of leukemic clones [43]. Although it has widely been presumed to be the case that these phenotypes all devolve from DNA repair defects in FA cells, the cause‐and‐effect relationship has not been established. First, defects in stem cell replication [44] and repopulation [45] are quantifiable early in development [46] and can involve signaling pathways uninvolved in DNA repair [47, 48]. Second, hematopoietic stem and progenitor cells are highly sensitive to inflammatory cytokines, including TNF‐α, IL‐1β, and MIP‐1α [8, 10, 15, 43, 49], and oxidative stress [5, 7, 33, 50, 51–52]. The cytokine hypersensitivity of FA hematopoietic cells is additionally exacerbated by unique abnormalities in FA macrophages. Specifically, human FA complementation group A‐, FANCC‐, and murine Fancc‐deficient mononuclear phagocytes challenged with selected TLR activators overproduce the very inflammatory cytokines that uniquely suppress the replicative activity of their progenitors [14, 15–16]. These findings are compatible with the idea that the self‐replicative defects of HSCs might result in exhaustion of the stem cell pool under conditions of inflammation and oxidative stress. HSC function is severely perturbed in LPS‐treated Fancc‐deficient mice [12], and HSC exhaustion results from repeated rounds of induced inflammation in Fancc‐deficient mice [11, 13].
Given that the FA proteins interact in nuclear complexes that govern responses to exogenous and endogenous DNA cross‐linking agents, one potential explanation for all of the phenotypes in FA hematopoietic cells is that they occur solely as a result of accumulated DNA damage. Alternatively, the innate immune and hematopoietic abnormalities might reflect noncanonical functions of the multifunctional FA proteins [53, 54–55]. Such alternative functions likely vary biochemically among the individual proteins; however, all members of the core complex might function together to protect HSCs from exhaustion during environmental stress and, in the case of FANCC‐ and FA complementation group A‐deficient mononuclear phagocytes, to suppress exaggerated responses to TLR7/8 agonists [14, 15–16].
The signaling pathways responsible for this aberrant response include inhibitor of NF‐κB kinase and p38 MAPK‐dependent prolonged activation of its key substrate MK2, a protein that enhances the stability and translation of mRNAs encoding inflammatory cytokines [14, 15, 56] and is an essential signaling protein for tissue damage after environmental stress [57]. Nonetheless, p38 MAPK is known to be activated by cisplatin [58], and MK2 is also known to be critically important for prolonged checkpoint maintenance after DNA damage [59]. Moreover, production of inflammatory cytokines can be directly influenced by ROS [60, 61]. Theoretically, then, it remained possible that DNA strand breaks in mononuclear phagocytes occur in response to canonical TLR pathway activation and that the DNA damage is a direct and sufficient cause of the observed exaggerated cytokine response of FA cells. However, the results of our studies suggest otherwise.
We quantified DNA strand breaks, chromosomal breakage, and TNF‐α production in primary murine M1 and M2 cells. The first evidence suggesting that crosslinker‐induced DNA damage was not involved in the aberrant cytokine response was that FA M2 macrophages maintained the MMC‐induced chromosomal breakage phenotype (Table 1) but did not overproduce cytokines when stimulated with TLR agonists. That MMC failed to enhance R848‐induced TNF‐α production in normal or FANCC‐deficient THP‐1 cells (Fig. 4C) confirmed this notion.
We also quantified phosphorylated histone H2AX nuclear foci in normal and FANCC‐deficient THP‐1 cells. We found that although unstimulated FANCC‐deficient cells contain more foci than control cells, focus formation was not enhanced by treatment with R848 (Fig. 5A and B). MMC‐induced focus formation was equivalent in FANCC‐deficient and control cells (Fig. 5A and B). It is unclear whether H2AX foci formation in unstimulated cells is DNA damage related or is instead mediated by the cell cycle checkpoint protein 2 pathway during mitosis or via phosphatidylinositol‐3‐kinase‐related kinase or other pathways [35], given that the specificity of phospho‐histone H2AX nuclear foci as a marker of DNA damage has been called into question [35]. In the ground state, when nuclear foci are increased in FANCC‐deficient cells (Fig. 5A and B), TNF‐α is not produced (Fig. 5C) and the addition of R848, although robustly inducing TNF‐α overproduction (Fig. 5C), did not result in an increase in the number of nuclear foci (Fig. 5B).
