

Short abstract
HCV subject monocytes‐derived macrophages increased gal‐9, with the highest levels of non‐classical monocytes.
Keywords: JFH‐1, non‐classical monocyte, exosome
Abstract
The lectin galectin‐9 may help establish and maintain chronic hepatitis C virus infection. Galectin‐9 is elevated in the liver and sera of hepatitis C virus patients, induces apoptosis of hepatitis C virus‐specific T cells, and increases inhibitory regulatory T cells. Kupffer cells stain strongly for galectin‐9 protein in hepatitis C virus patients. In the current study, we determined stimuli that induce galectin‐9 production by monocytes and macrophages in hepatitis C virus infection. With the use of real‐time PCR and flow cytometry, we analyzed galectin‐9 mRNA and protein from human monocytes cocultured with hepatitis C virus‐infected cells or noninfectious hepatitis C virus subgenomic replicon cells. We focused on finding the stimuli for galectin‐9 production. Additionally, we measured galectin‐9 during monocyte‐to‐macrophage maturation. Finally, we examined galectin‐9 in peripheral monocytes from hepatitis C virus patients using flow cytometry. Galectin‐9 mRNA increased 8‐fold when primary monocytes were exposed to hepatitis C virus‐‐infected cells. Maximum induction required proximity or contact and did not require IFN‐γ or hepatitis C virus virions. Coculture of monocytes with subgenomic replicon cells increased galectin‐9 5‐fold, and purified exosomes from infected cells stimulated galectin‐9 production. Stimulation of monocyte TLR3, ‐7, and ‐8 increased galectin‐9 production. Differentiation of monocytes to macrophages increased galectin‐9, and nonclassic monocytes from hepatitis C virus patients had the highest levels of galectin‐9. Hepatitis C virus‐infected cells stimulated monocytes to produce galectin‐9 in close proximity, possibly, in part, as a result of exosomes and endosomal TLRs. Differentiation of monocytes to macrophages increased galectin‐9. Nonclassic monocytes from hepatitis C virus patients express the highest galectin‐9 levels, suggesting they may contribute to elevated galectin‐9 and adaptive immune inhibition in hepatitis C virus infection.
Abbreviations
- APC
allophycocyanin
- gal‐9
galectin‐9
- HBV/HCV
hepatitis B/C virus
- JFH‐1
Japanese fulminant hepatitis‐1
- MFI
median fluorescence intensity
- MOI
multiplicity of infection
- ORN02
oligoribonucleotide 02
- pDC
plasmacytoid dendritic cell
- poly(I:C)
polyinosinic:polycytidylic acid
- SGR
subgenomic replicon
- Tim‐3
T cell Ig and mucin domain‐containing molecule 3
- Treg
regulatory T cell
Introduction
HCV chronically infects 3% of the world's population, ∼180 million people worldwide [1]. Acute HCV infection progresses to chronic hepatitis in up to 80% and leads to irreversible liver fibrosis (cirrhosis) in 20% over 20 yr [1]. The complications of cirrhosis include liver cancer and fatal liver decompensation; HCV remains a leading indication for liver transplantation. How HCV establishes and maintains chronic liver infection remains incompletely understood, and knowledge of the mechanism may aid vaccine development.
A variety of mechanisms enable HCV to infect hepatocytes persistently despite immune activation. These include direct inhibition of the hepatocyte innate immune system that recognizes the pathogen‐associated molecular patterns and inhibition of the antigen‐specific adaptive immune response [2, 3–4]. Alterations in adaptive immunity described in chronic HCV infection include T cell exhaustion and increased inhibitory Tregs, leading to an ineffective immune response [2]. In acute and chronic hepatitis C infection, increased levels of the glycosylated membrane protein Tim‐3 are found on the surface of exhausted T cells that are associated with the development of chronic infection [5, 6].
A ligand of Tim‐3, gal‐9 is a tandem repeat lectin that binds β‐d‐galactosides and has immunoregulatory properties [7]. Upon binding of gal‐9 to Tim‐3, T cells proliferate less, make less IFN‐γ, and undergo apoptosis [8]. Normally, gal‐9/Tim‐3 activity may serve as a brake to the adaptive immune response, inhibiting autoimmunity [7, 9, 10–11]. gal‐9 treatment increases the number of inhibitory Tregs, and a decrease of gal‐9/Tim‐3 activity is associated with autoimmune disease, including autoimmune hepatitis [10, 12, 13]. Elevations of gal‐9 have been found in a variety of cancers, including hepatocellular carcinoma [14]. gal‐9 is emerging as a critical regulator of the immune responses to a number of viruses, including HCV [6, 12, 15], HSV [16, 17], HBV [14, 18], influenza A [19], and HIV [20]. For example, HSV‐1 reactivation from latency was reduced in gal‐9 knockout mice [17].
We found that gal‐9 levels were increased in the blood and livers of patients with chronic HCV infection [12]. Immunostaining localized high gal‐9 production to Kupffer cells from HCV patients. gal‐9 induced apoptosis of antigen‐specific T cells and increased inhibitory Tregs. These experiments suggest that HCV may stimulate chronic gal‐9/Tim‐3 activity leading to immune suppression and chronic infection. The understanding of why gal‐9 levels are elevated in HCV infection could improve our knowledge of the mechanisms leading to chronic infection.
We sought to characterize further the stimuli leading to increased gal‐9 expression in Kupffer cells by use of the JFH‐1 HCV culture system [21, 22] and monocytes isolated from the blood of donors. We hypothesized that HCV‐infected cells could stimulate gal‐9 production in macrophages via pattern recognition receptors. We found that gal‐9 levels increase during differentiation of monocytes to macrophages and that HCV‐infected cells triggered gal‐9 production. Hepatocytes harboring SGRs of HCV that do not secrete infectious particles but do secrete HCV exosomes also triggered gal‐9 production and proximity, or contact was required for maximum response. Furthermore, purified exosomes from HCV‐infected cells stimulated gal‐9 production. Finally, we found that stimulation of TLR3, ‐7, and ‐8 increased gal‐9 in monocytes, suggesting that HCV ssRNA and dsRNA could stimulate endosomal TLRs during the immune response to infection.
The dominant monocyte species are CD14posCD16neg classic and CD14low/highCD16pos nonclassic monocytes [23]. Nonclassic monocytes are thought to be involved in inflammatory responses to viral infection [24] and have been implicated in the pathogenesis of chronic liver disease and HBV infection [25]. The role played by nonclassic monocytes in immunity to HCV infection is unknown. We found that nonclassic monocytes had elevated levels of gal‐9 compared with classic monocytes and that HCV patients had the highest levels, confirming that mononuclear cells are a source of gal‐9. Increased gal‐9 in HCV may lead to immune dampening and viral persistence.
MATERIALS AND METHODS
Ethics statement
The study protocol was approved by the Institutional Review Boards at the University of Colorado School of Medicine and the Denver VA Medical Center. Written and oral informed consent was obtained before samples were collected. De‐identified samples were created and studied.
Primary cell isolation and storage
PBMCs were isolated by apheresis or Ficoll separation and viably frozen in 80% FBS (BioWhittaker, Walkersville, MD, USA), 10% DMSO, and 10% RPMI 1640 (Life Technologies, Thermo Fisher Scientific, Grand Island, NY, USA). Samples were stored in liquid nitrogen. Monocytes were isolated from PBMCs by magnetic bead negative selection by use of the EasySep Human Monocyte Enrichment Kit (Stemcell Technologies, Vancouver, BC, Canada). Purity >90% was confirmed by flow cytometry.
Cell lines and culture conditions
The THP‐1 human monocytic cell line (Sigma, St. Louis, MO, USA) and primary human monocytes were cultured in RPMI 1640 (Life Technologies, Thermo Fisher Scientific), supplemented with 10% FBS and 1× penicillin, streptomycin, and l‐glutamine (Life Technologies, Thermo Fisher Scientific). Huh7.5 hepatocytes (Apath, New York, NY, USA) were grown in DMEM (Life Technologies, Thermo Fisher Scientific), supplemented with 10% FBS and 1× penicillin, streptomycin, l‐glutamine, and nonessential amino acids. Infection of Huh 7.5 hepatocytes was performed in the above complete DMEM containing only 2% FBS. The APC140 cell line (Apath) [26], Huh7.5 cells stably transfected with genotype 2 HCV SGR, were cultured in complete DMEM with 0.5 g/L G418 (Sigma). Cocultures with monocytes were performed in RPMI 10% FBS. All cultures were incubated at 37°C and 5% CO2. To mature monocytes to macrophages, 5 × 105 monocytes were cultured for 1 wk in complete RPMI or complete RPMI supplemented with 10 ng/ml GM‐CSF (inflammatory macrophage, M1) or 150 ng/ml M‐CSF (alternative‐regulatory macrophage, M2) [27, 28].
JFH‐1 HCV production and infection
JFH‐1 HCV was produced and quantitated as described previously [22]. JFH‐1 stocks were aliquoted and frozen at −80°C until future use. Before coculture, 2 × 106 Huh7.5s were plated in a T75 flask and allowed to adhere. Then, cells were infected with JFH‐1 at a MOI of 0.1 for 2–3 d before harvesting with 0.05% Trypsin/EDTA.
Cocultures
Monocytes (2.5 × 105) were placed with 1.25 × 105 uninfected Huh7.5s, HCV‐infected Huh7.5s, or Huh7.5/SGRs in each well of a 24‐well plate. For the IFN‐γ blockade, monocytes were preincubated for 1 h with 0.5 μg/ml anti‐ IFN‐γ IgA (H7WM120; InvivoGen, San Diego, CA, USA) or 0.5 μg/ml isotype control (T9C6; InvivoGen). Next, 2.5 × 105 uninfected or HCV‐infected Huh7.5s were added. Total RNA was harvested after 4 d. Where indicated, 5 μM spiroepoxide (Sigma) was added for the duration of the coculture. THP‐1/Huh7.5 cocultures were performed similarly; however, 5 × 105 cells of each type were cultured together, and RNA was harvested after 24 h. Stimulation with IFN‐γ (25 ng/m; R&D Systems, Minneapolis, MN, USA) served as a positive control for all experiments.
Transwell cocultures
Monocytes (2.5 × 105) were placed in the bottom chamber of a 24‐well transwell plate with 0.4 μm pores (Corning, Corning, NY, USA). Uninfected (1.25 × 105) or HCV‐infected Huh7.5s were placed in the upper chamber. RNA was harvested from monocytes in the lower chamber after 4 d.
Coculture flow cytometry
All antibodies were from BioLegend (San Diego, CA, USA). Cultured cells were removed from vessels with 0.05% Trypsin/EDTA and washed with FACS wash [PBS, Ca+2, Mg+2 (Life Technologies, Thermo Fisher Scientific), supplemented with 1% BSA (Amresco, Solon, OH, USA), 0.01% EDTA (Sigma), and 0.016% sodium azide (Sigma)] before incubation with human FcR Blocking Reagent (Miltenyi Biotec, San Diego, CA, USA). Cells were again washed and surface stained with anti‐CD14‐FITC (M5E2) and anti‐HLA‐DR‐Pacific Blue (L243). Cells were then stained for intracellular antigens by use of Fix & Perm (Life Technologies, Thermo Fisher Scientific), anti‐gal‐9‐PE (9M1‐3), or PE‐mouse IgG isotype control (MOPC‐21), according to the manufacturers’ protocols. Compensation was performed by use of anti‐mouse Ig, κ CompBeads (BD Biosciences, San Jose, CA, USA) before acquisition on an LSRII flow cytometer (BD Biosciences) and analysis with FlowJo software (Tree Star, Ashland, OR, USA).
Ex vivo flow cytometry
Multiparameter flow cytometry was used to determine gal‐9 expression on monocyte populations in normal control (n = 9) and chronic HCV subjects (n = 11). Acquisition was performed by use of a BD FACSCanto II instrument (BD Biosciences), compensated with single fluorochromes, and data were analyzed by use of Diva software (BD Biosciences). Monocytes were distinguished by positive staining for CD14 and/or CD16 and high levels of HLA‐DR within a low forward‐/side‐scatter gate. Monocyte subsets were distinguished by CD16 expression. HLA‐DR FITC clone LN3 was from eBioscience (San Diego, CA, USA), CD14 APC‐Cy7 clone MφP9 was from BD PharMingen (San Diego, CA, USA). BioLegend antibodies used were the following: CD16 Pacific Blue clone 3G8, gal‐9‐PE clone 9M1‐3, and IgG‐PE clone MOPC‐21.
Real‐time quantitative PCR
Total RNA was isolated by use of QIAshredder columns (Qiagen, Germantown, MD, USA) in combination with the PureLink RNA Mini Kit (Life Technologies, Thermo Fisher Scientific). cDNA was then reverse transcribed by use of the QuantiTect Reverse Transcription Kit (Qiagen) or ProtoScript (New England Biolabs, Ipswich, MA, USA), according to the manufacturers’ protocols. Real‐time PCR was performed by use of TaqMan Gene Expression Master Mix (Life Technologies, Thermo Fisher Scientific) and prevalidated primer/probe sets for gal‐9 (Hs00247135) or GAPDH (Hs99999902_m1) on a 7500 Fast or StepOne thermal cycler (Life Technologies, Thermo Fisher Scientific). Experiments were done in biologic triplicate and technical duplicate, and data were analyzed by use of the comparative ΔΔ comparative threshold method [29] or a gal‐9 cDNA standard curve. The gal‐9 standard curve encompassing the Hs00247135_m1 assay location was made by PCR amplifying monocytic cDNA by use of the following primers under standard conditions: gal‐9 forward, 5′ CAA GGT GAT GGT GAA CGG GA 3′; gal‐9 reverse, 5′ ACC TTG AGG CAG TGA GCT TC 3′. The resulting product was confirmed by sequencing before being quantified, serially diluted, and frozen.