Having uncoupled the crosslinker hypersensitivity and cytokine phenotypes, we next tested the potential linkage between ROS‐induced DNA strand breaks and TNF‐α by quantifying NAC‐sensitive LPS‐induced breaks in both murine and human cells. DNA strand breaks were induced in both wild‐type and mutant M1 and M2 cells in response to LPS (Fig. 1D), albeit excessively only in Fancc‐deficient M1 cells. We also found that LPS did not induce DNA breaks in THP‐1 cells (Supplemental Fig. 4). The LPS/DNA strand break/TNF‐α phenotype was not irreversibly fixed in FA M1 cells because repolarization of Fancc‐deficient M1 cells to the M2 phenotype reduced both differential TNF‐α production (Fig. 1C) and DNA strand breaks (Fig. 1E). These findings are of potential therapeutic relevance and also indicate that TLR‐induced TNF‐α overproduction was not caused by accumulation of unrepairable DNA lesions in ex vivo cultured macrophages from Fancc −/− mice. NAC suppressed LPS‐induced TNF‐α overproduction (Fig. 2A) and DNA strand breaks (Fig. 2B), compatible with the view that, in FA cells, the TLR4‐dependent cytokine response pathway does depend on ROS‐induced DNA damage.
Although we have demonstrated that cross‐linking agent‐induced DNA damage is not related to the TLR hyperactive state of FANCC‐deficient cells, we cannot be certain that ROS‐induced damage is unrelated to that state. Although the NAC effects we observed are suggestive, others have shown that suppression of ROS with NAC can suppresses NF‐κB activation by blocking TNF‐α from binding to its receptor [34]; thus, NAC might partially suppress TNF‐α production by blocking a TNF‐α–induced TNF‐α positive feedback pathway. Therefore, it remains possible that strand breaks and TLR/cytokine responses were either independent events in Fancc‐deficient cells or were linked but only uniquely to the LPS/TLR4 response and not to other TLR pathways. When TLR7/8 was activated by R848, the FA TLR/cytokine response was again seen in M1 cells (Fig. 3A); however, R848 did not induce strand breaks (Fig. 4A and Supplemental Fig. 3). Moreover, the addition of NAC did not suppress R848‐induced TNF‐α production (Fig. 3D). Consequently, unlike the TLR4 response, the aberrant TLR7/8 response was independent of DNA strand breaks, observations confirmed in FANCC‐deficient human mononuclear phagocytes (Fig. 4A).
We also questioned whether Fancc might function to suppress expression of TLR4, thereby explaining why Fancc −/− M1 cells overproduce TNF‐α and sustain more DNA breaks in response to LPS. However, we found that a higher percentage of M2 cells expressed TLR4 than did M1 cells (Supplemental Fig. 5). More importantly, wild‐type and Fancc −/− M1 cells expressed similar levels of TLR4 (Supplemental Fig. 5A), as did wild‐type M2 and Fancc−/− M2 cells (Supplemental Fig. 5B), ruling out an effect of Fancc on TLR4 gene expression in M1 cells.
Additionally, TLR ligands have been shown to activate noncanonical cytokine production through the inflammasome, independent of the TLR pathway [62, 63]. In recent studies [15], to determine whether inflammasome activation contributes to TNF‐α production, we used the inflammasome inhibitor glyburide, combined with the TLR7/8 agonist R848, in cultures of T‐shNT and T‐shFC cells. Although glyburide suppressed TLR‐induced IL‐1β production, it did not suppress TNF‐α production [15]. We, therefore, concluded that noncanonical inflammasome activation by R848 does not contribute to TNF‐α production in these cell types.
Finally, we also investigated whether R848‐induced cytokines other than TNF‐α or IL‐1β [15] were overproduced by FANCC‐deficient cells and whether anti‐inflammatory cytokines such as IL‐10 were produced by control cells and not FANCC‐deficient cells. Other cytokines overproduced by T‐shFC cells include basic fibroblast growth factor, G‐CSF, IL‐15, hepatocyte growth factor, vascular endothelial growth factor, IFN‐α, IL‐2R, IL‐6, RANTES, IL‐12, MCP‐1, and IL‐1 receptor antagonist (Supplemental Fig. 6). We found that IL‐10 was also overproduced by T‐shFC cells, by 3‐fold (Supplemental Fig. 6). Therefore, a lack of IL‐10 production in response to TLR stimulation does not explain why FANCC‐deficient cells overproduce inflammatory cytokines.
Taken together, our studies have demonstrated clearly that DNA damage per se is insufficient to account for cytokine overproduction in FA macrophages and that TLR pathway activation is absolutely required. Although cross‐linking agent‐induced damage is clearly uninvolved, we cannot state that DNA damage, in particular, ROS‐induced double strand breaks, is entirely unlinked to the cytokine response. This is especially true for the case of TLR4 activation with LPS, in which we found a significant early increase in double strand breaks only in Fancc‐deficient monocytes (Fig. 1D). Two findings suggest that excessive double strand breaks might enhance TNF‐α production ignited by TLR agonists. First, consistent with the findings of Sejas et al. [12], LPS likely induces high‐level double strand breaks in Fancc‐deficient cells through a ROS‐dependent mechanism, because NAC abrogates both the differential strand breaks and the differential production of TNF‐α in those cells (Fig. 2). Although chemically induced double strand breaks induced by exposure to CPT alone did not induce TNF‐α expression at all, it did result in a greater amount of DNA in the comet tail in FANCC‐deficient monocytes (Fig. 4B). Also, when combined with R848 at high doses, it did enhance TNF‐α gene expression and did so specifically in FANCC‐deficient cells (Fig. 4C). However, the results of our studies using the specific TLR7/8 activator R848 have indicated that the overproduction of TNF‐α in FANCC‐deficient monocytes treated with R848 is not associated with a greater than normal load of double strand breaks (Fig. 4A and Supplemental Fig. 3), suggesting that the noncanonical functions of FANCC (e.g., modulation of p38 MAPK and MK2 activity [14, 15–16]) might play a major role in the exaggerated inflammatory response of FA macrophages.
In FA, the inefficiently constrained innate immune response very likely contributes to the onset and progression of bone marrow failure and clonal selection [13, 43, 64]. Identifying aberrant signaling pathways in FA monocytes is clinically important, because potential pharmacological approaches to treatment pivot importantly on a better molecular biologic determination of tractable pathways and the careful avoidance of harm. If the hyperactive inflammatory state evolves in FA hematopoietic tissues in a way that effectively targets only the most genomically unstable stem cells for destruction (allowing the more fit ones to survive), pharmacological correction of the aberrant cytokine response might permit the undesirable survival of mutant stem cell clones. In contrast, if the proinflammatory condition of FA macrophages stems from noncanonical FA protein functions and if that condition drives stem cell exhaustion indiscriminately in individual stem cells, interdicting the aberrant inflammatory response would have some appeal as a therapeutic option to prevent both marrow failure and the selection of cytokine‐resistant stem cell clones. Future experiments in newly described murine FA models of inflammation‐induced stem cell exhaustion [11, 12–13] should seek to address the effect of pharmacological suppression of the inflammatory response on stem cell quiescence and exhaustion and must also critically focus on quantifying the load of somatic mutations that might accumulate as a result.
AUTHORSHIP
M.R.G., L.E.H., and G.C.B. designed the project, developed the assays, and analyzed the primary data. M.R.G. and L.E.H. harvested and cultured murine cells. L.E.H. and K.C. maintained Fancc‐deficient mice and performed macrophage repolarization experiments. M.R.G. maintained THP‐1 cell cultures, performed ELISA experiments, Western blotting, immunofluorescence, flow cytometry, and analyzed primary data. R.K.R. and N.J. performed comet assays. M.R.G. and G.C.B. performed microscopy and counting of phosphorylated histone H2AX foci. M.A.‐D., A.E.H.N., and S.B.O. performed chromosomal breakage assays. A.A. performed the cytokine array. M.R.G., G.C.B., and L.E.H. wrote the manuscript.
DISCLOSURE
The authors declare no conflicts of interest.