Data were analyzed and P values calculated by use of GraphPad Prism, as described in the figure legends. Data shown are biologic replicates; 3 replicates were done for each patient and included in the statistical analysis.
Monocyte stimulation
All TLR reagents were from InvivoGen. Monocytes (5 × 105) were stimulated for 4 d with TLR3 agonist poly(I:C) (10 μg/ml) or TLR7 agonist imiquimod (5 μg/ml). TLR8 stimulation was achieved with the ssRNA ORN02 (1 μg/ml) complexed to LyoVec as a carrier at a 1:2 ratio (weight: weight) or an equivalent amount of DNA complexed to LyoVec as a negative control.
Exosome isolation and monocyte coculture
In 3 independent experiments, 1.25 × 105 Huh7.5.1 cells were seeded in a 24‐well plate and cultured ± JFH1 (Takaji Wakita, National Institute of Infectious Diseases, Japan) at a MOI of 0.1 on the following day. Five days after infection, supernatants were harvested and exosomes isolated by use of the ExoQuick‐TC (System Biosciences, Mountain View, CA, USA), per the manufacturer's protocol [30]. Isolated monocytes from 2 individuals were treated with exosomes isolated from infected and uninfected supernatants from the 3 independent isolations.
RESULTS
HCV‐infected hepatocytes stimulate gal‐9 in monocytes
Our previous work demonstrated that the immunoregulatory protein gal‐9 was increased in HCV patients and localized to Kupffer cells [12]. Monocytes from the peripheral blood are a source of liver macrophages [31]. We hypothesized that recognition of HCV‐infected cells by monocytes could trigger gal‐9 production. To test this hypothesis, we cultured CD14pos monocytes negatively isolated from PBMCs of normal donors with Huh7.5 hepatocytes or Huh7.5 hepatocytes infected with the genotype 2a JFH‐1 strain of HCV [32]. Levels of gal‐9 mRNA were assessed with real‐time PCR. Mean gal‐9 mRNA increased by 8.5‐fold in cocultures of CD14pos monocytes with JFH‐1‐infected Huh7.5s compared with uninfected Huh7.5s ( Fig. 1A and B ). Basal levels of gal‐9 were 32‐fold higher in chronically HCV‐infected patients and also increased 8‐fold when exposed to infected Huh7.5s (Fig. 1A and B). Importantly, gal‐9 mRNA levels are 6 × 103‐fold higher in monocytes than Huh7.5s (Supplemental Fig. 1), and HCV infection does not alter the level of gal‐9 in hepatocytes (data not shown). To confirm that gal‐9 protein expression correlates with gal‐9 mRNA levels and to confirm gal‐9 expression was found predominantly in monocytes, we examined intracellular gal‐9 by flow cytometry. The MFI of gal‐9 in CD14pos, HLA‐DRpos monocytes increased by 60% when cultured with infected versus uninfected Huh7.5s (Fig. 1C). Immunofluorescence staining of cocultures also demonstrated gal‐9 protein expression in monocytes but not hepatocytes (Supplemental Fig. 1).
Figure 1.
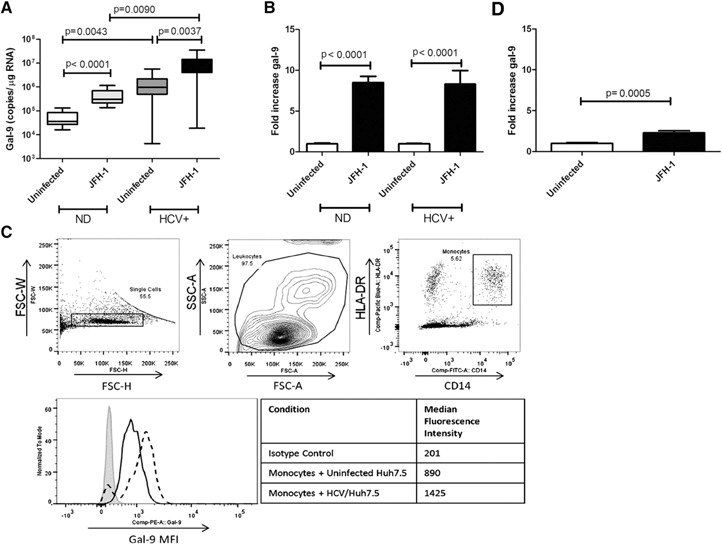
Monocyte gal‐9 mRNA and protein are elevated in cocultures of primary CD14pos monocytes and HCV‐infected Huh7.5 cells. CD14pos monocytes were isolated by negative selection and cocultured for 4 d with uninfected or JFH‐1‐infected hepatocytes. gal‐9 mRNA expression was analyzed by quantitative real‐time PCR by use of a standard curve. Four normal donors (ND) and 5 HCV patients were analyzed in triplicate. Pooled triplicate data are shown; P values are calculated by use of a 2‐tailed Mann‐Whitney test. (A) Whisker plot of absolute gal‐9 levels, with bars showing the range of values and boxes the 25th–75th percentile, with the horizontal line showing the mean. (B) Pooled fold induction data with means and sem. (C) Gating strategy and intracellular gal‐9 protein expression in CD14pos, HLA‐DRpos monocytes after 4 d coculture of PBMCs with uninfected (black line) or HCV‐infected (dashed line) hepatocytes compared with an IgG isotype control (gray shading). (D) gal‐9 in monocytes from 3 normal donors cultured on opposite sides of a transwell membrane, with 0.4 μm pores, from uninfected or JFH‐1‐infected hepatocytes. FSC‐W, Forward‐scatter‐width; FSC‐H, FSC‐height; SSC‐A, side‐scatter‐area; Comp, compensation.
Full gal‐9 induction requires cell contact or proximity
We next examined the mechanism by which HCV‐infected cells induce gal‐9 in monocytes. To determine whether gal‐9 increases in response to soluble factors, such as cytokines and viral particles, or via direct contact, we cultured uninfected or HCV‐infected Huh7.5s on opposite sides of a transwell membrane with 0.4 μm pores. In this model, monocytes up‐regulated gal‐9 mRNA by 2‐fold (Fig. 1D), substantially less than the 8.5‐fold seen in our direct coculture model, indicating that cell contact or proximity is an important factor. Further suggesting that contact is necessary, challenging monocytes with concentrated JFH‐1 virions failed to induce gal‐9 mRNA ( Fig. 2A ). IFN‐γ plays an important role in the immune response and is elevated in HCV patients [33]. We demonstrated previously that IFN‐γ is a potent stimulator of gal‐9 in macrophages [12]. However, neutralization of IFN‐γ in our coculture model did not affect gal‐9 levels significantly (P = 0.7980; Fig. 2B), indicating that gal‐9 induction proceeds through an IFN‐γ‐free mechanism.