Supporting information
Supplementary data
ACKNOWLEDGMENTS
This study was supported by the U.S. National Institutes of Health National Heart, Lung, and Blood Institute (Grant P01 HL048546; to G.C.B. and S.B.O.), the Department of Veterans Affairs (Merit Review; to G.C.B.), and the Fanconi Anemia Research Fund (to M.R.G.).
REFERENCES
- 1. Bagby, G.C. , Alter, B.P. (2006) Fanconi anemia. Semin. Hematol. 43, 147–156. [DOI] [PubMed] [Google Scholar]
- 2. De Winter, J.P. , Joenje, H. (2009) The genetic and molecular basis of Fanconi anemia. Mutat. Res. 668, 11–19. [DOI] [PubMed] [Google Scholar]
- 3. Sawyers, C.L. (1998) Molecular abnormalities in myeloid leukemias and myelodysplastic syndromes. Leuk. Res. 22, 1113–1122. [DOI] [PubMed] [Google Scholar]
- 4. Kottemann, M.C. , Smogorzewska, A. (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mukhopadhyay, S.S. , Leung, K.S. , Hicks, M.J. , Hastings, P.J. , Youssoufian, H. , Plon, S.E. (2006) Defective mitochondrial peroxiredoxin‐3 results in sensitivity to oxidative stress in Fanconi anemia. J. Cell Biol. 175, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pang, Q. , Keeble, W. , Christianson, T.A. , Faulkner, G.R. , Bagby, G.C. (2001) FANCC interacts with Hsp70 to protect hematopoietic cells from IFN‐gamma/TNF‐alpha‐mediated cytotoxicity. EMBO J. 20, 4478–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Du, W. , Rani, R. , Sipple, J. , Schick, J. , Myers, K.C. , Mehta, P. , Andreassen, P.R. , Davies, S.M. , Pang, Q. (2012) The FA pathway counteracts oxidative stress through selective protection of antioxidant defense gene promoters. Blood 119, 4142–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haneline, L.S. , Broxmeyer, H.E. , Cooper, S. , Hangoc, G. , Carreau, M. , Buchwald, M. , Clapp, D.W. (1998) Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac‐/‐ mice. Blood 91, 4092–4098. [PubMed] [Google Scholar]
- 9. Dufour, C. , Corcione, A. , Svahn, J. , Haupt, R. , Poggi, V. , Béka'ssy, A.N. , Scimè, R. , Pistorio, A. , Pistoia, V. (2003) TNF‐alpha and IFN‐gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF‐alpha suppresses erythropoiesis in vitro. Blood 102, 2053–2059. [DOI] [PubMed] [Google Scholar]
- 10. Li, X. , Yang, Y. , Yuan, J. , Hong, P. , Freie, B. , Orazi, A. , Haneline, L.S. , Clapp, D.W. (2004) Continuous in vivo infusion of interferon‐gamma (IFN‐gamma) preferentially reduces myeloid progenitor numbers and enhances engraftment of syngeneic wild‐type cells in Fancc‐/‐ mice. Blood 104, 1204–1209. [DOI] [PubMed] [Google Scholar]
- 11. Hu, L. , Huang, W. , Hjort, E. , Eklund, E.A. (2013) Increased Fanconi C expression contributes to the emergency granulopoiesis response. J. Clin. Invest. 123, 3952–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sejas, D.P. , Rani, R. , Qiu, Y. , Zhang, X. , Fagerlie, S.R. , Nakano, H. , Williams, D.A. , Pang, Q. (2007) Inflammatory reactive oxygen species‐mediated hemopoietic suppression in Fancc‐deficient mice. J. Immunol. 178, 5277–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walter, D. , Lier, A. , Geiselhart, A. , Thalheimer, F.B. , Huntscha, S. , Sobotta, M.C. , Moehrle, B. , Brocks, D. , Bayindir, I. , Kaschutnig, P. , Muedder, K. , Klein, C. , Jauch, A. , Schroeder, T. , Geiger, H. , Dick, T.P. , Holland‐Letz, T. , Schmezer, P. , Lane, S.W. , Rieger, M.A. , Essers, M.A. , Williams, D.A. , Trumpp, A. , Milsom, M.D. (2015) Exit from dormancy provokes DNA‐damage‐induced attrition in haematopoietic stem cells. Nature 520, 549–552. [DOI] [PubMed] [Google Scholar]