Figure 2.
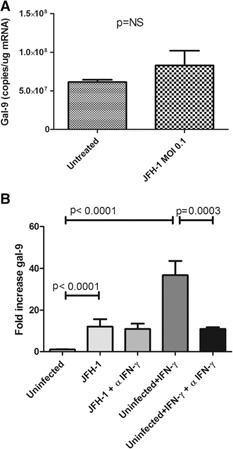
gal‐9 induction is independent of IFN‐γ or infectious HCV particles. (A) CD14pos monocytes (5 × 105) were incubated with JFH‐1 HCV virions (MOI 0.1) for 4 d in triplicate. gal‐9 mRNA was quantified by use of real‐time PCR with a standard curve; P values are calculated with a 2‐tailed t‐test. (B) CD14pos monocytes from 3 normal donors were incubated in triplicate with an anti‐IFN‐γ‐neutralizing antibody or an isotype control throughout a 4‐d coculture with uninfected or HCV‐infected hepatocytes. gal‐9 mRNA levels were determined by quantitative real‐time PCR by use of a gal‐9 standard curve. The positive control IFN‐γ was used at a concentration of 25 ng/ml with uninfected Huh 7.5 cells, with or without an IFN‐γ blocking antibody. Pooled data are shown with means and sem, and P values are calculated by use of a 2‐tailed Mann‐Whitney test.
Noninfectious JFH‐1 SGRs and purified HCV exosomes induce gal‐9 in monocytes
SGRs of HCV in Huh 7.5 cells allow for the study of cytoplasmic HCV RNA effects without infectious particles [34]. Additionally, recent work suggests that transfer of HCV RNA by short‐range exosomes released from HCV‐infected cells or SGRs to pDCs stimulates the innate immune response [35]. To examine whether HCV‐containing exosomes or intracellular HCV RNA may be responsible for gal‐9 induction in monocytes, we cultured monocytes with SGRs or control Huh7.5 cells. The SGRs also increased gal‐9 mRNA levels in cocultures, stimulating levels by 4.8‐fold ( Fig. 3A ). Separation of SGR cells from monocytes by a 0.4 μM transwell membrane abolished induction (Fig. 3B). To test whether exosome secretion was involved in stimulation of monocytes, we used the neutral sphingomyelinase inhibitor spiroepoxide, which inhibits HCV SGR exosome release [35]. We found that spiroepoxide significantly inhibited gal‐9 induction (Fig. 3C), suggesting a role for exosomes in gal‐9 stimulation. Purified exosomes from JFH‐1 infected cells slightly (Fig. 3D) but significantly (P = 0.0164) stimulated gal‐9 in monocytes after 24 h of culture, further suggesting that they could be part of the gal‐9 response of monocytes to JFH‐1‐infected cells.
Figure 3.
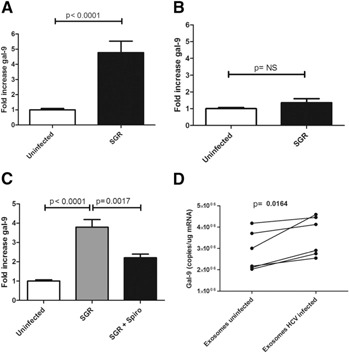
HCV SGRs increase gal‐9 mRNA in CD14pos monocytes, and this is blocked by Spiroepoxide. Monocytes from normal donors incubated in triplicate with Huh7.5s or HCV genotype‐2 SGRs. gal‐9 mRNA levels were analyzed by quantitative real‐time PCR and a gal‐9 standard curve. Pooled data are shown with means and sem; P values are calculated by use of a 2‐tailed Mann‐Whitney test. (A) Monocytes from 3 patients cocultured in triplicate for 4 d with Huh7.5s or Huh7.5 SGRs. (B) Monocytes from 3 patients cultured in triplicate for 4 d across a 0.4 μM transwell from SGR cells. (C) The exosome release inhibitor Spiroepoxide (Spiro; 5 μM) or DMSO control was used in 60 h coculture with 4 patients in triplicate. (D) In each well, 100 μg purified exosomes from 3 separate isolations was incubated with 106 monocytes for from 2 different patients for 24 h. mRNA was isolated and analyzed with quantitative real‐time PCR by use of a standard curve; P values are calculated with a 2‐tailed paired t‐test.
TLR3, ‐7, and ‐8 activation up‐regulates gal‐9 in monocytes
Macrophages contain a number of pathogen‐recognition receptors involved in the innate immune response [36]. As HCV produces a ssRNA genome and a dsRNA intermediate [37, 38], we questioned whether endosomal TLR3 (dsRNA), ‐7 (ssRNA), and ‐8 (ssRNA) could trigger gal‐9 in monocytes. Stimulation of CD14+ monocytes with poly(I:C), a TLR3 agonist, increased gal‐9 mRNA levels 5‐fold. The TLR7 agonist imiquimod [39] increased gal‐9 3 fold, whereas ORN02, a specific TLR8 agonist [40], elevated gal‐9 mRNA levels 10‐fold ( Fig. 4 ), confirming that TLR stimulation can increase gal‐9.
Figure 4.
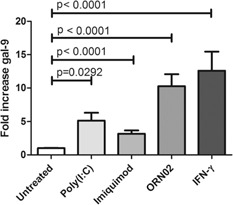
Activation of TLR3, ‐7, and ‐8 significantly induces gal‐9 in monocytes. gal‐9 levels in CD14pos monocytes from 3 normal donors incubated for 3 d in the presence of TLR agonists in triplicate. TLR3 agonist poly(I:C) (10 μg/ml), TLR7 agonist imiquimod (5 μg/ml) or TLR8 agonist ORN02 (1 μg/ml) complexed to a LyoVec carrier. IFN‐γ serves as a positive control. gal‐9 mRNA levels were determined by real‐time PCR by use of a standard curve. Pooled data are shown with means and sem; P values are calculated by use of a 2‐tailed Mann‐Whitney test.
gal‐9 increases with monocyte‐to‐macrophage differentiation
The gal‐9 increase seen in monocytes after exposure to HCV‐infected cells and SGRs takes 3–4 d (data not shown), suggesting that gal‐9 expression might correlate with monocyte‐to‐macrophage differentiation. To differentiate monocytes, we cultured THP‐1 monocytes with PMA [41] and then cocultured cells with uninfected or HCV‐infected Huh7.5s. PMA maturation to a macrophage‐like phenotype increased basal gal‐9 nearly 4‐fold ( Fig. 5A ). Coculture with HCV‐infected Huh7.5s increased gal‐9 2.6‐fold to nearly 10‐fold greater than that seen in cocultures between undifferentiated THP‐1 monocytes and uninfected Huh7.5s (Fig. 5A). Additionally we cultured primary human monocytes from 3 normal patients in the presence of GM‐CSF to create adherent M1 inflammatory macrophages or M‐CSF to create adherent M2 regulatory macrophages for 7 d [27, 28]. gal‐9 significantly increased in both macrophage subtypes with a larger increase seen in GM‐CSF‐treated cells. Taken together, these data suggest that gal‐9 expression increases as monocytes mature to macrophages. M1 and M2 differentiated macrophages both responded to JFH‐1‐infected cells with increased gal‐9 production (Fig. 5C), although the response was less than the response of monocytes (Fig. 1A and B), likely because they were differentiated before coculture.
Figure 5.
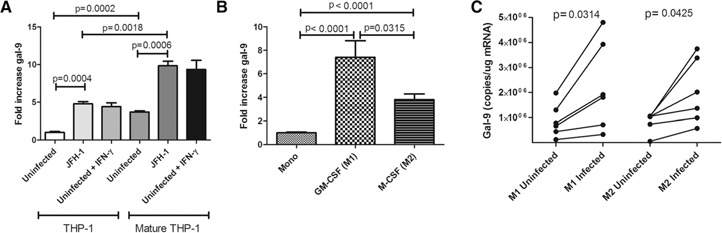
gal‐9 increases with monocyte‐to‐macrophage differentiation and increases further with exposure to HCV‐infected hepatocytes. gal‐9 mRNA levels were analyzed by quantitative real‐time PCR. (A) Naïve THP‐1 monocytes or PMA‐matured THP‐1 macrophages cocultured with uninfected or HCV‐infected Huh7.5s for 24 h. IFN‐γ served as the positive control at 25 ng/ml. (B) Relative gal‐9 mRNA in primary human CD14pos monocytes (Mono), GM‐CSF‐matured M1 macrophages, or M‐CSF‐matured M2 macrophages [27, 28] after 7 d culture. Three normal donors were analyzed in triplicate; P values are calculated by use of a 2‐tailed t‐test. (C) Monocytes from 5 different normal donors were incubated for 7 d with GM‐CSF (M1) or M‐CSF (M2) and then incubated 4 additional d with uninfected or HCV‐infected Huh 7.5 cells. gal‐9 levels were measured with real‐time PCR and a standard curve; P values are calculated with a 2‐tailed paired t‐test.
Nonclassic peripheral monocytes from HCV patients contain the highest levels of gal‐9
Monocytes can be divided into classic (CD16negCD14high HLA‐DRpos) and nonclassic (CD16pos CD14high/low HLA‐DRpos) [23]. With the use of flow cytometry to study PBMC directly ex vivo, HCV patients had higher levels of gal‐9 in both types of monocytes compared with normal controls ( Fig. 6 ). Nonclassic monocytes from both types of patients had higher levels of gal‐9 frequency, and HCV patients showed the greatest frequency overall compared with normal donors.
Figure 6.
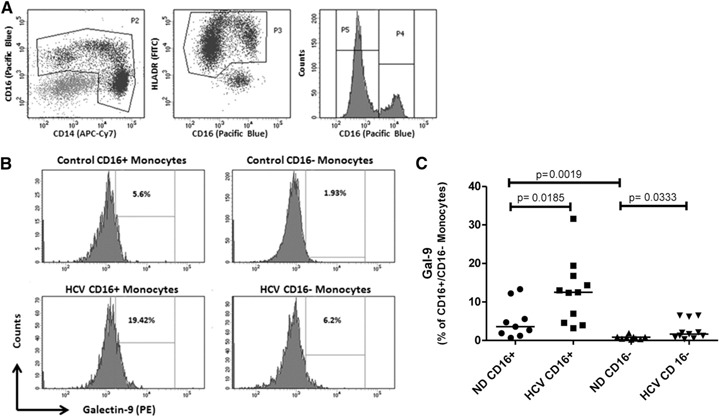
Elevated gal‐9 in monocytes from patients with chronic HCV infection. Nine normal donors and 11 chronically HCV‐infected donors were analyzed. Monocytes were identified as CD14pos and/or CD16pos cells (P2), coexpressing high levels of HLA‐DR (P3). Nonclassic monocytes also express CD16 (P4), whereas classic monocytes (P5) are negative for CD16 (A). Surface gal‐9 expression was determined on monocyte subsets from control and chronic HCV subjects, as shown in representative histograms. Positive gates are set on isotype control stains (B). Higher levels of gal‐9 were detected in chronic HCV on classic (CD16neg) and nonclassic (CD16pos) monocyte subsets (C). P Values are calculated by use of the 2‐tailed Mann‐Whitney test.
DISCUSSION
We demonstrated previously that gal‐9 is increased in the sera and liver macrophages of chronically infected HCV patients and may regulate adaptive immunity [12]. gal‐9 also inhibits NK cell function [15]. In this study, we sought to explore the triggers of gal‐9 production in monocytes and macrophages during HCV infection. We discovered that HCV‐infected hepatocytes directly stimulate up‐regulation of gal‐9 mRNA and protein in primary monocytes. We also found that peripheral nonclassic monocytes from HCV patients had higher basal levels of gal‐9. Next, we studied the mechanism of gal‐9 stimulation. We found that close proximity or contact was required for a maximal response. Previously, we found that IFN‐γ strongly stimulated gal‐9 [12] in macrophages; however, the blocking of IFN‐γ (Fig. 2) did not significantly reduce gal‐9. Direct stimulation of macrophages by HCV virions occurs [42, 43], but we found that SGR still stimulated gal‐9 (Fig. 3), and virions did not (Fig. 2A). Overall, this indicates that HCV‐triggered gal‐9 induction proceeds through contact or close proximity and may be an IFN‐γ‐free mechanism, i.e., related to direct HCV RNA sensing without the need for full‐length virus.