- 14. Anur, P. , Yates, J. , Garbati, M.R. , Vanderwerf, S. , Keeble, W. , Rathbun, K. , Hays, L.E. , Tyner, J.W. , Svahn, J. , Cappelli, E. , Dufour, C. , Bagby, G.C. (2012) p38 MAPK inhibition suppresses the TLR‐hypersensitive phenotype in FANCC‐ and FANCA‐deficient mononuclear phagocytes. Blood 119, 1992–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garbati, M.R. , Hays, L.E. , Keeble, W. , Yates, J.E. , Rathbun, R.K. , Bagby, G.C. (2013) FANCA and FANCC modulate TLR and p38 MAPK‐dependent expression of IL‐1β in macrophages. Blood 122, 3197–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vanderwerf, S.M. , Svahn, J. , Olson, S. , Rathbun, R.K. , Harrington, C. , Yates, J. , Keeble, W. , Anderson, D.C. , Anur, P. , Pereira, N.F. , Pilonetto, D.V. , Pasquini, R. , Bagby, G.C. (2009) TLR8‐dependent TNF‐(alpha) overexpression in Fanconi anemia group C cells. Blood 114, 5290–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garaycoechea, J.I. , Crossan, G.P. , Langevin, F. , Daly, M. , Arends, M.J. , Patel, K.J. (2012) Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575. [DOI] [PubMed] [Google Scholar]
- 18. Guinan, E.C. , Barbon, C.M. , Kalish, L.A. , Parmar, K. , Kutok, J. , Mancuso, C.J. , Stoler‐Barak, L. , Suter, E.E. , Russell, J.D. , Palmer, C.D. , Gallington, L.C. , Voskertchian, A. , Vergilio, J.A. , Cole, G. , Zhu, K. , D'Andrea, A. , Soiffer, R. , Weiss, J.P. , Levy, O. (2011) Bactericidal/permeability‐increasing protein (rBPI21) and fluoroquinolone mitigate radiation‐induced bone marrow aplasia and death. Sci. Transl. Med. 3, 110ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Becker, L. , Liu, N.C. , Averill, M.M. , Yuan, W. , Pamir, N. , Peng, Y. , Irwin, A.D. , Fu, X. , Bornfeldt, K.E. , Heinecke, J.W. (2012) Unique proteomic signatures distinguish macrophages and dendritic cells. PLoS One 7, e33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumaravel, T.S. , Vilhar, B. , Faux, S.P. , Jha, A.N. (2009) Comet Assay measurements: a perspective. Cell Biol. Toxicol. 25, 53–64. [DOI] [PubMed] [Google Scholar]
- 21. Lensch, M.W. , Tischkowitz, M. , Christianson, T.A. , Reifsteck, C.A. , Speckhart, S.A. , Jakobs, P.M. , O'Dwyer, M.E. , Olson, S.B. , Le Beau, M.M. , Hodgson, S.V. , Mathew, C.G. , Larson, R.A. , Bagby, G.C., Jr. (2003) Acquired FANCA dysfunction and cytogenetic instability in adult acute myelogenous leukemia. Blood 102, 7–16. [DOI] [PubMed] [Google Scholar]
- 22. Heaton, P.R. , Ransley, R. , Charlton, C.J. , Mann, S.J. , Stevenson, J. , Smith, B.H. , Rawlings, J.M. , Harper, E.J. (2002) Application of single‐cell gel electrophoresis (comet) assay for assessing levels of DNA damage in canine and feline leukocytes. J. Nutr. 132 (6:, Suppl 2)1598S–1603S. [DOI] [PubMed] [Google Scholar]
- 23. Noz, K.C. , Bauwens, M. , van Buul, P.P. , Vrolijk, H. , Schothorst, A.A. , Pavel, S. , Tanke, H.J. , Vermeer, B.J. (1996) Comet assay demonstrates a higher ultraviolet B sensitivity to DNA damage in dysplastic nevus cells than in common melanocytic nevus cells and foreskin melanocytes. J. Invest. Dermatol. 106, 1198–1202. [DOI] [PubMed] [Google Scholar]