Exosomes are small (40–120 nm), membrane‐bound vesicles that contain nucleic acids and proteins. They facilitate cell‐to‐cell communication and modulation of the immune response, often between cells in close proximity [30, 44, 45]. Their role in the viral immune response is an exciting, new area of study, and HCV RNA‐containing exosomes that transmit infection were recently found in the sera of chronically infected patients [46]. Additionally Dreux et al. [35] demonstrated that HCV‐RNA‐containing exosomes from HCV‐infected cells and HCV SGR activate pDCs and trigger IFN‐α release. Consistent with the findings of Dreux et al. [35], we found that HCV genotype‐2 SGRs, which produce RNA‐containing exosomes but do not produce infectious viral particles, are sufficient to trigger a 3.8‐fold increase of gal‐9 in monocytes (Fig. 3). Spiroepoxide, which inhibits neutral sphingomyelinase and exosome release [35], inhibited gal‐9 induction by SGR cells (Fig. 3). Finally, purified exosomes released from HCV‐infected cells also increased gal‐9 levels. However, as transwell separation of SGR cells with 400 nm pores, large enough for exosomes to pass, decreased the gal‐9 response in monocytes (Fig. 3), direct contact, and uptake of infected cells by phagocytic monocytes/macrophages, or close transmission of exosomes leading to endosomal TLR stimulation could be part of the mechanism triggering gal‐9 expression. Consistent with our results, Dreux et al. [35] found that exosome transfer of HCV to pDCs occurred only when cells were in close proximity. Further experiments are needed to elucidate the steps required.
Our prior work demonstrated that LPS (TLR4), IL‐1β, and the HCV core protein (TLR2 stimulator) did not stimulate gal‐9 production in macrophages [12]. As monocytes and macrophages contain endosomal TLRs that recognize viral RNA [36, 47], we hypothesized that TLR3 (dsRNA), TLR7 (ssRNA), and TLR8 (ssRNA), might sense HCV RNA‐triggering gal‐9 production. Agonists of all 3 TLRs stimulated gal‐9 mRNA in monocytes. The ssRNA TLR8 agonist ORN02 proved to be the most potent gal‐9 stimulator, suggesting that TLR8 may be a target for HCV viral RNA. TLR8 binds to uridine‐rich regions [48, 49], and HCV viral RNA contains a 3′ uridine‐rich region that could be a binding target.
gal‐9 increased with differentiation from monocytes to macrophages in the THP‐1 cell line and primary human monocytes (Fig. 5A). Classic, inflammatory M1 macrophages, created in vitro from normal patients, displayed the highest levels of gal‐9, and nonclassic macrophages also had elevated levels compared with monocytes. Basal levels of gal‐9 were significantly higher in cultured monocytes from HCV patients compared with normal donors (Fig. 1A). Macrophages still responded to HCV‐infected cells, but the fold increase was lower, possibly because the cells are already expressing gal‐9 after differentiation (Fig. 5B).
In vivo, 90% of monocytes can be categorized as classic, defined as CD16neg CD14pos [50]. In contrast, nonclassic CD16posCD14pos monocytes are elevated in the peripheral blood and livers of patients with cirrhosis, may have regulatory functions, and can activate profibrotic stellate cells [50, 51]. Additionally, patients with chronic HBV have elevated levels of nonclassic monocytes in their peripheral blood and livers [25], and higher levels correlated with increased liver injury. Chronic activation of monocytes and macrophages is seen in HCV and correlates with liver damage [33, 47]. Analysis of monocyte and macrophage subtypes in HCV has been limited. Our finding that nonclassic monocytes had the highest gal‐9 levels in chronically infected HCV patients suggests that they may be a source of T cell inhibitory gal‐9 in HCV infection. Intriguingly, Coquillard and Patterson [52] used ultrasensitive PCR and flow cytometry of monocyte subtypes, and they identified negative strands from HCV replication in nonclassic monocytes, suggesting that they could serve as a reservoir of infection. Given our in vitro findings, this suggests that HCV RNA could be stimulating nonclassic monocyte gal‐9 production in vivo, leading to T cell dysfunction.
In summary, we found that HCV‐infected cells and TLR3, ‐7, and ‐8 are capable of stimulating immunoregulatory gal‐9 production in monocytes. This mechanism may involve close proximity and exosome release from cells. We also found that differentiation of monocytes to macrophages increases gal‐9, and nonclassic monocytes from HCV patients have the highest levels.
AUTHORSHIP
N.M.K.H. and L.G.‐M. designed experiments, performed experiments, and contributed to the manuscript. L.C. performed experiments. H.R.R. provided patient samples and contributed to the manuscript. J.A.M. designed and performed experiments and wrote and contributed to the manuscript.
J.A.M. is supported by the Denver Research Institute and a Liver Scholar Award from the American Association for the Study of Liver Disease and the American Liver Foundation. H.R.R. is funded by U.S. National Institutes of Health National Institute of Allergy and Infectious Diseases Grant R21‐AI103361 and a VA Merit Review Grant. The authors thank our leukocyte donors for their generous contribution and Diana Ir and Katelyn Leahy for tissue‐culture work. Additionally, the authors thank Harsh Pratap for flow cytometry assistance and Ron Bouchard for immunofluorescence guidance. Amy Stone provided valuable advice regarding TLR stimulation.
DISCLOSURES
The authors declare no conflicts of interest.
Supporting information
Supplementary data
Supplementary data
REFERENCES
- 1. Rosen, H.R. (2011) Clinical practice. Chronic hepatitis C infection. N. Engl. J. Med. 364, 2429–2438. [DOI] [PubMed] [Google Scholar]
- 2. Rosen, H.R. (2013) Emerging concepts in immunity to hepatitis C virus infection. J. Clin. Invest. 123, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mengshol, J.A. , Golden‐Mason, L. , Castelblanco, N. , Im, K.A. , Dillon, S.M. , Wilson, C.C. , Rosen, H.R. ; Virahep‐C Study Group. (2009) Impaired plasmacytoid dendritic cell maturation and differential chemotaxis in chronic hepatitis C virus: associations with antiviral treatment outcomes. Gut 58, 964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mengshol, J.A. , Golden‐Mason, L. , Rosen, H.R. (2007) Mechanisms of disease: HCV‐induced liver injury. Nat. Clin. Pract. Gastroenterol. Hepatol. 4, 622–634. [DOI] [PubMed] [Google Scholar]
- 5. Golden‐Mason, L. , Palmer, B.E. , Kassam, N. , Townshend‐Bulson, L. , Livingston, S. , McMahon, B.J. , Castelblanco, N. , Kuchroo, V. , Gretch, D.R. , Rosen, H.R. (2009) Negative immune regulator Tim‐3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 83, 9122–9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McMahan, R.H. , Golden‐Mason, L. , Nishimura, M.I. , McMahon, B.J. , Kemper, M. , Allen, T.M. , Gretch, D.R. , Rosen, H.R. (2010) Tim‐3 expression on PD‐1+ HCV‐specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte‐directed in vitro cytotoxicity. J. Clin. Invest. 120, 4546–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rabinovich, G.A. , Toscano, M.A. (2009) Turning ‘sweet’ on immunity: galectin‐glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 9, 338–352. [DOI] [PubMed] [Google Scholar]
- 8. Haining, W.N. (2012) Thinking inside the box: how T cell inhibitory receptors signal. Nat. Med. 18, 1338–1339. [DOI] [PubMed] [Google Scholar]
- 9. Johnson, J.L. , Jones, M.B. , Ryan, S.O. , Cobb, B.A. (2013) The regulatory power of glycans and their binding partners in immunity. Trends Immunol. 34, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rabinovich, G.A. , Croci, D.O. (2012) Regulatory circuits mediated by lectin‐glycan interactions in autoimmunity and cancer. Immunity 36, 322–335. [DOI] [PubMed] [Google Scholar]
- 11. Sánchez‐Fueyo, A. , Tian, J. , Picarella, D. , Domenig, C. , Zheng, X.X. , Sabatos, C.A. , Manlongat, N. , Bender, O. , Kamradt, T. , Kuchroo, V.K. , Gutiérrez‐Ramos, J.C. , Coyle, A.J. , Strom, T.B. (2003) Tim‐3 inhibits T helper type 1‐mediated auto‐ and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 4, 1093–1101. [DOI] [PubMed] [Google Scholar]
- 12. Mengshol, J.A. , Golden‐Mason, L. , Arikawa, T. , Smith, M. , Niki, T. , McWilliams, R. , Randall, J.A. , McMahan, R. , Zimmerman, M.A. , Rangachari, M. , Dobrinskikh, E. , Busson, P. , Polyak, S.J. , Hirashima, M. , Rosen, H.R. (2010) A crucial role for Kupffer cell‐derived galectin‐9 in regulation of T cell immunity in hepatitis C infection. PLoS One 5, e9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liberal, R. , Grant, C.R. , Holder, B.S. , Ma, Y. , Mieli‐Vergani, G. , Vergani, D. , Longhi, M.S. (2012) The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin‐9/tim‐3 pathway. Hepatology 56, 677–686. [DOI] [PubMed] [Google Scholar]
- 14. Li, H. , Wu, K. , Tao, K. , Chen, L. , Zheng, Q. , Lu, X. , Liu, J. , Shi, L. , Liu, C. , Wang, G. , Zou, W. (2012) Tim‐3/galectin‐9 signaling pathway mediates T cell dysfunction and predicts poor prognosis in patients with HBV‐associated hepatocellular carcinoma. Hepatology 56, 1342–1351. [DOI] [PubMed] [Google Scholar]
- 15. Golden‐Mason, L. , McMahan, R.H. , Strong, M. , Reisdorph, R. , Mahaffey, S. , Palmer, B.E. , Cheng, L. , Kulesza, C. , Hirashima, M. , Niki, T. , Rosen, H.R. (2013) Galectin‐9 functionally impairs natural killer cells in humans and mice. J. Virol. 87, 4835–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sehrawat, S. , Reddy, P.B. , Rajasagi, N. , Suryawanshi, A. , Hirashima, M. , Rouse, B.T. (2010) Galectin‐9/TIM‐3 interaction regulates virus‐specific primary and memory CD8 T cell response. PLoS Pathog. 6, e1000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reddy, P.B. , Sehrawat, S. , Suryawanshi, A. , Rajasagi, N.K. , Mulik, S. , Hirashima, M. , Rouse, B.T. (2011) Influence of galectin‐9/Tim‐3 interaction on herpes simplex virus‐1 latency. J. Immunol. 187, 5745–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nebbia, G. , Peppa, D. , Schurich, A. , Khanna, P. , Singh, H.D. , Cheng, Y. , Rosenberg, W. , Dusheiko, G. , Gilson, R. , ChinAleong, J. , Kennedy, P. , Maini, M.K. (2012) Upregulation of the Tim‐3/galectin‐9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One 7, e47648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharma, S. , Sundararajan, A. , Suryawanshi, A. , Kumar, N. , Veiga‐Parga, T. , Kuchroo, V.K. , Thomas, P.G. , Sangster, M.Y. , Rouse, B.T. (2011) T cell immunoglobulin and mucin protein‐3 (Tim‐3)/galectin‐9 interaction regulates influenza A virus‐specific humoral and CD8 T‐cell responses. Proc. Natl. Acad. Sci. USA 108, 19001–19006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jost, S. , Moreno‐Nieves, U.Y. , Garcia‐Beltran, W.F. , Rands, K. , Reardon, J. , Toth, I. , Piechocka‐Trocha, A. , Altfeld, M. , Addo, M.M. (2013) Dysregulated Tim‐3 expression on natural killer cells is associated with increased galectin‐9 levels in HIV‐1 infection. Retrovirology 10, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang, Z.Y. , Dong, J.H. , Chen, Y.W. , Wang, X.Q. , Li, C.H. , Wang, J. , Wang, G.Q. , Li, H.L. , Wang, X.D. (2012) Galectin‐9 acts as a prognostic factor with antimetastatic potential in hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 13, 2503–2509. [DOI] [PubMed] [Google Scholar]
- 22. Kato, T. , Date, T. , Murayama, A. , Morikawa, K. , Akazawa, D. , Wakita, T. (2006) Cell culture and infection system for hepatitis C virus. Nat. Protoc. 1, 2334–2339. [DOI] [PubMed] [Google Scholar]
- 23. Abeles, R.D. , McPhail, M.J. , Sowter, D. , Antoniades, C.G. , Vergis, N. , Vijay, G.K. , Xystrakis, E. , Khamri, W. , Shawcross, D.L. , Ma, Y. , Wendon, J.A. , Vergani, D. (2012) CD14, CD16 and HLA‐DR reliably identifies human monocytes and their subsets in the context of pathologically reduced HLA‐DR expression by CD14(hi) /CD16(neg) monocytes: expansion of CD14(hi) /CD16(pos) and contraction of CD14(lo) /CD16(pos) monocytes in acute liver failure. Cytometry A 81, 823–834. [DOI] [PubMed] [Google Scholar]
- 24. Belge, K.U. , Dayyani, F. , Horelt, A. , Siedlar, M. , Frankenberger, M. , Frankenberger, B. , Espevik, T. , Ziegler‐Heitbrock, L. (2002) The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J. Immunol. 168, 3536–3542. [DOI] [PubMed] [Google Scholar]