- 24. Chen, B.Y. , Huang, C.H. , Lin, Y.H. , Huang, C.C. , Deng, C.X. , Hsu, L.C. (2014) The K898E germline variant in the PP1‐binding motif of BRCA1 causes defects in DNA Repair. Sci. Rep. 4, 5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Swain, U. , Subba Rao, K. (2011) Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mech. Ageing Dev. 132, 374–381. [DOI] [PubMed] [Google Scholar]
- 26. Wynn, T.A. , Chawla, A. , Pollard, J.W. (2013) Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lavin, Y. , Winter, D. , Blecher‐Gonen, R. , David, E. , Keren‐Shaul, H. , Merad, M. , Jung, S. , Amit, I. (2014) Tissue‐resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lawrence, T. , Natoli, G. (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761. [DOI] [PubMed] [Google Scholar]
- 29. Jaguin, M. , Houlbert, N. , Fardel, O. , Lecureur, V. (2013) Polarization profiles of human M‐CSF‐generated macrophages and comparison of M1‐markers in classically activated macrophages from GM‐CSF and M‐CSF origin. Cell. Immunol. 281, 51–61. [DOI] [PubMed] [Google Scholar]
- 30. Fleetwood, A.J. , Lawrence, T. , Hamilton, J.A. , Cook, A.D. (2007) Granulocyte‐macrophage colony‐stimulating factor (CSF) and macrophage CSF‐dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J. Immunol. 178, 5245–5252. [DOI] [PubMed] [Google Scholar]
- 31. Davis, M.J. , Tsang, T.M. , Qiu, Y. , Dayrit, J.K. , Freij, J.B. , Huffnagle, G.B. , Olszewski, M.A. (2013) Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio 4, e00264–e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Glukhov, I.L. , Sirota, N.P. , Kuznetsova, E.A. (2008) DNA damage in human mononuclear cells induced by bacterial endotoxin. Bull. Exp. Biol. Med. 146, 301–303. [DOI] [PubMed] [Google Scholar]
- 33. Joenje, H. , Arwert, F. , Eriksson, A.W. , de Koning, H. , Oostra, A.B. (1981) Oxygen‐dependence of chromosomal aberrations in Fanconi's anaemia. Nature 290, 142–143. [DOI] [PubMed] [Google Scholar]
- 34. Hayakawa, M. , Miyashita, H. , Sakamoto, I. , Kitagawa, M. , Tanaka, H. , Yasuda, H. , Karin, M. , Kikugawa, K. (2003) Evidence that reactive oxygen species do not mediate NF‐kappaB activation. EMBO J. 22, 3356–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cleaver, J.E. , Feeney, L. , Revet, I. (2011) Phosphorylated H2Ax is not an unambiguous marker for DNA double‐strand breaks. Cell Cycle 10, 3223–3224. [DOI] [PubMed] [Google Scholar]
- 36. Heinrich, M.C. , Hoatlin, M.E. , Zigler, A.J. , Silvey, K.V. , Bakke, A.C. , Keeble, W.W. , Zhi, Y. , Reifsteck, C.A. , Grompe, M. , Brown, M.G. , Magenis, R.E. , Olson, S.B. , Bagby, G.C. (1998) DNA cross‐linker‐induced G2/M arrest in group C Fanconi anemia lymphoblasts reflects normal checkpoint function. Blood 91, 275–287. [PubMed] [Google Scholar]
- 37. Sasaki, M.S. , Tonomura, A. (1973) A high susceptibility of Fanconi's anemia to chromosome breakage by DNA cross‐linking agents. Cancer Res. 33, 1829–1836. [PubMed] [Google Scholar]
- 38. Garaycoechea, J.I. , Patel, K.J. (2014) Why does the bone marrow fail in Fanconi anemia? Blood 123, 26–34. [DOI] [PubMed] [Google Scholar]