- 25. Zhang, J.Y. , Zou, Z.S. , Huang, A. , Zhang, Z. , Fu, J.L. , Xu, X.S. , Chen, L.M. , Li, B.S. , Wang, F.S. (2011) Hyper‐activated pro‐inflammatory CD16 monocytes correlate with the severity of liver injury and fibrosis in patients with chronic hepatitis B. PLoS One 6, e17484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dufner‐Beattie, J. , O'Guin, A. , O'Guin, S. , Briley, A. , Wang, B. , Balsarotti, J. , Roth, R. , Starkey, G. , Slomczynska, U. , Noueiry, A. , Olivo, P.D. , Rice, C.M. (2014) Identification of AP80978, a novel small‐molecule inhibitor of hepatitis C virus replication that targets NS4B. Antimicrob. Agents Chemother. 58, 3399–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lacey, D.C. , Achuthan, A. , Fleetwood, A.J. , Dinh, H. , Roiniotis, J. , Scholz, G.M. , Chang, M.W. , Beckman, S.K. , Cook, A.D. , Hamilton, J.A. (2012) Defining GM‐CSF‐ and macrophage‐CSF‐dependent macrophage responses by in vitro models. J. Immunol. 188, 5752–5765. [DOI] [PubMed] [Google Scholar]
- 28. Fleetwood, A.J. , Lawrence, T. , Hamilton, J.A. , Cook, A.D. (2007) Granulocyte‐macrophage colony‐stimulating factor (CSF) and macrophage CSF‐dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J. Immunol. 178, 5245–5252. [DOI] [PubMed] [Google Scholar]
- 29. Livak, K.J. , Schmittgen, T.D. (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 30. Giugliano, S. , Kriss, M. , Golden‐Mason, L. , Dobrinskikh, E. , Stone, A.E. , Soto‐Gutierrez, A. , Mitchell, A. , Khetani, S.R. , Yamane, D. , Stoddard, M. , Li, H. , Shaw, G.M. , Edwards, M.G. , Lemon, S.M. , Gale, M., Jr. , Shah, V.H. , Rosen, H.R. (2015) Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 148, 392–402 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dixon, L.J. , Barnes, M. , Tang, H. , Pritchard, M.T. , Nagy, L.E. (2013) Kupffer cells in the liver. Compr. Physiol. 3, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhong, J. , Gastaminza, P. , Cheng, G. , Kapadia, S. , Kato, T. , Burton, D.R. , Wieland, S.F. , Uprichard, S.L. , Wakita, T. , Chisari, F.V. (2005) Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102, 9294–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dolganiuc, A. , Norkina, O. , Kodys, K. , Catalano, D. , Bakis, G. , Marshall, C. , Mandrekar, P. , Szabo, G. (2007) Viral and host factors induce macrophage activation and loss of toll‐like receptor tolerance in chronic HCV infection. Gastroenterology 133, 1627–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kato, T. , Date, T. , Miyamoto, M. , Furusaka, A. , Tokushige, K. , Mizokami, M. , Wakita, T. (2003) Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125, 1808–1817. [DOI] [PubMed] [Google Scholar]
- 35. Dreux, M. , Garaigorta, U. , Boyd, B. , Décembre, E. , Chung, J. , Whitten‐Bauer, C. , Wieland, S. , Chisari, F.V. (2012) Short‐range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zarember, K.A. , Godowski, P.J. (2002) Tissue expression of human Toll‐like receptors and differential regulation of Toll‐like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 168, 554–561. [DOI] [PubMed] [Google Scholar]
- 37. Choo, Q.L. , Richman, K.H. , Han, J.H. , Berger, K. , Lee, C. , Dong, C. , Gallegos, C. , Coit, D. , Medina‐Selby, R. , Barr, P.J. (1991) Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 88, 2451–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Targett‐Adams, P. , Boulant, S. , McLauchlan, J. (2008) Visualization of double‐stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 82, 2182–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hemmi, H. , Kaisho, T. , Takeuchi, O. , Sato, S. , Sanjo, H. , Hoshino, K. , Horiuchi, T. , Tomizawa, H. , Takeda, K. , Akira, S. (2002) Small anti‐viral compounds activate immune cells via the TLR7 MyD88‐dependent signaling pathway. Nat. Immunol. 3, 196–200. [DOI] [PubMed] [Google Scholar]
- 40. Forsbach, A. , Nemorin, J.G. , Montino, C. , Muller, C. , Samulowitz, U. , Vicari, A.P. , Jurk, M. , Mutwiri, G.K. , Krieg, A.M. , Lipford, G.B. , Vollmer, J. (2008) Identification of RNA sequence motifs stimulating sequence‐specific TLR8‐dependent immune responses. J. Immunol. 180, 3729–3738. [DOI] [PubMed] [Google Scholar]
- 41. Maess, M.B. , Sendelbach, S. , Lorkowski, S. (2010) Selection of reliable reference genes during THP‐1 monocyte differentiation into macrophages. BMC Mol. Biol. 11, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Negash, A.A. , Ramos, H.J. , Crochet, N. , Lau, D.T. , Doehle, B. , Papic, N. , Delker, D.A. , Jo, J. , Bertoletti, A. , Hagedorn, C.H. , Gale, M., Jr. (2013) IL‐1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 9, e1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shrivastava, S. , Mukherjee, A. , Ray, R. , Ray, R.B. (2013) Hepatitis C virus induces interleukin‐1β (IL‐1β)/IL‐18 in circulatory and resident liver macrophages. J. Virol. 87, 12284–12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stoorvogel, W. (2012) Functional transfer of microRNA by exosomes. Blood 119, 646–648. [DOI] [PubMed] [Google Scholar]
- 45. El Andaloussi, S. , Mäger, I. , Breakefield, X.O. , Wood, M.J. (2013) Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357. [DOI] [PubMed] [Google Scholar]
- 46. Momen‐Heravi, F. , Bala, S. , Bukong, T. , Szabo, G. (2014) Exosome‐mediated delivery of functionally active miRNA‐155 inhibitor to macrophages. Nanomedicine (Lond.) 10, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heydtmann, M. (2009) Macrophages in hepatitis B and hepatitis C virus infections. J. Virol. 83, 2796–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heil, F. , Hemmi, H. , Hochrein, H. , Ampenberger, F. , Kirschning, C. , Akira, S. , Lipford, G. , Wagner, H. , Bauer, S. (2004) Species‐specific recognition of single‐stranded RNA via Toll‐like receptor 7 and 8. Science 303, 1526–1529. [DOI] [PubMed] [Google Scholar]
- 49. Tanji, H. , Ohto, U. , Shibata, T. , Miyake, K. , Shimizu, T. (2013) Structural reorganization of the Toll‐like receptor 8 dimer induced by agonistic ligands. Science 339, 1426–1429. [DOI] [PubMed] [Google Scholar]
- 50. Zimmermann, H.W. , Seidler, S. , Nattermann, J. , Gassler, N. , Hellerbrand, C. , Zernecke, A. , Tischendorf, J.J. , Luedde, T. , Weiskirchen, R. , Trautwein, C. , Tacke, F. (2010) Functional contribution of elevated circulating and hepatic non‐classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS One 5, e11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tacke, F. , Randolph, G.J. (2006) Migratory fate and differentiation of blood monocyte subsets. Immunobiology 211, 609–618. [DOI] [PubMed] [Google Scholar]
- 52. Coquillard, G. , Patterson, B.K. (2009) Determination of hepatitis C virus‐infected, monocyte lineage reservoirs in individuals with or without HIV coinfection. J. Infect. Dis. 200, 947–954. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data