- 39. Medhurst, A.L. , Laghmani, H. , Steltenpool, J. , Ferrer, M. , Fontaine, C. , de Groot, J. , Rooimans, M.A. , Scheper, R.J. , Meetei, A.R. , Wang, W. , Joenje, H. , de Winter, J.P. (2006) Evidence for subcomplexes in the Fanconi anemia pathway. Blood 108, 2072–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Briot, D. , Macé‐Aimé, G. , Subra, F. , Rosselli, F. (2008) Aberrant activation of stress‐response pathways leads to TNF‐alpha oversecretion in Fanconi anemia. Blood 111, 1913–1923. [DOI] [PubMed] [Google Scholar]
- 41. Futaki, M. , Igarashi, T. , Watanabe, S. , Kajigaya, S. , Tatsuguchi, A. , Wang, J. , Liu, J.M. (2002) The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis 23, 67–72. [DOI] [PubMed] [Google Scholar]
- 42. Zhang, X. , Li, J. , Sejas, D.P. , Rathbun, K.R. , Bagby, G.C. , Pang, Q. (2004) The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR). J. Biol. Chem. 279, 43910–43919. [DOI] [PubMed] [Google Scholar]
- 43. Li, J. , Sejas, D.P. , Zhang, X. , Qiu, Y. , Nattamai, K.J. , Rani, R. , Rathbun, K.R. , Geiger, H. , Williams, D.A. , Bagby, G.C. , Pang, Q. (2007) TNF‐alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J. Clin. Invest. 117, 3283–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haneline, L.S. , Gobbett, T.A. , Ramani, R. , Carreau, M. , Buchwald, M. , Yoder, M.C. , Clapp, D.W. (1999) Loss of FancC function results in decreased hematopoietic stem cell repopulating ability. Blood 94, 1–8. [PubMed] [Google Scholar]
- 45. Carreau, M. , Gan, O.I. , Liu, L. , Doedens, M. , Dick, J.E. , Buchwald, M. (1999) Hematopoietic compartment of Fanconi anemia group C null mice contains fewer lineage‐negative CD34+ primitive hematopoietic cells and shows reduced reconstruction ability. Exp. Hematol. 27, 1667–1674. [DOI] [PubMed] [Google Scholar]
- 46. Mankad, A. , Taniguchi, T. , Cox, B. , Akkari, Y. , Rathbun, R.K. , Lucas, L. , Bagby, G. , Olson, S. , D'Andrea, A. , Grompe, M. (2006) Natural gene therapy in monozygotic twins with Fanconi anemia. Blood 107, 3084–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dao, K.H. , Rotelli, M.D. , Petersen, C.L. , Kaech, S. , Nelson, W.D. , Yates, J.E. , Hanlon Newell, A.E. , Olson, S.B. , Druker, B.J. , Bagby, G.C. (2012) FANCL ubiquitinates β‐catenin and enhances its nuclear function. Blood 120, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huard, C.C. , Tremblay, C.S. , Helsper, K. , Delisle, M.C. , Schindler, D. , Lévesque, G. , Carreau, M. (2013) Fanconi anemia proteins interact with CtBP1 and modulate the expression of the Wnt antagonist Dickkopf‐1. Blood 121, 1729–1739. [DOI] [PubMed] [Google Scholar]
- 49. Whitney, M.A. , Royle, G. , Low, M.J. , Kelly, M.A. , Axthelm, M.K. , Reifsteck, C. , Olson, S. , Braun, R.E. , Heinrich, M.C. , Rathbun, R.K. , Bagby, G.C. , Grompe, M. (1996) Germ cell defects and hematopoietic hypersensitivity to gamma‐interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood 88, 49–58. [PubMed] [Google Scholar]
- 50. Cumming, R.C. , Lightfoot, J. , Beard, K. , Youssoufian, H. , O'Brien, P.J. , Buchwald, M. (2001) Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat. Med. 7, 814–820. [DOI] [PubMed] [Google Scholar]
- 51. Li, J. , Du, W. , Maynard, S. , Andreassen, P.R. , Pang, Q. (2010) Oxidative stress‐specific interaction between FANCD2 and FOXO3a. Blood 115, 1545–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pearl‐Yafe, M. , Halperin, D. , Scheuerman, O. , Fabian, I. (2004) The p38 pathway partially mediates caspase‐3 activation induced by reactive oxygen species in Fanconi anemia C cells. Biochem. Pharmacol. 67, 539–546. [DOI] [PubMed] [Google Scholar]
- 53. Kim, Y. , Spitz, G.S. , Veturi, U. , Lach, F.P. , Auerbach, A.D. , Smogorzewska, A. (2013) Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 121, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pang, Q. , Christianson, T.A. , Keeble, W. , Diaz, J. , Faulkner, G.R. , Reifsteck, C. , Olson, S. , Bagby, G.C. (2001) The Fanconi anemia complementation group C gene product: structural evidence of multifunctionality. Blood 98, 1392–1401. [DOI] [PubMed] [Google Scholar]
- 55. Hays, L.E. , Keeble, W.W. , Yates, J.E. , Rathbun, R.K. , Koretsky, T. , Olson, S.B. , Sun, Z. , Clapp, D.W. , Bagby, G.C., Jr. (2010) Human FANCC is hypomorphic in murine Fancc‐deficient cells. Blood 116, 2057–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Neininger, A. , Kontoyiannis, D. , Kotlyarov, A. , Winzen, R. , Eckert, R. , Volk, H.D. , Holtmann, H. , Kollias, G. , Gaestel, M. (2002) MK2 targets AU‐rich elements and regulates biosynthesis of tumor necrosis factor and interleukin‐6 independently at different post‐transcriptional levels. J. Biol. Chem. 277, 3065–3068. [DOI] [PubMed] [Google Scholar]
- 57. Kotlyarov, A. , Neininger, A. , Schubert, C. , Eckert, R. , Birchmeier, C. , Volk, H.D. , Gaestel, M. (1999) MAPKAP kinase 2 is essential for LPS‐induced TNF‐alpha biosynthesis. Nat. Cell Biol. 1, 94–97. [DOI] [PubMed] [Google Scholar]
- 58. Hernández Losa, J. , Parada Cobo, C. , Guinea Viniegra, J. , Sánchez‐Arevalo Lobo, V.J. , Ramón y Cajal, S. , Sánchez‐Prieto, R. (2003) Role of the p38 MAPK pathway in cisplatin‐based therapy. Oncogene 22, 3998–4006. [DOI] [PubMed] [Google Scholar]
- 59. Reinhardt, H.C. , Hasskamp, P. , Schmedding, I. , Morandell, S. , van Vugt, M.A. , Wang, X. , Linding, R. , Ong, S.E. , Weaver, D. , Carr, S.A. , Yaffe, M.B. (2010) DNA damage activates a spatially distinct late cytoplasmic cell‐cycle checkpoint network controlled by MK2‐mediated RNA stabilization. Mol. Cell 40, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xiao, Y.Q. , Freire‐de‐Lima, C.G. , Janssen, W.J. , Morimoto, K. , Lyu, D. , Bratton, D.L. , Henson, P.M. (2006) Oxidants selectively reverse TGF‐beta suppression of proinflammatory mediator production. J. Immunol. 176, 1209–1217. [DOI] [PubMed] [Google Scholar]
- 61. Matsuzawa, A. , Saegusa, K. , Noguchi, T. , Sadamitsu, C. , Nishitoh, H. , Nagai, S. , Koyasu, S. , Matsumoto, K. , Takeda, K. , Ichijo, H. (2005) ROS‐dependent activation of the TRAF6‐ASK1‐p38 pathway is selectively required for TLR4‐mediated innate immunity. Nat. Immunol. 6, 587–592. [DOI] [PubMed] [Google Scholar]
- 62. Kayagaki, N. , Wong, M.T. , Stowe, I.B. , Ramani, S.R. , Gonzalez, L.C. , Akashi‐Takamura, S. , Miyake, K. , Zhang, J. , Lee, W.P. , Muszyński, A. , Forsberg, L.S. , Carlson, R.W. , Dixit, V.M. (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- 63. Hagar, J.A. , Powell, D.A. , Aachoui, Y. , Ernst, R.K. , Miao, E.A. (2013) Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 341, 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bagby, G.C. , Meyers, G. (2009) Myelodysplasia and acute leukemia as late complications of marrow failure: future prospects for leukemia prevention. Hematol. Oncol. Clin. North Am. 23, 361–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data